

Abstract
Major histocompatibility complex (MHC) class II molecules can present peptides derived from two different sources. The predominant source of peptide in uninfected antigen presenting cells (APCs) is from self-proteins that are synthesized within the cell and traffic through the MHC class II compartment. The other source of antigen is endocytosed proteins, which includes both self- and foreign proteins. Foreign protein antigens generate adaptive immune responses, whereas self-peptides stabilize the MHC class II heterodimer on the cell surface, allowing positive and negative selection of thymocytes. Therefore, self-antigens play an important normal role in shaping the T cell receptor repertoire as well as a pathological role in autoimmunity. To determine whether processing and presentation of self-antigens by MHC class II molecules differs depending on whether the antigen is supplied through synthesis within the cell or by endocytosis, we used a T cell clone against an Eα peptide presented by I-Ab to show that processing through these two routes can differ. We also show that mice can be tolerant to the epitope formed through the endogenous route, but responsive to the epitope that can be formed through endocytosis. This suggests that negative selection occurs primarily against antigens that are synthesized within the APC, and that endocytosed self-antigens could serve as autoantigens. Finally, we also demonstrate that lipopolysaccharide-activated B cells are defective for uptake, processing, and presentation of this self-antigen, and that this correlates with the increased expression of the costimulatory molecules B7.1 and B7.2. This may provide a model for studying the onset of an autoimmune response.
Peptides that are presented on MHC class II molecules are derived from two different sources, and they serve two different functions. Under normal conditions, most MHC class II molecules are occupied by peptides derived from antigens that are synthesized within the APC itself. Peptides that are derived from endogenously synthesized proteins traffic through the endocytic compartment and are primarily derived from other MHC class I and class II molecules (1). These self-peptides allow stable expression of the MHC class II molecules on the cell surface (2), so that these complexes can be used for positive and negative selection of thymocytes (3). The other protein source for MHC class II associated peptides are proteins that must be internalized via endocytosis. Pathogens provide a source of antigen which must be endocytosed, and peptides of this type are also presented by MHC class II molecules to naive T cells, so that the organism can mount an appropriate adaptive immune response against the pathogen.
This implies that two types of antigen can be loaded through the endocytic pathway. The first type is pathogen derived, whereas the second is endocytosed self-antigens. This raises the question of whether processing of proteins that gain access to the MHC class II compartment after endogenous synthesis differs from processing of the same proteins which gain access to the MHC class II compartment through endocytosis. Although some data exists that suggests that processing of endogenously synthesized and endocytosed exogenous antigen may differ (4), essentially no data exist that specifically address this question. Such differences could have important implications both for positive and negative selection of the TCR repertoire and for autoimmunity.
Studies in recent years have focused on identifying the compartment(s) in which antigen processing and peptide loading onto MHC class II molecules occurs, referred to as the MHC class II compartment (MIIC)1 or the class II loading vesicle (CIIV) (5, 6). From these studies and others examining mechanisms of MHC trafficking, proteolysis of the MHC class II invariant chain (Ii), and peptide loading has emerged a more complete model for MHC class II trafficking. MHC class II molecules are transported as a complex with Ii to an endosomal compartment. Ii directs this localization and retains the complex in this compartment until Ii is cleaved to a form called the class II Ii peptide (CLIP) that is still bound in the groove of the MHC molecule but is no longer anchored to the membrane (7–9). At this point, Ii can still prevent binding of other peptides to MHC class II, but can no longer direct its trafficking. MHC class II itself, however, contains signals that direct it to the endocytic compartment (10). These signals may then direct the localization of MHC class II to MIIC/CIIV, perhaps even directing its formation (11). In the CIIV, DM can catalyze the removal of CLIP and the binding of peptide (12). Rapid transport of the MHC–peptide complex to the cell surface then occurs. This rapid transport is suggested by the kinetics of transport of SDS stable MHC– peptide complexes from CIIV to the cell surface (5), and the fact that large intracellular pools of MHC class II are not detected in early endosomal compartments under normal conditions (13). Rapid transport to the cell surface may preclude further processing of the MHC–peptide complex under the variable conditions potentially available along the endosomal pathway.
In any case, the finding that endogenously synthesized antigen is presented by MHC class II has raised the question of whether endogenous and endocytosed exogenous antigens are processed and loaded onto MHC class II molecules in the same form (5, 14). A study using the antibody Y-Ae, which can detect a specific MHC–peptide complex (I-Ab + Eα 52–68), shows that this complex forms from endogenously synthesized molecules in the MIIC (15), as can the same epitope from exogenously derived peptides. Thus, what appears to be the same MHC–peptide complex can form in the same specialized compartment. However, it is not clear from this analysis if the same product is produced, as T cell recognition of the Y-Ae epitope was not determined. It is not yet clear, furthermore, that endogenously synthesized protein antigens have access to all of the antigen-processing compartments. To examine this question, we prepared T cell clones by immunization of C57BL/6J mice that express I-Ab but lack Eα. We also immunized B10.A(5R) and B10.A(3R) mice that are similarly I-Ab positive but possess the Eα chain and express the Y-Ae epitope (16, 17). These studies illustrate three important points. First, that endocytosed antigen is processed distinctly from endogenously synthesized antigen. Second, that tolerance exists to endogenously synthesized antigen but not to antigen internalized by endocytosis. Third, that the ability of APCs to take up and present antigen is reciprocally regulated with the induction of costimulatory molecules, which may serve to prevent autoimmunization of existing potentially autoreactive T cells.
Materials and Methods
Peptides.
The peptides were synthesized as previously described (16), purified by HPLC, and checked for accuracy by mass spectroscopy and amino acid analysis by the W.M. Keck Biotechnology Resource Center (Yale University, New Haven, CT). The sequence of Eα 52–68, the longest peptide used here, is: A S F E A Q G A L A N I A V D K A, whereas peptide Ea 52–66 is truncated two amino acid residues (-KA) at its COOH-terminus.
Mice.
C57BL/6J(B6), B10.A(3R) (3R), B10.A(5R) (5R), or 107.1 (B6–I-E transgenic) mice were used in the experiments shown here. B6 and 5R mice were obtained from The Jackson Laboratory (Bar Harbor, ME). 3R mice were bred and maintained within our own animal facility. 107.1 mice were obtained from Richard Flavell (Yale University).
T Cells.
Eα6, the T cell clone used in these experiments, was raised by footpad immunization of C57BL6/J (B6) mice with 30 μg of Eα 52–68 in CFA. After 8 d, popliteal lymph nodes were removed and placed in bulk culture with 10 μg/ml Eα 52–68, ∼10 U/ml of IL-2 and 10% FCS. After three weekly restimulations the cultures were cloned by limiting dilution. Clones were expanded from plates that contained much less than 1 cell/well by Poisson distribution. Two of the clones were subcloned, but parental clones were found to give better responses, so they were used for these studies. Anti-Vβ FACS® staining and anti-Vα PCR analysis confirmed that the parental lines were monoclonal (Viret, C., unpublished data). The clones were maintained by restimulation every 2 wk with fivefold excess irradiated (2,000 rads) or mitomycin C–treated (50 μg/ml; Boehringer Mannheim, Indianapolis, IN) C57BL6/J spleen cells, 3 μg/ml Eα 52–68 or Eα 52–66, <5 U/ml IL-2, and 5% FCS. Unless otherwise stated, all assays and cultures were done in Click's medium (Irvine Scientific Co., Santa Ana, CA) containing 5% FCS, 3 × 10−5 M 2-ME, 2 × 10−4 M l-glutamine, and antibiotics. The T cell hybridoma, 1H3.1, has been previously described (16). Finally, to examine tolerance to Eα52–68 and Eα52–66, we immunized 3R, 5R, and B6 mice with either peptide and measured their responses to the immunizing and the alternative form of the peptide. These responses were inhibitable with Y-Ae monoclonal antibodies added to the cultures.
T Cell Clone and Hybrid Assays.
All proliferation assays were set up using 2 × 104 cloned T cells/well in 96-well plates. 2–3 × 105 irradiated or mitomycin C–treated splenocytes/well were used as APCs. All proliferation assays were cultured for 40–48 h at 37°C, and then pulsed with 1 μCi per well [3H]thymidine (6.7 Ci/mmol; Amersham Corp., Arlington Heights, IL) for 6–12 h. The plates were then harvested and counted. Hybrid assays, measuring IL-2 production by CTLL assay, were performed as described (16). All T cell and hybrid assays were performed in duplicate or triplicate. Each experiment shown is representative of multiple assays. For LPS assays, 4 × 106 splenocytes/ml were cultured with 10 μg/ml LPS (Difco Labs., Detroit, MI) for 24 h and were then harvested, irradiated (2,000 rads), washed, counted, and cultured with peptides and T cells.
Antibodies.
The antibodies used in these experiments were Y-Ae (17), the anti–I-Ab antibody Y3JP (18), and the anti–I-E antibody Y17 (19). FACS® (Becton Dickinson, Mountain View, CA) staining and analysis was performed as previously described (16). Y-Ae was detected using FITC-labeled Fc-specific goat anti–mouse IgG (1:500; Sigma Chemical Co., St. Louis, MO). The anti-CD28 antibody 37N51was used to provide costimulation in some experiments. Culture supernatant was added to T cells and APCs at the optimal dilution of 1:30 (final concentration) before the addition of the cross-linking antibody, goat anti– hamster IgG (Caltag Labs., San Francisco, CA) at 1 μg/ml.
Antibody Stimulation.
In mAb stimulation assays, mAbs were added to the APCs as purified mAb or tissue culture supernatants 30–60 min before the addition of the T cells.
Inhibitors of Antigen Processing.
100 μM chloroquine, 100 μM primaquine, or 1.5 mM ammonium chloride was used to block antigen processing. The inhibitors were added to irradiated splenic APCs at twice the concentration listed above for 30–45 min at 37°C before dilution to the concentration listed above by peptide antigen. The APCs were then cultured overnight in the presence of the inhibitors and the peptide before we washed the APCs and added T cells. Anti-CD28 and the cross-linking antibody goat anti–hamster were added to all wells to provide costimulation. The APCs, having been irradiated on the previous day, probably would not be able to provide the necessary costimulation for the T cells. The APCs were treated overnight because long pulses with Eα 52–68 were necessary to obtain a strong proliferative response from Eα6.
Fixation.
APCs were fixed at <3 × 107 cells/ml in 0.2% paraformaldehyde for 5 min at room temperature. The cells were then diluted threefold with 0.2 M glycine in Click's medium, pelleted, and washed two times with complete media. Anti-CD28 was added in combination with the cross-linking antibody goat anti–hamster to provide costimulation, as described below.
Purification of I-E Protein.
Whole I-E protein was isolated as described for I-Ab, from 100 (B6 × C3H) F2 mice (provided by Richard Flavell). In brief, for this procedure a Y17 sepharose column was added between a nonspecific control column and a Y-Ae column. I-E was eluted from the Y17 column with high pH buffer, and neutralized immediately with 1 M Tris, pH7. This material was dialyzed at 4°C against PBS, and stored at 4°C until use in the assays described above.
Immunization of B6 Mice against I-E.
To generate an antigen-specific, receptor-mediated uptake mechanism to enhance presentation of limited quantities of I-E protein, B6 mice were repeatedly immunized with B6/I-E transgenic (strain 107.1, provided by Richard Flavell) splenocytes. Before experiment 1, which is shown in Fig. 8, two B6 mice were immunized intraperitoneally with 2 × 107 107.1 splenocytes. After 5 d the mice were reimmunized intravascularly via retroorbital sinus with 107 107.1 splenocytes. After another 4 d, a B6 spleen was removed from one of the immunized animals, irradiated with 1,000 rads to maintain the APC function of B cells, and used for experiment 1 (Fig. 8, A and B). After ∼1 wk, the other mouse was reimmunized intravascularly with 2 × 107 107.1 splenocytes, except this time the 107.1 cells were treated with LPS for 20 h before injection. After another 4 d the B6 spleen was removed, irradiated (1,000 rads), and used for experiment 2 (Fig. 8 C). Spleen from unimmunized B6 was also used.
Figure 8.

The ability of Y-Ae to inhibit B10A (3R) T cell responses to Eα52–66. The proliferative response of popliteal lymph node cells immunized with Eα52–66 and restimulated with Eα52–66 is specifically inhibited with the Y-Ae monoclonal antibody. The mean response in three separate experiments is shown with the SEM.
Results
Clone Eα6 Does Not Respond to 3R APCs unless Eα 52–68 Peptide Is Added.
To compare processing of endogenously synthesized antigen with the processing of exogenously supplied antigen, T cell clones were raised in C57BL/6J (B6) mice by immunization with a synthetic peptide derived from the sequence of MHC class II Eα chain residues 52–68. B6 mice express the MHC class II molecule I-Ab, but do not express the peptide donor molecule I-E. This peptide was chosen because it is known to be presented by the MHC class II molecule I-Ab when I-E is also synthesized within the cell (1). Additionally, a monoclonal antibody, Y-Ae, specifically recognizes this MHC–peptide complex, allowing its easy detection (1, 17).
All of the clones responded to synthetic peptides and were restricted by I-Ab as shown by antibody blocking experiments; their response was also blocked by the antibody Y-Ae (Fig. 1 A). Surprisingly, none of the clones responded to the I-Ab–Eα peptide complex presented on 3R splenocytes that express I-E molecules and the Y-Ae epitope (Fig. 1 B). Although the experiment shown here has an unusually strong response of Eα6 to 3R APCs in the absence of synthetic peptide, this response was weak relative to the response of Eα6 to APCs plus synthetic peptide. Typically, this response was no greater than the background response of Eα6 to B6 APCs (see Fig. 5). This suggested that processing of endogenously synthesized antigen differed from processing of exogenously supplied antigen. It may be that the synthetic peptide which was added to the culture had been processed to a shorter form that is required for stimulating the Eα6 clone. A control hybrid was also raised by immunization of B6 mice with the peptide Eα52–68; this hybrid is also restricted by I-Ab, and blocked by Y-Ae, but this hybrid also responded to the endogenously synthesized and processed ligand on 3R spleen cells (Fig. 1 B).
Figure 1.
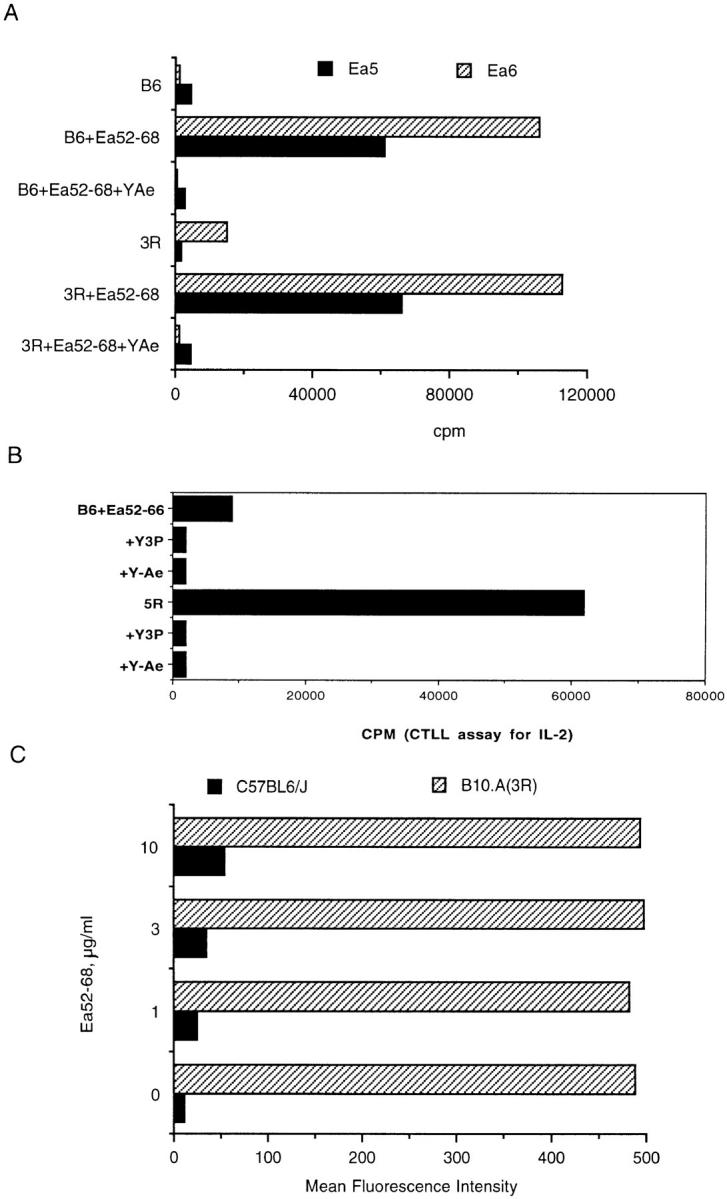
Eα6 responds to Eα52–68 presented by I-Ab but not to endogenous ligand on B10.A (3R), whereas control hybrid 1H3.1 responds to both exogenous and endogenous ligand. (A) The proliferative response of the T cell clone Eα6 to APCs which did (3R) or did not (B6) express endogenous ligand, in the presence or absence of the peptide Eα52–68. The ability of the monoclonal antibody Y-Ae, which also recognizes the I-Ab–Eα peptide complex, to block the response of the clone is also shown. (B) The response of 1H3.1 was measured by culture of 105 hybrids with 2 × 105 splenocytes along with the indicated antibodies and peptide. Culture supernatant was removed after 20 h and tested for the presence of IL-2 using the IL-2–dependent cell line CTLL. In A, the response to B6 splenocyte + the synthetic peptide Eα52–66 is shown. Responses to Eα52–68 were similar. In B, the response of 1H3.1 cells to the endogenous ligand on 3R cells is also shown. No synthetic peptide was added since 1H3.1 responds to the endogenous ligand. The antibodies were added to the culture wells to show that the response of 1H3.1 is restricted by I-Ab and is also blocked by Y-Ae. Y3P is an anti–I-Ab antibody. Y-Ae did not block the response of a control clone D10 (data not shown), which recognizes an I-Ab ligand different from that recognized by Y-Ae. Y-17, an anti–I-E antibody, does not block the response of either 1H3.1 or Eα6 (data not shown). (C) Flow cytometric staining with the antibody Y-Ae. Staining of B6 and 3R splenocytes in the presence of a titration of the peptide Eα52–68 is compared.
Figure 5.
Anti–I-Ab mAb created an I-E–dependent epitope for Eα6 on splenocytes. The proliferative response of Eα6 to mAb treatment of APCs which did not (A) or did (B and C) express endogenous ligand is shown. No synthetic peptide was added to this experiment. 107.1 is a B6 mouse which expresses an Eα transgene. The APCs were cultured with mAb for 30–60 min at 37°C and then T cells were added. The mixture was then cultured for 48–60 h and pulsed overnight with 1 μCi per well of [3H]thymidine, as with a proliferation assay to test the response to synthetic peptide.
The Presence of Too Much or Too Little Ligand Can Not Explain the Lack of Response of Eα6 to 3R Cells.
The possibility that either too much or too little ligand was responsible for the lack of response of the cloned T cell line Eα6 to the endogenously generated ligand was ruled out in several initial experiments. Since the Y-Ae antibody recognizes the synthetic Eα peptide bound to I-Ab as well as the endogenously generated complex, FACS® staining with Y-Ae was used to compare the level of ligand created by addition of the synthetic peptide to the level of endogenous ligand. Fig. 1 C shows that even at doses of synthetic peptide sufficient to give plateau stimulation of Eα6 (10 μg/ml), the staining with Y-Ae was much lower than that on 3R splenocytes. Therefore, the lack of response of Eα6 was not due simply to overly low levels of the ligand on the APCs.
However, too much ligand can suppress the response of T cell clones, as is seen in Fig. 2 A. As shown in Fig. 1 A, Eα6 T cells responded similarly to synthetic peptide added to B6, which does not express the endogenous ligand, and 3R, which does express the endogenous ligand. Dose– response curves of the response of Eα6 to peptide on B6 or 3R completely overlap (data not shown). Since addition of antigen allowed the clones to respond, their lack of response to the endogenously synthesized protein could not be explained by the phenomenon of high antigen dose suppression.
Figure 2.
The response of Eα6 and control hybrid 1H3.1 to length variants of Eα peptide. (A) The proliferative response (measured by incorporation of [3H]thymidine) of Eα6 to two synthetic length variants of the Eα peptide, Eα52–68 and Eα52–66. (B) The response of control hybrid 1H3.1 to the same Eα peptide length variants. The response of 1H3.1 was measured by IL-2 production that was detected by the proliferative response of the IL-2–dependent cell line CTLL.
The Clones Were Not Raised against a Contaminant in the Peptide Preparation.
The possibility that the clones were responding to a contaminant in the peptide preparation was examined. The peptide, Eα52–68, was synthesized on several separate occasions, purified by HPLC, and checked for accuracy by amino acid analysis and mass spectroscopy. The main species was determined to have the correct sequence, but, minor contaminants were still present. However, blocking of the response of the clones by the antibody Y-Ae suggests that they are specific for the Eα peptide, since this antibody specifically recognizes this peptide when bound to I-Ab (Fig. 1, A–C). Additional HPLC purifications enhanced the response of Eα6, indicating that Eα6 does respond to the correct peptide and not to a contaminant (not shown).
Eα6 Required a Truncated Eα Peptide which Was Not Processed from the Endogenous Protein.
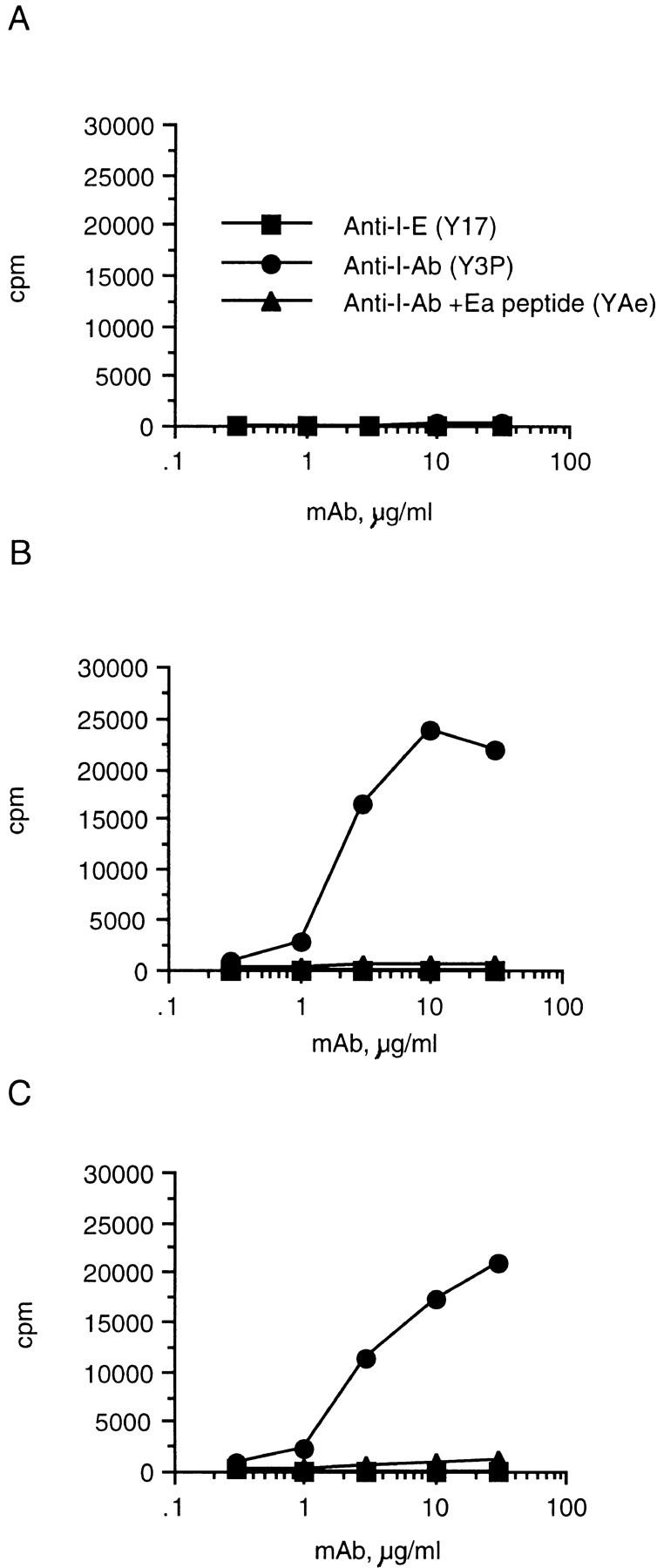
We next asked if Eα6 responded to a different length of the Eα peptide than did 1H3.1. As shown in Fig. 2 A, 10-fold less Eα52–66 than Eα52–68 was required to give the same stimulation of Eα6, whereas 1H3.1 responded equally to both peptides (Fig. 2 B). Further truncation on either end of the peptide decreased stimulatory capacity for Eα6, whereas 1H3.1 could respond to a nonapeptide epitope Eα56–64 (Fig. 2 A and data not shown). This suggested that Eα6 required processing of Eα52–68 to Eα52–66 in order to respond.
To investigate this further, two experiments were performed to determine whether Eα6 requires that Eα52–68 be processed to a shorter form. The first is shown in Fig. 3. Splenocytes were either irradiated (designated live), or fixed in paraformaldehyde before addition of the synthetic peptides. Eα6 could only respond to Eα52–68 when presented by live APCs that could still endocytose and process the peptide. The peptide Eα52–66 did not need processing to stimulate Eα6, since it could be presented by either live or formaldehyde-fixed APCs (Fig. 3 A). The control hybrid, 1H3.1, could respond to both peptides on either live or fixed cells, showing that neither peptide required processing to stimulate 1H3.1, and that Eα52–68 could bind to I-Ab on fixed cells (data not shown). Flow cytometric staining with Y-Ae, shown in Fig. 3 B, confirmed that loading of Eα52–68 onto I-Ab on live or fixed cells was not less than loading of the other two peptides. Thus, the inability of Eα6 to respond to the longer peptide on fixed cells could not be explained as an inability of this peptide to bind I-Ab on fixed cells. This supported the hypothesis that Eα52–68 was the endogenous ligand, to which only 1H3.1 could respond, and that Eα52–66 was not produced from the endogenously synthesized and processed protein.
Figure 3.

Eα6 responds to Eα52–66 and Eα52–68 on live APCs, but does not respond to the longer peptide presented by fixed APCs. (A) Live APCs. The proliferative response of Eα6 to length variants of the Eα peptide presented on irradiated splenic APCs that could still take up antigen for processing (Live). (B) Fixed APCs + cross-linked anti-CD28. A similar proliferation assay, except the APCs were paraformaldehyde fixed splenocytes, which could no longer take up and process antigen. Anti-CD28 and goat anti–hamster IgG (Caltag Labs.) were added to the experiment in B to provide costimulatory signals, which could not be provided by the fixed APCs. C shows Y-Ae flow cytometric staining of live APCs after peptide loading, and D shows Y-Ae staining of APCs that were fixed before loading peptides. Y-Ae detects the loading of Eα peptides on I-Ab. The mean fluorescence intensity of unstained cells was subtracted from all samples to give a shift in mean fluorescence intensity. The mean fluorescence intensity of B6 APCs stained with Y-Ae in the absence of added Eα peptide was subtracted from the mean fluorescence intensity of samples stained after peptide loading in order to control for background staining by Y-Ae, which does not normally stain B6 APCs. The staining was performed in triplicate.
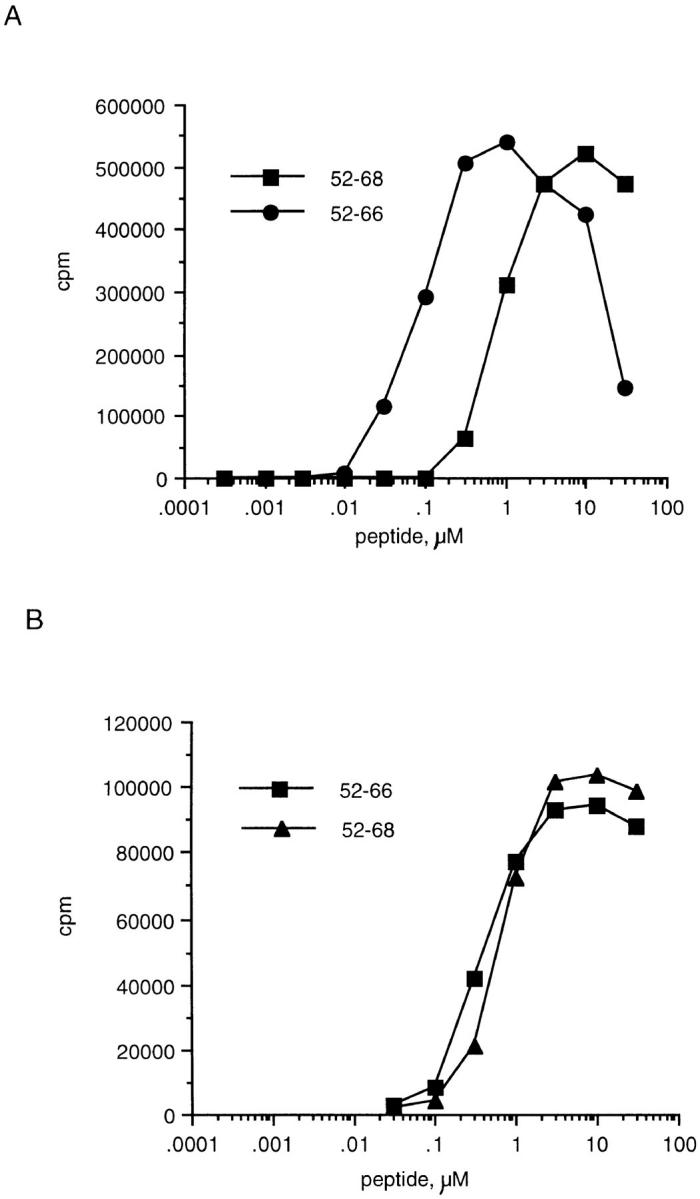
We next asked whether drugs that block the acidification of the endocytic pathway and are known to inhibit processing of exogenous peptides and proteins could also inhibit the response of Eα6 to Eα52–68. APCs were treated for 30 min with the lysosomotropic agents chloroquine or ammonium chloride. Peptides were then pulsed onto the APCs overnight in the presence of the inhibitors. The APCs were finally washed and added to T cells in the presence of cross-linked anti-CD28, added to all wells to provide costimulation needed for T cell responses. Fig. 4 shows that both chloroquine and ammonium chloride inhibited presentation of Eα52–68 to Eα6, but could not completely block presentation of Eα 52–66 to Eα6 or presentation of either peptide to 1H3.1. These inhibitors decreased the overall proliferation of the clones to all of the peptides. However, the background response was also lower, so the fold response was actually higher in the presence of inhibitors. The flow cytometric analysis with Y-Ae (data not shown) confirmed that loading of the peptides onto I-Ab was equivalent in the presence and absence of the inhibitors. Thus, the lack of response of Eα6 to the longer peptide was not due to a decrease in the ability to load the longer peptide in the presence of the inhibitors. Therefore, Eα6 appeared to respond selectively to Eα52– 66, and could not recognize the naturally processed peptide Eα52–68 in the absence of further processing.
Figure 4.
Comparison of the responses of Eα6 and control 1H3.1 to Eα52–66 and Eα52–68 in the presence or absence of inhibitors of processing. (A) Proliferative responses of Eα6. (B) IL-2 production by 1H3.1 as measured by CTLL assay. Both panels show the fold response to Eα peptides loaded onto APCs in the presence of inhibitors of processing as a percentage of the fold response to Eα peptides loaded onto APCs in the absence of inhibitors of processing. Some of the responses shown are >100%, because the background response was lower in the samples in which inhibitors of processing were included. Thus, although maximum cpm achieved in the presence of the inhibitors was lower than in the absence of the inhibitors, the fold response was greater in the presence of the inhibitors. □, ▵, chloroquine; ▪,▴, ammonium chloride. □, ▪, Eα52–66; ▴, ▵, Eα52–68.
Forced Internalization of I-Ab on 3R or I-E Transgenic Spleen Cells Created a Ligand for Eα6.
The ability of live 3R splenocytes to create the ligand for Eα6 from the synthetic peptide Eα52–68 (see Fig. 1 A) suggested that these APCs may also be capable of creating the correct ligand from the endogenously synthesized proteins if these proteins were processed in the correct compartment(s). To test this hypothesis, anti–I-Ab antibodies were added to 3R or control B6 splenocytes before addition of Eα6 in an attempt to force the recycling and reprocessing of I-Ab–peptide complexes. The presence of anti–I-Ab antibodies allowed Eα6 to respond to the endogenous ligand in the absence of added exogenous synthetic peptide. This response was specific for the Eα peptide–I-Ab complex, since the clones only responded when both I-Ab and I-E were expressed in the APCs, either as 3R or as a transgenic Eα chain in the B6/I-E transgenic line called 107.1 (Fig. 5, A–C). Several anti–I-Ab antibodies including both culture supernatants and purified mAb could mediate this response (data not shown), indicating that the response was not due to contamination by other potentially stimulatory antibodies. Additionally, cultured bone marrow dendritic cells from 3R mice only stimulated Eα6 after treatment with anti–I-Ab antibody culture supernatants (data not shown).
The possibility that the antibodies lead to proliferation of Eα6 by stimulating the APCs to express costimulatory signals was tested and ruled out. Providing costimulation in conjunction with the endogenous ligand, by using 3R LPS blasts, or cross-linked anti-CD28 with 3R APCs, was not sufficient to cause proliferation of Eα6 (data not shown). Thus, the most likely explanation for the ability of the anti–I-Ab antibodies to cause proliferation of Eα6 to endogenous ligand was that the antibodies force internalization of I-Ab and thus allow reprocessing of the ligand.
APCs Generated the Epitope for Eα6 from Whole I-E Protein Added Exogenously.
APCs were capable of generating the epitope required for the Eα6 response from both exogenously added synthetic peptide and endogenously generated peptide–MHC complexes that were forced to recycle. Thus, it seemed likely that the lack of response of Eα6 to the unmanipulated endogenous ligand was due to an inability of the complex or individual components to traffic through the compartment required to generate the ligand for Eα6. Alternatively, APCs may have been able to generate the ligand for Eα6 from the peptide Eα52–68, but they may not have been able to generate the ligand for Eα6 from whole protein. This possibility was actually already partially excluded. As discussed in the preceding section, the APCs could generate the ligand for Eα6 from the endogenous ligand when it was forced to recycle. In this case, the APC was able to perform all of the required processing steps. However, the antibody-forced recycling is an artificial system and did not necessarily indicate that whole antigen added exogenously would gain access to the compartment(s) required to generate the epitope for Eα6. Therefore, it was of interest to determine whether APCs could process the whole I-E protein to the epitope required for Eα6 stimulation when the protein was added exogenously.
I-E was purified from (C57BL/6J × C3H) F2 mice by immunoaffinity chromatography using the anti–I-E antibody Y17. Fig. 6 A shows that splenic APCs processed the whole I-E molecule into an epitope that Eα6 could recognize. The response was very weak compared with the response to synthetic peptide (Fig. 6 B); however, 1H3.1 makes an even weaker response to the exogenous I-E protein. The weak response of 1H3.1 to the whole protein indicated that the weak response of Eα6 was due to low concentrations of the antigen and not to differences in the ability of APCs to create the longer versus the shorter peptide. The response of both Eα6 and 1H3.1 to the whole protein was weaker than the response to the peptide if the response was plotted against the micromolar concentration of antigen rather than its mass. This was not unexpected as far fewer steps are required to generate a T cell epitope from a peptide than to generate a T cell epitope from whole protein.
Figure 6.
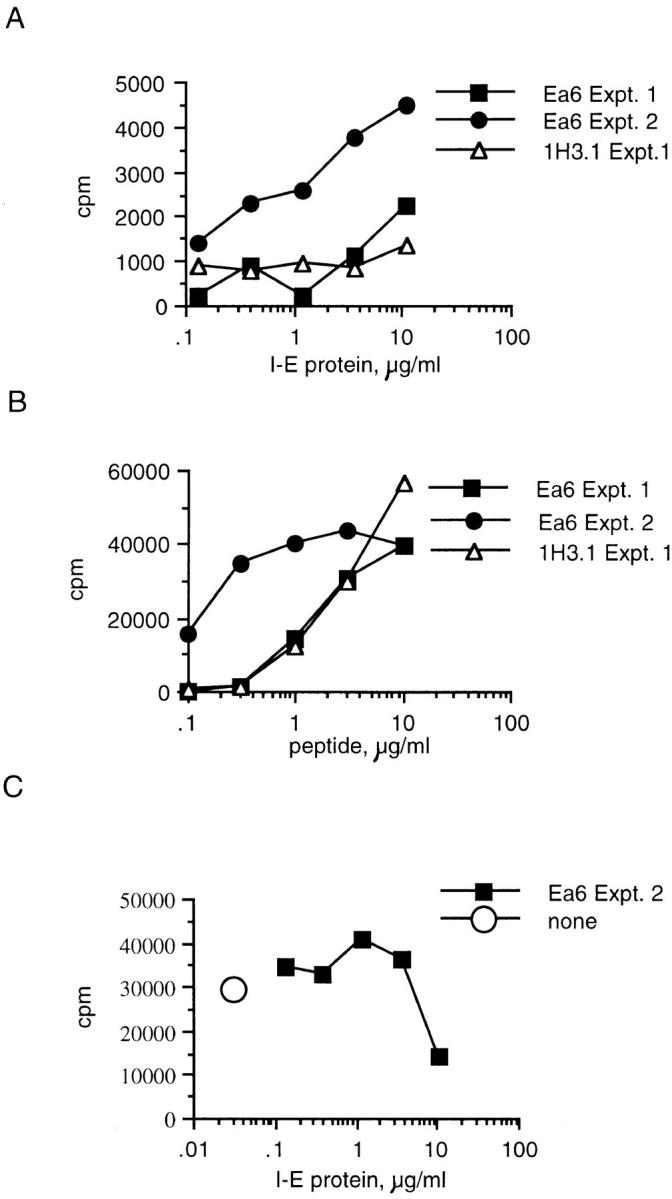
APCs can process whole I-E protein into the epitope required for Eα6 response when the protein was added exogenously. The response of Eα6 and 1H3.1 to whole I-E protein is shown. Fig. 8 B shows the control response to Eα52–66 peptide. Two independent experiments are shown for Eα6. I-E was obtained by immunoaffinity purification from (B6 × C3H)F2 splenocytes using the anti–I-E antibody Y17. Some of the B6 mice used for APCs in this experiment were immunized with splenocytes from the B6, I-E transgenic strain 107.1 in order to try to expand I-E–specific B6 B cells to enhance the efficiency of presentation of the whole I-E protein. The data shown for experiment (Expt.) 1 in A and B are from cultures that had APCs from mice immunized twice with fresh 107.1 splenocytes (see text for details of immunization). The data shown for experiment 2 in A and B are from cultures that had APCs from unimmunized mice. Note that in experiment 1, this immunization protocol did not enhance I-E presentation over that seen using APCs from unimmunized mice. The data shown for experiment 2 in C are from cultures that had APCs from mice immunized a third time, intravascularly, with 107.1 LPS blasts. Splenocytes from the immunized mouse were used as APCs 4 d after the last immunization. No additional exogenous antigen was needed at this time to stimulate a response by Eα6. none, without I-E antigen, but with Eα6 T cells and APCs from immunized B6 mice.
To enhance the efficiency of presentation of the limited quantities of whole I-E protein available, an attempt was made to obtain APCs with the ability to take up the antigen specifically. To accomplish this, B6 mice were repeatedly immunized with splenocytes from the mouse strain 107.1, which, as discussed earlier, is a B6 mouse with an I-E transgene. Before experiment 1, shown in Fig. 6, B6 mice were immunized intraperitoneally with 2 × 107 107.1 splenocytes. After 5 d the mice were reimmunized intravascularly via the retroorbital sinus with 107 107.1 splenocytes. After another 4 d, a B6 spleen was removed from one of the immunized animals, irradiated with 1,000 rads to maintain the APC function of B cells, and used for experiment 1 in Fig. 6. APCs from unimmunized mice were also used (data not shown), but no significant difference in the ability to present I-E protein or peptide was noted in experiment 1. After ∼1 week, another mouse was reimmunized with 2 × 107 107.1 splenocytes, except that this time the 107.1 cells were treated with LPS for 20 h before injection. After another 4 d, the B6 spleen was removed and used for the experiment shown in Fig. 6 C (the response to APCs from unimmunized mice is shown for experiment 2 in Fig. 6, A and B). Interestingly, Eα6 responded to these APCs even without the addition of peptide or protein antigen (Fig. 6 C). The APCs must still have been presenting antigen that was taken up and processed from the 107.1 splenocytes. The caveat to the experiments shown in Fig. 6, A and B, is that the preparation of purified I-E may contain undetectable peptide species. However, the ability of APCs from B6 mice immunized with I-E expressing splenocytes to stimulate Eα6 suggested that APCs could process the whole I-E molecule to the epitope required to activate Eα6.
These data suggest that bona fide mechanisms of antigen uptake, such as the surface Ig receptors of B cells, can lead to the generation of the epitope required for Eα6 activation. Importantly, this indicates that the MHC class II epitopes generated from endogenously synthesized antigen can be different than epitopes that can be created from exogenously provided, endocytosed antigen.
Responses of 3R and B6 Mice to Priming with Eα52–68 and Eα52–66 Peptides.
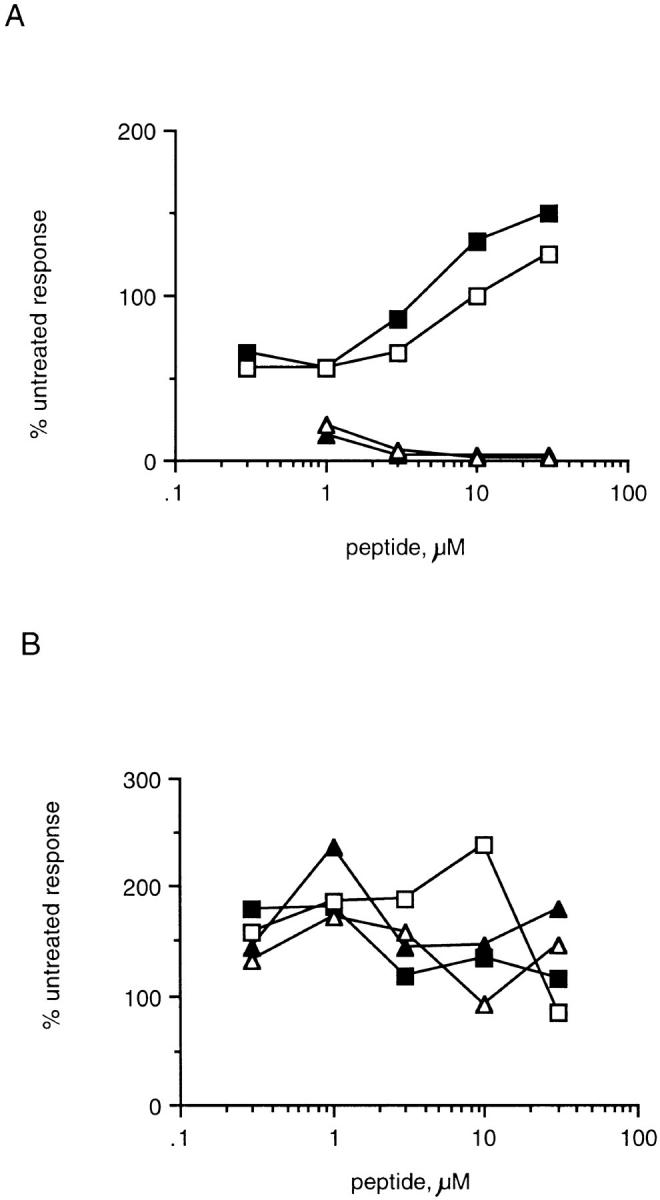
To determine whether B10.A(3R) mice fail to induce tolerance to Eα52–66, 3R mice that express the endogenous Y-Ae ligand (I-Ab + Eα52–68) or control B6 mice (I-Ab only) were immunized with either Eα52–66 or Eα52–68. Fig. 7 A shows that despite the fact that 3R mice express large quantities of the Y-Ae epitope (I-Ab + Eα52–68), they are still able to respond to the peptide Eα52–66. These mice, as expected, do not make a response to the Eα52–68 peptide (Fig. 7 B). This peptide, although processed efficiently in vitro to Eα52–66, may not be processed efficiently enough in vivo to prime a response to Eα52–66 in 3R mice. Fig. 7, C and D, shows that, as expected, B6 mice can respond to either Eα52–68 or Eα52–66. Thus, 3R (and 5R; data not shown) mice are tolerant to Eα52–68 but are not tolerant to Eα52–66. The response of 3R mice to Eα52–66 was inhibitable by Y-Ae, proving that it involved the same epitope that was recognized by Eα6 (Fig. 8); it was also shown to be mediated by CD4 T cells (data not shown).
Figure 7.

B10.A(3R) mice are tolerant to Eα52–68, but not to Eα52–66, whereas C57BL/6 mice can be primed with both peptides. The proliferative response of whole lymph node cultures from 3R mice immunized with 30 μg Eα52–68 (A) or Eα52–66 (B). Popliteal lymph node cells from two mice per experiment were pooled. 4 × 105 cells were cultured with various doses of peptide for 60–72 h before pulsing overnight with 1 μCi/well of [3H]thymidine. The data are representative of several experiments. However, the response of mice immunized with Eα52–66 was low, comparable to that of mice immunized with Eα52–68, in two of five experiments. The response of mice immunized with Eα52–68 was very weak in all experiments. The response of B6 mice immunized with Eα52–68 (C) and Eα52–66 (D) is shown. B6 mice responded similarly in response to both peptides in all experiments, although typically, as here, the response to Eα52–66 was greater than the response to Eα52–68.
One implication of the above results is that autoantigen processing is likely to occur only when an autoantigen is acquired from the exogenous milieu and not from altered processing of endogenous proteins in APCs, as central tolerance exists to such proteins. Autoimmune disease could only result from the uptake and processing of extracellular antigen, as happens in various disease models. To test this hypothesis, we carried out the following experiments.
Cells Rendered Costimulator Positive Cannot Take Up and/ or Process Antigen.
In the experiment shown in Fig. 9, single cell suspensions of spleen were cultured for 24 h with 10 μg/ml LPS in order to induce costimulatory molecules. In this experiment, macrophages would be depleted by overnight culture on tissue culture–treated plastic, but dendritic cells would dissociate from the plastic after overnight culture (20). The cultures were subsequently harvested, irradiated, and washed before addition to peptide and T cells. The harvested APCs should have been primarily B cells with some dendritic cells. The peptide Eα52–68, which requires processing to stimulate Eα6, cannot be presented by splenic LPS blasts. The control peptide, Eα52–66, is presented to Eα6 by LPS blasts, thus indicating that the lack of response to the longer peptide is not due to a non-specific toxic effect of LPS. Additionally, the lack of response to Eα52–68 presented by LPS blasts is not due to a decrease in the ability of this peptide to bind to surface MHC class II, since this peptide is presented to the control processing-independent hybrid 1H3.1 (Fig. 9 B). Y-Ae staining of both of these peptides loaded onto LPS blasts is also comparable (Fig. 9 C).
Figure 9.
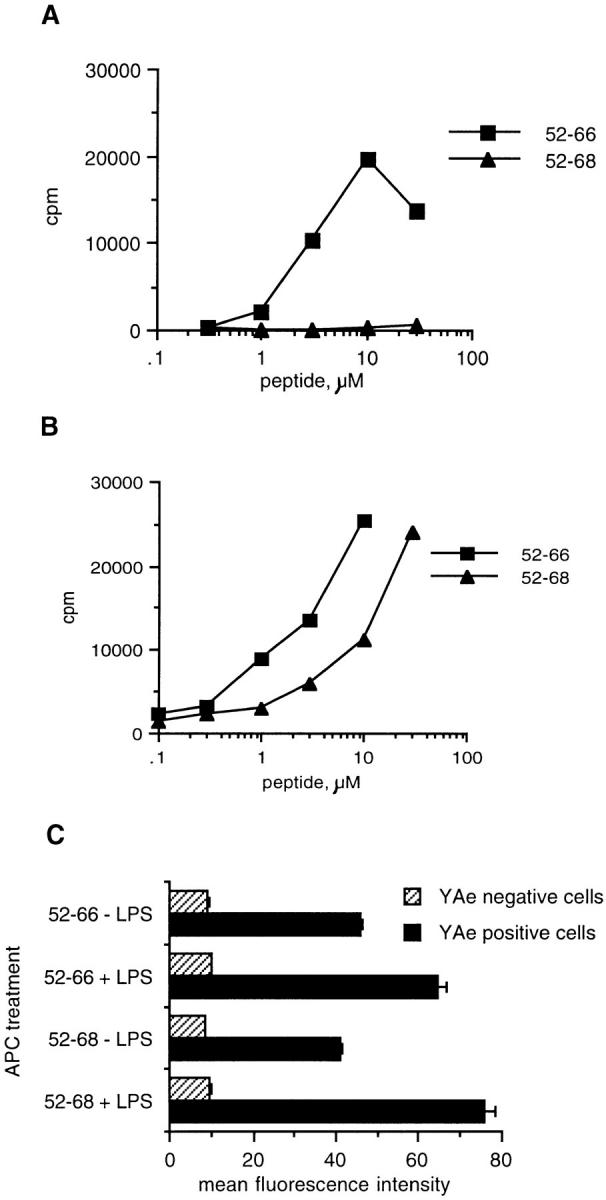
LPS stimulation of splenocytes blocks the processing-dependent presentation of Eα52–68 to Eα6, but does not block the processing-independent presentation of Eα52–66 to Eα6 or the processing-independent presentation of Eα52–68 and Eα52–66 to 1H3.1: (A) The proliferative response of Eα6 to synthetic Eα peptides presented by B6 splenocytes that were treated for 18 h with 10 μg/ml LPS. (B) The response of 1H3.1, measured by CTLL assay, to synthetic Eα peptides presented by B6 LPS blasts. (C) Flow cytometric analysis using Y-Ae to detect binding of Eα52–66 and Eα52–68 onto I-Ab on LPS or untreated B6 splenocytes. In C, Y-Ae positive cells represents the mean fluorescence intensity of cells that stained with Y-Ae. Y-Ae negative cells indicate internal negative controls, showing that the fluorescence intensity of unstained cells in each sample was equivalent.
This general rule of reciprocal regulation of antigen processing and the ability to costimulate T cell growth was further tested by a time course experiment in which the ability of APCs to present processing-independent (Eα52– 66) versus processing-dependent (Eα52–68) antigen is compared at various times after treatment with LPS. APCs were treated with LPS for 0 or 4 h. After 4 h, the LPS was washed out and the APCs were incubated for the indicated time period before addition of antigen. Antigen was then added for 2 h before the APCs were washed, irradiated, and added to culture wells with T cells or hybrids. The incubations on tissue culture–treated plastic would be expected to deplete the splenocytes of adherent cells (macrophages and dendritic cells), so the APCs studied in this experiment were primarily B cells (20).
Fig. 10 A shows that presentation of Eα52–66 to Eα6 peaks at 8 h. Presentation of the processing-dependent peptide Eα52–68 to Eα6 is greatly decreased at this time (Fig. 10 B). In Fig. 10 C we show flow cytometric analysis of the expression of the costimulatory molecule B7-2 at various time points after LPS stimulation. Expression of this molecule also peaks at 8 h with this protocol of LPS treatment. Thus, there is a reciprocal correlation between the ability of splenic B cells to present processing-dependent antigen and the expression of the costimulatory molecule B7-2. Although the significance of the difference in responses between time points is questionable, the correlation is repeatable. This experiment does show that the ability of B cells to present processing-dependent antigen can be negatively modulated by LPS treatment.
Figure 10.
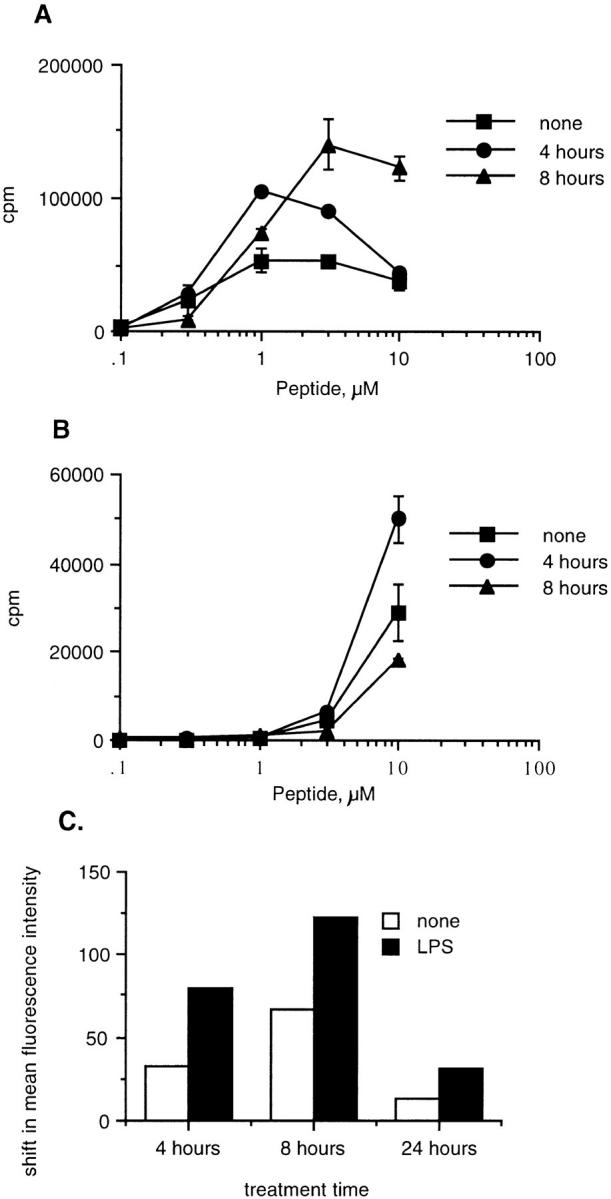
Presentation of processing-dependent antigen is diminished at peak expression of B7. The proliferative response of Eα6 to processing-independent (A; Eα52–66) and processing-dependent (B; Eα52–68) epitopes, which were added to APCs at various time points after LPS induction of costimulation. The APCs were treated for 4 h with LPS under conditions which would deplete adherent dendritic cells and macrophages. The APCs were washed and peptide was added immediately (4 h) or after reculture until the time point indicated (8 h). APCs were irradiated immediately before addition of peptide. The peptide was pulsed onto the APCs for 2 h and then the unbound peptide was washed away and T cells were added. Error bars show the SEM calculated on Cricket Graph. (C) Flow cytometric analysis B7.2 (09272D; PharMingen, San Diego, CA) expression after treatment with LPS in order to induce costimulatory molecule expression. Splenic APCs were cultured in flasks for 4 h in the presence (LPS) or absence (none) of LPS. The cells were then harvested (this procedure should have removed dendritic cells and macrophages due to plastic adherence), washed, and recultured at 37°C for various time points before staining (8 and 24 h), or stained immediately (4 h). No significant differences in Y-Ae staining were detected on the APCs loaded with Eα52–66 or Eα52–68 at 4, 8, or 24 h after initiation of LPS treatment (data not shown).
Discussion
Eα6, a T cell clone raised against a peptide derived from MHC class II Eα chain bound to I-Ab, served as a useful reagent for studying processing of endogenously synthesized versus exogenously provided antigen for presentation by MHC class II molecules. The fine specificity of the T cell epitope was determined. The T cell required a 15-amino acid peptide, Eα52–66, and was unresponsive to the longer peptide Eα52–68 unless Eα52–68 was processed. The Eα52–68 epitope that was created from endogenously synthesized antigen had already been identified (1). The evidence that Eα6 could not recognize Eα52–68, despite the fact that the clone was produced by immunization with this peptide, came from several sources. First, Eα6 prefers the shorter peptide by about a factor of 10 when they are presented by viable APCs (Fig. 2 A). Second, Eα6 responds to the longer peptide only when it is presented by viable, processing-competent APCs (Fig. 3 A), and known inhibitors of antigen processing can block the response to the longer peptide (Fig 4 A). Third, forced recycling with monoclonal anti–I-Ab antibody produced the epitope for Eα6 in cells that expressed the Eα chain and I-Ab (3R, 5R, and B6-107), but not in B6 cells that do not express the Eα chain. Moreover, only antibodies directed at I-Ab could produce this effect; the I-E–specific antibodies Y-17 and 14.4.4S (data not shown) were unable to produce the Eα6 epitope, presumably because they forced internalization only of I-E and not that of I-Ab (Fig. 5). Finally, when B6 mice were used to present purified I-E molecules, they could produce the epitope recognized by Eα6 (Fig. 6). Thus, the evidence is overwhelming that the shorter peptide was involved in the epitope recognized by Eα6.
To confirm this, we then immunized both B6 and 5R mice with the peptides Eα52–68 and Eα52–66. The results, shown in Fig. 7, demonstrate that 5R mice, as previously shown (Rudensky, A.Y., and C.A. Janeway, Jr., unpublished data), were tolerant of the Eα52–68 peptide, even when it was injected in strong adjuvant. Likewise, 5R mice immunized with Eα52–68 did not mount a significant response to Eα52–66. However, 5R mice primed with the shorter peptide Eα52–66 mounted a vigorous response to Eα52–66 as well as a weaker, but still significant, response to Eα52–68. This is presumably due to the processing of the longer Eα52–68 to the shorter Eα52–66 in the culture. Thus, 5R mice, which express the endogenously processed Eα52–68, are tolerant to it, whereas they are nontolerant to the shorter peptide Eα52–66. When B6 mice were immunized with either peptide, they showed vigorous responses to both peptides, although the response to priming with the shorter peptide gave a stronger response, and priming with the longer peptide was recalled more powerfully with the shorter of the two peptides. This could reflect some difference in the TCR repertoire in B6 mice, as suggested by the earlier finding that most T cells raised by immunization of B6 mice with Eα52–68 could not respond to 5R spleen APCs (Rudensky, A.Y., P. Preston-Hurlburt, and C.A. Janeway, Jr., unpublished observations). This response was specifically inhibitable with the Y-Ae monoclonal antibody, proving its specificity (Fig. 8).
Finally, we used the requirement for processing of Eα52–68 to examine the relationship between the acquisition of costimulatory capacity and the ability to take up and process antigen. This revealed that LPS, used to induce B7 expression on spleen-derived APCs, could inhibit the antigen uptake and processing by such APCs (Figs. 9 and 10). This reciprocal regulation of costimulatory molecules that are required for naive T cell activation, and processing of antigen required for antigen presentation to the TCR, may be a crucially important defense against autoimmune disease. APCs take up self-antigens continuously and are especially likely to do so when stimulated by infection, which can induce APCs to become costimulatory. Nevertheless, the ability of an APC to take up and process antigen declines as it becomes B7 positive, perhaps to the point that the level of autoantigen presented to naive T cells is insufficient for breaking self-tolerance. Although the evidence in Figs. 9 and 10 is suggestive, more work clearly needs to be done to explore this result.
An interesting result that emerged from this work was the role of the two COOH-terminal amino acids in presentation of Eα52–66 to the T cell clone Eα6. This clone is representative of many cloned T cell lines we have prepared over the years that reacted to the immunizing peptide but not to the naturally processed Y-Ae epitope expressed on 3R and 5R spleen cells. Once we had worked out that Eα6 is specific for the Eα52–66 peptide, and in fact ignores the longer, naturally processed peptide Eα52– 68, we realized that most of our cloned lines and hybridomas reacted exclusively to the shorter peptide Eα52–66. The problem posed by this result is twofold: first, how are the two COOH-terminal amino acid residues perceived by the TCR on the majority of our clones and hybridomas, when they lie at the extreme end of the peptide and are thus unlikely to contact the TCR at all? Second, why do most T cells from B6 mice prefer this shorter peptide, even though immunized with the longer peptide? We will address these two questions separately.
The processing of peptides for presentation by MHC class II molecules has been studied by many authors. It is generally concluded that peptides can range in size from a minimum of 13 amino acids to a maximum of ∼25 amino acids or longer (1, 25, 26). The modal value for peptide length is 17, exactly the length of Eα52–68. In truncation studies carried out by many investigators over the years, the minimal peptide length that can stimulate most T cell clones and hybrids is 8–9 amino acids, and indeed, the T cell hybrid 1H3.1 can respond to the minimal peptide Eα56–64 (AQGALANIA) (Barlow, A.K., R. Medzhitov, and C.A. Janeway, Jr., data not shown). Nonetheless, the TCR on Eα6, and most clones we have derived from immunization of B6 mice with Eα52–68, ignores this longer peptide in favor of the shorter Eα52–66, and can not recognize the minimal peptide epitope AQGALANIA. Recent studies have demonstrated that the COOH terminus of a peptide makes contact with the TCR Vβ region (21– 23), and thus its structure should contain clues as to the recognition of the two residues that have to be removed from the endogenously processed form. However, the TCR-β chains of Eα6 and 1H3.1 are both encoded in Vβ6, so it is unlikely that the terminal residues at the very extreme end of the peptide can make contact with the only variable residues of the TCR-β chain, those encoded in the CDR3 region. We are planning to make β chain–only transgenic mice from Eα6 and 1H3.1 in order to attempt to resolve this mystery (3).
The second mystery is why most of the T cell lines and clones we have analyzed appear to prefer the shorter peptide, as they are unresponsive to 5R spleen cells that present the naturally processed epitope known to consist primarily of the 17-amino acid peptide Eα52–68. There are two possibilities to explain this. The first is that the shorter peptide has a higher affinity for the I-Ab molecule than the longer peptide, as suggested by its greater immunogenicity in B6 mice (Fig. 7 B) and its greater potency in eliciting proliferative responses from T cell clones (Fig. 2). However, the T cell hybrid, 1H3.1, does not distinguish these peptides on a molar basis, so the avidity for I-Ab is probably not the explanation for this difference. The second possibility is based on our recent finding that the MHC class II–specific repertoire is selected on the basis of recognition of self-peptide–self-MHC class II complexes during its development in the thymus (3, 24). The self-peptide repertoire affects both positive and negative intrathymic selection, with the main effect being on positive selection. We hypothesize that there is a ligand in the B6 thymus that more closely resembles the Eα52–66–I-Ab complex than the Eα52–68–I-Ab complex, and that this ligand selects a majority of T cells that favor recognition of the former over the latter complex. Evidence for this being true comes from the ability of Y-Ae treatment of newborn B6 mice, which are conventionally thought to lack the Y-Ae epitope altogether, to inhibit development of T cells specific for the Eα52–68 and, by inference, Eα52–66 specific cells as well (Rudensky, A.Y., and C.A. Janeway, Jr., unpublished observations). Now that we have analyzed the nature of the difference between these two cloned T cell lines, one of which shows an absolute preference for the shorter peptide, whereas the other can not distinguish peptide length, we are in the process of preparing TCR transgenic mice to analyze selection both in B6 and in I-E+ mice. We predict that the Eα6 TCR will show positive selection in B6 and 5R mice, and we have already observed profound deletion of the 1H3.1 TCR when it is bred to 5R mice.2
In summary, we have deduced the specificity of two cloned T cell lines, Eα6 and 1H3.1, and shown them to differ by the two COOH-terminal amino acids (−KA) of the peptide Eα52–68. The origin of the differences in recognition by these two clones remains to be determined by further studies, perhaps using immunization of single TCR chain transgenic mice. As the phenotype of Eα6-like clones appears to be dominant in mice primed with the peptide Eα52–68, it remains to be determined whether this is due to events occurring in the thymus before the immunization event. However, this seems likely to be the case (3, 24). Finally, we have used this system to infer that costimulation is regulated reciprocally with the ability to take up and process antigen for potentially autoreactive T cells. This may be yet another mechanism to avoid the induction of autoimmune responses.
Acknowledgments
The authors wish to thank their many colleagues for helpful discussions, especially Alexander Rudensky, Alexander Chervonsky, Ruslan Medzhitov, Christophe Viret, Peter Cresswell, Ira Mellman, Kim Bottomly, Nick Crispe, and Richard Flavell. We thank Kara McCarthy for secretarial assistance.
This study was supported in part by the Howard Hughes Medical Institute and by National Institutes of Health (NIH) grant R37 AI-14579 to C.A. Janeway, Jr. A.K. Barlow was supported in part by NIH training grant T32 AI-07019 and by a fellowship from Miles Laboratories (West Haven, CT).
Abbreviations used in this paper
- CIIV
class II loading vesicle
- Ii
invariant chain
- MIIC
MHC class II compartment
Footnotes
A.K. Barlow's present address is Albert Einstein College of Medicine, 1300 Morris Park Ave., Bronx, NY 10461.
Viret, C., H. Ramaswamy, D.B. Sant'Angelo, and C.A. Janeway, Jr., manuscript submitted for publication.
References
- 1.Rudensky AY, Preston-Hurlburt P, Hong S-C, Barlow A, Janeway CA., Jr Sequence analysis of peptides bound to MHC class II molecules. Nature. 1991;353:622–624. doi: 10.1038/353622a0. [DOI] [PubMed] [Google Scholar]
- 2.Germain RN, Rinker AG., Jr Peptide binding inhibits protein aggregation of invariant-chain free class II dimers and promotes surface expression of occupied molecules. Nature. 1993;363:725–728. doi: 10.1038/363725a0. [DOI] [PubMed] [Google Scholar]
- 3.Sant'Angelo DB, Waterbury PG, Cohen BE, Martin WD, VanKaer L, Hayday AC, Janeway CA., Jr Impact of intrathymic self peptides on the mature T cell receptor repertoire. Immunity. 1997;7:517–524. doi: 10.1016/s1074-7613(00)80373-8. [DOI] [PubMed] [Google Scholar]
- 4.Bodmer H, Viville S, Benoist C, Mathis D. Diversity of endogenous epitopes bound to MHC class II molecules limited by invariant chain. Science. 1994;263:1284–1286. doi: 10.1126/science.7510069. [DOI] [PubMed] [Google Scholar]
- 5.Amigorena S, Drake JR, Webster P, Mellman I. Transient accumulation of new class II MHC molecules in a novel endocytic compartment in B lymphocytes. Nature. 1994;369:113–120. doi: 10.1038/369113a0. [DOI] [PubMed] [Google Scholar]
- 6.Peters PJ, Neefjes JJ, Oorschot V, Ploegh HL, Geuze HJ. Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature. 1991;349:669–676. doi: 10.1038/349669a0. [DOI] [PubMed] [Google Scholar]
- 7.Lamb CA, Yewdell JW, Bennink JR. Invariant chain targets HLA class II molecules to acidic endosomes containing internalized influenza virus. Proc Natl Acad Sci USA. 1991;88:5998–6002. doi: 10.1073/pnas.88.14.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lotteau V, Teyton L, Peleraux A, Nilsson T, Karlsson L, Schmid SL, Quaranta V, Peterson PA. Intracellular transport of class II MHC molecules directed by invariant chain. Nature. 1990;348:600–605. doi: 10.1038/348600a0. [DOI] [PubMed] [Google Scholar]
- 9.Denzin LK, Hammond C, Cresswell P. HLA-DM interactions with intermediates in HLA-DR maturation and a role for HLA-DM in stabilizing empty HLA-DR molecules. J Exp Med. 1996;184:2153–2165. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chervonsky AV, Gordon L, Sant AJ. A segment of the MHC class II beta chain plays a critical role in targeting class II molecules to the endocytic pathway. Int Immunol. 1994;6:973–982. doi: 10.1093/intimm/6.7.973. [DOI] [PubMed] [Google Scholar]
- 11.Calafat J, Nijenhuis M, Janssen H, Tulp A, Dusseljee S, Wubbolts R, Neefjes J. Major histocompatibility complex class II molecules induce the formation of endocytic MIIC-like structures. J Cell Biol. 1994;126:967–977. doi: 10.1083/jcb.126.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amigorena S, Webster P, Drake J, Newcomb J, Cresswell P, Mellman I. Invariant chain cleavage and peptide loading in major histocompatibility complex class II vesicles. J Exp Med. 1995;181:1729–1741. doi: 10.1084/jem.181.5.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neefjes JJ, Stollorz V, Peters PJ, Geuze HJ, Ploegh HL. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell. 1990;61:171–183. doi: 10.1016/0092-8674(90)90224-3. [DOI] [PubMed] [Google Scholar]
- 14.West MA, Lucocq JM, Watts C. Antigen processing and class II MHC peptide-loading compartments in human B-lymphoblastoid cells. Nature. 1994;369:147–150. doi: 10.1038/369147a0. [DOI] [PubMed] [Google Scholar]
- 15.Rudensky AY, Maric M, Eastman S, Shoemaker L, DeRoos PC, Blum JS. Intracellular assembly and transport of endogenous peptide-MHC class II complexes. Immunity. 1994;1:585–594. doi: 10.1016/1074-7613(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 16.Rudensky AY, Rath S, Preston-Hurlburt P, Murphy D, Janeway CA., Jr On the complexity of self. Nature. 1991;353:660–662. doi: 10.1038/353660a0. [DOI] [PubMed] [Google Scholar]
- 17.Murphy DB, Lo D, Rath S, Brinster RL, Flavell RA, Slanetz A, Janeway CA., Jr A novel MHC class II epitope expressed in thymic medulla but not cortex. Nature. 1989;338:765–768. doi: 10.1038/338765a0. [DOI] [PubMed] [Google Scholar]
- 18.Janeway CA, Jr, Conrad PS, Lerner EA, Babich J, Wettstein P, Murphy DB. Monoclonal antibodies specific for Ia glycoproteins raised by immunization with activated T cells: possible role of T cell bound Ia antigens as targets of immunoregulatory T cells. J Immunol. 1984;132:662–667. [PubMed] [Google Scholar]
- 19.Lerner EA, Matis LA, Janeway CA, Jr, Jones P. Monoclonal antibody against an Ir gene product. J Exp Med. 1980;152:1085–1101. doi: 10.1084/jem.152.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, and W. Strober, editors. 1992. Enrichment of dendritic cells by plastic adherence and EA rosetting, and depletion of monocytes/macrophages from mononuclear cells using adherence method. In Current Protocols in Immunology, Units 3.7 and 7.1. pp. 3.7.1–3.7.11, 7.1.3.
- 21.Garcia KC, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA. An ab T cell receptor structure at 2.5 Å and its orientation in the TCR–MHC complex. Science. 1996;274:209–219. doi: 10.1126/science.274.5285.209. [DOI] [PubMed] [Google Scholar]
- 22.Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–139. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 23.Hong S-C, Sant'Angelo DB, Dittel BN, Medzhitov R, Yoon ST, Waterbury PG, Janeway CA., Jr The orientation of a T cell receptor to its MHC class II:peptide ligands. J Immunol. 1997;159:4395–4402. [PubMed] [Google Scholar]
- 24.Sant'Angelo, D.B., B.E. Cohen, T. Brabb, P.G. Waterbury, J. Goverman, and C.A. Janeway, Jr. 1997. The selection of the T cell receptor repertoire by self peptides in the thymus. In HLA and Disease: The Molecular Basis. A. Svejgaard, S. Busse, and L. Fugger, editors. Alfred Benzon Symposium 40, Munksgaard, Copenhagen. pp 204–213.
- 25.Chicz RM, Urban RG, Gorga JC, Vignali DAA, Lane WS, Strominger JL. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med. 1993;178:27–47. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudensky AY, Preston-Hurlburt P, Al-Ramadi B, Rothbard J, Janeway CA., Jr Truncation variants of peptides isolated from MHC class II molecules suggest sequence motifs. Nature. 1992;359:429–431. doi: 10.1038/359429a0. [DOI] [PubMed] [Google Scholar]