

Abstract
The NH2-terminal peptide of myelin basic protein (MBP) bound to the class II major histocompatibility complex (MHC) protein I-Au is an immunodominant epitope in experimental autoimmune encephalomyelitis, a murine model of multiple sclerosis. However, the MBP–I-Au complex is very unstable. To investigate this, we performed site-directed mutagenesis of the I-Au MHC protein and the MBP peptide. Biochemical, T cell activation, and molecular modeling studies of mutant complexes demonstrate that the MBP peptide's key residue for MHC binding, lysine 4, is buried in the P6 pocket of I-Au, which is predominantly hydrophobic. This implies that the MBP–I-Au complex differs from more stable complexes in two respects: (a) the peptide leaves the NH2-terminal region of the MHC peptide-binding cleft unoccupied; (b) the peptide is not anchored by typical favorable interactions between peptide side chains and MHC pockets. To test these hypotheses, a modified MBP peptide was designed based on molecular modeling, with the aim of producing strong I-Au binding. Extension of the NH2 terminus of MBP with six amino acids from the ova peptide, and replacement of the lysine side chain in the P6 pocket with an aromatic anchor, results in >1,000-fold increased binding stability. These results provide an explanation for the unusual peptide–MHC-binding kinetics of MBP, and should facilitate an understanding of why mice are not tolerant to this self-peptide– MHC complex.
X-ray diffraction studies of MHC–peptide complexes have provided a structural picture of how peptide antigens are presented by the MHC (1). A striking feature of these structures is the conservation of peptide conformation. In crystal structures of class II MHC–peptide complexes (2–5), the backbone conformations of the bound peptides are very similar, suggesting a basic pattern that other structures may be expected to follow: an extended peptide conformation spanning the MHC cleft from end to end, with burial of individual sidechains (especially in the P1 and P9 pockets) resulting in stable, selective binding (1, 6).
However, some aspects of this picture may not be applicable to all antigens. So far, structures have been solved only for stable peptide–MHC complexes. Short-lived and low-affinity complexes are also important in mediating immune responses (7, 8) and in autoimmune disease (9–13). Stable antigen–MHC complexes have dissociation half-times of tens to hundreds of hours (14). By contrast, short-lived complexes dissociate in minutes (9, 15, 16), suggesting that their structures may be significantly different.
An interesting example is the short-lived peptide–MHC complex involved in the murine model of multiple sclerosis, formed by the NH2-terminal fragment of mouse myelin basic protein (MBP)1 and the class II MHC proteins I-Au or I-Ak. This fragment of MBP binds only weakly to I-Au and I-Ak with a dissociation half-time of ∼15 min from I-Au and a dissociation half-time of <3 min from I-Ak (15). Nevertheless, it appears to be the immunodominant epitope in the disease model experimental autoimmune encephalomyelitis (EAE; references 9, 17–21).
There is evidence that the NH2-terminal peptide of MBP binds within the classical MHC peptide-binding cleft. It competes for MHC binding with invariant chain peptide (22), which has been shown to bind in the cleft of a class II MHC (3). MBP also competes with other peptide antigens such as Ova 323-339 (15, 19) and HEL 46-61 (15). MBP binding is strongly influenced by several residues (23) that have been observed to be peptide contacts in crystal structures of MHC.
We have previously constructed molecular models of the class II MHC alleles I-Au and I-Ak that present the MBP Ac1-9 peptide (23). These models did not include any antigenic peptide. In analyzing these models, we originally focused on two polymorphic MHC residues, β38 and β61, which appeared to modulate T cell recognition of MBP in both I-Au and I-Ak. Substitutions at these positions had quite different effects in I-Au vs. I-Ak. Our molecular models suggested that these differences might be due to neighboring polymorphisms, especially residue β9 (23). This polymorphism (histidine in K vs. valine in U) appeared to shift surrounding sidechains significantly, and, because of its position in an important specificity pocket (P6), also seemed likely to create a strong difference in peptide binding selectivity.
Here, we report the results of site-directed mutagenesis of I-A residue β9 and the application of these results to obtain a molecular model of the MBP–I-Au complex. In addition, how the model has been used to design an analogue of MBP that binds very strongly to I-Au is described.
Materials and Methods
Peptide Synthesis.
All peptides were synthesized using standard Fmoc chemistry and purified by reverse phase HPLC. The identify and purity of each peptide was confirmed by a combination of analytical HPLC, amino acid analysis, and mass spectrometry. For FACS® binding assays, biotinylated peptides based on the rat MBP sequence, MBP Ac1-13 (AcASQKRPSQRHGSK-biotin) and mutants (K4A, K4Y, K4E) were synthesized. For studies of the stability of isolated peptide–MHC complexes, the following peptides and mutated variants as described in the text were synthesized: CLIP (86-100) (KPVSQMRMATPLLMR) and variants; mouse MBP Ac1-12 (AcASQKRPSQRSKY) and MBP Ac1-12 K4Y (AcASQYRPSQRSKY); and ova-MBP (ISQAVAASQKRPSQRSKY) and ova-MBP K4Y (ISQAVAASQYRPSQRSKY) (MBP position 4 indicated in bold). A cysteine was added to the COOH-terminal end of MBP Ac1-12 and MBP Ac1-12 K4Y, and the free thiol side chain of the cysteine was labeled with 5-iodoacetamidofluorescein. All other peptides were labeled at their NH2 terminus using the N-hydroxysuccinimide ester of 5,6-carboxyfluorescein (Molecular Probes, Eugene, OR).
Cell Cultures and Conditions.
Cells were grown in RPMI 1640 supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 × 10−5 M β-mercaptoethanol. Selection medium for the L cell transfectants was prepared by supplementing with 100 μM hypoxanthine, 400 μM aminopterin, and 16 μM thymidine (HAT; GIBCO BRL, Gaithersburg, MD).
MHC Protein Purification.
Procedures for class II MHC protein isolation have been described previously (24). I-Ak and I-Au were obtained from BW5147.G.1.4 cells transfected with the appropriate α and β chain cDNA (16) and purified by affinity chromatography on a 10-2.16 anti–I-A antibody column. Protein was eluted with solutions of either 0.2 or 1.0 mM dodecyl maltoside (DM)/0.5 M NaCl/0.1 M Na2CO3 pH 11.5. Protein concentrations were determined using a micro BCA assay (Pierce Chemical Co., Rockford, IL).
Peptide Binding to Live L Cell Transfectants.
Flow cytometric analysis was performed on nontransfected, wild-type, and mutated L cell transfectants incubated with biotinylated-MBP Ac1-13 (4A, 4E, or 4Y) as described previously (23, 25). L cell transfectants (5 × 105) were washed once and incubated with 200 μM biotinylated MBP or MBP analogue peptide in 200 μl final volume of RPMI 1640 with 1% BSA for 4 h at 37°C, 7% CO2, followed by two washes. After washing, the cells were immediately incubated with streptavidin–Quantum red/PE conjugate (Sigma Chemical Co., St. Louis, MO) for 20 min on ice and analyzed. FITC-conjugated mouse anti-Aβk,u–specific monoclonal antibody 10-2.16 (26) was used to determine the MHC expression level; these cells were also stained for viability with propidium iodide. Samples were analyzed on an EPICS 753 flow cytometer for dual-color fluorescence as previously described (23). Net peptide binding was calculated as streptavidin–Quantum red or streptavidin–phycoerythrin linear mean fluorescence–background linear mean fluorescence (nontransfected L cells plus peptide), and divided by the FITC mean fluorescence to give the ratio of bound peptide over the level of MHC expression. Normalized peptide binding to the various transfectants was calculated as the ratio of net peptide binding to the transfectant, divided by the net peptide binding of the MBP 4A peptide to the wild-type transfectant.
Peptide Dissociation Kinetics.
MHC protein was incubated with excess fluorescein-labeled peptide for various times (depending upon the time required to get the maximal amount of binding). Excess peptide was removed from the sample with Sephadex G50-SF spin columns pretreated with a 10 mg/ml BSA solution to minimize the amount of nonspecific binding to the column. The complex was isolated in a pH 5.3 sodium citrate buffer at 4°C, and the dissociation reaction was initiated by raising the temperature to 37°C. Periodically, aliquots were taken from the heated sample and analyzed by high performance size exclusion chromatography (HPSEC) using a 7.5 mm × 60-cm TSK G3000SW column (Toso Haas, Montgomeryville, PA) with a guardcolumn (7.5 × 75mm). Fluorescence of the peptide–MHC complex was monitored by a Shimadzu RF-551 spectrofluorometric detector and a standard UV detector connected in series (27). The HPLC solvent was PBS/0.2 mM DM (pH 7.0) running at room temperature at 1.0 ml/min. The protein–peptide complex eluted at ∼15 min. In the dissociation experiments where time points were necessarily <30 min apart, aliquots were taken and placed immediately at 4°C. Samples were sitting on ice no longer than 20 min before injection onto the HPSEC apparatus. In control experiments, it was found that no significant dissociation of complex occurs in less than 10 h at 4°C (data not shown).
T Cell Stimulation Assays.
The MBP-specific T cell hybridoma BR4 was obtained from B10.BR mice as described (23). The MBP-specific T cell clone E3 was obtained from a B10.A mouse immunized with MBP 1–11 as described (28). For measurement of T cell hybridoma IL-2 production, 4 × 104 liters of cell transfectants were incubated with 2 × 104 T cell hybridoma cells and the indicated peptide in a volume of 200 μl. After 18–24 h, this mixture was frozen at −80°C for at least 2 h before harvesting supernatants, and IL-2 production was measured using the IL-2–dependent cell line HT-2. For measurement of T cell clone IL-3 production, 5 × 104 I-Au–transfected BW5147.G.1.4 cells were incubated with 5 × 104 E3 clone T cells and the indicated peptide in a volume of 200 μl. Supernatants were harvested at 24 h and assayed for IL-3 production using an Endogen (Cambridge, MA) ELISA assay as described by the manufacturer. MBP 4A 5A was used as a negative control; this peptide has been found to produce no stimulation of the MBP-specific T cells tested (data not shown).
Molecular Modeling.
Models of the murine MHC proteins I-Au and I-Ak from previous work (23) were used as the basis for modeling their complexes with the MBP Ac1-9 peptide. Peptide backbone coordinates of the hemagglutinin 306-318 peptide, from the crystal structure of its complex with the class II MHC protein HLA-DR1 (29), were superimposed on our I-Au and I-Ak models, as a canonical framework for assessing different MBP-binding modes. To model each hypothetical alignment of the MBP Ac1-9 sequence onto the canonical peptide framework, the MBP sidechains and MHC sidechains contacting the peptide were predicted ab initio by self-consistent ensemble optimization (30, 31), as previously applied to MHC modeling (23, 32).
The final MBP–MHC models were modified to refine the region surrounding a two-residue deletion in the β-chain helix in the I-Au and I-Ak alleles at β65-66. Tyr β65 was rebuilt in the context of the existing model by the program SEGMOD (33), and the whole model refined by constrained minimization in the program ENCAD (34), using the LOOK software suite (Molecular Applications Group, Palo Alto, CA) with default settings and parameters.
An NH2-terminally extended N-ova–MBP peptide (ISQAVAASQKRPSQRHG) was modeled starting from the MBP Ac1-9–I-Au model. To construct the full NH2-terminal extension, the crystal structure coordinates for the complex of CLIP bound to HLA-DR3 (3) were superimposed on our model using the Cαs of the first 90 amino acids of α and β chains, and the P1 and P9 positions of the bound peptides. N-ova–MBP was based on the first six amino acids of CLIP, plus the MBP and I-Au coordinates of our original model, and modeled using the same procedures and parameters as described above.
Results
Interaction of MBP Residue 4 With MHC Residue β9.
We focused initially on one MBP residue (position 4) shown to interact strongly with I-Au (9, 21) and one MHC residue, β9, predicted to be a key determinant of the P6 pocket (23). I-A β9 is polymorphic: it is valine in I-Au vs. histidine in I-Ak. This suggested that β9 could create a preference for interacting with a hydrophobic peptide sidechain in I-Au, vs. a polar or negatively charged sidechain in I-Ak. We synthesized an MBP Ac1-11 mutant with peptide residue 4 lysine replaced by a negatively charged glutamate (4E), and tested its binding to I-Ak and I-Au by FACS® analysis (Fig. 1 a). This substitution improves binding to I-Ak by >20-fold over wild-type, and ∼2-fold over MBP Ac1-11 4A, an altered MBP peptide in which replacement of peptide residue 4 lysine with alanine enhances MHC affinity (17, 19). In contrast, binding to I-Au is reduced at least 10-fold (Fig. 1 a; for comparison with wild-type MBP Ac1-11, Tate, K.M., and P.P. Jones, unpublished results).
Figure 1.


I-A residue β9 interacts with MBP residue 4. (a) Binding of biotinylated MBP peptides with residue 4 lysine replaced with either glutamate (4E), tyrosine (4Y), or alanine (4A) to the surface of L cells transfected with β9 mutants of I-Au and I-Ak. Binding for each mutant is compared with MBP 4A binding to wild-type I-Au and I-Ak (normalized to 1.0 for each allele). (b) T cell hybridoma BR4 response to the altered MBP peptides described above presented by L cells transfected with β9 mutants of I-Au and I-Ak. T cell responses to MBP 4A were normalized to 1.0 for each allele.
Evidence for a direct interaction between MBP peptide residue 4 and I-A residue β9 is provided by studies of the ability of site-directed mutants of β9 to bind a series of MBP Ac1-11 peptides substituted at position 4 with alanine (4A), glutamate (4E), or tyrosine (4Y). We have constructed a mutant of I-Au with β9 valine replaced by histidine as found in I-Ak (V9H), and a mutant of I-Ak with β9 histidine replaced by valine (H9V). FACS® peptide binding studies show that replacement of β9 switches the specificity of the MHC from that characteristic of one allele to that of the other (Fig. 1 a). Thus, although I-Ak prefers 4E and I-Au prefers 4Y, I-Ak H9V prefers 4Y and I-Au V9H prefers 4E. In contrast, binding of the 4A peptide is relatively insensitive to these β9 substitutions (Fig. 1 a), indicating little interaction between β9 and the short sidechain of MBP 4A.
The I-Au,k β9 mutants can present MBP peptides to I-Au– and I-Ak–restricted T cells (Fig. 1 b). The pattern of T cell stimulation by the MBP residue 4 substituted peptides mirrors their pattern of MHC binding (Fig. 1 a). These data show that the specific structural complex recognized by the T cells does contain the MBP 4–MHC β9 interaction demonstrated by the binding data.
Structural Modeling of the MBP Ac1-9–I-Au Complex.
To assess the structural implications of the MBP 4–I-A β9 interaction, we sought to model MBP Ac1-9 bound to I-Au. Our approach is based on the most conserved feature of MHC–peptide binding, the network of backbone hydrogen bonds observed in all known MHC crystal structures, which enforces an extended conformation on the peptide (1). This feature simplifies the problem of solving the structure of the MHC-bound peptide to determining the “alignment” of the peptide's sequence in the peptide-binding groove; that is, which peptide sidechains should be placed in which MHC pockets. This strategy has been successful in using x-ray crystallographic data obtained for the hemagglutinin 306-318 peptide to model the structure of an invariant chain peptide (CLIP) binding to several class II MHCs (32), independent of x-ray crystallographic data concerning CLIP binding (3).
We used self-consistent ensemble optimization sidechain modeling (30, 31) to compare several possible alignments of MBP Ac1-9 (the native peptide is acetylated on its NH2 terminus) within the peptide-binding cleft (for a detailed description of these results, see Appendix). Most of these modes diverge from the pattern of TCR- vs. MHC-contact residues suggested by experimental studies of MBP Ac1-9 and its analogues, suggesting that MBP 4 is unlikely to be in any pocket other than P6. In addition to resulting in logical MHC and TCR contacts, the placement of MBP residue 4 in the P6 pocket results in a direct contact between MBP 4 and I-A β9, consistent with the site-directed mutagenesis data.
Our model of MBP Ac1-9 complexed with I-Au is shown in Fig. 2, compared with the crystal structure of hemagglutinin 306-318 peptide complexed with HLA-DR1 (29). Based on existing class II MHC–peptide complex crystal structures, only the first nine amino acids of MBP could be modeled; however, the model should be applicable to longer MBP peptides since they bind to I-Au in the same registry as Ac1-9 (as shown by T cell stimulation). Unlike known MHC–peptide structures, in which the peptide spans the full length of the MHC cleft, MBP binds in only part of the cleft, leaving the NH2-terminal side (as defined by the orientation of the antigenic peptide) unoccupied (Fig. 2 b). The first amino acid of the peptide lies midway in the cleft, in the P3 position. Whereas nearly all peptides used to study antigen presentation in vitro are fragments from the middle of a protein sequence, and may in vivo be presented with variable NH2- and COOH-terminal extensions, MBP Ac1-9 constitutes the first nine amino acids of the protein's NH2 terminus.
Figure 2.
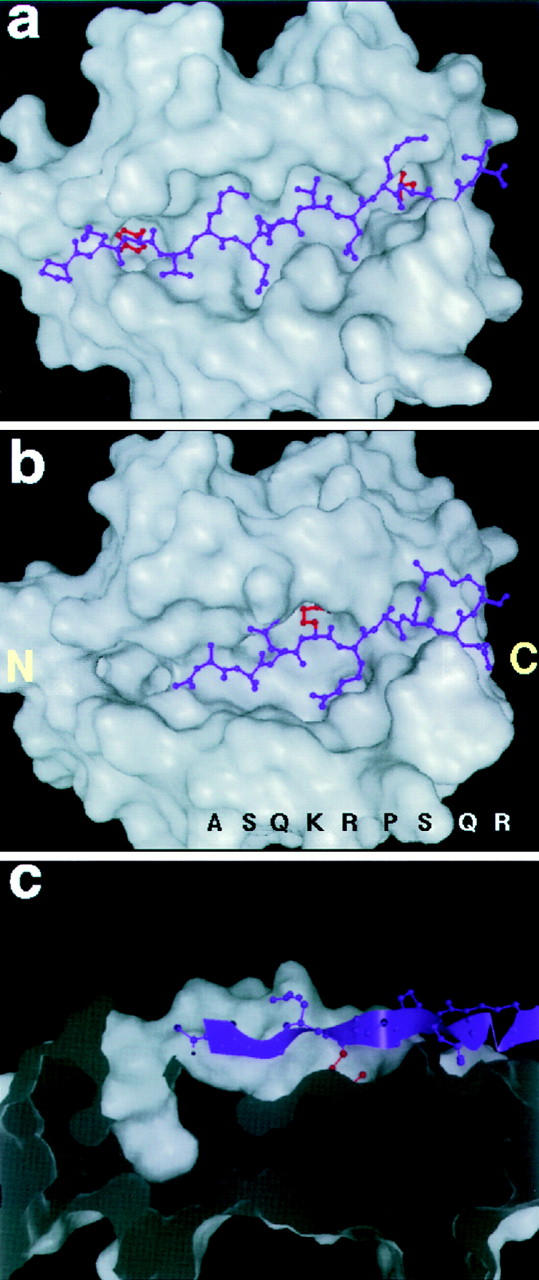
Molecular model of the MBP Ac1-9–I-Au complex compared with the crystal structure of a stable peptide–MHC complex. (a) The crystal structure of a hemagglutinin (purple)-DR1 (white) complex, with the hydrophobic anchor residues P1 (tyrosine) and P9 (leucine) marked in red. (b) The model of MBP Ac1-9 (Ac-ASQKRPSQR, purple) complexed with I-Au (white); the MBP 4 Lys sidechain is marked in red. The NH2- and COOH-terminal ends of the cleft (as defined by the orientation of the antigenic peptide) are indicated by letters. (c) Cross-section side view of the MBP Ac1-9–I-Au complex, showing the empty P1 pocket (left) and general absence of anchoring interactions. The MBP peptide backbone is shown as a ribbon (purple), with the MBP 4 Lys sidechain in red.
The model also diverges from known peptide–MHC structures by its lack of anchor residues in the deep specificity-determining pockets of the cleft. In HLA-DR1, for example, large hydrophobic sidechains near the peptide's NH2 and COOH termini are entirely buried in deep pockets (P1 and P9, shown in red in Fig. 2 a). In the MBP–I-Au model, by contrast, MBP does not even reach the P1 end of the cleft, leaving the large P1 pocket empty. MBP places a serine sidechain at P9, which enters the pocket only superficially (Fig. 1 b). Indeed, the only substantial sidechain buried in an MHC pocket is MBP residue 4 lysine (shown in red in Fig. 1 b), in the largely hydrophobic P6 pocket. In the model, the 4 lysine sidechain is deeply buried like a typical anchor, but is unfavorable to binding because of the cost in free energy of desolvating its charged amine group. A longitudinal cross-section view of the MBP–I-Au model shows the striking absence of MBP anchoring residues from the cleft's pockets (Fig. 2 c).
The Specificity of the I-Ak,u P6 Pocket Is Consistent with MBP Residue 4.
The evidence for a direct interaction between the I-A β9 and MBP residue 4 (see first section of Results) is an important element of our model. In our models of class II alleles I-Ak,d,u, the β9 sidechain points towards the P6 pocket (23, 32), and is predicted to contact the MBP residue 4 sidechain there. However, the β9 mainchain atoms lie between pockets P6 and P9, and in recent class II crystal structures (HLA-DR1, HLA-DR3, I-Ek) the β9 sidechain forms one side of the P9 pocket (3, 4, 29).
To test the location of the β9 sidechain in I-Ak,u, we have sought to relate our peptide substitution experiments to a known crystal structure. CLIP binds to all class II MHCs, and a crystal structure of its complex with HLA-DR3 has been reported (3), which closely matches our models of CLIP bound to several different MHCs including I-Ak,u (32). We made substitutions in CLIP at the positions (residues 96, 99) found in the P6 and P9 pockets (3), and measured their effects on binding to I-Au and I-Ak. Our results show that I-A P6 pocket specificity, as assayed by CLIP 96 proline → tyrosine or proline → glutamate substitutions, closely matches the MBP residue 4 amino acid preferences, whereas P9 does not. In I-Ak, glutamate is preferred at CLIP position 96 (Fig. 3 a) whereas in I-Au tyrosine is preferred (Fig. 3 b). A similar pair of substitutions at CLIP 99 (P9 position), methionine → phenylalanine or methionine → glutamate produce effects unlike the MBP residue 4 amino acid preferences (both decreased stability of binding, see Fig. 3). Substitution studies of the CLIP residues that bind in P1, P5, and P7 show no pattern of preferences similar to that of P6 (36).
Figure 3.

Dissociation kinetics of P6- and P9-substituted CLIP peptides. Dissociation of the wild-type CLIP 86-100 peptide (−) and peptide analogues CLIP 96 Pro → Tyr (filled circles), CLIP 96 Pro → Glu (filled triangles), CLIP 99 Met → Phe (open circles), CLIP 99 Met → Glu (open triangles), and CLIP 95 Thr → Tyr (×) from (a) I-Ak; (b) I-Au. The CLIP 95 tyrosine substitution is a negative control showing that the stabilizing effect of tyrosine at CLIP 96 is P6 position specific.
Design of an NH2-terminal Extension of MBP That Greatly Stabilizes Its Binding.
We have used the model to attempt to design a variant of the MBP peptide with enhanced binding stability. We sought to fill the NH2-terminal side of the cleft and place a favorable anchoring sidechain (valine) in the P1 pocket, by adding a six amino acid sequence (323–328) from the NH2 terminus of the ova peptide. The model suggests several stabilizing interactions, including three new hydrogen bonds to the peptide backbone from I-Au residues α55–57, and burial of the valine sidechain within the P1 pocket (Fig. 4 a).
Figure 4.



Design, dissociation kinetics, and biological activity of an NH2-terminal extension of MBP. (a) Molecular model of ova-MBP K4Y (ISQAVAASQYRPSQRSK, shown in ball and stick) bound to I-Au (ribbon). MBP residues 1–9 are colored blue, with the NH2-terminal extension in lighter bonds. Three hydrogen bonds from I-Au to the NH2-terminal extension are shown as dashed green lines. (b) Dissociation of the wild-type MBP Ac1-12 (open diamonds), ova-MBP (filled diamonds), MBP Ac1-12 K4Y (open circles), and ova-MBP K4Y (filled circles) complexes with I-Au. (c) IL-3 production of the MBP-specific E3 T cell clone in response to the following peptides presented by I-Au–transfected BW5147.G.1.4 cells: MBP Ac1-14 (open triangles), MBP Ac1-14 K4A (inverted triangles), fluorescently-labeled ova-MBP (open squares), fluorescently labeled ova-MBP K4Y (filled squares), and negative control peptide (see Materials and Methods) MBP Ac1-14 K4A R5A (filled circles). In studies of T cell proliferation, wild-type and 4A MBP, but not either of the extended peptides, triggered full T cell clone proliferation when presented by I-Au–expressing splenocytes (data not shown).
We measured the binding and dissociation of this NH2-terminally extended MBP peptide (ova-MBP) with I-Au (Fig. 4 b). Ova–MBP binds with higher affinity than MBP, and dissociates ∼80-fold more slowly than wild-type MBP Ac1-12. This large increase in the stability of the complex by addition of the six-residue NH2-terminal extension strongly suggests that the wild-type MBP peptide does not fill the binding groove, as shown in Fig. 2. Furthermore, the enhanced stability of the ova–MBP complex is not due to ova–MBP binding in a different registry from MBP, as ova-MBP can bind to I-Au to trigger IL-3 production from an MBP-specific T cell clone (Fig. 4 c).
To stabilize further the extended MBP peptide, we introduced the substitution 4 lysine → tyrosine (MBP residue numbering). This mutation increases stability of binding to I-Au for both the original MBP Ac1-11 peptide (9, 15) and for the extended ova–MBP peptide (Fig. 4 b). This result suggests both bind to I-Au the same way, placing residue 4 (MBP residue numbering) in the P6 pocket. Moreover, the extended peptide is a potent activator of IL-3 production by an MBP-specific T cell clone (Fig. 4 c), clearly indicating an identical registry as wild-type MBP. Thus, by making two changes in MBP suggested by the model extending its NH2 terminus and adding favorable anchor residues (Fig. 4 a) we have increased the stability of binding to I-Au by >1,000-fold, producing slow dissociation kinetics (t 1/2 >200 h) characteristic of high affinity MHC–antigen complexes.
Discussion
Evidence That the MBP Ac1-9 Peptide Binds Towards the COOH-terminal End of the I-Au Cleft.
The function of MHC proteins is to present a diverse array of antigenic peptides to T cells. X-ray crystallographic studies of stable peptide– MHC complexes have shown how the MHC structure accomplishes this. A series of hydrogen bonds from MHC amino acid side chains holds the peptide backbone in a conserved extended conformation, while deep pockets in the MHC protein accept specific peptide sidechains (especially at P1 and P9 positions; 1, 3–5). Although this design allows stable binding of many peptides, some important antigens, in particular some involved in autoimmune disease, do not bind stably to MHC (7–13). To investigate the structure of the short-lived peptide–MHC complex formed by the NH2-terminal peptide of MBP bound to the class II MHC protein I-Au, we have combined the approaches of molecular modeling and site-directed mutagenesis. The results of these studies suggest that the MBP–I-Au complex differs from more stable complexes in that the peptide occupies only part of the MHC–peptide-binding cleft, failing to occupy the important P1 anchor pocket and also lacking favorable stabilizing anchors in the other MHC pockets (Fig. 2). Addition of either a single strong anchor (tyrosine in the P6 pocket) or extension of the peptide to fill the P1 pocket (the designed ova–MBP peptide) greatly stabilizes this peptide–MHC complex. Extension of the peptide combined with addition of the strong P6 anchor results in a greater than 1,000-fold stabilization of the complex compared with the wild-type complex.
Our proposed wild-type MBP–I-Au structure does contain several stabilizing interactions (Fig. 5). The MBP 4 lysine terminal amine group makes contact with two aromatic sidechains (β11 Phe, β30 Tyr); aromatic rings have been shown to stabilize both charged and polar groups (37). The model also indicates that the MBP 4 lysine amino group may hydrogen bond to the I-Au residue β30 tyrosine hydroxyl. It is not certain whether the MBP 4 lysine amino group is protonated (in this connection see reference 38). Another potential stabilization is a salt bridge between MBP 5 Arg and I-Au β72 glutamate from the β1 helix (Fig. 5); mutating β72 blocks binding (39). For the seven residues of MBP that lie within the cleft there are a total of seven backbone hydrogen bonds from I-Au, including all the conserved hydrogen bonds observed in this region in the HLA–DR1 complexes (2). Among these hydrogen bonds is one from I-Au β80 asparagine (I-Au numbering) to the MBP N-acetyl carbonyl, the only specific interaction to the first two residues of MBP, which may be important for positioning the NH2 terminus.
Figure 5.

Interactions of MBP Ac1-9 with I-Au. MBP Ac1-9 is shown in skeleton bonds (thick lines, labeled in bold); I-Au residues (thin lines) making hydrogen bonds and a salt bridge (β72 Glu–MBP 5 Arg) to MBP are also shown (straight dotted lines). There is only one I-Au pocket (P6) holding a deeply buried MBP sidechain (dashed circle). I-Au residues contacting MBP are indicated by small labels. The NH2- and COOH-terminal ends of the peptide are indicated by letters. Figure generated using LOOK.
Comparison of the MBP–I-A Model with Published Data.
We have compared our model with available experimental evidence. First, the model specifies the individual role of each MBP sidechain in binding to the MHC and potentially contacting the TCR (Fig. 6 a). Two residues are predicted to point directly upwards towards the TCR, MBP 3 glutamine and MBP 6 proline, consistent with experimental evidence that these positions are critical for stimulation of both Au- and Ak-restricted T cell clones (17, 19, 20, 40). MBP 5 arginine is also predicted to be exposed at the surface of the complex, and could contact the TCR. According to our model, it could also make a contribution to binding, via hydrophobic burial in the P7 pocket and a salt bridge to I-Au β72 glutamic acid. Replacement of this residue by alanine reduces binding several-fold in competition assays (19). In the model, residues 2 and 7–11 appear to have only minor interaction with the MHC and TCR; experimentally, they can be replaced with alanine with little effect on binding, T cell stimulation, or induction of the MBP-mediated autoimmune disease EAE (19). Our model indicates that the NH2-terminal acetyl group of MBP is placed at the P2 position, a known TCR contact site.
Figure 6.
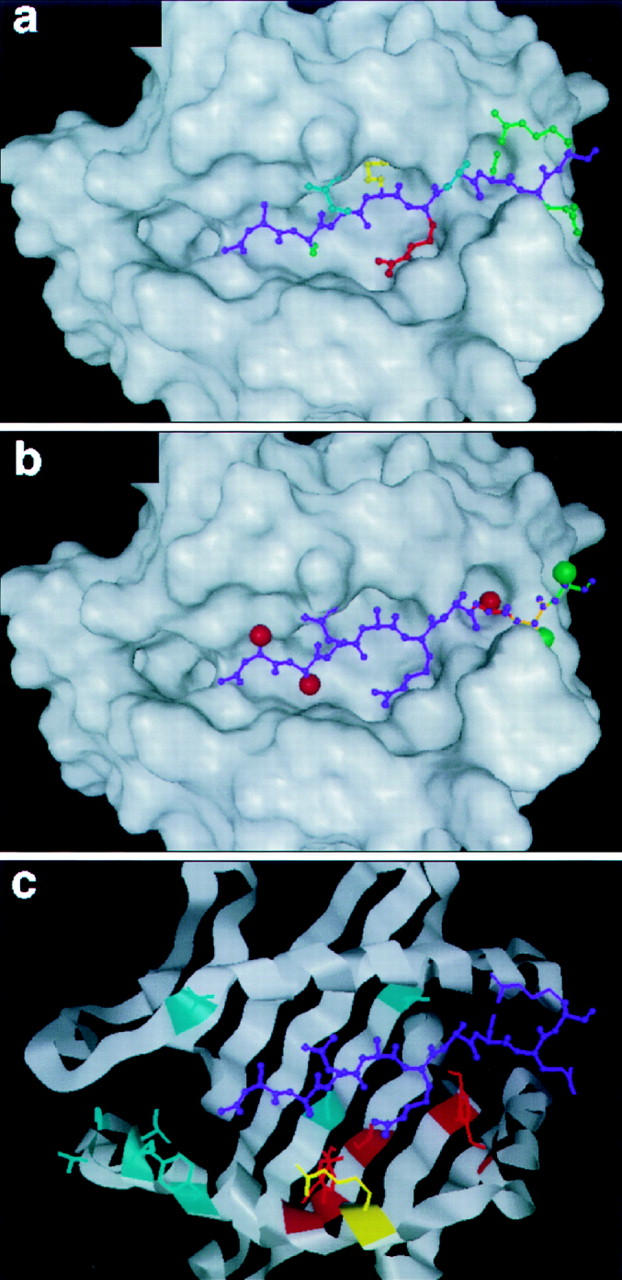
Comparison of the MBP Ac1-9–I-Au model with published data. MBP sidechains pointing towards the T cell receptor (blue), intermediate between T cell receptor and the MHC (red), buried in an MHC pocket (yellow), or interacting relatively little with either (green). These features closely match the pattern of MBP interactions with TCR–MHC as mapped by alanine-scanning substitutions (see text). Structural analysis of d-ala substitutions and COOH-terminal deletions of MBP. MBP residues 1, 2, and 7 can not accept d-ala sterically (red spheres), while residue 8 and beyond have little constraint against d-ala (green spheres). Residue 9 and beyond (green backbone) have no mainchain hydrogen bonds to the MHC, whereas deletion of residues 8 (orange) and 7 (red) would remove one and two hydrogen bonds, respectively. This is in good agreement with experimental assay of the effects of d-ala substitutions or COOH-terminal deletions on MBP–I-Au binding (see text). Amino acid substitution sites in I-Au that have no effect on MBP binding (blue), cause diminished binding (red), or improved binding (yellow). This fits the model's predicted pattern of MBP–MHC interactions well (see text).
The predicted MBP–MHC contacts (Fig. 6 b) appear consistent with reported data on COOH-terminal truncations (21). The large reduction in binding caused by truncating the peptide at residues 7 or 8, compared with the relatively slight effect of replacing these residues with alanine, indicates hydrogen bonds to the mainchain of residues 7 and 8, consistent with the model (Fig. 5). The absence of such large reductions for truncations of MBP 9–11 (21) suggests an absence of hydrogen bonding to these residues, which can be explained most directly by the placement of MBP shifted towards the COOH-terminal end of the cleft, with residues 9–11 outside the cleft.
Data on d-alanine (d-ala) substitutions in MBP (Fig. 6 b) also appear consistent with the model. At positions 1 and 2, d-ala does not fit Ramachandran map constraints in the model, clashing with the adjacent carbonyl group. At position 7 in our model, the d-ala Cβ makes bad contacts with β57 Asp and β 61 Tyr. At positions 8–11, outside the MHC cleft, the model indicates few constraining interactions disfavoring the d-amino acid. Experimentally, replacing Ala 8–11 with d-ala reduces binding by only half; by contrast, a single d-ala substitution at position 1, 2, or 7 renders binding undetectable (6).
There is also extensive data on the effects of MHC mutations and polymorphisms on MBP binding (Fig. 6 c). MBP's asymmetric binding, which leaves the NH2-terminal region of the cleft empty (see Fig. 2 b), implies that binding of MBP Ac1-9 should be insensitive to mutations in the NH2-terminal pockets, in agreement with experimental data. For example, β86 is an important polymorphic position in the P1 pocket that strongly affects peptide binding to HLA-DR (2, 42–44). I-A alleles also exhibit significant polymorphism at this position, and peptide binding to I-Au has been shown to be sensitive to mutation of this residue. The substitution β86 threonine → leucine reduces binding of CLIP, but not of MBP (22). Similarly, mutation of α59 → aspartate (lysine), adjacent to the P1 and P2 peptide positions, reduces CLIP binding to I-Au, but has no effect on MBP binding (22). According to our model, this position is too distant from MBP to alter its binding. In general, the positions reported not to affect MBP binding (blue in Fig. 6 c) in our model are distant from the peptide.
By contrast, mutations in the COOH-terminal side of the cleft influence MBP–peptide binding. For example, β38 and β61 are polymorphic residues in the side and floor of the MHC cleft, and contact MBP in the model. Mutations at these sites alter MBP binding and T cell stimulation in both I-Au and I-Ak (23). Several other positions have been observed to influence MBP binding (39, Tate, K.M., unpublished results). Of these, I-Au β72 glutamate makes a potential salt-bridge to MBP 5 arginine in the model; mutating it to alanine greatly reduces binding (39). β68 arginine also is close enough to interact with MBP 5 arginine; mutating it to alanine improves binding, potentially by alleviating electrostatic repulsion. Overall, the positions that affect MBP binding cluster strongly on the COOH-terminal side of the cleft (red), with no effects on the NH2-terminal side (Fig. 6 c). There is evidence that peptides as short as five amino acids can bind to the class Ib MHC H2-M3 (which has an occluded A pocket and appears to bind peptides shifted one residue relative to class Ia MHCs; references 46–48), or to I-Ad (49), so filling the entire binding groove is not a prerequisite for binding to the MHC.
Implications for T Cell Selection.
In the absence of excess amounts of peptide, the rapid dissociation of MBP (half-time of minutes; reference 15) leads to a low number of functional MBP–I-Au complexes on the surface of antigen presenting cells. Recent microphysiometer studies have shown that the duration of T cell stimulation then correlates with the lifetime of the peptide–MHC complexes (8). MBP Ac1-11, which dissociates rapidly from the MHC, produces transient T cell responses, whereas peptides that dissociate slowly produce long-lived responses (8). Kumar et al. (11) reported that substitutions at MBP residue 4 that confer tight binding preferentially produce a Th1-like response (characterized by production of IFN-γ), that is not triggered by wild-type MBP peptide and MBP variants with similarly rapid dissociation. These results may potentially be related to the observation that rapid dissociation of TCR-MHC-antigen complexes appears to favor an antagonist (as opposed to agonist) response (50, 51). However, whether short-lived peptide–MHC complexes tend to form TCR–MHC-peptide ternary complexes of lower than average stability is currently unknown. It has been suggested that the half-time of the MBP–I-Au complex may be too rapid for effective induction of tolerance mechanisms (9).
The placement of MBP's NH2 terminus within the region of the cleft known to be contacted by the TCR (52) raises questions about whether alternatively spliced transcripts of MBP could affect thymic selection of MBP-responsive T cell populations. MBP has been shown to be expressed as a fusion protein (with the golli polypeptide attached to its NH2 terminus) during development, and at some later stages, in the spleen, thymus and other tissues (53–55). If the MBP epitope spanned the entire MHC cleft like known antigens (2–5), the golli residues attached to its NH2 terminus in the fusion protein would be outside of the cleft, and would likely have little effect on T cell selection. By contrast, with the NH2 terminus of MBP placed directly in the region contacted by the T cell receptor, the addition of golli residues during thymic selection would likely alter T cell responses, potentially producing autoreactive T cells. We have observed that NH2-terminal extension of MBP (both ova-MBP and ova-MBP K4Y) abrogated proliferation responses of a series of MBP-responsive T cell lines tested in this laboratory (Rabinowitz, J.D., unpublished data), despite allowing some IL-3 production (Fig. 4 c). Thus, some MBP-specific T cells appear to respond to the NH2-terminally extended MBP as a partial agonist (56–58), consistent with the possibility that the golli-MBP fusion peptide may fail to negatively select, and instead positively select (50, 59), some MBP-responsive T cells.
Acknowledgments
We thank Drs. J. Goverman and E. Sercarz for bringing the golli protein to our attention. We also wish to thank Drs. M. Davis, H. McDevitt, L. Steinman, T. Anderson, and L. Schmitt for valuable discussions and critical reading of this manuscript.
This work was supported by the Public Health Service (National Institutes of Health grant [NIH] 5R37 AI13587-20 to H.M. McConnell, and NIH grant AI15732 to P.P. Jones). C. Lee is a postdoctoral fellow of the American Cancer Society (grant PF-4220). M.N. Liang is supported by a Franklin Veatch Fellowship. K.M. Tate was supported by NIH training grant AI07290. J.D. Rabinowitz is supported by the Medical Scientist Training Program. C. Beeson was supported by a postdoctoral fellowship from the Cancer Research Institute.
Abbreviations used in this paper
- CLIP
class II–associated invariant chain peptide
- d-ala
d-alanine
- DM
dodecyl maltoside
- EAE
experimental autoimmune encephalomyelitis
- HPSEC
high-performance size exclusion chromatography
- MBP
myelin basic protein
Appendix
Evaluation of Possible Registries for MBP Ac1-9 Binding to I-Au
Models containing MBP Ac1-9 in all likely registries were evaluated by comparison with experimental data, to identify features of the models that were consistent versus inconsistent with the data. The results are enumerated below for each alignment model (listed according to the MHC pocket in which the model places MBP residue 4, the most thoroughly studied MHC contact residue of the NH2-terminal MBP peptide). No discussion of the models with MBP residue 4 in the P2, P5, or P8 protein pockets is included as residues at these positions are TCR contacts but not MHC contacts. MBP residue 4 is known to be an important MHC contact and is not a TCR contact.
Model 1: MBP Residue 4 in MHC Pocket P1
Inconsistent.
Places MBP residue 4 in P1. I-Au residue β86 threonine in the P1 pocket is likely to disfavor large aromatic peptide sidechains there (2, 42–44). A large aromatic sidechain (tyrosine) at MBP 4 greatly stabilizes MBP binding to I-Au (9). Places MBP 5 arginine in a position (P2) with very little interaction with the MHC. There is evidence it has a significant interaction with I-Au (19). Places MBP 6 proline in a position (P3) not commonly considered a major TCR contact. Proline 6 is a key TCR contact residue (17, 19, 20, 40). However, TCR contact may be possible here. Places MBP 8 glutamine at P5, a major TCR contact position. MBP 8 glutamine can be replaced by alanine with little effect on T cell stimulation (19, 21). Places MBP residue 4 far from I-A residue β9. Our site-directed mutagenesis data suggest a direct interaction between these residues.
Model 2: MBP Residue 4 in MHC Pocket P3
Inconsistent.
Places MBP residue 4 in P3, a relatively shallow, exposed pocket in I-Au that seems unlikely to act as a strong anchor position favoring a large aromatic. A large aromatic sidechain (tyrosine) at MBP 4 greatly stabilizes MBP binding to I-Au (9). Places MBP 9 arginine at P8, a major TCR contact position. MBP 9 arginine can be replaced by alanine with little effect on T cell stimulation (19, 21). Places MBP 6 proline in a deep pocket (P6) mostly inaccessible to TCR contact. MBP 6 proline is a key TCR contact residue (17, 19, 20, 40). Places MBP residue 4 far from I-A residue β9. Our site-directed mutagenesis data suggest a direct interaction between these residues.
Model 3: MBP Residue 4 in MHC Pocket P4
Inconsistent.
Places MBP residue 4 in P4, a relatively shallow, exposed pocket in I-Au that seems unlikely to act as a strong anchor position favoring a large aromatic. A large aromatic sidechain (tyrosine) at MBP 4 greatly stabilizes MBP binding to I-Au (9). Places MBP 2 serine at P2 and MBP 8 glutamine at P8, known TCR contact positions. Both MBP 2 serine and MBP 8 glutamine can be replaced by alanine with little effect on T cell stimulation (19, 21). Places MBP residue 4 far from I-A residue β9. Our site-directed mutagenesis data suggest a direct interaction between these residues.
Model 4: MBP Residue 4 in MHC Pocket P6
Consistent.
Places MBP residue 4 in P6, a large, hydrophobic pocket in I-Au that has been shown to act as a strong anchor pocket favoring a large aromatic (36). A large aromatic sidechain (tyrosine) at MBP 4 greatly stabilizes MBP binding to I-Au (9). Places MBP 3 glutamine at P5 and MBP 6 proline at P8, the major TCR contact positions. These residues have been shown to be major TCR contacts in MBP Ac1-9 (17, 19, 20, 40). Places MBP residue 4 in direct contact with I-A residue β9, consistent with our β9 site-directed mutagenesis data.
Model 5: MBP Residue 4 in MHC Pocket P7
Inconsistent.
Places MBP residue 4 in P7, a relatively shallow, exposed pocket in I-Au that seems unlikely to act as a strong anchor position favoring a large aromatic. A large aromatic sidechain (tyrosine) at MBP 4 greatly stabilizes MBP binding to I-Au (9). Places MBP 3 glutamine buried in the P6 pocket, and MBP 6 proline buried in the P9 pocket, where they would be relatively inaccessible to the TCR. These residues have been shown to be major TCR contacts in MBP Ac1-9 (17, 19, 20, 40).
Model 6: MBP Residue 4 in MHC Pocket P9
Consistent.
Places MBP 4 in P9, a deep pocket that could act as a strong anchor position favoring a large aromatic. A large aromatic sidechain (tyrosine) at MBP 4 greatly stabilizes MBP binding to I-Au (9). Places MBP 3 glutamine at P8, a major TCR contact position. MBP 3 glutamine is a major TCR contact (17, 19, 20, 40). Places MBP residue 4 near to I-A residue β9. MHC residue β9 lies potentially between pockets P6 and P9, and in recent class II crystal structures its sidechain forms one side of the P9 pocket (3, 4, 29). The site-directed mutagenesis data suggest a direct interaction between MBP residue 4 and I-A residue β9.
Inconsistent.
Places MBP 5 arginine and MBP 6 proline beyond the COOH-terminal end of the peptide binding cleft, where they are unlikely to be major TCR contacts. Both are key TCR contact residues (17, 19, 20, 40; Tate, K.M., and C. Beeson, unpublished data). Leaves a majority of the peptide binding groove empty. Studies of mutated CLIP peptides binding to I-A alleles demonstrates that I-A residue β9 determines P6 not P9 pocket specificity.
Footnotes
Christopher Lee's present address is Department of Chemistry & Biochemistry, University of California, Los Angeles, Los Angeles, CA 90095. Michael N. Liang's present address is Department of Chemistry, Harvard University, Cambridge, MA 02138. Keri M. Tate's present address is Anergen Inc., Redwood City, CA 94603. Craig Beeson's present address is Department of Chemistry, University of Washington, Box 351700, Seattle, WA 98195-1700.
References
- 1.Stern LJ, Wiley DC. Antigenic peptide binding by class I and class II histocompatibility proteins. Structure. 1994;2:245–251. doi: 10.1016/s0969-2126(00)00026-5. [DOI] [PubMed] [Google Scholar]
- 2.Stern LJ, Brown JH, Jardetzky TJ, Gorga JC, Urban RG, Strominger JL, Wiley DC. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh P, Amaya M, Mellins E, Wiley DC. The structure of an intermediate in class II MHC maturation: CLIP bound to HLA-DR3. Nature. 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 4.Fremont DH, Hendrickson WA, Marrack P, Kappler J. Structures of an MHC class II molecule with covalently bound single peptides. Science. 1996;272:1001–1004. doi: 10.1126/science.272.5264.1001. [DOI] [PubMed] [Google Scholar]
- 5.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC. Crystallographic analysis of endogenous peptides associated with HLA-DR1 suggests a common, polypropylene II-like conformation for bound peptides. Proc Natl Acad Sci USA. 1996;93:734–738. doi: 10.1073/pnas.93.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson CA, Petzold SJ, Unanue ER. Peptides determine the lifespan of MHC class II molecules in the antigen-presenting cell. Nature. 1994;371:250–252. doi: 10.1038/371250a0. [DOI] [PubMed] [Google Scholar]
- 7.Bertoletti A, Sette A, Chisari FV, Penna A, Levrero M, De Carli M, Fiaccadori F, Ferrari C. Natural variants of cytotoxic epitopes are T-cell receptor antagonists for antiviral cytotoxic T cells. Nature. 1994;369:407–410. doi: 10.1038/369407a0. [DOI] [PubMed] [Google Scholar]
- 8.Beeson C, Rabinowitz J, Tate K, Gutgemann I, Chien Y-H, Jones PP, Davis MM, McConnell HM. Early biochemical signals arise from low affinity TCR-ligand reactions at the cell–cell interface. J Exp Med. 1996;184:777–782. doi: 10.1084/jem.184.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fairchild PJ, Wildgoose R, Atherton E, Webb S, Wraith DC. An autoantigenic T cell epitope forms unstable complexes with class II MHC: a novel route for escape from tolerance induction. Int Immunol. 1993;5:1151–1158. doi: 10.1093/intimm/5.9.1151. [DOI] [PubMed] [Google Scholar]
- 10.Joosten I, Wauben MHM, Holewijn MC, Reske K, Pederson LO, Roosenboom CFP, Hensen EJ, van Eden W, Buus S. Direct binding of autoimmune disease related T cell epitopes to purified Lewis rat MHC class II molecules. Int Immnol. 1994;6:751–759. doi: 10.1093/intimm/6.5.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar V, Bhardwaj V, Soares L, Alexander J, Sette A, Sercarz E. Major histocompatibility complex binding affinity of an antigenic determinant is crucial for the differential selection of interleukin 4/5 or interferon γ by T cells. Proc Natl Acad Sci USA. 1995;92:9510–9514. doi: 10.1073/pnas.92.21.9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu GY, Fairchild PJ, Smith RM, Prowie JR, Kioussis D, Wraith DC. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 13.Fairchild PJ, Wraith DC. Lowering the tone: mechanisms of immunodominance among epitopes with low affinity for MHC. Immunol Today. 1996;17:80–85. doi: 10.1016/0167-5699(96)80584-6. [DOI] [PubMed] [Google Scholar]
- 14.Rothbard JB, Gefter ML. Interactions between immunogenic peptides and MHC proteins. Annu Rev Immunol. 1991;9:527–565. doi: 10.1146/annurev.iy.09.040191.002523. [DOI] [PubMed] [Google Scholar]
- 15.Mason K, Denney DW, Jr, McConnell HM. Myelin basic protein peptide complexes with the class II MHC molecules I-Au and I-Akform and dissociate rapidly at neutral pH. J Immunol. 1995;154:5216–5227. [PubMed] [Google Scholar]
- 16.Mason K, Denney DW, Jr, McConnell HM. Kinetics of the reaction of a myelin basic protein peptide with soluble IAu . Biochemistry. 1995;34:14874–14878. doi: 10.1021/bi00045a031. [DOI] [PubMed] [Google Scholar]
- 17.Wraith DC, Smilek DE, Mitchell DJ, Steinman L, McDevitt HO. Antigen recognition in autoimmune encephalomyelitis and the potential for peptide-mediated immunotherapy. Cell. 1989;59:247–255. doi: 10.1016/0092-8674(89)90287-0. [DOI] [PubMed] [Google Scholar]
- 18.Smilek DE, Wraith DC, Hodgkinson S, Dwivedy S, Steinman L, McDevitt HO. A single amino acid change in a myelin basic protein peptide confers the capacity to prevent rather than induce experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 1991;88:9633–9637. doi: 10.1073/pnas.88.21.9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautam AM, Pearson CI, Smilek DE, Steinman L, McDevitt HO. A polyalanine peptide with only five native myelin basic protein residues induces autoimmune encephalomyelitis. J Exp Med. 1992;176:605–609. doi: 10.1084/jem.176.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wraith DC, Bruun B, Fairchild PJ. Cross-reactive antigen recognition by an encephalitogenic T cell receptor. Implications for T cell biology and autoimmunity. J Immunol. 1992;149:3765–3770. [PubMed] [Google Scholar]
- 21.Gautam AM, Lock CB, Smilek DE, Pearson CI, Steinman L, McDevitt HO. Minimum structural requirements for peptide presentation by major histocompatibility complex class II molecules: implications in induction of autoimmunity. Proc Natl Acad Sci USA. 1994;91:767–771. doi: 10.1073/pnas.91.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gautam AM, Pearson C, Quinn V, McDevitt HO, Milburn PJ. Binding of an invariant-chain peptide, CLIP, to I-A major histocompatibility complex class II molecules. Proc Natl Acad Sci USA. 1995;92:335–339. doi: 10.1073/pnas.92.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tate KM, Lee C, Edelman S, Carswell-Crumpton C, Liblau R, Jones PP. Interactions among polymorphic and conserved residues in MHC class II proteins affect MHC:peptide conformation and T cell recognition. Int Immunol. 1995;7:747–761. doi: 10.1093/intimm/7.5.747. [DOI] [PubMed] [Google Scholar]
- 24.Watts TH, Brian AA, Kappler JW, Marrack P, McConnell HM. Antigen presentation by supported planar membranes containing affinity-purified I-Ad . Proc Natl Acad Sci USA. 1984;81:7564–7568. doi: 10.1073/pnas.81.23.7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Busch R, Strang G, Howland K, Rothbard J. Degenerate binding of immunogenic peptides to HLA-DR proteins on B cell surfaces. Int Immunol. 1990;2:443–451. doi: 10.1093/intimm/2.5.443. [DOI] [PubMed] [Google Scholar]
- 26.Oi VT, Jones PP, Herzenberg LA, Herzenberg LA. Properties of monoclonal antibodies to mouse Ig allotypes, H-2, and Ia antigens. Curr Top Microbiol Immunol. 1978;81:115–129. doi: 10.1007/978-3-642-67448-8_18. [DOI] [PubMed] [Google Scholar]
- 27.Witt SN, McConnell HM. A first-order reaction controls the binding of antigenic peptides to major histocompatibility complex class II molecules. Proc Natl Acad Sci USA. 1991;88:8164–8168. doi: 10.1073/pnas.88.18.8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rabinowitz JD, Tate K, Lee C, Beeson C, McConnell HM. Specific T cell recognition of kinetic isomers in the binding of peptide to class II major histocompatibility complex. Proc Natl Acad Sci USA. 1997;94:8702–8707. doi: 10.1073/pnas.94.16.8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC. The three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature. 1993;364:33–39. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- 30.Lee C, Subbiah S. Prediction of protein side-chain conformation by packing optimization. J Mol Biol. 1991;217:373–388. doi: 10.1016/0022-2836(91)90550-p. [DOI] [PubMed] [Google Scholar]
- 31.Lee C. Predicting protein mutant energetics by self-consistent ensemble optimization. J Mol Biol. 1994;236:918–939. doi: 10.1006/jmbi.1994.1198. [DOI] [PubMed] [Google Scholar]
- 32.Lee C, McConnell HM. A general model of invariant chain association with class II major histocompatibility complex proteins. Proc Natl Acad Sci USA. 1995;92:8269–8273. doi: 10.1073/pnas.92.18.8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levitt M. Accurate modeling of protein conformation by automatic segment matching. J Mol Biol. 1992;226:507–533. doi: 10.1016/0022-2836(92)90964-l. [DOI] [PubMed] [Google Scholar]
- 34.Levitt M. Molecular dynamics of native protein: computer simulation of trajectories. J Mol Biol. 1983;168:595–620. doi: 10.1016/s0022-2836(83)80304-0. [DOI] [PubMed] [Google Scholar]
- 35.Deleted in proof.
- 36.Liang MN, Lee C, Xia Y, McConnell HM. Molecular modeling and design of invariant chain peptides with altered dissociation kinetics from class II MHC. Biochemistry. 1996;35:14734–14742. doi: 10.1021/bi961725b. [DOI] [PubMed] [Google Scholar]
- 37.Dougherty DA. Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr and Trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 38.Stites WE, Gittis AG, Lattman EE, Shortle D. In a staphylococcal nuclease mutant the side-chain of a lysine replacing valine 66 is fully buried in the hydrophobic core. J Mol Biol. 1991;221:7–14. doi: 10.1016/0022-2836(91)80195-z. [DOI] [PubMed] [Google Scholar]
- 39.Pearson, C.I. 1994. An analysis of peptide, MHC, and T cell receptor trimolecular complexes in experimental autoimmune encephalomyelitis (encephalomyelitis). Ph.D. thesis. Stanford University, Stanford, CA. 144 pp.
- 40.Davis CB, Mitchell DJ, Wraith DC, Todd JA, Zamvil SS, McDevitt HO, Steinman L, Jones PP. Polymorphic residues on the I-Aβ chain modulate the stimulation of T cell clones specific for the N-terminal peptide of the autoantigen myelin basic protein. J Immunol. 1989;143:2083–2093. [PubMed] [Google Scholar]
- 41.Mason, K. 1995. Interactions of antigenic peptides with class II major histocompatibility molecules. Ph.D. thesis. Stanford University, Stanford, CA. 102 pp.
- 42.Busch R, Hill CM, Hayball JD, Lamb JR, Rothbard JB. Effect of natural polymorphism at residue 86 of the HLA-DR β chain on peptide binding. J Immunol. 1991;147:1292–1298. [PubMed] [Google Scholar]
- 43.Demotz S, Barbey C, Corradin G, Amoroso A, Lanzavecchia A. The set of naturally processed peptides displayed by DR molecules is tuned by polymorphism of residue 86. Eur J Immunol. 1993;23:425–432. doi: 10.1002/eji.1830230219. [DOI] [PubMed] [Google Scholar]
- 44.Newton-Nash DK, Eckels DD. Differential effect of polymorphism at HLA-DR1 β-chain positions 85 and 86 on binding and recognition of DR1-restricted antigenic peptides. J Immunol. 1993;150:1813–1821. [PubMed] [Google Scholar]
- 45.Deleted in proof.
- 46.Gulden PH, Fischer PI, Engelhard VH, Shabanowitz J, Hunt DF, Pamer EG. A Listeria monocytogenes pentapeptide is presented to cytolytic T lymphocytes by the H2-M3 MHC class Ib molecule. Immunity. 1996;5:73–79. doi: 10.1016/s1074-7613(00)80311-8. [DOI] [PubMed] [Google Scholar]
- 47.Lenz LL, Dere B, Bevan MJ. Identification of an H2-M3-restricted Listeria epitope: implications for antigen presentation by M3. Immunity. 1996;5:63–72. doi: 10.1016/s1074-7613(00)80310-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindahl KF, Byers DE, Dabhi VM, Hovik R, Jones EP, Smith GP, Wang CR, Xiao H, Yoshino M. H2-M3, a full-service class Ib histocompatibility antigen. Annu Rev Immunol. 1997;15:851–879. doi: 10.1146/annurev.immunol.15.1.851. [DOI] [PubMed] [Google Scholar]
- 49.Dornmair K, Clark BR, McConnell HM. Binding of truncated peptides to the MHC molecule IAd . FEBS Lett. 1991;294:244–246. doi: 10.1016/0014-5793(91)81439-f. [DOI] [PubMed] [Google Scholar]
- 50.Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, Jameson SC, Gascoigne NRJ. T-cell-receptor affinity and thymocyte positive selection. Nature. 1996;381:616–620. doi: 10.1038/381616a0. [DOI] [PubMed] [Google Scholar]
- 51.Lyons DS, Lieberman SA, Hampl J, Boniface JJ, Chien Y, Berg LJ, Davis MM. A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 52.Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA. An αβ T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science. 1996;274:209–219. doi: 10.1126/science.274.5285.209. [DOI] [PubMed] [Google Scholar]
- 53.Pribyl TM, Campagnoni CW, Kampf K, Kashima T, Handley VW, McMahon J, Campagnoni AT. The human myelin basic protein gene is included within a 179-kilobase transcription unit: expression in the immune and central nervous systems. Proc Natl Acad Sci USA. 1993;90:10695–10699. doi: 10.1073/pnas.90.22.10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fritz RB, Kalvakolanu I. Thymic expression of the golli-myelin basic protein gene in the SJL/J mouse. J Neuroimmunol. 1995;57:93–99. doi: 10.1016/0165-5728(94)00167-m. [DOI] [PubMed] [Google Scholar]
- 55.Landry CF, Ellison JA, Pribyl TM, Campagnoni C, Kampf K, Campagnoni AT. Myelin basic protein gene expression in neurons: developmental and regional changes in protein targeting within neuronal nuclei, cell bodies, and processes. J Neurosci. 1996;16:2452–2462. doi: 10.1523/JNEUROSCI.16-08-02452.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evavold BD, Allen PM. Separation of IL-4 production from Th cell proliferation by an altered T cell receptor ligand. Science. 1991;252:1308–1310. doi: 10.1126/science.1833816. [DOI] [PubMed] [Google Scholar]
- 57.Racioppi L, Ronchese F, Matis LA, Germain RN. Peptide-major histocompatibility complex class II complexes with mixed agonist/antagonist properties provide evidence for ligand-related differences in T cell receptor-dependent intracellular signaling. J Exp Med. 1993;177:1047–1060. doi: 10.1084/jem.177.4.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kersh GJ, Allen PM. Essential flexibility in the T-cell recognition of antigen. Nature. 1996;380:495–498. doi: 10.1038/380495a0. [DOI] [PubMed] [Google Scholar]
- 59.Margulies DH. An affinity for learning. Nature. 1996;381:558–559. doi: 10.1038/381558a0. [DOI] [PubMed] [Google Scholar]