

Abstract
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, one subtype in the family of ionotropic glutamate receptors, are the main receptors responsible for excitatory signaling in the mammalian central nervous system. Previous studies utilitizing the isolated ligand binding domain of these receptors have provided insight into the role of specific ligand-protein interactions in mediating receptor activation. However, these studies relied heavily on the partial agonist kainate, in which the α-amine group is constrained in a pyrrolidine ring. Here we have studied a series of substituted and unsubstituted willardiines with primary α-amine groups similar to that of the full agonist glutamate whose activation can be varied depending on the size of the substituent. The specific ligand-protein interactions in the mechanism of partial agonism in this subtype were investigated using vibrational spectroscopy and the large scale conformational changes in the ligand binding domain were studied with fluorescence resonance energy transfer (FRET). These investigations show that the strength of the interaction at the α-amine group correlates with the extent of cleft closure and extent of activation, with the agonist of higher efficacy showing larger cleft closure and stronger interactions at this group, suggesting that this is one of the mechanisms by which the agonist controls receptor activation.
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, one subtype of ionotropic glutamate receptors, are responsible for mediating the majority of excitatory synaptic signaling in the central nervous system. Activation of these channels occurs when an agonist, such as glutamate, binds in the bilobed extracellular ligand binding domain of the receptor, initiating a series of conformational changes that lead to the formation of cation selective channels (1–4).
Recent vibrational spectroscopic investigations (5–8) in combination with fluorescence resonance energy transfer (FRET) studies (5, 8, 9) on the isolated ligand binding domain of the GluR2 subunit (GluR2-S1S2) have provided insight into the role of specific ligand-protein interactions in mediating AMPA receptor activation. By using these techniques to study activation by the three ligands kainate, AMPA, and glutamate on wild type and mutant proteins, which provide a wide spectrum of activations, a correlation was developed between the changes at specific ligand-protein interactions and extent of activation of the channel. These studies suggest that the strength of the interaction at the α-amine group correlates in most cases to the extent of activation (5, 6). However, it should be noted that these correlations depended heavily on conclusions from the partial agonist kainate, which is inherently different in structure compared to glutamate and AMPA because its α-amine group is constrained in a pyrrolidine ring while the α-amine groups of AMPA and glutamate are free and not restricted in a ring structure (Figure 1). Thus, it cannot be ruled out that some of these changes could be due to inherent differences in the structures of the agonists. For a more accurate comparison between the strength of the interaction at the α-amine group and the extent of activation, it is essential to study a spectrum of ligands with similar structures to glutamate and AMPA that cause a varied scale of activation through the systematic substitution of some part of that ligand. To that effect, a series of substituted willardiines that act as partial agonists of AMPA receptors are available, and they have a similar backbone structure to glutamate and AMPA (free α-carboxylate and α-amine groups) but allow for systematic substitution of the 5 position on the uracil ring with a halogen (10, 11) (Figure 1). Crystal structures of GluR2-S1S2 in complex with a series of 5-substituted willardiines with halogen substituents of increasing size have been determined and show that the α-carboxylate and α-amine groups of these ligands also bind in similar arrangements as glutamate and AMPA in the ligand binding core (12, 13). The systematic variation of activation by the willardiine compounds and shared binding configurations make them an excellent tool to use to draw correlations between ligand-protein interactions and activation. Hence we have used unsubstituted willardiine (HW), fluorowillardiine (FW), chlorowillardiine (ClW), and iodowillardiine (IW) and investigated the molecular level interactions of the GluR2-S1S2 protein in the presence of these partial agonists using vibrational spectroscopy.
Figure 1.
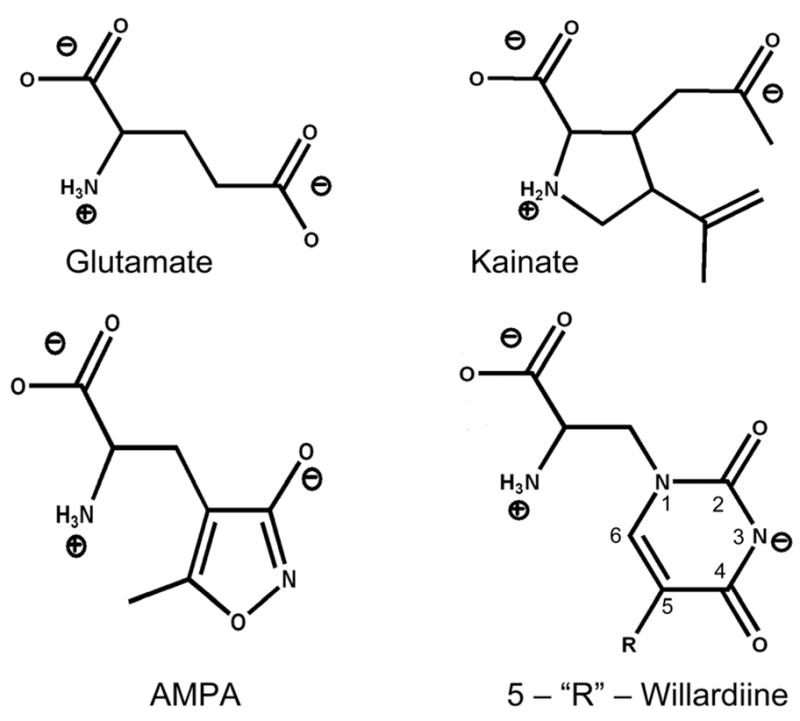
Structures of AMPA receptor full agonists glutamate and AMPA and partial agonists kainate and 5-‘R’-willardiine.
In addition to studying the changes in the agonist-protein interactions, we have also used fluorescence resonance energy transfer (FRET) to investigate the average degree of cleft closure in the ligand binding domain as a result of the binding of these willardiines. The degree of cleft closure in a number of agonist-bound states of the wild type and mutant GluR2-S1S2 proteins show a correlation between the extent of cleft closure due to the agonist binding and efficacy of the agonist. Hence it has been proposed that the cleft closure conformational change is a mechanism by which the agonist mediates and controls receptor activation (12, 14, 15). The cleft closures for the FW- and HW- bound forms follow the expected trends where extent of cleft closure correlates to extent of activation. However the crystal structures for IW show a large variation in the cleft closure conformational change ranging from 15 to 19 degrees (12). IW is a partial agonist mediating approximately 24% of the currents at saturating concentration relative to saturating concentrations of glutamate. Hence the larger cleft closure of 19 degrees, close to that observed for full agonists such as glutamate (19–21 degrees), is inconsistent with the proposed hypothesis where cleft closure is thought to be the primary control by which the agonist mediates receptor activation. Here we have used a FRET-based probe to determine the distance between the N-terminus and residue 652 and used this as a readout of the extent of cleft closure and then determined the average extent of cleft closure due to HW, FW, ClW, and IW binding. The extent of cleft closure and strength of the molecular level ligand-protein interactions were then correlated to the extent of activation to determine their relationship to channel activation and elucidate their role in the mechanism by which partial agonists act on AMPA receptors.
Materials and Methods
Chemicals
(S)-5-Fluorowillardiine (FW), (S)-5-Chlorowillardiine (ClW), and (S)-Willardiine (HW) were obtained from Ascent Scientific (Somerset, England, United Kingdom), and (S)-5-Iodowillardiine (IW) was obtained from Tocris Bioscience (Ellisville, MO).
Electrophysiology
The GluR2-flip plasmid was donated by Dr. Seeburg (Max Planck Institute, Heidelberg, Germany). The wild type GluR2 receptors were expressed in human embryonic kidney 293 (HEK 293) cells (ATCC CRL 1573) and cultured in Minimum Essential Medium with Earle’s Salts and 2 mM L-glutamine (Invitrogen, Carlsbad, CA) supplemented with 9–10% fetal bovine serum (Invitrogen). Transfections were performed using Fugene-6 transfection reagent (Roche, Indianapolis, IN) with 1 μg of plasmid encoding for the cDNA of GluR2 and 0.2 μg of plasmid encoding for the cDNA of green fluorescent protein. Cells were used for experiments 2 days after transfection.
For the electrophysiological measurements, the electrode solution consisted of 140 mM CaCl2, 2 mM MgCl2, 1 mM CaCl2, 10 mM EGTA, 2 mM Na2ATP, and 10 mM HEPES (pH 7.4). The extracellular bath solution consisted of 145 mM NaCl, 1.8 mM MgCl2, 3 mM KCl, 10 mM glucose, and 10 mM HEPES (pH 7.4). All ligand solutions were prepared in extracellular buffer and kept at pH 7.4. Cyclothiazide at a concentration of 100 μM was used for all the experiments.
The cells expressing the GluR2 receptors were voltage-clamped at a holding potential of −60 mV for the whole cell current recordings, and solutions were applied using a homemade U-tube mixing device with a 100 μm aperature. Currents were amplified using an Axon 200B amplifier (Molecular Devices, Sunnyvale, CA), with low-pass filtering at 1 kHz. The filtered signal was digitized with a LabMaster DMA digitizing board that was controlled by Axon PClamp. All measurements were done at room temperature.
Preparation and purification of GluR2-S1S2 and S1S2-T394C-S652C proteins
The GluR2 S1S2J construct was provided by Dr. Eric Gouaux (Oregon Health and Science University, Portland, OR), and the construct with cysteines at sites 394 and 652 used for the FRET measurements was prepared as previously detailed (9). The GluR2-S1S2 (for the FTIR experiments) and S1S2-T394C-S652C (for the FRET experiments) proteins were purified as described previously (9, 15–17). In brief, E. coli Origami B (DE3) cells were used to express the protein, which was then purified using a Ni-NTA HiTrap column (GE Healthcare Life Sciences, Piscataway, NJ). The histidine tag was removed by thrombin digestion, and the protein was subjected to further purification using a SP-Sepharose column (GE Healthcare Life Sciences).
Fluorophore Labeling
500 nM S1S2-T394C-S652C protein was dialyzed in phosphate-buffered saline containing 1 mM glutamate (Sigma-Aldrich, St. Louis, MO) and then labeled with a 1:1 ratio of the maleimide derivatives of fluorescein (Biotium, Hayward, CA) and triethylenetetraaminehexaacetic acid (TTHA) chelate of terbium (Invitrogen). The labeled protein was extensively dialyzed against phosphate-buffered saline before proceeding with the FRET measurements. Saturating concentrations of HW, FW, and IW were used for each of the experiments with these ligands. 10 μM ClW, which corresponds to 99% of the receptors being bound with ClW, was used for the experiments involving this ligand due to absorbance of the 337 nm Tb excitation light by saturating concentrations of this ligand.
Fluorescence Measurements
For the FRET measurements, a TimeMaster model TM-3/2003 instrument (Photon Technologies, Inc., Lawrenceville, NJ), a cuvette-based lifetime spectrofluorometer, was used. The data were collected using Felix 32 software (Photon Technologies, Inc.) and subsequently analyzed with Origin software (OriginLab Corp., Northampton, MA). The distance between the donor and acceptor fluorophores was calculated using Förster's theory of energy transfer, which is given by:
| (1) |
where τD is the lifetime of the decay of donor fluorescence in the absence of the acceptor, and τDA is the lifetime of the sensitized emission of the acceptor due to energy transfer from the donor. Ro was calculated using the overlap integral as described previously (9, 18).
FTIR Difference Spectroscopy
FTIR spectra were obtained using the protocol as outlined in refs (5, 6, 19, 20). 250 μM to 500 μM protein in 25 mM phosphate buffer containing 250 mM NaCl and 0.02% NaN3 (pH 7.4) was used to obtain spectra in the 1450–1800 cm−1 region using D2O as the solvent, while 500 μM to 1 mM protein in 10 mM HEPES buffer containing 100 mM NaCl (pH 7.0) was used for the S-H stretching vibration with water as the solvent. Excess ligand concentrations (≥10mM) were used in all cases. Excess ligand was confirmed by peaks arising from unbound ligand in all spectra and subtracted out using a spectrum of the ligand in buffer.
FTIR spectra were obtained using a Nicolet Nexus 870 spectrometer. An adjustable pathlength cell with CaF2 windows was used to obtain spectra. Spectra were collected at 4 cm−1 resolution and a constant temperature of 15°C. A 50μm pathlength was used to obtain spectra in the 1400–1800 cm−1 range, while a 75 μm pathlength was used for the S-H stretch. Data collection and analysis were done using Omnic and GRAMS/AI (7.01) (Thermoelectron, Waltham, MA).
Binding affinity measurements for ClW
The binding affinity of ClW to GluR2-S1S2 was determined as previously detailed (12, 21). Briefly, GluR2-S1S2 was dialyzed extensively against buffer consisting of 30 mM Tris-HCl, 100 mM KSCN, 2.5 mM CaCl2, and 10% glycerol (pH 7.2). For the competition binding experiments, 20 nM [3H]-AMPA was incubated with 500 ul of 0.2 ug/ml GluR2-S1S2 and varying concentrations of ClW for 60 min at 4°C. The protein solutions were filtered through wet membranes using a vacuum manifold. The membranes were washed with the binding buffer, and the radioactivity retained on the filter was counted. Two parallel experiments were performed, and the mean values and standard errors were calculated.
Results
Functional characterization
Whole cell currents mediated by saturating concentrations of glutamate (10 mM) and HW (1 mM), ClW (1 mM), FW (1 mM), and IW (1 mM) in wild type GluR2-flip receptors were measured under non-desensitizing conditions in the presence of 100 μM cyclothiazide. The peak currents evoked by the series of willardiines were normalized to the peak currents mediated by 10 mM glutamate and are shown in Figure 2. The extent of activation follows as HW~FW>ClW> IW, with HW, FW, ClW, and IW producing 62%, 60%, 53%, and 24% of the currents produced by glutamate, respectively. This trend is in agreement with previous electrophysiological investigations (12), which show that these compounds act as partial agonists with varying activations depending on their substituent, with the extent of activation correlating to the size of the substituent.
Figure 2. Activation profile of the series of willardiines.
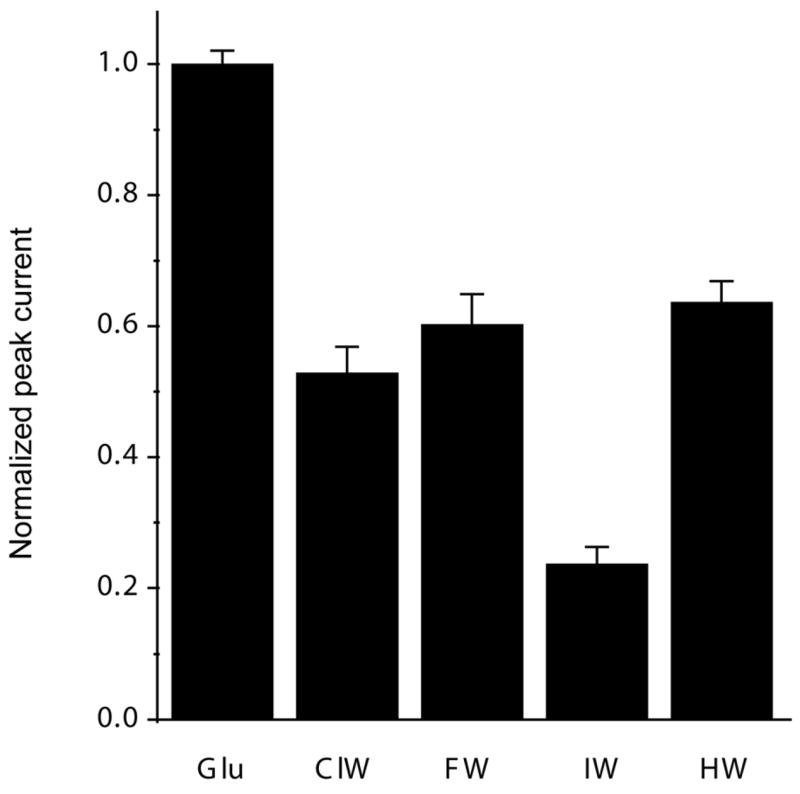
Maximum currents elicited under non-desensitizing conditions by saturating concentrations of glutamate (10 mM), ClW (1mM), FW (1mM), IW (1mM), and HW (1 mM), normalized to maximum response mediated by glutamate.
Degree of cleft closure
As the degree of cleft closure in the ligand binding domain has been proposed to be one of the major conformational changes leading to activation in these receptors (12, 14, 15), we used FRET to measure the extent of cleft closure (9) due to the binding of this series of willardiines. The distances calculated based on the sensitized fluorescence lifetimes for GluR2-S1S2 with cysteines at sites 394 and 652 (S1S2-T394C-S652C), sites that reflect the extent of cleft closure, tagged with a 1:1 ratio of the terbium chelate donor and the fluorescein acceptor in the apo, HW-, FW-, ClW- and IW-bound states are shown in Figure 3 and plotted against the extent of activation. The distance versus activation correlation for the series of willardiines follows the same trend as that observed for kainate, glutamate, and AMPA activation of the GluR2-S1S2 protein, where agonists exhibiting a larger activation have a larger degree of cleft closure, supporting the cleft closure hypothesis for activation of these receptors. To obtain an understanding of the molecular-level environment in the ligand binding pocket, vibrational spectroscopic experiments were performed to assess these interactions between the series of willardiine agonists and the GluR2-S1S2 protein.
Figure 3. FRET to measure the extent of cleft closure with various agonists.
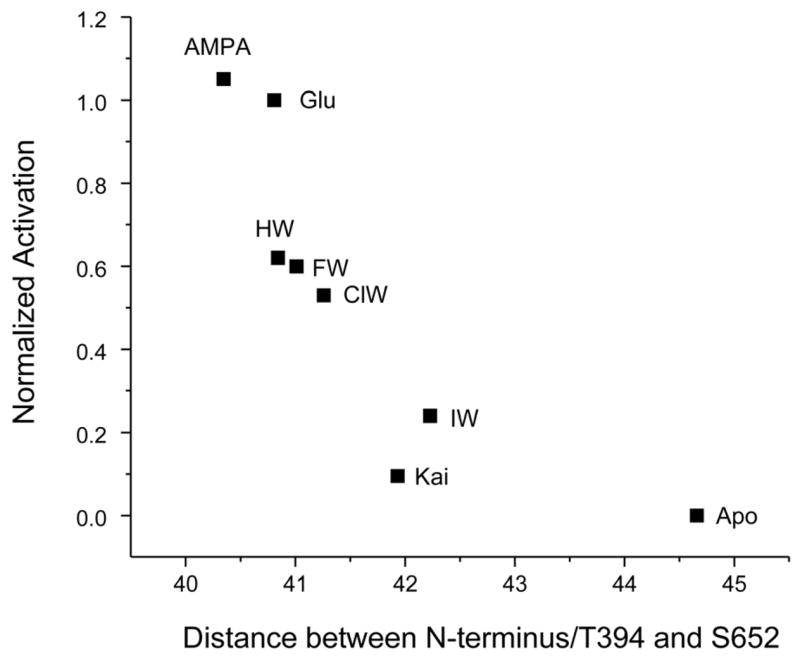
Extent of activation (normalized to glutamate activation) versus distances between residues 394 and 652 in S1S2-T394C-S652C as determined from the fluorescence lifetimes for the series of willardiines and agonists glutamate, AMPA, and kainate (9).
Environment of the willardiines free in solution
The difference FTIR spectra between the free willardiines and buffer are shown in Figure 4. The asymmetric carboxylate vibration for α-amino acids typically occurs in the region of 1610 to 1630 cm−1 (5–7, 19, 22), and hence the bands at 1629 cm−1 in the HW and FW spectra and at 1632 cm−1 in the ClW and IW spectra can be assigned to the asymmetric stretching vibration of the α-carboxylate, while the higher frequency modes can be assigned to the stretching vibrations of the two other carbonyl moieties present in the willardiine structures. Specifically, the vibrations at 1653 cm−1 (FW), 1655 cm−1 (HW), 1652 cm−1 (IW), and 1658 cm−1 (ClW) can be assigned to the carbonyl moiety at the 2 position on the ring (Figure 1) due to the fact that this carbonyl is further from the halogen substitution, and the vibrations due to this moiety are not significantly affected by the substituent. The bands at 1678 cm−1 (FW), 1690 cm−1 (HW), 1696 cm−1 (IW), and 1702 cm−1 (ClW), on the other hand, are significantly different between the four willardiines and hence arise from the carbonyl moiety at the 4 position on the ring (Figure 1), which is in close proximity to the substituents at the 5 position on the ring. Furthermore, the assignments for the carbonyl vibrations at the 4 position are consistent with the fact that HW, ClW, and IW have hydrogen, chlorine, and iodine substitutions which have electronegativities that cause their carbon-halogen bond at the 5 position on the ring to be less polar, hence having less delocalization of their electrons and exhibiting higher frequency, while fluorine, being highly electronegative, is expected to cause this carbon-halogen bond to be more polar and induce more delocalization; hence FW has a lower frequency vibration for the carbonyl at the 4 position.
Figure 4. Environment of willardiines free in solution.
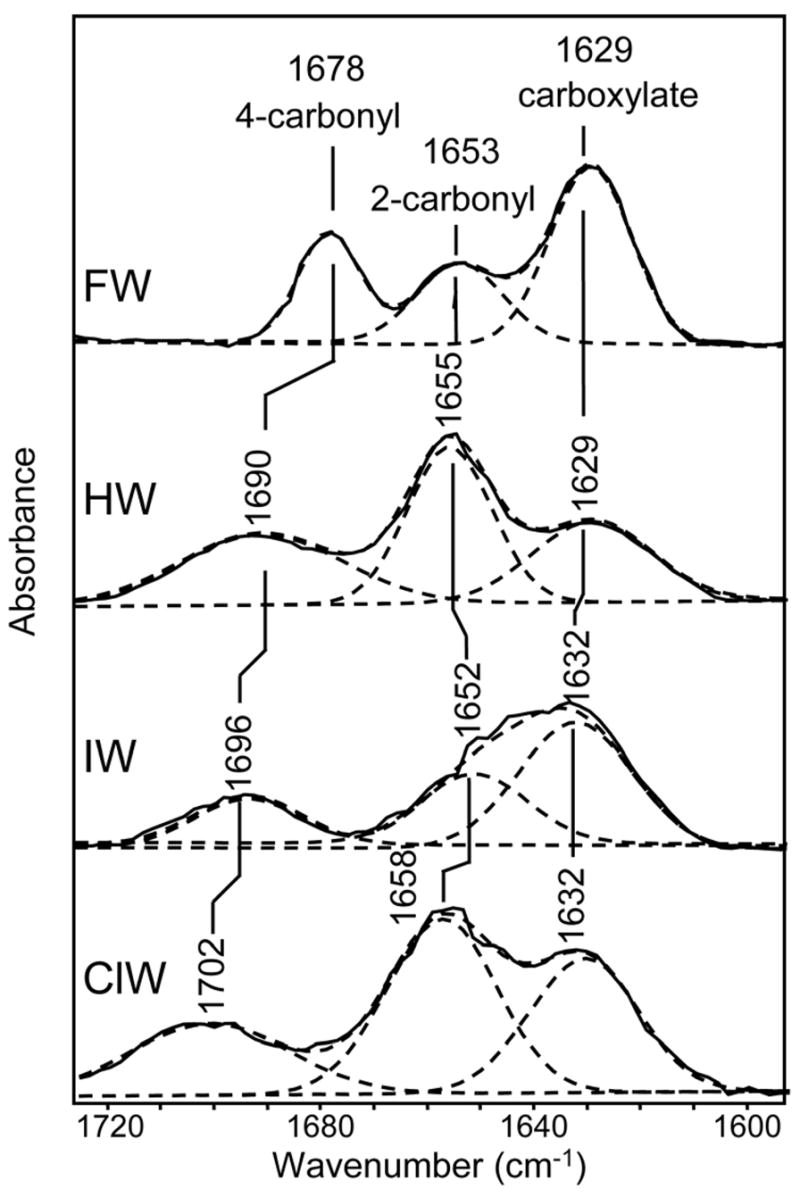
Difference FTIR spectra in the region of 1580–1740 cm−1 between FW and buffer, HW and buffer, IW and buffer, and ClW and buffer.
Willardiine environment when bound to the protein
The FTIR difference spectra between the willardiine-bound states and unligated state of GluR2-S1S2 are shown in Figure 5. Based on the frequency, the bands at 1618 cm−1 in these spectra can be assigned to the asymmetric carboxylate vibration of the willardiines. The difference spectra for the free willardiines and bound willardiines exhibit an 11 cm−1 downshift in the asymmetric carboxylate vibration for FW and HW upon binding to GluR2-S1S2 and a 14 cm−1 downshift in the asymmetric carboxylate vibration for ClW and IW upon binding to GluR2-S1S2. Since the frequency of the asymmetric carboxylate vibration can be directly correlated to the strength of the interaction at this moiety (22), these downshifts would correspond to a ΔH of −3.7 kcal/mol when FW and HW bind to GluR2-S1S2 and ΔH of −4.7 kcal/mol when ClW and IW bind to GluR2-S1S2 as compared to the ligands free in solution. The favorable interactions at the α-carboxylate for the four willardiines are consistent with the crystal structures that show ionic and hydrogen bonding interactions between this moiety and the R485 side chain of the protein (12, 13). Furthermore, the vibrational spectroscopic data show that the interactions are slightly more favorable at this α-carboxylate group for ClW and IW compared to FW and HW, which have lesser extents of activation than FW and HW (12). This is in agreement with the previous studies on the wild type protein looking at this interaction with the ligands glutamate, AMPA, and kainate, where a more favorable interaction between the α-carboxylate and the protein was observed for the partial agonist kainate than for full agonists glutamate and AMPA (6).
Figure 5. Changes in interaction at α-carboxylate and protein secondary structure.
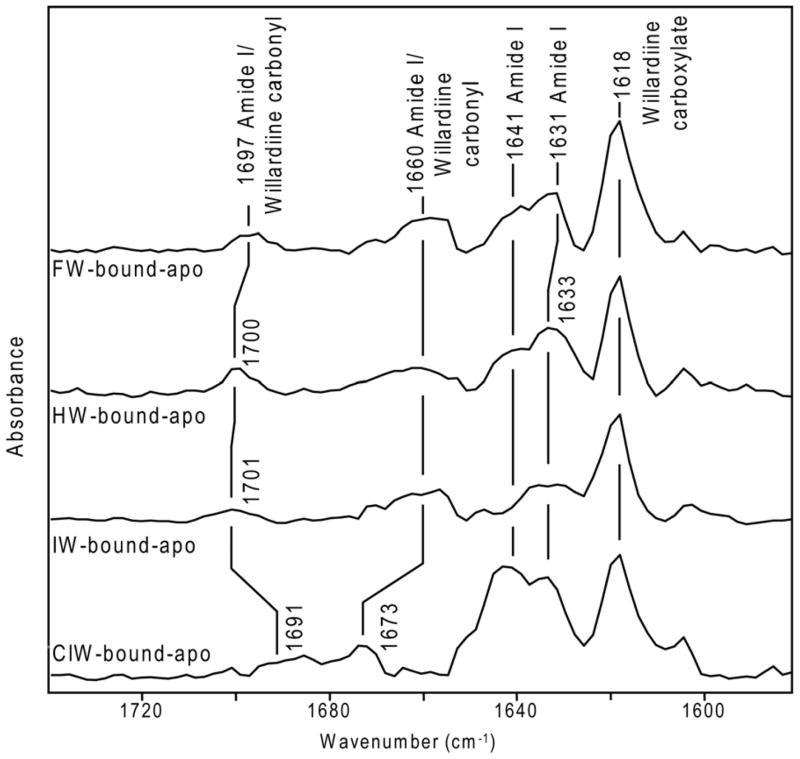
Difference FTIR spectra in region of 1580 cm−1–1740 cm−1 for FW-bound GluR2-S1S2 and apo GluR2-S1S2, HW-bound GluR2-S1S2 and apo GluR2-S1S2, IW-bound GluR2-S1S2 and apo GluR2-S1S2, and ClW-bound GluR2-S1S2 and apo GluR2-S1S2.
Secondary structure changes as characterized by amide I vibrations
Comparison of difference spectra for the series of willardiines in Figure 5 with the features observed in the difference FTIR spectrum between the glutamate-bound and apo state of the protein (6) allows for the bands at 1631/1633 cm−1 and 1641 cm−1 to be assigned to backbone amide I vibrations. Particularly, the frequency at 1631/1633 cm−1 can be assigned to changes in β-sheets, while the frequency at 1641 cm−1 can be attributed to changes in random structures, irregular β-sheets, and/or solvent-exposed helices (5, 6, 23). An increased intensity in the positive feature at 1641 cm−1 is a characteristic signature observed in this region for larger activation of the receptor by a particular agonist (5, 6) and consistent with HW, FW, and ClW exhibiting relatively larger activations as compared to IW. The difference spectrum for IW, on the other hand, exhibits no significant change in intensity at 1641 cm−1. This pattern in the amide I intensities has been observed in the similar band at 1643/1644 cm−1 for AMPA activation of the L650T mutant and kainate activation of the Y450F mutant. In both these cases, the activation is significantly lower as compared to activation of L650T by full agonist glutamate and Y450F by full agonist AMPA, 69% and 65% lower respectively, which correlates to a decrease in the intensity of the 1643/1644 cm−1 feature (5). These data show that while assignment of the amide I bands to specific protein conformational changes is not possible, their intensities to some degree provide a signature for extent of activation by a given ligand.
Interactions at the α-amine group through Cys 425
The vibrational spectra of GluR2-S1S2 in the apo state and in the HW-, FW-, ClW- and IW-bound states in the region of the S-H stretching vibration are shown in Figure 6. The band in this region could be peak-fit using a Gaussian function to two bands, arising from S-H stretching vibrations due to the two non-disulphide-bonded cysteines in GluR2-S1S2 at positions 425 and 436. The assignments of these two cysteines are based on the frequency of the S-H stretching vibration in GluR4-S1S2, which has a single non-disulphide bonded cysteine at a position similar to Cys 425 (19).
Figure 6. Interaction at the α-amine group as determined by the Cys 425 S-H stretching mode.
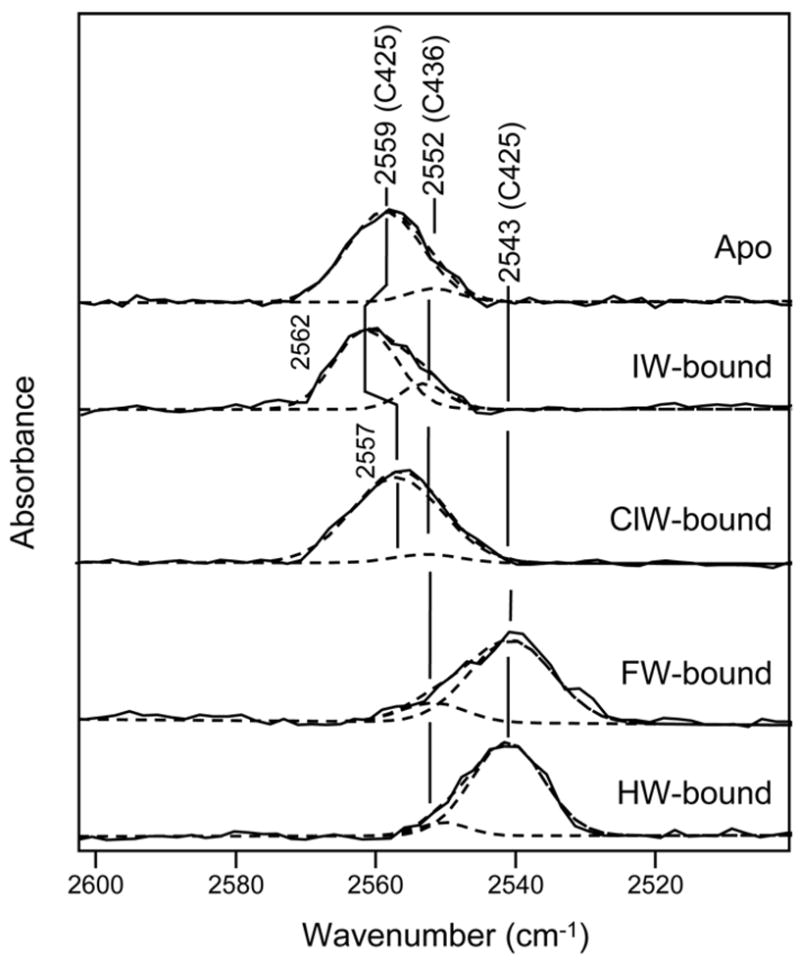
Difference FTIR spectra in region of 2500–2600 cm−1 between apo GluR2-S1S2 and buffer, IW-bound GluR2-S1S2 and buffer, ClW-bound GluR2-S1S2 and buffer, FW-bound GluR2-S1S2 and buffer, and HW-bound GluR2-S1S2 and buffer.
The frequency of the S-H stretching vibration of Cys 436 is unaltered between the apo and willardiine-bound states, consistent with previous observations that showed no shift in this band in the apo, glutamate-, kainate-, and AMPA-bound states (6). The absence of any changes in this mode is consistent with the fact that this residue is far from the ligand binding site and hence not affected by ligand binding. The frequency of the S-H stretching vibration of Cys 425, on the other hand, does change between the apo and ligated states. Due to the hydrogen bond between the α-amine group of the agonist and Pro 478 that is adjacent to Ala 477, which is in turn hydrogen bonded to Cys 425 (14), the changes in the S-H stretching vibration of Cys 425 act as an indicator of the strength of the interaction between the α-amine group of the agonist and GluR2-S1S2 (5, 6, 19, 20). The frequency of the S-H stretching vibration is at 2559 cm−1 for the apo state, 2562 cm−1 for the IW-bound state, 2557 cm−1 for the ClW-bound state, and at 2543 cm−1 for the HW- and FW-bound states. The 16 cm−1 downshift in the frequency of this mode in the HW- and FW-bound states combined with the smaller 2 cm−1 downshift for the ClW-bound state and 3 cm−1 upshift for the IW-bound state suggests stronger interactions between the α-amine group of the ligand and the protein in the HW- and FW-bound states than for the ClW- and IW-bound states, in agreement with our previous investigations (5, 6).
Discussion
Previous FTIR studies using mutant receptors and agonists AMPA, kainate, and glutamate have established a correlation between the strength of the interaction at the α-amine group of the agonist with the protein and the extent of activation, with a stronger interaction leading to a larger activation of the receptor (5, 6). Comparison of the extent of the full agonist interactions to those of the partial agonist was hindered previously by differences in structure between full agonists glutamate and AMPA and partial agonist kainate, specifically at the α-amine group of these ligands, which is constrained in a ring structure in kainate but not in glutamate and AMPA (Figure 1). Here, using a series of substituted willardiine agonists, which have α-amine groups in a similar structure to that of glutamate and AMPA and whose activation can be varied systematically depending on the halogen substituent, we have employed a multiscale approach using vibrational spectroscopy to characterize specific ligand-protein interactions and FRET to measure the large scale cleft closure conformational change and then correlated these results to the functional consequences in the full receptor to determine how the interactions in the ligand binding pocket lead to activation of the receptor by this series of partial agonists.
Correlation between cleft closure and extent of activation
Electrophysiological measurements using whole cell current recordings in HEK 293 cells and saturating concentrations of the willardiines showed a correlation between the size of the halogen substituent of the willardiine and extent of activation (Figure 3), with a larger substituent leading to a larger degree of activation, in agreement with previous results (12). Using FRET, we were able to determine the extent of cleft closure in the soluble ligand binding domain of GluR2 with the series of substituted willardiines bound. In agreement with previously published crystal structures (12), the extent of cleft closure correlated to the extent of activation, with a larger amount of cleft closure leading to a larger extent of activation. These trends exhibited by the FRET measurements show that the willardiines exhibit cleft closures that are in agreement with the cleft closure hypothesis. While the crystal structures showed a large variation in the extent of cleft closure by IW (12), the FRET data show that on average the extent of cleft closure for the IW-bound form of GluR2-S1S2 follows the same trend as that of the other willardiines.
Interactions in the ligand binding pocket as investigated with vibrational spectroscopy
While the FRET data are able to provide a picture of the large conformational changes occurring in the ligand binding domain as a result of the binding of the series of willardiines, vibrational spectroscopy was used to obtain a molecular-level view of the environment of the specific interactions occurring in the binding pocket. Of particular interest were interactions at the α-carboxylate and α-amine groups as well as the changes in dynamics of the protein backbone from the apo to the agonist-bound state. The vibrational spectroscopy data showed favorable interactions at the α-carboxylate for all of the four willardiines used in this study, consistent with the crystal structures (12, 14) and also our previous vibrational spectroscopic investigations of the wild type and mutant proteins using the agonists AMPA, kainate, and glutamate (5, 6).
In addition, the vibrational spectra also showed an increase in intensity of the amide I band at 1641 cm−1 with greater agonist activation, consistent with our studies using the mutant GluR2 receptors (Figure 5) (5). This band is a reflection of the change in secondary structure resulting from binding of the different ligands and can be assigned to changes in random structures, irregular β-sheets, and/or solvent-exposed helices (5, 6, 23). The increase in intensity of this feature due to the binding of more efficacious ligands to a particular protein shows that while the amide I features cannot be assigned to specific changes in secondary structure upon ligand binding, they can provide to some extent an indication of the extent of activation for a given ligand. Since the structure of the ligand binding domain does not show large changes in the secondary structure due to agonist binding, these increases in intensities observed in the amide I region are most likely indicative of the changes in the dynamics of the protein backbone when going from an apo to a ligated state and hence the dipole moment of the backbone carbonyl groups. Thus the signature changes observed for different extents of activation could suggest that the changes in dynamics of the ligated protein correlate to the extent of activation, indicating another mechanism by which the agonist controls the extent of activation of the receptor. While the vibrational spectroscopic data provides a picture of the changes in the protein backbone in the static end state structures when going from the apo to the ligated state, it is unable to illustrate these changes in a time-resolved manner. Nuclear magnetic resonance (NMR) investigations studying the changes in the dynamics due to different agonists being bound to the ligand binding domain should be able to shed further light into this mode of control (24, 25).
Additionally, and in agreement with our investigations of wild type and mutant GluR2 receptors (5, 6), this series of willardiines also exhibits a trend in the strength of the interactions at the α-amine group, in that a stronger interaction at the α-amine group of the agonist correlates to a larger activation by that particular agonist. This relationship is further clarified in Figure 7, which shows a plot of the frequency of the S-H stretching vibration vs. the extent of activation for the apo state and GluR2-S1S2 bound to the agonists AMPA, glutamate, and kainate (6, 9) and HW, FW, ClW, and IW. While an increase in activation correlates to an increase in the strength of the interactions at the α-amine group as monitored by the Cys 425 S-H stretching vibration, the relationship between the Cys 425 S-H stretching frequency and the extent of activation is not linear but sigmoidal. Larger changes in the S-H shift are observed at intermediate activations and smaller changes at the large and small activations. This suggests that the interactions at the α-amine group of the agonist with the protein play a large role in controlling the subtleties in the extent of activation, specifically at intermediate levels of activation.
Figure 7. Relationship between strength of interaction at α-amine group and extent of activation.

Plot of S-H stretching frequency vs. extent of activation for apo and agonist-bound GluR2-S1S2 (6, 9), illustrating the correlation between the strength of interaction at α-amine group and the extent of activation.
Relationship between affinities for willardiines, extent of activation, and interactions in the ligand binding pocket
Since the affinities of the willardiines for GluR2-S1S2 do not follow the same trends as extent of activation, this group of ligands allows us to establish if the strength of interactions at the α-amine groups of the agonists are a mere reflection of changes in affinity or if they truly reflect changes in activation. Previous competition binding studies using [3H]-AMPA and GluR2-S1S2 to determine the IC50 values for HW, FW, and IW (12), and our investigations of the IC50 for ClW (Supplementary Figure 1) show that the affinity follows the electronegativity of the halogen with the order being FW>ClW>IW>HW. The order for the strength of interaction at the α-amine group of HW FW>ClW>IW does not follow the trends observed for the affinities but clearly follows the order observed for the extent of activation thus establishing that the strength of the interaction at the α-amine group is not a reflection of changes in affinity of the agonist but that this interaction plays a critical role in controlling activation by the agonist.
Conclusions
While previous studies have shown a correlation between the strength of the interaction between the α-amine group of the agonist and GluR2-S1S2 and activation of the receptor, detailed partial agonist studies were limited by the use of kainate. Through analyzing this series of willardiine partial agonists that have similar structure to full agonists glutamate and AMPA and systematic variation of their extent of activation, we have further confirmed the correlation between the strength of the interaction at the α-amine group activation of the receptor and have also shown that there is a range in the strength of this interaction as there is a range of activation resulting from these particular agonists, supporting the multi-state model of allosteric activation (26) for these receptors.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Grant MCB-0444352 and National Institutes of Health Grant R01GM073102. K.A.M. was supported by National Institutes of Health Molecular Biophysics Training grant T32 GM008280.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- FRET
fluorescence resonance energy transfer
- HW
(S)-Willardiine (unsubstituted willardiine)
- FW
(S)-5-Fluorowillardiine
- ClW
(S)-5-Chlorowillardiine
- IW
(S)-5-Iodowillardiine
- CTZ
cyclothiazide
- GluR2
glutamate receptor subunit 2
- GluR4
glutamate receptor subunit 4
- FTIR
Fourier transform infrared
- GluR2-S1S2
isolated ligand binding domain of GluR2
- S1S2-T394C-S652C
isolated ligand binding domain of GluR2 with T394C and S652C mutations
- NMR
nuclear magnetic resonance
Footnotes
Supporting Information available. The ClW binding data is available as supplementary figure 1. This material is free of charge via the Internet at http://pubs.acs.org.
References
- 1.Oswald RE. Ionotropic glutamate receptor recognition and activation. Adv Protein Chem. 2004;68:313–349. doi: 10.1016/S0065-3233(04)68009-0. [DOI] [PubMed] [Google Scholar]
- 2.McFeeters RL, Oswald RE. Emerging structural explanations of ionotropic glutamate receptor function. Faseb J. 2004;18:428–438. doi: 10.1096/fj.03-0873rev. [DOI] [PubMed] [Google Scholar]
- 3.Madden DR. The structure and function of glutamate receptor ion channels. Nat Rev Neurosci. 2002;3:91–101. doi: 10.1038/nrn725. [DOI] [PubMed] [Google Scholar]
- 4.Palmer CL, Cotton L, Henley JM. The molecular pharmacology and cell biology of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol Rev. 2005;57:253–277. doi: 10.1124/pr.57.2.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mankiewicz KA, Rambhadran A, Du M, Ramanoudjame G, Jayaraman V. Role of the Chemical Interactions of the Agonist in Controlling alpha-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid Receptor Activation. Biochemistry. 2007;46:1343–1349. doi: 10.1021/bi062270l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng Q, Jayaraman V. Chemistry and conformation of the ligand-binding domain of GluR2 subtype of glutamate receptors. J Biol Chem. 2004;279:26346–26350. doi: 10.1074/jbc.M403111200. [DOI] [PubMed] [Google Scholar]
- 7.Cheng Q, Du M, Ramanoudjame G, Jayaraman V. Evolution of glutamate interactions during binding to a glutamate receptor. Nat Chem Biol. 2005;1:329–332. doi: 10.1038/nchembio738. [DOI] [PubMed] [Google Scholar]
- 8.Mankiewicz KA, Jayaraman V. Glutamate receptors as seen by light: spectroscopic studies of structure-function relationships. Braz J Med Biol Res. 2007;40:1419–1427. doi: 10.1590/s0100-879x2007001100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramanoudjame G, Du M, Mankiewicz KA, Jayaraman V. Allosteric mechanism in AMPA receptors: A FRET-based investigation of conformational changes. Proc Natl Acad Sci U S A. 2006;103:10473–10478. doi: 10.1073/pnas.0603225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong LA, Mayer ML, Jane DE, Watkins JC. Willardiines differentiate agonist binding sites for kainate- versus AMPA-preferring glutamate receptors in DRG and hippocampal neurons. J Neurosci. 1994;14:3881–3897. doi: 10.1523/JNEUROSCI.14-06-03881.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patneau DK, Mayer ML, Jane DE, Watkins JC. Activation and desensitization of AMPA/kainate receptors by novel derivatives of willardiine. Journal of Neuroscience. 1992;12:595–606. doi: 10.1523/JNEUROSCI.12-02-00595.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci. 2003;6:803–810. doi: 10.1038/nn1091. [DOI] [PubMed] [Google Scholar]
- 13.Jin R, Gouaux E. Probing the function, conformational plasticity, and dimer-dimer contacts of the GluR2 ligand-binding core: studies of 5-substituted willardiines and GluR2 S1S2 in the crystal. Biochemistry. 2003;42:5201–5213. doi: 10.1021/bi020632t. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong NA, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 15.Armstrong N, Mayer M, Gouaux E. Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc Natl Acad Sci U S A. 2003;100:5736–5741. doi: 10.1073/pnas.1037393100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armstrong N, Jasti J, Beich-Frandsen M, Gouaux E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 17.Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. Embo J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du M, Reid SA, Jayaraman V. Conformational changes in the ligand-binding domain of a functional ionotropic glutamate receptor. J Biol Chem. 2005;280:8633–6. doi: 10.1074/jbc.C400590200. [DOI] [PubMed] [Google Scholar]
- 19.Jayaraman V, Keesey R, Madden DR. Ligand--protein interactions in the glutamate receptor. Biochemistry. 2000;39:8693–8697. doi: 10.1021/bi000892f. [DOI] [PubMed] [Google Scholar]
- 20.Jayaraman V. Spectroscopic and kinetic methods for ligand-protein interactions of glutamate receptor. Methods Enzymol. 2004;380:170–187. doi: 10.1016/S0076-6879(04)80008-9. [DOI] [PubMed] [Google Scholar]
- 21.Chen GQ, Gouaux E. Overexpression of a glutamate receptor (GluR2) ligand binding domain in Escherichia coli: application of a novel protein folding screen. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13431–13436. doi: 10.1073/pnas.94.25.13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng Q, Thiran S, Yernool D, Gouaux E, Jayaraman V. A vibrational spectroscopic investigation of interactions of agonists with GluR0, a prokaryotic glutamate receptor. Biochemistry. 2002;41:1602–1608. doi: 10.1021/bi015729e. [DOI] [PubMed] [Google Scholar]
- 23.Chapman D, Jackson M, Haris PI. Investigation of Membrane-Protein Structure Using Fourier-Transform Infrared-Spectroscopy. Biochemical Society Transactions. 1989;17:617–619. doi: 10.1042/bst0170617. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed AH, Loh AP, Jane DE, Oswald RE. Dynamics of the S1S2 Glutamate Binding Domain of GluR2 Measured Using 19F NMR Spectroscopy. J Biol Chem. 2007;282:12773–12784. doi: 10.1074/jbc.M610077200. [DOI] [PubMed] [Google Scholar]
- 25.McFeeters RL, Oswald RE. Structural mobility of the extracellular ligand-binding core of an ionotropic glutamate receptor. Analysis of NMR relaxation dynamics. Biochemistry. 2002;41:10472–10481. doi: 10.1021/bi026010p. [DOI] [PubMed] [Google Scholar]
- 26.Koshland DE, Jr, Nemethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.