

Abstract
The Kit ligand (KL)/Kit receptor pair functions in hematopoiesis, gametogenesis, and melanogenesis. KL is encoded at the murine steel (Sl) locus and encodes a membrane growth factor which may be proteolytically processed to produce soluble KL. The membrane-associated form of KL is critical in mediating Kit function in vivo. Evidence for a role of cytoplasmic domain sequences of KL comes from the Sl 17H mutation, a splice site mutation that replaces the cytoplasmic domain with extraneous amino acids. Using deletion mutants and the Sl 17H allele, we have investigated the role of the cytoplasmic domain sequences of KL in biosynthetic processing and cell surface presentation. The normal KL protein products are processed for cell surface expression, where they form dimers. Both Sl 17H and the cytoplasmic deletion mutants of KL were processed to the cell surface; however, the rate of transport and protein stability were affected by the mutations. Deletion of cytoplasmic domain sequences of KL did not affect dimerization of KL. In contrast, dimerization of the Sl 17H protein was reduced substantially. In addition, we have characterized the hematopoietic cell compartment in Sl 17H mutant mice. The Sl 17H mutation has only minor effects on hematopoiesis. Tissue and peritoneal mast cell numbers were reduced in mutant mice as well as in myeloid progenitors. Interestingly, long-term bone marrow cultures from Sl 17H mice did not sustain the long-term production of hematopoietic cells. In addition, homing of normal hematopoietic progenitors to the spleen of irradiated Sl 17H/Sl 17H recipient mice was diminished in transplantation experiments, providing evidence for a role of Kit in homing or lodging. These results demonstrate that the membrane forms of KL exist as homodimers on the cell surface and that dimerization may play an important role in KL/Kit-mediated juxtacrine signaling.
Polypeptides which transmit extracellular signals are most often secreted as soluble factors. An increasing number of growth factors that derive from membrane-anchored precursors and growth factors which function both as soluble and as membrane growth factors have been described in the past several years. They include members of the epidermal growth factor family, e.g., epidermal growth factor, TGF-α, and c-erb/HER2 ligand (1), several hematopoietic growth factors, including the c-kit ligand (KL),1 CSF-1, IL-1, and TNF (2, 3). Transmembrane growth factors have also been identified in the nematode Caenorhabditis elegans (lin-3) (4) and in Drosophila, bride of sevenless (boss) (5, 6). Several of these transmembrane growth factors release soluble growth factors by proteolytic processing or shedding (7–9). The nonprocessed membrane-anchored growth factors themselves possess biological activities and promote cell adhesion and juxtacrine stimulation in adjacent cells expressing cognate receptors on their cell surface (3). Furthermore, the recently described ligands of the eph-family receptor tyrosine kinases with roles in axon guidance are obligate membrane growth factors (10).
The protooncogene c-kit encodes a receptor tyrosine kinase which is a member of the platelet-derived growth factor receptor subfamily (11). c-kit and its cognate ligand, KL, also called steel factor, are encoded at the murine white-spotting (W) (12, 13) and steel (Sl) loci (14–16), respectively. The phenotypes of mice with mutations at the W and Sl loci imply functions of Kit/KL in germ cell development, melanogenesis, and hematopoiesis (17–21). In target cells, KL promotes cell proliferation, survival, cell adhesion/migration, and differentiation. The steel gene encodes two KL protein products, KL-1 and KL-2, produced by alternative splicing (2, 22). Both the KL-1 and KL-2 proteins are synthesized as transmembrane proteins and are expressed on the cell surface (2). Both membrane-bound KL-1 and KL-2 can mediate direct cell–cell contact with c-kit–expressing cells, e.g., bone marrow–derived mast cells (BMMCs) (22, 23) and primordial germ cells (24, 25). Proteolytic processing of both membrane-bound KL-1 and KL-2 releases biologically active soluble KL proteins, although with different efficiencies and through distinct proteolytic cleavage mechanisms (2, 9, 26). Molecular characterization of various Sl alleles has provided important insight into the role of the soluble and membrane-anchored forms of KL (14, 15, 22, 27). Homozygotes for the Steel-Dickie mutation (Sld) are viable and less severely affected, implying some residual functional activity of KL, but they display all of the pleiotropic effects normally associated with steel mutations (17, 18). Molecular analysis showed that the Sld allele arose as a result of an intragenic deletion including the transmembrane domain and COOH terminus, generating a secreted KL protein product with normal biological activity (2, 22, 27). The biological characteristics of mice carrying the Sld mutation imply that the Sld KL protein sustains some activity but is largely defective in facilitating proliferation and survival of target cells, indicating that the membrane-anchored forms of KL play pivotal roles in c-kit function. The Sl 17H allele contains a splice donor site mutation resulting in the substitution of amino acids 239–273 in the cytoplasmic domain with 27 extraneous amino acids (28). The phenotypes of mice carrying the Sl 17H allele include white spotting with residual pigmentation on the ears and macrocytic anemia (28, 29). Interestingly, the Sl 17H allele affects male but not female fertility.
The importance of the cell membrane–anchored forms of KL revealed by the phenotypes of the Sld mutation and the intriguing phenotypes of mice carrying the Sl 17H allele raise questions about the role of the cytoplasmic domain sequences of KL. Comparison of the cytoplasmic domain sequences of murine and human KL reveals 91% shared identity. In contrast, the extracellular domain sequences of murine and human KL are 79% identical. The strong conservation of the cytoplasmic domain sequences of KL throughout evolution suggests a function for these sequences. The cytoplasmic domain sequences of KL may interact with itself or other molecule(s), and these interactions may play a role in processing to the cell surface, cell surface presentation, and juxtacrine signaling. We have investigated the role of cytoplasmic domain sequences of wild-type KL and the Sl 17H protein maturation and cell surface presentation. We have also investigated the characteristics of hematopoietic cells in mice carrying the Sl 17H allele. The results obtained in this study are discussed in the context of mechanisms governing juxtacrine signaling.
Materials and Methods
Construction of KL Deletion Mutants.
Deletion mutations in the cytoplasmic domain of KL were created by the primer-directed PCR mutagenesis method. One 5′ primer covering the initiation codon of KL (5′-GACGGAAAGGAATACTTCTCTGTGTT-3′) and several 3′ primers containing a termination codon located at the desired position were used in the PCR reaction. A plasmid containing the KL cDNA clone 19.1.1 (2) was used as template. The 3′ primers used to create the following mutants were 5′-CTCTCTAGATTACAACATACTTATCTC-3′, for KL-L263; 5′-ATTTCTAGATTAATTAATCTGTATATT-3′, for KL-N254; 5′-TATTCTAGAAACTGCCCTTGTAAGACT-3′, for KL-V248; and 5′-CCTTTCTAGACTTTACTGTTTCTTCTT-3′ for KL-Q241. For generation of the internal deletion mutation, KL-Δ241/254, and KL-Sl 17H, an M13 phage DNA containing the KL cDNA clone 19.1.1 and the following oligonucleotides were used: for KL-Δ241/254, 5′-ATTATCCTCTTCATTCTGTTTCTTCTTCCA-3′; and for KL-Sl 17H, 5′-TCTTTCTGTTGCAACATACTTCCAGTATAAGGCTCC-3′. All mutations were confirmed by sequence analysis. The mutant KL cDNAs were subsequently cloned into pCDM8 for expression in COS-1 cells.
Transient Expression of KL cDNAs in COS-1 Cells.
COS-1 cells were transfected with the DEAE-dextran method described previously (2, 30) with minor modifications. Briefly, COS-1 cells were grown to subconfluence 1 d before use, trypsinized, and reseeded on 150-mm petri dishes at a density of 6 × 106 cells per dish. After 24 h, the cells had reached ∼70% confluence and were transfected with 5 μg of plasmid DNA in the presence of 10% DEAE-dextran (Sigma Chemical Co., St. Louis, MO) for 6–12 h. The medium containing the plasmid DNA was removed, and the cells were shocked with 10% DMSO/PBS++ for 1 min. Residual DMSO was removed by washing the cells with PBS++ twice. Transfected COS-1 cells were grown in DME containing 10% FCS, 100 mg/ml L-glutamine, and antibiotics.
Pulse Chase and Immunoprecipitation Analyses of KL Proteins.
At 72 h after transfection, COS-1 cells expressing KL proteins were used for pulse chase experiments. Cells were incubated with methionine-free DME containing 10% dialyzed FCS for 30 min and labeled with 35S-methionine (DuPont-NEN, Boston, MA) at 0.5 mCi/ml. At the end of the labeling period, the labeling medium was replaced with regular medium containing an excess amount of methionine. Cell lysates and supernatants were collected at the indicated times. Cell lysates were prepared as described previously (31) in 1% Triton X-100, 20 mM Tris (pH 7.5), 150 mM NaCl, 20 mM EDTA, 10% glycerol, and protease inhibitors phenylmethyl sulfonyl chloride (1 mM) and leupeptin (20 μg/ml). For immunoprecipitation analysis of KL protein products, equal volumes of cell lysates or supernatants were immunoprecipitated with excess anti-KL antiserum. KL antiserum was obtained by immunization of rabbits with soluble native and recombinant murine KL, and both antisera were used with indistinguishable results. The anti-KL serum was conjugated to protein A–Sepharose (Pharmacia Biotech, Inc., Piscataway, NJ) and washed three times with wash A (0.1% Triton X-100, 20 mM Tris [pH 7.5], 150 mM NaCl, 10% glycerol). Anti-KL serum— protein A–Sepharose conjugate was incubated with supernatants and cell lysates at 4°C for at least 2 h. The immunoprecipitates were then washed once in wash B (50 mM Tris, 500 mM NaCl, 5 mM EDTA, 0.2% Triton X-100), three times in wash C (50 mM Tris, 150 mM NaCl, 0.1% Triton X-100, 0.1% SDS, 5 mM EDTA), and once in wash D (10 mM Tris, 0.1% Triton X-100). For gel analysis, immunoprecipitates were solubilized in SDS sample buffer by boiling for 5 min, and analyzed by SDS-PAGE (12%) and autoradiography. For endoglycosidase (endo) H treatment, digestion with endo H (Boehringer Mannheim Biochemicals, Indianapolis, IN) was performed by incubating immunoprecipitates for 16 h at 37°C with 5 mU of endo H in 50 μl of 0.1 M sodium phosphate buffer, pH 6.1, containing 0.1% Triton X-100, 0.03% SDS, and 20 mM EDTA.
Immunofluorescence Microscopy.
At 40 h after transfection, COS-1 cells were trypsinized and reseeded on 12-mm round coverslips. After 32 h, the cells were fixed for 15 min at 25°C with 2% (vol/vol) formaldehyde in PBS and rinsed twice with PBS. Cells were then either used directly for immunofluorescence staining or permeabilized by incubating for 15 min at 25°C with 0.1% (wt/vol) saponin in PBS. For staining, cells were incubated at 25°C for 30 min consecutively with 100 μl of normal goat serum, anti-KL serum (1:100 dilution) (32), or an ER marker antibody recognizing the signal sequence receptor (1:100 dilution) (33) and FITC-conjugated goat anti–rabbit antibody, sequentially. The unbound antibody was removed by washing in PBS, and the coverslips were mounted on microscope slides using Permount.
Covalent Cross-linking of KL Protein, Immunoprecipitation, and Western Blot Analysis.
At 72 h after transfection of COS-1 cells expressing KL proteins, the transfected cells were washed with HBSS++ and then incubated with 1 mmol/liter bis(sulfosuccinimidyl) substrate (BS3) (Pierce, Rockford, IL) for 1 h at 37°C. The reaction was terminated with 10 mmol/liter Tris-HCl (pH 7.5) for 10 min. Cell lysates were prepared by incubating equivalent amounts of cell lysate with anti–mouse stem cell factor rat mAb (2 μg per 107 cells) (Genzyme Corp., Cambridge, MA) for 3 h at 4°C. Protein G–Sepharose beads (Pharmacia Biotech, Inc.) were used to collect the antigen–antibody complexes. For gel analysis, immunoprecipitates were solubilized in SDS sample buffer by boiling for 5 min, and analyzed by SDS-PAGE (10%). Proteins were electrophoretically transferred onto nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA). After transfer, the membrane was incubated with 4% skim milk/TBST (50 mmol/liter Tris-HCl, pH 7.5, 150 mmol/liter NaCl, 0.05% Tween 20) for 12 h at room temperature, and Western blot analysis using anti–mouse stem cell factor polyclonal antibody (1:500 dilution; Genzyme Corp.) and enhanced chemiluminescence detection (Pierce Chemical Co., Rockford, IL) were used for identification.
Mast Cell Adhesion Assay.
Transfected COS cells were grown in 24-well plates (7.5 × 104 per well). BMMCs, obtained as described previously (34), were washed three times with RPMI complete medium and incubated for 1 h with transfected COS cells (1.5 × 105 cells per well). Nonadherent BMMCs were then removed by washing three times with medium, and the BMMCs attached to the COS cells were counted by visual inspection in the microscope. BMMCs were counted as total number in each well or the number attached to individual COS cells. To compete the adhesion of BMMCs to COS cells, the c-kit antagonistic antibody ACK2 (a gift from Dr. Nishikawa, Kyoto University, Kyoto, Japan) was used.
Analysis of Peripheral Blood Parameters and Hematopoietic Progenitor Assays.
Blood samples for platelet and white blood cell (WBC) count were drawn from the retroorbital plexus or the tail vein with a capillary pipette (Unopette; Becton Dickinson Labware, Rutherford, NJ). Platelet and WBC numbers were determined using a hemacytometer and phase–contrast microscopy. The hematocrit was determined using heparinized microhematocrit tubes (Fisher Scientific Co., Pittsburgh, PA).
For in vitro progenitor assays, bone marrow was harvested, and the cellularity was determined as described previously (35). 105 bone marrow cells were resuspended in 1 ml of IMDM/0.8% methylcellulose (Fisher Scientific Co.)/30% FBS (Hyclone Laboratories, Inc., Logan, UT)/0.2 mM hemin (Sigma Chemical Co.) containing 50 ng/ml mouse IL-3 (BioSource International, Camarillo, CA), 3 U/ml human erythropoietin (Amgen, Thousand Oaks, CA), and 20 ng/ml recombinant mouse KL. Cultures were incubated at 37°C in a 5% CO2 atmosphere. After 7 d of incubation, colonies were scored on the basis of gross morphology as erythroid burst (BFU-E), granulocyte/macrophage (CFU-GM), and mixed colonies (CFU-GEMM).
Splenetic CFU (CFU-S)–day 12 were assessed by injection of 105 donor bone marrow cells suspended in MEM supplemented with Mops and 10% fetal bovine serum into the lateral tail vein of lethally irradiated (9.5 Gy) recipient mice. Mice were killed 12 d later, their spleens were fixed in Bouin's solution and 10% formalin, and macroscopically visible colonies on the surface of the spleens were enumerated.
For long-term bone marrow cultures, bone marrow cells from one femur were flushed into a T-25 flask in Fisher's medium containing 20% horse serum and dexamethasone. Cultures were incubated at 33°C and semidepopulated at weekly intervals.
Determination of Mast Cell Numbers in the Skin and Peritoneal Cavity.
For the determination of mast cell numbers in the skin, pieces of dorsal skin were removed from different parts of the back, smoothed onto a piece of thick filter paper to keep them flat, and fixed in Carnoy's solution. Tissues were embedded in paraffin, and 5-μm sections were stained with 0.1% acidified toluidine blue (pH 4.0). In skin sections, mast cells between the epithelium and the paniculus carnosus were counted under the microscope. The number of mast cells per centimeter length of skin was determined by dividing the mast cell number by the length of each section of skin counted. For each sample, measurements were made in two separate histological sections and averaged to provide the sample mean.
Peritoneal mast cells were obtained from Sl KL2/Sl KL2 mice and control mice by lavage of the peritoneal cavity with 5 ml PBS. Mast cells were identified by staining with 0.1% toluidine blue (34).
Results
Construction of KL-1 Cytoplasmic Domain Variants and Analysis of Their Turnover Characteristics in COS-1 Cells.
The cytoplasmic domain sequences of KL are highly conserved in evolution. To study roles for cytoplasmic domain sequences of KL, COOH-terminal deletion mutations of varying lengths were constructed (Fig. 1). The wild-type KL-1 cDNA plasmid was used as a template for mutagenesis. The deletion mutations were named according to the position of the COOH-terminal amino acid, e.g., KL-L263, KL-N254, KL-V248, and KL-Q241. In addition, an internal deletion mutation, KL-Δ241/254, and KL-Sl 17H were constructed. All constructs were subcloned into the eukaryotic expression vector pCDM8 suitable for transient expression in COS-1 cells.
Figure 1.

Schematic representation of KL cytoplasmic variants and amino acid sequence of KL-Sl 17H. SP, Signal peptide. ECD, Extracellular domain. THS, Transmembrane segment. CD, Cytoplasmic domain.
The primary KL translation products are progressively processed primarily by carbohydrate modification as they are transported through the endoplasmic reticulum (ER) and Golgi compartments to the cell surface (2). Biosynthetic processing of the protein products of the KL cytoplasmic domain variants and KL-Sl 17H was investigated by performing pulse chase experiments in transiently transfected COS-1 cells by using 35S-methionine. Immunoprecipitation analysis using anti-KL antiserum and SDS-PAGE indicated that the biosynthetic processing pattern of all cytoplasmic domain mutants and KL-Sl 17H was similar to that of wild-type KL-1 (Fig. 2). Smaller molecular mass (Mr) proteins representing the unglycosylated forms of KL were synthesized: KL-1 (35,000), KL-L263 (33,000), KL-N254 (31,000), KL-V248 (33,000, not shown), KL-Δ241/254 and KL-Sl 17H (33,000), and KL-Q241 (30,000). These proteins were progressively modified by glycosylation to form more mature products representing the cell surface forms. The respective Mr for the mature proteins were as follows: KL-1, 45,000 daltons; KL-L263, 43,000 daltons; KL-N254, 41,000 daltons; KL-V248 (42,000 daltons, not shown), KL-Δ241/254 and KL-Sl 17H (43,000 daltons); and KL-Q241 (40,000 daltons) (Fig. 2). Previous cell surface I125-iodination experiments have shown that the 45-kD KL-1 protein represented the mature KL-1 protein on the cell membrane (2). Quantitation of the different KL protein products during the chase indicated that after 15 min, ∼50% of the immature 35-kD KL-1 protein was processed to the mature 45-kD form. With KL-Sl 17H, this process was somewhat delayed, in that 30–45 min was required for 50% of the smaller 33,000-Mr protein products to be processed to their higher 43,000-Mr forms. KL-L263, KL-N254, KL-V248, and KL-Δ241/254 were also delayed in their maturation to a similar extent as KL-Sl 17H. KL-Q241 displayed the most striking delay of maturation, as only a small fraction of the KL-Q241 protein products was processed to the mature 40-kD protein. In agreement with these findings, stromal cells derived from homozygous Sl 17H/Sl 17H mice express a reproducibly reduced level of cell surface KL (∼60% of normal levels) as determined by FACS® analysis compared with the low levels of cell surface KL seen in normal controls (Fig. 3).
Figure 2.

Biosynthetic characteristics of KL-1 cytoplasmic domain variants and KL-Sl 17H. COS-1 cells were transfected with various mutant constructs, KL-1, KL-L263, KL-N254, KL-Δ241/254, KL-Sl 17H, and KL-Q241, labeled with 35S-methionine for 30 min, and chased in regular medium. Cell lysates were collected at designated time points, immunoprecipitated with anti-KL antibody, and analyzed by SDS-PAGE (12%). Arrowheads, Mature glycosylated KL proteins. *Immature forms. Molecular mass (Mr) markers are indicated in kilodaltons.
Figure 3.
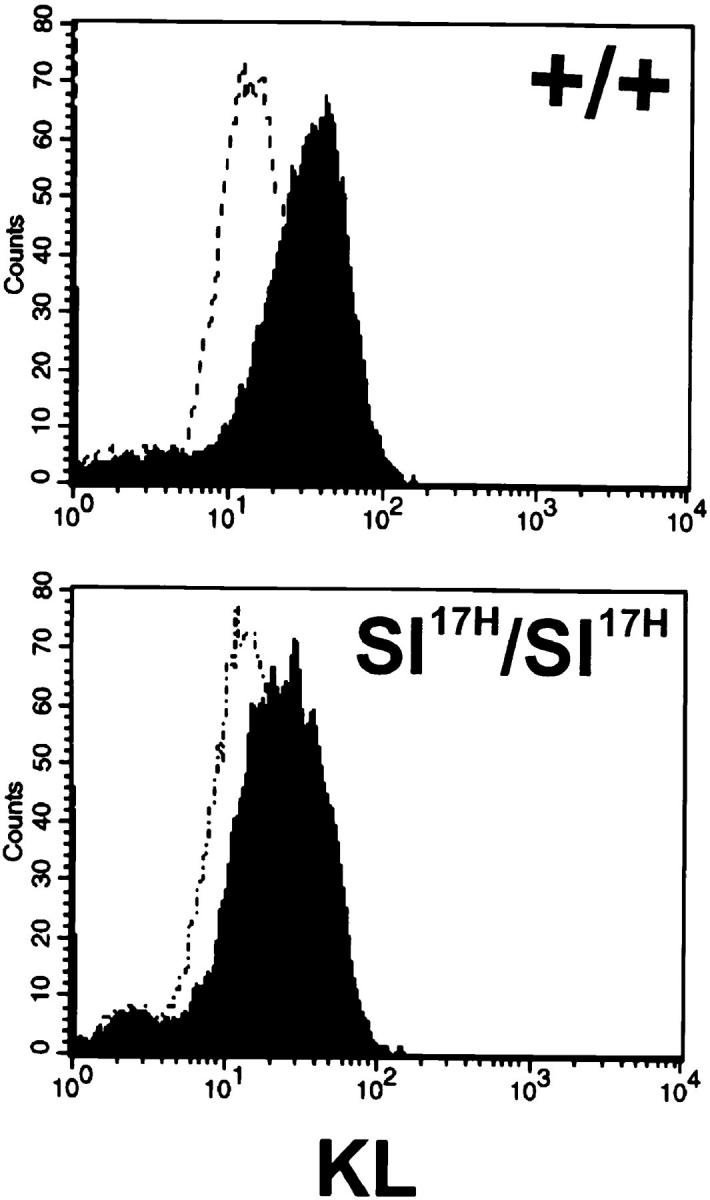
Determination of cell surface expression of KL in Sl 17H/Sl 17H fibroblasts by FACS®.
We then determined whether the wild-type KL-1 and the mutant KL protein products follow the same maturation process through the ER and the Golgi complex. Upon reaching the medial Golgi complex, N-acetylglucosamine is added to glycoproteins, rendering the carbohydrates resistant to the cleavage by endo H (36, 37). To characterize the maturation pathway of mutant KL proteins, we used sensitivity to endo H treatment. COS-1 cells transfected with various constructs were pulse labeled with 35S-methionine and then chased in the presence of excess nonradioactive methionine for 0, 1, and 2 h, and KL protein products were immunoprecipitated from cell lysates and digested with endo H. The wild-type KL-1 protein became resistant to endo H treatment at 1 and 2 h of chase, consistent with rapid transit through the ER Golgi complex. The KL-L263 and KL-N254 protein products displayed a slight persistence of endo H sensitivity at 1 h of chase compared with KL-1, indicating that passage of these proteins through the Golgi complex was delayed (Fig. 4 A). This is consistent with the delayed maturation of these proteins to their cell surface forms. Most of the KL-Q241 protein remained sensitive to endo H treatment at 1 h of chase, indicating that the protein was not properly transiting the Golgi complex. A more thorough time course comparing KL-1 and KL- Sl 17H showed that at 45 and 60 min of chase, KL-Sl 17H displayed some persistence of endo H sensitivity, whereas KL-1 had become fully resistant (Fig. 4 B). Thus, KL-Sl 17H is delayed in exiting the ER. Immunofluorescence staining of KL-1 expressing COS-1 cells revealed a pattern characteristic of a membrane protein which outlined the shape of the cytoplasm (Fig. 5). The KL-Sl 17H staining pattern was indistinguishable from that of KL-1. KL-Q241 staining was observed at the cell surface as well, but appeared less intense than KL-1 staining. Upon permeabilization, staining patterns of all KL-expressing cells included a reticular pattern surrounding the nucleus. Relative to cell surface staining, KL-Q241 immunofluorescence was more pronounced in this juxtanuclear region, and this intracellular staining pattern appears to overlap with that of an E.R. marker antibody. Therefore, these results are consistent with the idea that the delay in maturation of the KL-Q241 protein and to a lesser extent of the SL17H protein is a consequence of a delay in passage through a pre-Golgi compartment.
Figure 4.


Endo H treatment of KL-1, KL cytoplasmic domain variants, and KL-Sl 17H. COS-1 cells were transfected with different KL constructs and labeled with 35S-methionine. Cell lysates collected at different time points were immunoprecipitated and treated in the presence or absence of endo H. The reaction products were analyzed by SDS-PAGE (12%). Arrowheads, Immature KL protein products.
Figure 5.

KL protein expression detected by immunofluorescence in COS-1 cells expressing wild-type and mutant KL protein products. COS-1 cells transfected with KL-1, KL-Q241, and KL-Sl 17H grown on coverslips were fixed with 2% formaldehyde (NP, nonpermeabilized control) and permeabilized with 0.1% saponin (P). The cells were incubated with anti-KL antibody or an ER marker antibody and FITC-conjugated goat anti– rabbit antibody. Photographs were taken with fluorescent microscopy.
Efficient Formation of Homodimers of Cell Membrane–associated KL and Cytoplasmic Domain Deletion Variants, and Diminished Formation of Dimers of the Sl17H Protein.
Growth factors and cytokines commonly activate their receptors by mediating receptor dimerization. Recent studies (see references 50 and 51) with the Kit receptor suggest that bivalent binding of KL dimers is driving Kit receptor dimerization. Soluble KL is predominantly in a dimeric state. However, studies of KL dimerization kinetics imply that in vivo, 90% of KL circulating in serum exists as monomers. It is not known if the membrane-associated KL protein on the cell surface is monomeric or dimeric. To address this question, we set out to identify KL dimers by using chemical cross-linking methodology. COS-1 cells transiently transfected to express KL-1 and KL-2 were treated with a hydrophilic and a hydrophobic cross-linker (DSS and BS3), cell lysates were then fractionated by SDS-PAGE, and KL protein products were identified by Western blotting (Fig. 6). Both cross-linkers efficiently cross-linked the normal KL-1 and KL-2 transmembrane proteins to produce dimeric molecules of 80–85 kD (Fig. 6). These results suggest that a major fraction of the cell-associated KL-1 and KL-2 molecules are in a dimeric form. It was next of interest to determine whether the cytoplasmic domain sequences of KL and/or the Sl 17H cytoplasmic domain sequences affect the formation of cell-associated KL dimers. Whereas deletion of cytoplasmic domain sequences did not affect KL dimer formation as determined by cross-linking (Fig. 6), importantly, Sl 17H dimer formation was diminished significantly (Fig. 7), and removal of the Sl 17H cytoplasmic domain sequences by deletion restored the ability to form dimers (Fig. 7).
Figure 6.

Demonstration of KL-1, KL-2 dimers in transfected COS-1 cells. COS-1 cells were transfected with various normal and mutant KL-1 and KL-2 constructs: (A) KL-1, KL-2, and KL-S; (B) KL-1: L263, V248, Δ241/254; KL-2: L263, N254, Q241. 72 h after transfection, they were treated with the cross-linkers DSS and BS3 for 1 h at 37°C. Cell lysates were collected and fractionated by SDS-PAGE (12%), and KL protein products were identified by Western blotting. Arrowheads, KL dimers. *Monomers. Molecular mass (Mr) markers are indicated in kilodaltons.
Figure 7.

The Sl 17H mutation affects dimer formation on the cell surface. COS-1 cells were transfected with normal KL-1 constructs and mutant Sl17H constructs containing various COOH-terminal deletions: Sl 17H-T257, Sl 17H-R249, and Sl 17H-A240. 72 h after transfection, cell surface proteins were biotinylated, and cells were treated with the cross-linkers DSS and BS3 (as indicated) for 1 h at 37°C. Cell lysates were fractionated on streptavidin-sepharose columns, the biotinylated fraction was electrophoretically fractionated by SDS-PAGE (12%,) and KL protein products were identified by Western blotting. Arrowheads, KL dimers. *Monomers. Molecular mass (Mr) markers are indicated in kilodaltons.
Cell Adhesion Properties of KL Cytoplasmic Domain Variants.
In an earlier study, Cheng and Flanagan found a significant reduction in binding of a Kit ectodomain alkaline phosphatase fusion protein to COS cells expressing the KL Sl 17H protein compared with COS cells expressing the normal KL protein (26). The reduction in KL Sl 17H dimerization we observed in COS cells may contribute to the reduced binding of Kit ectodomain. KL/Kit interactions may also be important in cell–cell adhesion. Therefore, we explored the functional consequences of the cytoplasmic domain deletion and Sl 17H mutations on cell–cell adhesion. Cell adhesion between kit receptor–expressing mast cells and KL-expressing COS-1 cells or fibroblasts had been demonstrated previously (22, 23). In 24-well dishes, BMMCs (1.5 × 105 cells per well) were plated on top of COS-1– expressing KL-1, KL-N254, KL-Δ241/254, KL-Q241, and KL-Sl 17H. 72 h after transfection, 1.5 × 105 BMMCs were plated per well. After an incubation period of 1 h at 37°C, nonattached BMMCs were removed, and adherent BMMCs were counted by visual inspection in a microscope. COS cells expressing KL-1, KL-N254, and KL-Δ241/254 had equivalent numbers of BMMCs attached to the cell surface, and attachment was inhibited completely by the antagonistic anti–c-kit mAb ACK2 (38) (Fig. 8). Adhesion of BMMCs to COS-1 cells expressing KL-Q241 was reduced dramatically (Fig. 8). Similar numbers of BMMCs attached to COS-1 cells transfected with either KL-Q241, the secretory KL-1S (2), or a control plasmid. Interestingly, the number of BMMCs attached to COS cells expressing KL-Sl 17H was only about half of that attached to COS cells expressing KL-1 (Fig. 8).
Figure 8.

Adhesion of BMMCs to COS-1 cells expressing KL-1, secretory KL-1S, different KL cytoplasmic variants, and KL-Sl 17H. COS-1 cells were transferred to 24-well plates 24 h after transfection at a density of 7.5 × 104 cells per well. BMMCs (1.5 × 105 cells per well) were incubated with transfected COS-1 cells for 1 h in the presence and absence of anti–c-kit antibody. Nonattached BMMCs were washed away three times with medium, and the number of attached BMMCs was counted by microscopic inspection. The number of BMMCs attached to individual COS cells is shown.
Effects of the Sl17H Mutation on Hematopoiesis In Vivo and In Vitro.
Previous analysis of mutant Sl 17H/Sl 17H mice suggested mild effects in the hematopoietic system, i.e., a mild macrocytic anemia was observed (29). We have reexamined the effects of the Sl 17H mutation on the hematopoietic system in vivo as well as in vitro. Careful analysis of the in vivo hematopoietic parameters indicates that hematocrit values, WBC, neutrophil, and platelet numbers, as well as bone marrow cellularity are normal in homozygous mutant mice (Table 1). In contrast, the numbers of skin mast cells were reduced by 45% and peritoneal mast cells by 85%, indicating an effect of the Sl 17H mutation on mast cell development and/or number (Table 1). In agreement with a lack of an erythroid deficiency, in vitro analysis of hematopoietic progenitors revealed normal numbers of BFU-E. However, mixed colonies (CFU-GEMM) and CFU-GM were reduced slightly, by ≈33%. We also established long term bone marrow cultures and followed output of hematopoietic cells at weekly intervals. These experiments showed that cultures from Sl 17H/Sl 17H bone marrow were not able to sustain long-term production of hematopoietic cells (Fig. 9). This result is reminiscent of the defect observed in cultures derived from bone marrow of Sl/Sld mice, which produce only the soluble form of KL and no membrane-associated KL.
Table 1.
Mast Cells and Hematopoietic Progenitors in Sl17H/Sl17H and C3H Control Mice
| Genotype | Skin mast cells | Peritoneal mast cells | Bone marrow cellularity | CFU-GEMM | CFU-GM | BFU-E | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| +/+ | ||||||||||||
| Exp. 1 | 162.5 ± 4.5 | 49.5 ± 9.9 | 11.54 ± 0.77 | 2,626 ± 335 | 29,003 ± 2,871 | 1,955 ± 430 | ||||||
| Exp. 2 | 770 ± 191 | 9,717 ± 1,215 | 2,677 ± 204 | |||||||||
| Sl 17H/Sl 17H | ||||||||||||
| Exp. 1 | 91.6 ± 12.8 | 8.0 ± 1.6 | 12.42 ± 1.67 | 1,634 ± 608 | 19,923 ± 3,163 | 1,925 ± 515 | ||||||
| Exp. 2 | 583 ± 210 | 10,266 ± 1,811 | 2,391 ± 657 |
Figure 9.

Long-term culture of bone marrow cells from Sl 17H/Sl 17H mice. Nonadherent cells were counted at weekly intervals. Results from three separate experiments are shown.
Kit-mediated adhesion of hematopoietic cells is known to be mediated by at least two distinct mechanisms: (a) tethering via the membrane growth factor–receptor interaction, and/or (b) by inside-out activation of integrin-mediated adhesion (very late antigen [VLA]-4 and VLA-5) (22, 23, 39, 40). Furthermore, it has been established that VLA-4–mediated adhesion is critical for homing or lodging of early hematopoietic progenitors to the bone marrow in transplantation experiments (41, 42). Therefore, we determined whether Sl 17H/Sl 17H mice were suitable recipients in spleen colony assays using normal donor bone marrow. Interestingly, we observed a reduced number of spleen colonies (65% of normal) in Sl 17H/Sl 17H recipient mice (Table 2). This may suggest that the Sl 17H mutation impairs lodging/homing of transplanted CFU-S progenitors to the spleen of irradiated mice.
Table 2.
Reciprocal Transplantation of Bone Marrow from Sl17H/Sl17H and Control C3H Mice into Irradiated Control and Sl17H/Sl17H Mice
| Donor | Recipient | CFU-S/105 cells | ||
|---|---|---|---|---|
| +/+ | +/+ | 25.3 ± 2.1 | ||
| Sl 17H/Sl 17H | +/+ | 36.9 ± 2.0 P = 0.0008 | ||
| +/+ | +/+ | 25.1 ± 1.7 | ||
| +/+ | Sl 17H/Sl 17H | 16.5 ± 1.0 P = 0.0024 |
Discussion
The Sld allele arose as a result of an intragenic deletion including the transmembrane domain and COOH terminus, generating a secreted KL protein product with normal biological activity (2, 22, 27). Analysis of the Sld phenotype has been of great value in understanding the differential biological roles of membrane-associated and soluble forms of KL. The biological characteristics of homozygous Sld/Sld mice and of Sl/Sld mice indicate that the Sld protein supports some level of KL function. However, Sld mice display major defects in facilitating proliferation and survival of target cells. Therefore, the cell-associated form of KL plays a critical role in c-kit function, and the cytoplasmic domain of KL is potentially important to the processes mediated by juxtacrine signaling. This notion is supported by the mutant phenotypes of the Sl 17H allele, a splice site mutation that results in the substitution of amino acids 239–273 in the KL cytoplasmic domain with 27 extraneous amino acids (28). Therefore, the Sl 17H allele provided an opportunity to analyze the in vitro and in vivo consequences of cytoplasmic domain modification.
The cytoplasmic domain of KL is highly conserved in evolution (Fig. 1), yet very little is known about its function. We have attempted to elucidate the roles of KL cytoplasmic domain sequences as they relate to biosynthetic processing, cell adhesion, and juxtacrine signaling by using in vitro and in vivo genetic approaches. The major conclusions of our study are (a) that cytoplasmic domain sequences are important for biosynthetic processing of KL through the ER and Golgi complex and to the cell surface; (b) that the membrane forms of KL exist as homodimers on the cell surface and that dimerization may be an essential step in KL/Kit-mediated juxtacrine signaling; and (c) that analysis of in vivo phenotypes of Sl 17H /Sl 17H mice revealed Kit-dependent processes in hematopoiesis in which membrane KL is limiting, and they suggest a role for Kit in homing of hematopoietic progenitors to spleen.
Our findings that the KL cytoplasmic domain is required for normal processing to the cell surface are consistent with reports on a variety of secreted or membrane-anchored proteins in which cytoplasmic domain mutations disrupted intracellular trafficking and maturation. For example, mutations removing the four COOH-terminal cytoplasmic residues of α-1 proteinase inhibitor or the COOH-terminal 22 amino acids of thyroxine-binding globulin caused nascent protein to be retained in the ER with resultant lack of secretion (43, 44). Additionally, single point mutations of glycines in the cytoplasmic domain of P-glycoprotein or a single point mutation in the cytoplasmic kinase domain of the Kit receptor caused these proteins which are normally membrane-anchored to be inefficiently glycosylated and retained in the ER (45, 46). Cytoplasmic domain mutant proteins may not interact properly with molecules involved with transport from the ER to the cell surface. Also, incorrect folding as a consequence of cytoplasmic domain mutations may be responsible for the maturation defects (47).
Soluble KL forms noncovalent dimers in solution (15, 48, 49), and it is thought that dimerization of the ligand as well as of the receptor are essential steps for receptor activation. Therefore, it seemed reasonable that juxtacrine signaling by cell-associated forms of KL may involve membrane KL dimers. Our finding of efficient dimer formation of cell-associated KL-1 and KL-2 molecules on transfected COS cells supports this conjecture. Importantly, the apparent large fraction of KL dimers on the cell surface in the absence of cognate receptor implies that dimer formation may not be a rate-limiting event in juxtacrine signaling between KL and Kit. This is in contrast to receptor tyrosine kinases such as Kit which primarily exist as monomers on the cell surface and which form dimers in response to engagement with the cognate ligand (50, 51). Our analysis of various cytoplasmic domain deletion mutations suggests that the cytoplasmic domain of KL does not have an active role in dimer formation. The decreased efficiency of dimer formation of the SL 17H protein suggests that the nonsense cytoplasmic domain sequence of SL 17H prevents normal KL dimerization (possibly by steric hindrance), and this may contribute to the phenotypic defects of SL 17H mice.
Cell adhesion assays indicated that Kit-mediated attachment of BMMCs to COS cells expressing KL-Sl 17H was ≈50% of the KL-1 control. This was in contrast to KL-N254 and KL-Δ241-254, where no significant reduction in adhesion was seen. Interestingly, while a similar delay in the processing of Sl 17H, KL-N254, and KL-Δ241-254 to the cell surface is observed, Sl 17H specifically displays reduced cell surface dimerization. This may suggest that the adhesion defect of Sl 17H is a consequence of reduced Sl 17H cell surface dimer formation. Therefore, the KL-Sl 17H protein is expressed on the cell surface, but the abnormal cytoplasmic domain sequences appeared to interfere with membrane KL–mediated adhesion of BMMCs and juxtacrine signaling.
The Sl 17H mutation affects melanogenesis and gametogenesis (28, 29). A mild effect on hematopoiesis of Sl 17H has also been reported. We have now investigated the hematopoietic parameters in Sl 17H /Sl 17H mice in detail. Although bone marrow cellularity, hematocrit, and WBC, neutrophil, and platelet counts appeared to be normal, the number of tissue mast cells was appreciably affected by the Sl 17H mutation. In addition, hematopoietic progenitor numbers in the bone marrow of Sl 17H /Sl 17H mice did not deviate significantly from normal controls, although slightly reduced numbers of mixed colonies (CFU-GEMM) and slightly increased numbers of CFU-S were observed. Evidence that the hematopoietic microenvironment is compromised in Sl 17H /Sl 17H mice comes from two different experiments. First, reciprocal transplantation of normal bone marrow into Sl 17H /Sl 17H and normal mice revealed a reduced number of spleen colonies in the Sl 17H /Sl 17H recipients, suggesting that homing or lodging to the spleen is affected by the mutation. Second, the bone marrow from Sl 17H /Sl 17H mice does not support long-term culture of hematopoietic progenitors. The biochemical defects we observed for the Sl 17H protein, including delayed processing and reduced dimer formation, may contribute to these hematopoietic deficiencies. The observation that homing or lodging of progenitors to the spleen of irradiated mice is affected is of great interest. KL had been shown to mediate adhesion processes in hematopoietic cells and mast cells by means of an inside-out activation of the integrins VLA-4 and VLA-5 (39, 40, 52, 53). Also, VLA-4–mediated adhesion has been demonstrated to play a role in homing or lodging of hematopoietic progenitors to the spleen (41). In addition, tethering via the membrane growth factor–receptor interaction is thought to provide a mechanism for KL/Kit-mediated cell–cell adhesion (2, 22). Although our results provide evidence that supports a role for KL/Kit in homing or lodging of hematopoietic progenitors to the spleen possibly by an integrin-mediated mechanism, a role for KL/ Kit in homing to the bone marrow as suggested by Papayannopoulou and Craddock (54) remains to be investigated.
The number of germ cells in neonatal gonads is reduced similarly in male and female Sl 17H /Sl 17H mice as a result of an effect on primordial germ cells; however, the Sl 17H allele affects male but not female fertility (28, 55). In postnatal spermatogenesis, KL-2 is more abundant than KL-1, whereas in oogenesis, KL-1 and KL-2 are equally abundant (2, 32). Since the KL-2 protein is more resistant to proteolytic processing and therefore is more stable on the cell surface, the preferred expression of KL-2 in testis development implies a more selective role for transmembrane KL in spermatogenesis. Therefore, the defect in dimerization of Sl 17H would more severely affect spermatogenesis and may in part account for the sterility of Sl 17H males, while females remain fertile. In addition, immunohistochemical studies showed that the transmembrane KL protein accumulates in the basolateral region of the seminal epithelium in Sertoli cells at the time when c-kit–expressing spermatogonia begin to mature (32). KL-expressing Sertoli cells supporting KL/c- kit–dependent spermatogonial proliferation have the morphology of a polarized epithelium. Possibly, the cytoplasmic domain of KL may direct sorting of this protein to the basolateral region of Sertoli cells and/or affect its stability in the basolateral region of these cells. In the adult ovary, follicle cells surrounding the oocyte are less polarized than Sertoli cells. Therefore, although a 50% decrease in Sl 17H protein may be tolerated in oogenesis, misdirected expression of KL-Sl 17H in polarized Sertoli cells may affect spermatogonial proliferation and survival and result in male sterility.
Acknowledgments
We would like to thank Harry Satterwhite and Maureen Sullivan for expert assistance with hematological assays, and Dr. Jeffrey Ravetch for the use of his cell sorter and for helpful discussions. We would also like to thank Drs. Martin Wiedmann, Marcus Bosenberg, Atanasio Pandiella, Joan Massague, and Rosemary Bachvarova for many discussions, and Dr. Shin-ichi Nishikawa for generously providing ACK2 antibody. ER marker–specific antibody was generously provided by Tom Rapoport (Harvard Medical School, Boston, MA) and Martin Wiedmann (Memorial Sloan-Kettering Cancer Center, New York).
Abbreviations used in this paper
- BFU-E
burst-forming unit–erythroid
- BMMC
bone marrow–derived mast cell
- CFU-GEMM
CFU-granulocyte erythroid megakaryocyte macrophage
- CFU-S
CFU-spleen
- endo
endoglycosidase
- ER
endoplasmic reticulum
- KL
Kit ligand
- Sl
steel
- VLA
very late antigen
- W
white-spotting
- WBC
white blood cell
Footnotes
Support by grants from the American Cancer Society and the National Institutes of Health is acknowledged.
References
- 1.Lee DC, Fenton SE, Berkowitz EA, Hissong MA. Transforming growth factor: expression, regulation, and biological activities. Pharmacol Rev. 1995;47:51–85. [PubMed] [Google Scholar]
- 2.Huang EJ, Nocka KH, Buck J, Besmer P. Differential expression and processing of two cell associated forms of the kit-ligand: KL-1 and KL-2. Mol Biol Cell. 1992;3:349–362. doi: 10.1091/mbc.3.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosenberg MW, Massague J. Juxtacrine cell signaling molecules. Curr Opin Cell Biol. 1993;5:823–838. doi: 10.1016/0955-0674(93)90032-l. [DOI] [PubMed] [Google Scholar]
- 4.Hill RJ, Sternberg PW. The gene lin-3 encodes an inductive signal for vulval development in C. elegans. . Nature. 1992;358:470–476. doi: 10.1038/358470a0. [DOI] [PubMed] [Google Scholar]
- 5.Hart AC, Kramer H, Van Vacter DL, Jr, Paidhungat M, Zipursky SL. Induction of cell fate in the Drosophilaretina: the bride of sevenless protein is predicted to contain a large extracellular domain and seven transmembrane segments. Genes Dev. 1990;4:1835–1847. doi: 10.1101/gad.4.11.1835. [DOI] [PubMed] [Google Scholar]
- 6.Kramer H, Cagan RL, Zipursky SL. Interaction of bride of sevenless membrane-bound ligand and the sevenless tyrosine-kinase receptor. Nature. 1991;352:207–212. doi: 10.1038/352207a0. [DOI] [PubMed] [Google Scholar]
- 7.Rettenmier CW. Biosynthesis of macrophage colony-stimulating factor (CSF-1): differential processing of CSF-1 precursors suggests alternative mechanisms for stimulating CSF-1 receptors. Curr Top Microbiol Immunol. 1989;149:129–141. doi: 10.1007/978-3-642-74623-9_12. [DOI] [PubMed] [Google Scholar]
- 8.Pandiella A, Massague J. Cleavage of the membrane precursor for transforming growth factor-α is a regulated process. Proc Natl Acad Sci USA. 1991;88:1726–1730. doi: 10.1073/pnas.88.5.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandiella A, Bosenberg MW, Huang EJ, Besmer P, Massague J. Cleavage of membrane-anchored growth factors involves distinct protease activities regulated through common mechanisms. J Biol Chem. 1992;267:24028–24033. [PubMed] [Google Scholar]
- 10.Pandey A, Lindberg RA, Dixit VM. Cell signalling. Receptor orphans find a family. Curr Biol. 1995;5:986–989. doi: 10.1016/s0960-9822(95)00195-3. [DOI] [PubMed] [Google Scholar]
- 11.Besmer P, Murphy PC, George PC, Qui F, Bergold PJ, Lederman L, Snyder HW, Brodeur D, Zuckerman EE, Hardy WD. A new acute transforming feline retrovirus and relation of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320:415–421. doi: 10.1038/320415a0. [DOI] [PubMed] [Google Scholar]
- 12.Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- 13.Geissler EN, Ryan MA, Housman DE. The dominant white-spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- 14.Copeland NG, Gilbert DJ, Cho BC, Donovan PJ, Jenkins NA, Cosman D, Anderson D, Lyman SD, Williams DE. Mast cell growth factor maps near the steel locus on mouse chromosome 10 and is deleted in a number of steel alleles. Cell. 1990;63:175–183. doi: 10.1016/0092-8674(90)90298-s. [DOI] [PubMed] [Google Scholar]
- 15.Nocka K, Huang E, Beier DR, Chu TY, Buck J, Lahm HW, Wellner D, Leder P, Besmer P. The hematopoietic growth factor KL is encoded by the SI locus and is the ligand of the c-kit receptor, the gene product of the W locus. Cell. 1990;63:225–233. doi: 10.1016/0092-8674(90)90303-v. [DOI] [PubMed] [Google Scholar]
- 16.Zsebo KM, William DA, Geissler EN, Broudy VC, Martin FH, Atkins HL, Hsu RY, Birkett NC, Okino KH, Murdock DC, et al. Stem cell factor is encoded at the SI locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63:213–224. doi: 10.1016/0092-8674(90)90302-u. [DOI] [PubMed] [Google Scholar]
- 17.Russell ES. Hereditary anemias of the mouse. Adv Genet. 1979;20:357–459. [PubMed] [Google Scholar]
- 18.Silvers, W.K. 1979. The Coat Colors of Mice. Springer-Verlag New York Inc., New York. 378 pp.
- 19.Besmer P. The kitligand encoded at the murine Steel locus: a pleiotropic growth and differentiation factor. Curr Opin Cell Biol. 1991;3:939–946. doi: 10.1016/0955-0674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 20.Besmer, P., K. Manova, R. Duttlinger, E. Huang, A.I. Packer, C. Gyssler, and R. Bachvarova. 1993. The kit-ligand (steel factor) and its receptor c-kit/W: pleiotropic roles in gametogenesis and melanogenesis. Dev. Suppl. 125–137. [PubMed]
- 21.Besmer, P. 1997. Kit ligand – stem cell factor. In Colony Stimulating Factors: Molecular and Cellular Biology, 2nd ed. J.M. Garland, P.J. Quesenberry, and D.J. Hilton, editors. Marcel Dekker, Inc., New York. 369–403.
- 22.Flanagan JG, Chan D, Leder P. Transmembrane form of the c-kit ligand growth factor is determined by alternative splicing and is missing in the SIdmutation. Cell. 1991;64:1025–1035. doi: 10.1016/0092-8674(91)90326-t. [DOI] [PubMed] [Google Scholar]
- 23.Adachi S, Ebi Y, Nishikawa SI, Yamazaki M, Kasugai T, Yamamura T, Nomura S, Kitamura Y. Necessity of extracellular domain of W (c-kit) receptors for attachment of murine cultured mast cells to fibroblasts. Blood. 1992;79:650–656. [PubMed] [Google Scholar]
- 24.Matsui Y, Zsebo KM, Hogan BLM. Derivation of pluripotent embryonic stem cells from murine primordial germ cells in culture. Cell. 1992;70:841–847. doi: 10.1016/0092-8674(92)90317-6. [DOI] [PubMed] [Google Scholar]
- 25.Resnick, J.L., L.S. Bixler, L. Cheng, and P.J. Donovan. 1992. Long-term proliferation of mouse primordial germ cells in culture. Nature. 359:550–551. [DOI] [PubMed]
- 26.Cheng HJ, Flanagan JG. Transmembrane kit ligand cleavage does not require a signal in the cytoplasmic domain and occurs at a site dependent on spacing from the membrane. Mol Biol Cell. 1994;9:943–953. doi: 10.1091/mbc.5.9.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brannan CI, Lyman SD, Williams DE, Eisenman J, Anderson DM, Cosman D, Bedell MA, Jenkins NA, Copeland NG. Steel-Dickie mutation encodes a c-kit ligand lacking transmembrane and cytoplasmic domains. Proc Natl Acad Sci USA. 1991;88:4671–4674. doi: 10.1073/pnas.88.11.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brannan CI, Bedell MA, Resnick JL, Eppig JJ, Handel MA, Williams DE, Lyman SD, Donovan PJ, Jenkins NA, Copeland NG. Developmental abnormalities in Steel 17Hmice result from a splicing defect in the steel factor cytoplasmic tail. Genes Dev. 1992;6:1832–1842. doi: 10.1101/gad.6.10.1832. [DOI] [PubMed] [Google Scholar]
- 29.Peter J, Ball ST, Loutit JF. A new steel allele which does not lead to dilution of coat color. Mouse News Lett. 1987;77:125–126. [Google Scholar]
- 30.Kriegler, M. 1990. Gene Transfer and Expression: A Laboratory Manual. Stockton Press, New York.
- 31.Nocka K, Majumder S, Chabot B, Ray P, Cervone M, Bernstein A, Besmer P. Expression of c-kit gene products in cellular targets of W mutations in normal and W mutant mice. Evidence for an impaired c-kit kinase in mutant mice. Genes Dev. 1989;3:816–826. doi: 10.1101/gad.3.6.816. [DOI] [PubMed] [Google Scholar]
- 32.Manova K, Huang EJ, Angeles M, De Leon V, Sanchez S, Pronovost SH, Besmer P, Bachvarova RF. The expression of the c-kit ligand in gonads of mice support a role for the c-kit receptor in oocyte growth and in proliferation of spermatogonia. Dev Biol. 1993;157:85–99. doi: 10.1006/dbio.1993.1114. [DOI] [PubMed] [Google Scholar]
- 33.Vogel F, Hartmann E, Gorlich D, Rapoport TA. Segregation of the signal sequence receptor protein in the rough endoplasmic reticulum membrane. Eur J Cell Biol. 1990;53:197–202. [PubMed] [Google Scholar]
- 34.Nocka K, Buck J, Levi E, Besmer P. Candidate ligand for the c-kit transmembrane kinase receptor: KL, a fibroblast-derived growth factor, stimulates mast cells and erythroid progenitors. EMBO (Eur Mol Biol Organ) J. 1990;9:3287–3294. doi: 10.1002/j.1460-2075.1990.tb07528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muench MO, Firpo MY, Moore MAS. Bone marrow transplantation with interleukin-1 plus kit-ligand ex vivo expanded bone marrow accelerates hematopoietic reconstitution in mice without loss of stem cell lineage and proliferative potential. Blood. 1993;81:3463–3473. [PubMed] [Google Scholar]
- 36.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 37.Chen C, Bonifacino JS, Yuan LC, Klausner RD. Selective degradation of T cell antigen receptor chains retained in a pre-Golgi compartment. J Cell Biol. 1988;107:2149–2161. doi: 10.1083/jcb.107.6.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishikawa S, Kusakabe M, Yoshinaga K, Ogawa M, Hayashi SI, Kunisada T, Era T, Sakakura T, Nishikawa SI. In utero manipulation of coat color formation by a monoclonal anti-c-kit antibody: two distinct waves of c-kit-dependency during melanocyte development. EMBO (Eur Mol Biol Organ) J. 1991;10:2111–2118. doi: 10.1002/j.1460-2075.1991.tb07744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinashi T, Springer TA. Steel factor and c-kit regulate cell-matrix adhesion. Blood. 1994;83:1033–1038. [PubMed] [Google Scholar]
- 40.Kovach NL, Lin N, Yednock T, Harlan JM, Broudy VC. Stem cell factor modulates avidity of alpha 4 beta 1 and alpha 5 beta 1 integrins expressed on hematopoietic cell lines. Blood. 1995;85:159–167. [PubMed] [Google Scholar]
- 41.Williams DE, De Vries P, Namen AE, Widmer MB, Lyman SD. The Steel factor. Dev Biol. 1992;151:368–376. doi: 10.1016/0012-1606(92)90176-h. [DOI] [PubMed] [Google Scholar]
- 42.Papayannopoulou T, Craddock C, Nakamoto B, Priestley GV, Wolf NS. The VLA4/VCAM-1 adhesion pathway defines contrasting mechanisms of lodgement of transplanted murine hemopoietic progenitors between bone marrow and spleen. Proc Natl Acad Sci USA. 1995;92:9647–9651. doi: 10.1073/pnas.92.21.9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brodbeck RM, Brown JL. Secretion of alpha-1-proteinase inhibitor requires an almost full length molecule. J Biol Chem. 1992;267:294–297. [PubMed] [Google Scholar]
- 44.Miura Y, Kambe F, Yamamori I, Mori Y, Tani Y, Murata Y, Oisom Y, Seo H. A truncated thyroxine-binding globulin due to a frameshift mutation is retained within the rough endoplasmic reticulum: a possible mechanism of complete thyroxine-binding globulin deficiency in Japanese. JClin Endocrinol Metab. 1994;78:283–287. doi: 10.1210/jcem.78.2.8106612. [DOI] [PubMed] [Google Scholar]
- 45.Loo TW, Clarke DM. Functional consequences of glycine mutations in the predicted cytoplasmic loops of P-glycoprotein. J Biol Chem. 1994;269:7243–7248. [PubMed] [Google Scholar]
- 46.Koshimizu Y, Tsufimura T, Isozaki K, Nomura S, Furitsu T, Kanakura Y, Kitamura Y, Nishimune Y. Wnmutation of c-kit receptor affects its post-translational processing and extra-cellular expression. Oncogene. 1994;9:157–162. [PubMed] [Google Scholar]
- 47.Doyle C, Sambrook J, Gething MJ. Analysis of progressive deletions of the transmembrane and cytoplasmic domains of influenza hemagglutinin. J Cell Biol. 1986;103:1193–1204. doi: 10.1083/jcb.103.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arakawa T, Yphantis DA, Lary JW, Narhi LO, Lu HS, Prestrelski SJ, Clogston CL, Zsebo KM, Mendiaz EA, Wypych J, Langley KE. Glycosylated and unglycosylated recombinant-derived human stem cell factors are dimeric and have extensive regular secondary structure. J Biol Chem. 1991;266:18942–18948. [PubMed] [Google Scholar]
- 49.Lu HS, Chang WC, Mendiaz EA, Mann MB, Langley KE, Hsu YR. Spontaneous dissociation-association of monomers of the human-stem-cell-factor dimer. Biochem J. 1995;305:563–568. doi: 10.1042/bj3050563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Philo J, Wen J, Wypych J, Schwartza MG, Mendiaz EA, Langley KE. Human stem cell factor dimer forms a complex with two molecules of the extracellular domain of its receptor, Kit. J Biol Chem. 1996;271:6895–6902. doi: 10.1074/jbc.271.12.6895. [DOI] [PubMed] [Google Scholar]
- 51.Lemmon MA, Pinchasi D, Zhou M, Lax I, Schlessinger J. Kit receptor dimerization is driven by bivalent binding of stem cell factor. J Biol Chem. 1997;272:6311–6317. doi: 10.1074/jbc.272.10.6311. [DOI] [PubMed] [Google Scholar]
- 52.Serve H, Yee NS, Stella G, Sepp-Lorenzino L, Tan JC, Besmer P. Differential roles of P13-kinase and Kit tyrosine 821 in Kit receptor-mediated proliferation, survival and cell adhesion in mast cells. EMBO (Eur Mol Biol Organ) J. 1995;14:473–483. doi: 10.1002/j.1460-2075.1995.tb07023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vosseller K, Stella G, Yee NS, Besmer P. c-kit receptor signaling through its phosphatidylinositide-3′-kinase binding site and protein kinase C: role in mast cell enhancement of degranulation, adhesion, and membrane ruffling. Mol Biol Cell. 1997;5:909–922. doi: 10.1091/mbc.8.5.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Papayannopoulou T, Craddock C. Homing and trafficking of hemopoietic progenitor cells. Acta Haematol. 1997;97:97–104. doi: 10.1159/000203665. [DOI] [PubMed] [Google Scholar]
- 55.Huang EJ, Manova K, Packer AI, Sanchez S, Bachvarova RF, Besmer P. The murine Steel panda mutation affects kit-ligand expression and growth of early ovarian follicles. Dev Biol. 1993;157:100–109. doi: 10.1006/dbio.1993.1115. [DOI] [PubMed] [Google Scholar]