

Abstract
Infection of C57BL/6 mice with lymphocytic choriomeningitis virus (LCMV) stimulates major histocompatibility complex class I–restricted cytotoxic T cells (CTLs), which normally resolve the infection. Three peptide epitopes derived from LCMV have been shown to bind the mouse class I molecule H-2 Db and to stimulate CTL responses in LCMV-infected mice. This report describes the identity and abundance of each CTL epitope after their elution from LCMV-infected cells. Based on this information, peptide abundance was found to correlate with the magnitude of each CTL response generated after infection with LCMV. Subsequent experiments, performed to determine the antiviral capacity of each CTL specificity, indicate that the quantitative hierarchy of CTL activity does not correlate with the ability to protect against LCMV infection. This report, therefore, indicates that immunodominant epitopes should be defined, not only by the strength of the CTL response that they stimulate, but also by the ability of the CTLs to protect against infection.
Keywords:
cytotoxic T cell • lymphocytic choriomeningitis virus • antiviral protection • peptide elution • adoptive transfer
Through lysis of cells infected with viruses or other intracellular pathogens, CTLs play an essential role in the control of infections. Recognition of infected cells by CTLs is mediated by MHC class I molecules, which behave as receptors for peptides derived from cytoplasmic proteins (1, 2). Generation of peptide epitopes occurs by proteolysis of cytoplasmic proteins by the 20S proteosome (3), and it is likely that the overall spectrum of peptide products generated in this way is influenced by the specificity of this proteolytic unit. The specificity of the proteosome is thought to be further enhanced by IFN-γ, which mediates the substitution of certain housekeeping subunits by MHC-encoded subunits that are thought to favor the generation of class I–binding peptides (4–8). After protein degradation, the resulting peptides are transported into the lumen of the endoplasmic reticulum (ER)1 by the Tap molecule (9–13). Since the Tap molecule has been shown to transport peptides of different lengths and amino acid compositions with different efficiencies (14), it is likely that this molecule also imposes a selection on the spectrum of peptides that reach the ER. On arrival in the ER, peptides may bind to nascent class I molecules that mature as they traffic through the Golgi to the cell surface. The rules that govern peptide binding to class I molecules reflect complementarity between the structure of the α1 and α2 domains of the class I molecule and the presence of certain key amino acids within the peptide, which behave as anchor residues (15–19). Sequencing of MHC-eluted peptides, which predicted the anchor pocket interactions, elucidated numerous class I allele-specific amino acid motifs that have proven to be extremely useful in the search for peptide epitopes (20–22). Therefore, it is clear that peptide epitopes that are ultimately presented on the cell surface are selected by the antigen processing pathway and by the class I molecules to which they bind. Although the number of class I–peptide complexes required to stimulate naive CTLs to proliferate and become effector cells remains unknown, evidence does exist that suggests that the magnitude of a given CTL response is influenced by the abundance of antigen on the cell surface.
CTL responses to three H-2b (Db)-restricted CTL epitopes have been identified in lymphocytic choriomeningitis virus (LCMV)-infected C57BL/6 (B6) mice. The putative optimal sequences representing each epitope have thus far been determined by measuring the efficiency of peptide binding to H-2 Db and by measuring CTL killing of target cells pulsed with decreasing concentrations of peptide. Two of these epitopes derive from the viral glycoprotein ([GP]33-41 and GP276-286), whereas one has been identified in the viral nucleoprotein ([NP]396-404; references 23–28). In this study we confirm, after the elution of H-2 Db–binding peptides from LCMV-infected cells, the precise identity of the CTL epitopes and determine their relative abundance in LCMV-infected cells. We have examined the relationship between antigen abundance and the magnitude of each peptide-specific CTL response stimulated after infection with LCMV as well as the relationship between antigen abundance and the capacity of CTLs of each peptide specificity to protect against LCMV infection.
Materials and Methods
Mice.
C57BL/6 (H-2b), wild-type 129 (H-2b), and IFN-γR– deficient (IFN-γR−/−) mice (G129, H-2b) were obtained from the Institut für Zuchthygiene (Tierspital Zürich, Zürich, Switzerland). All mice were kept in a specific pathogen–free mouse house facility. The generation of IFN-γR−/− mice has been described previously (29).
Peptides.
The LCMV peptides NP396-404, GP276-286, and GP33-41 were purchased from Neosystem Laboratoire (Strasburg, France).
Virus.
The LCMV-WE strain was originally obtained from Dr. F. Lehmann-Grube (Hamburg, Germany) and was grown using L929 fibroblast cells (30). The nucleotide sequence of LCMV-WE has been described previously (31).
Cells and Media.
Cultures of the methylcholantrene-induced mouse fibroblast line MC57 (H-2b) were maintained in MEM. Cultures of the human Tap-defective cell line T2 (32) transfected with Db and the Rauscher virus–transformed mouse T cell line RMA-S, which has a defective Tap-2 gene (33), were maintained in RPMI. Media were supplemented with 5 and 10% fetal calf serum, respectively, penicillin-streptomycin, and L-glutamine.
Detection of Virus-specific Cytotoxic T Cells.
Spleen cell suspensions were prepared from mice infected intravenously with 200 PFU LCMV or 2 × 106 PFU recombinant vaccinia viruses at the indicated time points. Cells were plated at 4 × 106 cells/well (24-well plate) in 1 ml of IMDM supplemented with 10% fetal calf serum, penicillin-streptomycin, and 2-ME. The cultures were supplemented with 1 ml of LCMV-infected, irradiated (2,000 rads) macrophages at a concentration of 2 × 105/ml. Macrophages were recovered from peritoneal washes of B6 mice that had been infected 4 d before with 200 PFU LCMV and 6 d before with 2 ml thioglycollate. The spleen cells were cultured in the presence of 10% Con A–induced rat spleen cell supernatant. After 5 d of culture, restimulated spleen cells were resuspended in 0.7 ml of medium/culture well and threefold dilutions of the effector cells were performed (referred to as dilution of culture). Cytotoxicity assays were carried out as described previously (34, 35). The target cells used were either MC57 cells that had been infected by incubation with 0.1 PFU LCMV/cell 48 h before the experiment or T2-Db pulsed with the indicated concentrations of peptide. For detection of ex vivo CTL activity, spleen cell suspensions from mice infected intravenously with 200 PFU LCMV 8 d before testing were prepared in complete MEM and used directly in cytotoxicity assays. For testing peptide fractions obtained by HPLC separation, synthetic and natural peptides were titrated and incubated with 104 51Cr-labeled T2-Db cells for 90 min at 37°C. CTL lines were added at an effector/target ratio of 10:1 and the cultures were incubated for a further 5 h before 100-μl supernatants were harvested and radioactivity was measured in a beta counter. Specific lysis was determined as described previously.
Generation of Polyclonal CTL Lines.
Spleen cell suspensions were prepared from mice that had been infected intravenously with 200 PFU LCMV-WE at least 3 mo previously. Cells were plated at 4 × 106 cells/well (24-well plate) in 1 ml IMDM/well supplemented with 10% fetal calf serum, penicillin-streptomycin, 2-ME, and 10% Con A supernatant. The cultures were supplemented with 1 ml of peptide-pulsed irradiated T2-Db cells at a concentration of 4 × 105/ml. Before irradiation, T2-Db cells were incubated with 100 μl of peptide at a concentration of 10 ng/ml for 1 h at 37°C before extensive washing to remove any unbound peptide. Cultures were restimulated at 10-d intervals using irradiated peptide-pulsed T2-Db cells as APCs at a responder/APC ratio of 10:1. These CTL lines were found to be of a single specificity after three rounds of restimulation.
Virus Infections.
MC57 cells were grown in triple layer tissue culture flasks to near confluent density. Subsequently, cells were infected by incubation with 0.1 PFU LCMV/cell and incubated for a further 48 h. After trypsinization, the cells were spun down and the pellets were stored at −70°C. Success of infection was monitored by staining for cell-surface expression of the LCMV-GP using the mAb KL25 (36). KL25 staining was detected using goat anti–mouse IgG1-FITC and analyzed by flow cytometry.
Extraction of Viral Peptides from Infected Cells.
109 virus-infected and uninfected cells were resuspended in 20 ml lysis buffer (PBS + 1% NP-40, 0.1 mM PMSF, 2 μg/ml leupeptin, 2 μg/ml aprotinin, and 0.0002% [wt/vol] pepstatin) and disrupted using a hand-held glass homogenizer and sonication. The suspensions were stirred at 4°C for 1 h before centrifugation at 4,000 rpm for 10 min. The supernatants were transferred to new tubes and spun in an ultracentrifuge at 40,000 rpm for 1 h. After ultracentrifugation, the supernatants were passed through prefilters before loading onto glycine-coupled cyanogen bromide–activated Sepharose 4B columns as a preclearing step. The Db complexes were then purified by immunoaffinity chromatography using mAb B22.249 coupled to cyanogen bromide-activated Sepharose 4B. After elution of the Db complexes using 0.1% TFA (pH 2), the eluted material was transferred to a Centricon 10 and centrifuged at 3,000 rpm for 30 min. Material <10 kD was concentrated to 1 ml by vacuum centrifugation.
Fractionation by HPLC.
Peptide separations were carried out on a reversed-phase prepacked column ([μRPC] C2/C18, 2.1 × 100 mm; Pharmacia LKB, Piscataway, NJ) using the Pharmacia LKB SMART system. Samples were injected in a volume of 500 μl. The following elution procedure was used: solvent A, 0.1% TFA in H2O; solvent B, 0.081% TFA in 80% acetonitrile; 0–5 min, 100% A; 5–10 min, 2%/min increase to 10% B; 10–15 min, constant 10% B; 15–55 min, 0.75%/ml increase to 40% B; 55–65 min, 2%/min increase to 60% B; and 65–70 min, 3%/min increase to 75% B. Flow rate was 150 μl/min. Fractions were collected by peak fractionation and elution was monitored by measuring UV light absorption at 214 nm in a continuous flow detector. Acetonitrile was removed from eluted material by vacuum centrifugation before samples were made up to a standard volume of 250 μl using PBS and stored at −70°C. Fractions containing peptide GP33-41 were supplemented with dithiothreitol (DTT) to a final concentration of 0.01 mM to prevent dimerization of the peptide via disulphide bond formation at the COOH-terminal cysteine residue.
Protection Assays and Virus Titration.
Mice were infected intravenously with 20 PFU LCMV and reinjected intravenously 5 h or 3 d later with the indicated numbers of peptide-specific CTL lines. Virus titers were assessed in spleens 4 d later. LCMV titers in spleens were determined as previously described (37).
Flow Cytometry.
For detection of cell-surface markers CD44, CD25, CD69, and CD62 ligand (CD62L), CTL lines were incubated on ice with FITC-labeled anti-CD8 and biotinylated anti-CD44, -CD25, -CD69, and -CD62L antibodies (PharMingen, San Diego, CA). The cells were subsequently incubated with streptavidin-PE. After washing, the stained cells were resuspended in PBS containing 2% FCS and 0.5 mM EDTA (PBS/ FCS/EDTA), and analyzed by FACS® (Becton Dickinson, Mountain View, CA) using Cellquest software (Becton Dickinson). Intracellular cytokine staining was carried out using CTLs that had been incubated at 37°C overnight with LCMV-WE–infected peritoneal macrophages at a macrophage/T cell ratio of 2:1. The next day, the cells were incubated for a further 2 h in the presence of 2 μM monensin. After washing with PBS/FCS/EDTA, the cells were stained as described above using tricolor-conjugated anti-CD8 antibodies. Subsequently, the cells were fixed in 100 μl PBS containing 2% (wt/vol) paraformaldehyde and permeabilized using PBS containing 1% FCS, 0.1% (wt/vol) sodium azide, and 0.1% (wt/vol) saponin (permeabilization buffer). Permeabilized cells were stained with FITC-conjugated anti–IFN-γ antibodies. The stained cells were washed twice in permeabilization buffer, resuspended in PBS/FCS/EDTA, and analyzed by FACS®. In all cases, spleen cells from naive B6 mice were used as negative controls.
Results
CTL Responses to Epitopes GP33-41, GP276-286, and NP396-404.
To determine the relative hierarchy of CTL activity against epitopes GP33, GP276, and NP396, B6 mice were infected intravenously with 200 PFU of LCMV, and cytotoxicity was measured at two different time points. First, CTL activity was measured in direct ex vivo assays 8 d after infection with LCMV. Second, CTL activity was measured after in vitro restimulation of spleen cells isolated from memory mice that had been infected at least 1 mo previously with LCMV. As shown in Fig. 1, A and B, CTL responses where target cells were pulsed with 10 ng of each peptide were strongest against those pulsed with GP33 followed by NP396 and then GP276. Thus, values of specific lysis obtained using bulk cultures correlate well with the estimated CTL precursor frequencies that have been calculated in LCMV-immune mice (data not shown). Lysis of target cells pulsed with titrated concentrations of each peptide was also measured using the same effector cells as described above. The results, shown in Fig. 1, C and D, indicate that NP396-specific CTLs recognize peptide-pulsed target cells 10–1,000 times more efficiently than CTLs specific for peptides GP276 and GP33, respectively. Polyclonal CTL lines specific for each peptide epitope were set up using spleen cells from mice that had been infected at least 3 mo before with LCMV. After three rounds of restimulation using peptide-pulsed T2-Db cells, all cells were found to be CD8+ and of a single specificity.
Figure 1.

Recognition of Db-restricted peptides by LCMV-specific CTLs. Spleen cells were isolated from B6 mice at day 8 after infection (A and C) and tested for cytotoxicity against unpulsed T2-Db cells (open triangles) or T2-Db cells pulsed with peptides GP33 (open circles), GP276 (closed triangles), and NP396 (closed circles). Cytotoxicity against the same target cells was tested using spleen cells isolated from LCMV-immune mice that were restimulated in vitro using LCMV-infected peritoneal macrophages (B and D). CTL assays shown in C were carried out at an effector/target ratio of 90:1, whereas assays shown in D were carried out at an effector/ target ratio of 20:1.
HPLC of Natural and Synthetic Peptides.
To ascertain the identity of the natural Db-restricted LCMV peptides, naturally processed peptides were eluted from Db molecules purified from both LCMV-infected and uninfected MC57 cells. Eluted peptides were separated by reversed-phase HPLC as described previously (38, 39). Each fraction was made up to a final volume of 250 μl, and between 0.1 and 10% of each fraction was used to pulse chromium-labeled T2-Db cells. Fractions containing active peptides were identified using polyclonal CTL lines specific for GP33-41, NP396-404, and GP276-286 (Fig. 2). When the synthetic peptides were chromatographed under the same conditions, the peptides were found to coelute with their corresponding active fractions (data not shown), thus confirming that each peptide is naturally presented in LCMV-infected cells. Neither CTL line recognized fractions derived from uninfected MC57 cells (data not shown). Each peptide fraction was also tested for its ability to sensitize target cells to lysis using spleen cells isolated from mice that had been infected 8 d before with LCMV-WE. Although LCMV-specific lysis was not observed using fractions other than those described above (data not shown), it remains possible that other LCMV-derived peptide epitopes, present at a copy number too low to detect from 109 infected MC57 cells, are processed and presented. This experiment was carried out on two separate occasions.
Figure 2.

Identification of naturally processed LCMV peptides from infected MC57 cells. Active peptide fractions eluted from immunopurified Db molecules from LCMV-infected MC57 cells were identified using polyclonal CTL lines. Dilutions of each peptide fraction were incubated with target cells, T2-Db, and tested for their ability to sensitize polyclonal CTL lines specific for peptides GP33-41, GP276-286, and NP396-404 to lysis. Fractions eluted from glycine columns or from immunopurified Db molecules from uninfected MC57 cells did not sensitize T2-Db cells to lysis by the same CTL lines. CTL assays were carried out using an E/T ratio of 5:1.
Quantitation of Peptides in Infected Cells.
To determine the extraction efficiency of each of the three naturally processed peptide epitopes, 109 uninfected MC57 cells were mixed with 10 ng of each peptide immediately before extraction and fractionation. Active fractions were identified as described above for naturally processed LCMV peptides. Chromium release assays were carried out whereby precisely quantitated synthetic peptides were titrated in 10-fold dilutions in the wells of round-bottomed 96-well plates (0.0002–2,000 pg peptide/well). Target cells, T2-Db, were added at 104 cells/well. Subsequently, CTL lines of corresponding peptide specificity were added to each well and cytotoxic activity was determined 5 h later. In the same assay, active fractions containing admixed peptides were also titrated in 10-fold dilutions and tested for their ability to sensitize target cells, T2-Db, to lysis by the appropriate CTL line. Fig. 3 shows the titration curves generated using synthetic peptides GP33-41 (A), GP276-286 (C), and NP396-404 (E) and the corresponding admixed peptide fractions (Fig. 3, B, D, and F). By comparison of the titration curves, it was determined that 0.6% of the GP33-41 active fraction contained 10 pg of the peptide (Fig. 3, A and B), 0.13% of the GP276-286 active fraction contained 5 pg of the peptide (Fig. 3, C and D), and 0.0006% of the NP396-404 active fraction contained 0.03 pg of the peptide (Fig. 3, E and F). Thus, ∼1.7 ng of admixed peptide GP33-41, ∼3.8 ng of admixed peptide GP276-286, and ∼5 ng of admixed peptide NP396-404 were recovered after the peptide extraction procedure. Since 10 ng of each peptide was initially used in the spiking procedure, the binding plus extraction efficiencies for peptides GP33-41, GP276-286, and NP396-404 were thus estimated as 17, 38, and 50%, respectively.
Figure 3.
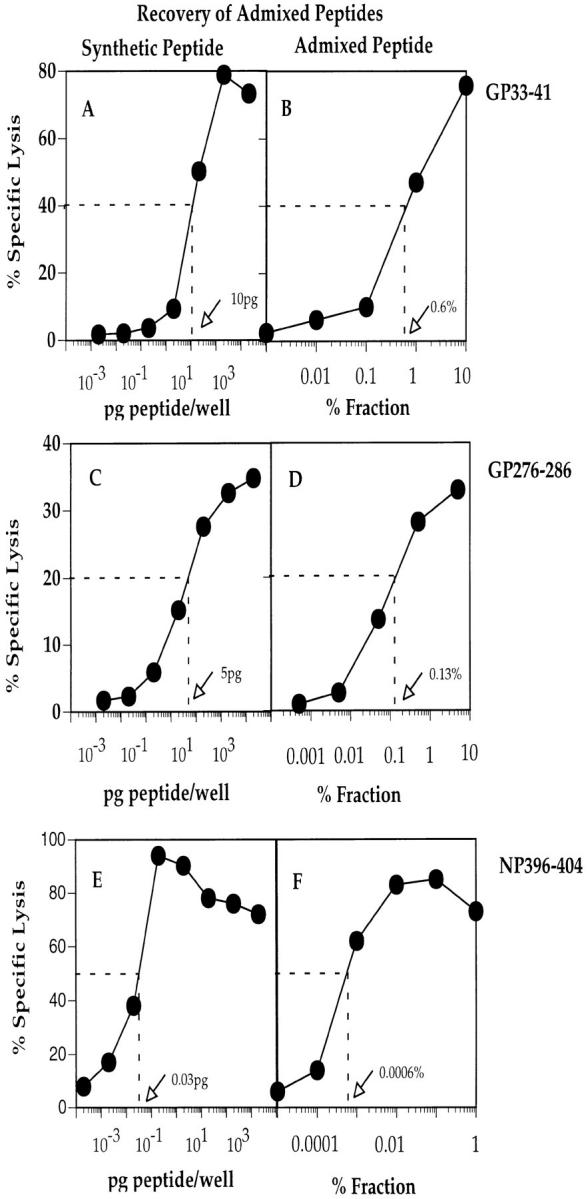
Quantitation of admixed synthetic peptides GP33-41, GP276-286, and NP396-404. Synthetic peptides were incubated in titrated concentrations with T2-Db target cells and tested for lysis by the appropriate CTL line. In addition, between 0.0001 and 1% of the admixed synthetic peptides extracted from 109 uninfected MC57 cells were incubated with T2-Db cells and tested simultaneously for lysis by the same CTL lines. The amount of peptide recovered after the elution procedure was determined by calculating the amount of peptide required for 40% specific lysis (A and B, GP33-41), 20% specific lysis (C and D, GP276-286), or 50% specific lysis (E and F, NP396-404). CTL assays were carried out using an E/T ratio of 5:1.
After another round of restimulation, each peptide-specific CTL line was used to determine the amount of naturally processed LCMV peptides eluted from virus-infected MC57 cells. This analysis was carried out as described above for the admixed synthetic peptides. Fig. 4, A and B show that 4.4% of the GP33-41 active fraction contained 15 pg of the peptide. Similarly, Fig. 4, C and D show that 3.9 and 5.9% of the two GP276-286 active fractions contained 1.5 pg of peptide each, whereas Fig. 4, E and F show that 0.13 of the NP396-404 active fraction contained 0.2 pg of peptide. Thus, a total of 341, 64, and 154 pg of peptides GP33-41, GP276-286, and NP396-404 was recovered, respectively, from 109 infected MC57 cells. By assuming the same recovery rate for admixed and naturally processed peptides (17% for GP33-41, 38% for GP276-286, and 50% for NP396-404), the amount of each naturally processed peptide was calculated as 2 ng of GP33-41, 0.16 ng of GP276-286, and 0.3 ng of NP396-404. Since 1 ng of peptide extracted from 109 cells corresponds to ∼540 molecules/cell, the amount of naturally processed peptide recovered corresponds to 1,080 molecules of peptide GP33-41, 162 molecules of peptide NP396-404, and 92 molecules of peptide GP276-286 per cell.
Figure 4.
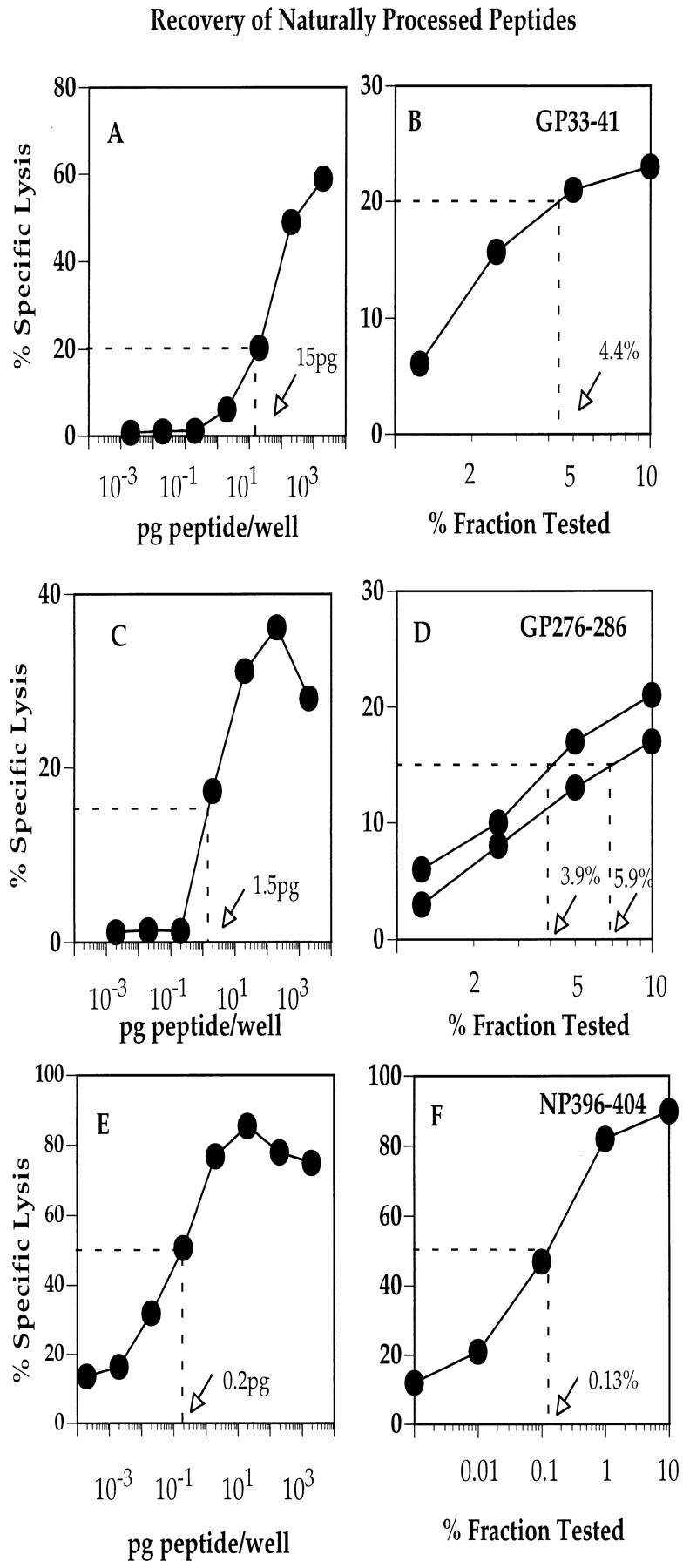
Quantitation of naturally processed peptides GP33-41, GP276-286, and NP396-404. Synthetic peptides were incubated in titrated concentrations with T2-Db target cells and tested for lysis by the appropriate CTL line. In addition, between 0.001 and 10% of each active peptide fraction recovered from 109 LCMV-infected MC57 cells were incubated with T2-Db cells and tested simultaneously for lysis by the same CTL lines. The amount of peptide recovered after the elution procedure was determined by calculating the amount of peptide required for 20% specific lysis (A and B, GP33-41), 15% specific lysis (C and D, GP276-286), or 50% specific lysis (E and F, NP396-404). CTL assays were carried out using an E/T ratio of 5:1.
Adoptive Transfer of CTL Lines in LCMV-infected Mice.
The CTL lines used in the experiments described above were used in adoptive transfer experiments whereby titrated numbers of each CTL line were injected intravenously into mice that had been infected 5 h before with 20 PFU of LCMV. Viral titers were determined 4 d later. The results, shown in Fig. 5, indicate that the protective capacity of each CTL line was different. The NP396-specific CTL line was most efficient at controlling infection with LCMV, the GP276-specific line controlled the infection with intermediate efficiency, and the GP33-specific line was the least efficient. Similar results were obtained after adoptive transfer of two other independently generated CTL lines of each specificity (data not shown). The experiment was repeated using mice that had been infected 48 h before with LCMV to ascertain that the protective effects observed in the above experiment were not due to an earlier expression of the viral nucleoprotein than the viral glycoprotein (40). The results, shown in Fig. 6, indicate that NP396-specific CTL were more protective than GP33-specific CTLs, even at a time point when all viral gene products would be expected to be expressed within infected cells. Thus, the qualitative hierarchy, as defined by the ability of CTLs of each specificity to control LCMV infection, is different from the quantitative hierarchy that was determined by measuring the frequencies of CTLs generated in response to LCMV infection.
Figure 5.

Adoptive transfer of GP33-, GP276-, and NP396-specific CTL lines into LCMV-infected C57BL/6 mice. C57BL/6 mice were infected intravenously with 20 PFU LCMV-WE. 6 h later, groups of three mice received 2 × 106 (A), 2 × 105 (B), or 2 × 104 (C) NP396- (open circles), GP33- (open triangles), or GP276- (closed triangles) specific CTLs intravenously. One group of three mice received no CTLs (open squares). Splenic virus titers were evaluated 4 d later. Dashed line, the limit of sensitivity of the assay.
Figure 6.
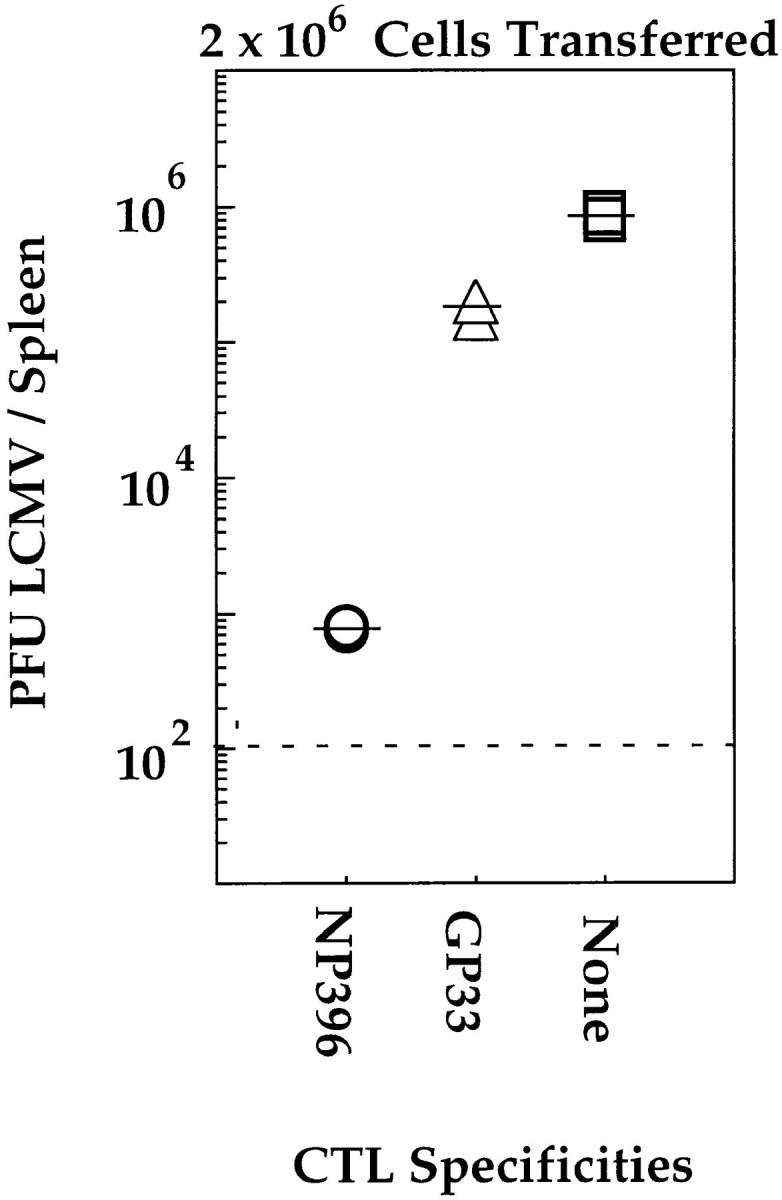
Adoptive transfer of GP33- and NP396-specific CTL lines into C57BL/6 mice infected 48 h before with LCMV. C57BL/6 mice were infected intravenously with 20 PFU LCMV-WE. 48 h later, groups of three mice intravenously received 2 × 106 NP396- (open circles) or GP33- (open triangles) specific CTLs. One group of two mice received no CTLs (open squares). Splenic virus titers were evaluated 2 d later. Dashed line, the limit of sensitivity of the assay.
Expression of Cell-surface Molecules on GP33-, GP276-, and NP396-specific CTL Lines.
To exclude the possibility that the different protective capacity of each CTL line was not attributable to differences in the expression of cell-surface markers known to be associated with memory T cells, each line was compared for expression of the activation markers CD44, CD69, and CD25 (IL-2R). In addition, the cells were compared for the expression of TCR and the lymphocyte homing receptor L-selectin (CD62L). The histograms (Fig. 7) show that expression of CD44, CD69, and CD25 were upregulated to the same extent in each CTL line in comparison to naive CD8+ cells taken from the spleen of a C57BL/6 mouse. CD62L, which is expressed on most naive CD8+ cells, was reduced to the same degree on each CTL line. Surface expression of TCRs was also similar on each CTL line. Thus, the protective potential of each CTL line was not attributable to differences in the expression of the cell-surface molecules described above.
Figure 7.

Surface expression of CD8, TCR, CD25, CD44, CD62L, and CD69 on GP33-, GP276-, and NP396-specific CTL lines. FACS® profiles demonstrating expression of cell-surface molecules on the three different CTL lines are shown in B. Naive C57BL/6 CD8+ splenic T cells were also stained (A).
Expression of IFN-γ in GP33-, GP276-, and NP396-specific CTL Lines.
To investigate the possible role of IFN-γ in the protective effects observed above, IFN-γ production by each CD8+ CTL line was monitored after overnight incubation of the T cells with LCMV-infected macrophages. After permeabilization of the cells and subsequent staining with an anti–IFN-γ antibody, expression of IFN-γ was monitored by FACS® analysis. Fig. 8 shows that although all three CTL lines expressed significant levels of IFN-γ after stimulation with infected cells, the highest level of expression was observed using NP396-specific CTLs. The same hierarchy was observed after measurement of IFN-γ in the supernatants of the cultures described above (data not shown). Expression of IFN-γ was barely detectable in CD8+ cells from the spleen of a naive mouse. To address the role of IFN-γ in vivo, each CTL line was adoptively transferred as described above into IFN-γR−/− mice. The results (Fig. 9) show that the hierarchy, in terms of antiviral protection, was the same. The NP396-specific CTL line was the most protective, followed by the GP276-specific line, and subsequently the GP33-specific line. These results show that the protective capacity of NP396-specific CTLs is not due to the production of IFN-γ. Although the hierarchy of protection was the same as that observed for wild-type mice, the results show that LCMV-titers were, on the whole, higher in IFN-γR−/− mice, thus indicating a role for IFN-γ in the initial clearance of LCMV. It is not clear from these experiments whether the results obtained reflect a direct antiviral effect of IFN-γ or indirect effects relating to the inability of APCs to upregulate MHC class I expression in response to IFN-γ. In the latter case, both the development of the endogenous LCMV-specific CTL response and recognition of infected APCs by the adoptively transferred CTL would be impaired. Interestingly, the reduction in the protective capacity of each CTL line in LCMV-infected IFN-γR−/− mice was more pronounced for the GP33- and GP276-specific CTL lines. Since these CTL lines are less sensitive to antigen than the NP396-specific line, their ability to recognize infected target cells may also be more sensitive to changes in the degree of antigen presentation. Based on the results obtained in the adoptive transfer experiments, it appears that the protective potential of CTLs depends on the avidity of the interaction between the CTLs and their target cells. Figs. 3 and 4 show that NP396- and GP276-specific CTLs are ∼1,000 and ∼10 times more sensitive to antigen, respectively, than are GP33-specific CTLs. The same hierarchy is seen when peptide titration curves are generated using ex vivo CTLs isolated during the primary phase of LCMV infection or in secondary cultures after in vitro restimulation of spleen cells from LCMV-immune mice (data not shown). As an additional test to determine the importance of avidity in the protective potential of virus-specific CTLs, two peptide GP33-specific CTL lines were generated using APCs pulsed with different concentrations of peptide. One CTL line was generated as described above, whereas the other was generated by weekly restimulation with RMA-S cells pulsed with 100 μl of peptide at a concentration of 1 μg/ml for 1 h at 37°C. As described previously by Alexander-Miller et al. (41), this methodology resulted in a lower avidity CTL line that required target cells to be pulsed with higher concentrations of peptide for recognition than the high-avidity CTL line of the same specificity (Fig. 10 A). In addition, although the low-avidity CTL line recognized LCMV-infected target cells less efficiently than the high-avidity line (data not shown), both lines recognized target cells pulsed with high concentrations of peptide (100– 1,000 nM) equally well (Fig. 10 A). Cell surface expression of CD8 and TCR was comparable on both high- and low-avidity CTL lines (data not shown). Upon adoptive transfer of each type of CTL line into mice that had been infected 4 d before with LCMV, the high-avidity CTL line was found to be more protective than an equivalent number of the low-avidity CTL line (Fig. 10 B). These data, therefore, further support the hypothesis that the avidity of the interaction between CTLs and their target cells plays an important role in determining the efficiency with which CTLs can recognize and lyse their specific targets.
Figure 8.

Intracellular staining of IFN-γ in GP33-, GP276-, and NP396-specific CTL lines. Expression of IFN-γ was analyzed by flow cytometry. CTL lines were incubated overnight with uninfected macrophages (A) or LCMV-infected macrophages (2:1 ratio of macrophages to T cells) (B), permeabilized, and stained with anti–IFN-γ antibodies. Naive C57BL/6 CD8+ splenic T cells were also stained.
Figure 9.
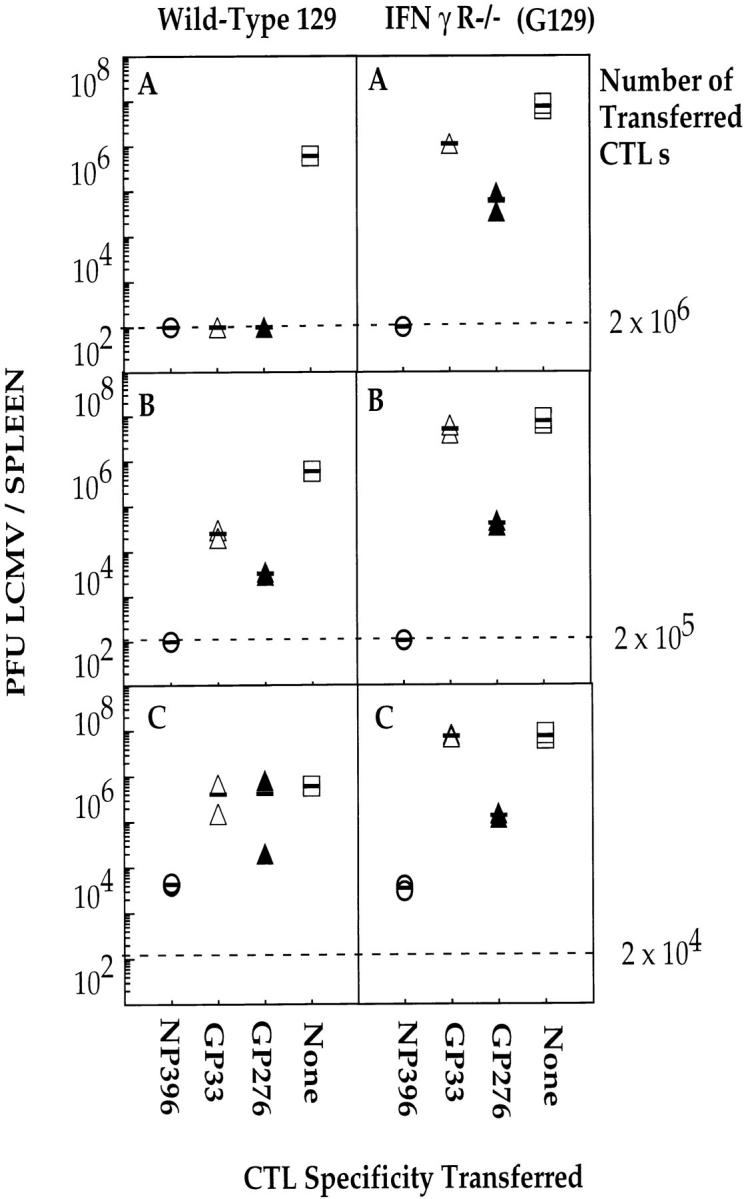
Adoptive transfer of GP33-, GP276-, and NP396-specific CTL lines into LCMV-infected wild-type 129 (left) and IFN-γR−/− mice (right). Mice were infected intravenously with 20 PFU LCMV-WE. 6 h later, groups of two mice received intravenously 2 × 106 (A), 2 × 105 (B), or 2 × 104 (C) of NP396- (open circles), GP33- (open triangles), or GP276- (closed triangles) specific CTLs. One group of two mice received no CTLs (open squares). Splenic virus titers were evaluated 4 d later. Dashed line, the limit of sensitivity of the assay.
Figure 10.

Recognition of peptide-pulsed target cells by two GP33-specific CTL lines of different avidities and adoptive transfer of each CTL line into LCMV-infected B6 mice. (A) Both high- (open triangles) and low- (closed triangles) avidity GP33-specific CTL lines were tested for cytotoxicity against unpulsed T2-Db cells pulsed with titrated concentrations of peptide GP33-41. (B) Mice were infected intravenously with 20 PFU LCMV-WE. 6 h later, groups of two mice received intravenously 2 × 106 (open triangles) or 2 × 105 (open circles) of the high-avidity GP33-specific CTL line or 2 × 106 (closed triangles) or 2 × 105 of the low-avidity (closed circles) GP33-specific CTL line. One group of two mice received no CTLs (open squares). Splenic virus titers were evaluated 4 d later. Dashed line, the limit of sensitivity of the assay.
Discussion
We have confirmed, by elution of Db-binding peptides from LCMV-infected cells, the identity of three previously described CTL epitopes. Thus, two epitopes from the viral glycoprotein are the 9-mer GP33-41 (KAVYNFATC) and the 11-mer GP276-286 (SGVENPGGYCL), and one epitope derived from the viral nucleoprotein is the 9-mer NP396-404 (FQPQNGQFI). Two reports have shown that the GP33 peptide can also be presented to CTL by the Kb molecule (42, 43). Since the presence of phenylalanine at position 5 has been identified as an anchor residue for Kb-binding peptides, it is possible that the naturally processed peptide is GP34-41. It would therefore be of interest to determine the precise nature of this epitope as well as the nature of the precursor peptide that is transported into the ER for binding to both Db and Kb.
Quantitation of each peptide epitope revealed that GP33-41 was present at ∼1,000 copies/cell, NP396-404 was present at ∼160 copies/cell, and GP276-286 was present at ∼90 copies/cell. We cannot, however, exclude the possibility that differential regulation of viral gene expression might occur in different cell types or that different amounts of the viral NP or GP are produced at different stages in the viral life cycle. Nevertheless, the relative density of each antigen corresponds to the relative magnitude of each CTL response stimulated after infection with virus. Interestingly, the relative antigen densities also correlate with the degree of TCR Vβ usage observed after staining the polyclonal CTL lines used in this study with a panel of anti-Vβ antibody supernatants. Expansions of Vβ7, Vβ6, and Vβ8 were observed in four GP33-specific lines, expansions of Vβ6 and Vβ9 were observed in four NP396-specific lines, but an expansion of Vβ10 only was observed in each of four GP276-specific lines (data not shown). This highly restricted TCR V gene usage has been described previously for GP276-specific CTLs cloned from the spleens of LCMV-infected mice (44).
Elution and subsequent quantitation of peptides from cells infected with EBV and HIV have produced similar estimates of peptide abundance (8–1,300 molecules/cell; references 45, 46). In both cases, the relative abundance of peptide epitopes recovered was shown to correlate with the magnitude of the CTL response. When three peptide epitopes were eluted from cells infected with the bacterium Listeria monocytogenes, a reciprocal relationship was found between their relative abundance and the magnitude of the CTL responses (47, 48). This observation has been attributed in part to the short half-life of the most abundant peptide epitope, which could limit its capacity to activate T cells. It is also possible that since the per cell copy number of each peptide was rather high (800, 3,000, and 10,000) compared with the relatively lower estimates of viral peptide copy numbers, the density of each complex is well over the threshold required for T cell activation and their immunogenicity might be limited by other factors such as the availability of T cells with appropriate TCRs. Studies by Harding et al. (49) and Demotz et al. (50) have shown that between 100 and 400 class II–peptide complexes are required to stimulate proliferation of CD4+ T cell hybridomas. Similar studies that have quantitated the number of class I–peptide complexes that are needed for recognition and lysis of target cells by CTLs have shown that as few as one molecule can be sufficient for lysis (51). Since the measurements described above have used either T cell hybridomas, T cell clones, or polyclonal T cell lines, it is not known if such low ligand densities would be sufficient for the activation of naive CTLs. Memory CTLs, possibly through the upregulation of costimulatory and adhesion molecules (52, 53), are able to respond to lower concentrations of antigen than primary effector cells. Whether this means that the number of ligands required for the stimulation of naive T cells is even higher remains unproven. The low density of peptide ligands estimated to be present in the virus-infected cells described above may argue against this hypothesis. In addition, a study by Malarkannan et al. has shown that a self-peptide recognized by alloreactive CTLs can be present at <10 copies/cell (54). Several recent papers have shown that encounter with MHC–peptide complexes results in serial downregulation of specific TCRs (55–57). This observation has been attributed to the very fast off-rate of the MHC + peptide–TCR interaction so that after disengagement, the MHC–peptide complex would be free to engage with other TCRs. It has been proposed that through the serial triggering of TCRs, very few MHC–peptide complexes would be required to sustain the correct signals that a T cell must receive in order for activation to occur.
The results presented in this study also show that the immunogenicities of CTL epitopes, determined by both ex vivo and in vitro analyses of peptide-specific CTL responses generated in LCMV-infected mice, do not correlate with the protective capacities of the different T cell specificities (summarized in Table 1). The protective capacity of the CTLs described here do, however, correlate with how little antigen CTLs require for recognition of LCMV-infected cells. The difference between quantitative and qualitative characteristics of CTLs may impinge upon the success of antiviral therapies using adoptively transferred CTLs.
Table 1.
Magnitude and Protective Capacity of H-2 Db–restricted CTLs Specific for LCMV Peptides NP396-404, GP33-41, and GP276-286
| Characteristics of LCMV-specific CTLs | Hierarchy | |
|---|---|---|
| Peptide density on infected MC57 cells | GP33-41 > NP396-404 > GP276-286 | |
| Magnitude of CTL activity stimulated by LCMV infection | GP33-41 > NP396-404 > GP276-286 | |
| Protective capacity of peptide-specific CTLs in C57BL/6 mice | NP396-404 > GP276-286 > GP33-41 | |
| Protective capacity of peptide-specific CTLs in IFN-γR−/− Mice | NP396-404 > GP33-41 ≥ GP276-286 |
Abbreviations used in this paper
- ER
endoplasmic reticulum
- GP
glycoprotein
- L
ligand
- LCMV
lymphocytic choriomeningitis virus
- NP
nucleoprotein
Footnotes
The authors would like to acknowledge Alana Althage, Stefan Stevanovic, Kevin Maloy, Peter Aichele, and Karin Brduscha-Reim for helpful assistance and discussion.
Awen Gallimore is supported by The Wellcome Trust Foundation, Great Britain. This work was also supported by grants from the Deutsch Forschungsgemeinschaft (Leibnizprogramm Ra 369/4-1), the Bundesminister für Bildung, wissenschaft, Forschung und Technologie, the European Union (Biotech2), and the Swiss National Science Foundation (grants 31-32179.91, 31-50884.97, 31-32195.91, and 31-50900.97).
References
- 1.Townsend A, Rothbard J, Gotch FM, Bahadur G, Wraith D, McMichael AJ. The epitopes of influenza nucleoprotein recognized by cytotoxic T lymphocytes can be defined with short synthetic peptides. Cell. 1986;44:959–968. doi: 10.1016/0092-8674(86)90019-x. [DOI] [PubMed] [Google Scholar]
- 2.Maryanski JL, Pala P, Corradin G, Jordan BR, Cerottini JC. H-2 restricted cytolytic T cells specific for HLA can recognize a synthetic HLA peptide. Nature. 1986;324:578–579. doi: 10.1038/324578a0. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg AL, Rock KL. Proteolysis, proteosomes and antigen presentation. Nature. 1992;357:375–379. doi: 10.1038/357375a0. [DOI] [PubMed] [Google Scholar]
- 4.Dick T, Ruppert T, Groettrup M, Kloetzel P, Kuehn L, Koszinowski U, Stevanovic S, Schild H, Rammensee H. Coordinated dual cleavages induced by the proteosome regulator PA28 lead to dominant MHC ligands. Cell. 1996;86:253–262. doi: 10.1016/s0092-8674(00)80097-5. [DOI] [PubMed] [Google Scholar]
- 5.Aki M, Shimbara N, Takashina M, Akiyama K, Kagawa S, Tamura T, Tanahashi M, Yoshimura T, Tanaka K, Ichihara A. Interferon-γ induces different subunit organizations and functional diversity of proteosomes. J Biochem. 1993;115:257–269. doi: 10.1093/oxfordjournals.jbchem.a124327. [DOI] [PubMed] [Google Scholar]
- 6.Gaczynska M, Rock KL, Goldberg AL. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteosomes. Nature. 1993;365:264–267. doi: 10.1038/365264a0. [DOI] [PubMed] [Google Scholar]
- 7.Driscoll J, Brown MG, Finley D, Monaco JJ. MHC-linked LMP gene products specifically alter peptidase activities of the proteosome. Nature. 1993;365:262–264. doi: 10.1038/365262a0. [DOI] [PubMed] [Google Scholar]
- 8.Groettrup M, Soza A, Eggers M, Kuehn L, Dick TP, Schild H, Rammensee H-G, Kosinowski UH, Kloetzel P-M. A role for the proteosome regulator PA28α in antigen presentation. Nature. 1996;381:166–168. doi: 10.1038/381166a0. [DOI] [PubMed] [Google Scholar]
- 9.Androlewicz MJ, Anderson KS, Cresswell P. Evidence that transporters associated with antigen processing translocate a major histocompatibility complex class I–binding peptide into the endoplasmic reticulum in an ATP-dependent manner. Proc Natl Acad Sci USA. 1993;90:9130–9134. doi: 10.1073/pnas.90.19.9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly A, Powis SH, Kerr LA, Mockridge I, Elliott T, Bastin J, Uchanska ZB, Ziegler A, Trowsdale J, Townsend A. Assembly and function of the two ABC transporter proteins encoded in the human major histocompatibility complex. Nature. 1992;355:641–644. doi: 10.1038/355641a0. [DOI] [PubMed] [Google Scholar]
- 11.Neefjes JJ, Momburg F, Hammerling GJ. Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science. 1993;261:769–771. doi: 10.1126/science.8342042. [DOI] [PubMed] [Google Scholar]
- 12.Shepherd JC, Schumacher TNM, Ashton-Rickardt PG, Imaeda S, Ploegh HL, Janeway CA, Tonegawa S. TAP-1–dependent peptide translocation in vitro is ATP dependent and peptide selective. Cell. 1993;74:577–584. doi: 10.1016/0092-8674(93)80058-m. [DOI] [PubMed] [Google Scholar]
- 13.Spies T, Cerundolo V, Colonna M, Cresswell P, Townsend A, DeMars R. Presentation of viral antigen by MHC class I molecules is dependent on a putative peptide transporter heterodimer. Nature. 1992;355:644–646. doi: 10.1038/355644a0. [DOI] [PubMed] [Google Scholar]
- 14.Schumacher TNM, Kantesaria DV, Heemels M-T, Ashton-Rickardt PG, Shepherd JC, Fruh K, Yang Y, Peterson PA, Tonegawa S, Ploegh HL. Peptide length and sequence specificity of the mouse TAP1/TAP2 translocator. J Exp Med. 1994;179:533–540. doi: 10.1084/jem.179.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garrett T, Saper MA, Bjorkman PJ, Strominger JL, Wiley DC. Specificity pockets for the side chains of peptide antigens in HLA-Aw68. Nature. 1989;342:692–696. doi: 10.1038/342692a0. [DOI] [PubMed] [Google Scholar]
- 16.Madden DR, Gorga JC, Strominger JL, Wiley DC. The three-dimensional structure of HLA-B27 at 2.1 Å resolution suggests a general mechanism for tight peptide binding to MHC. Cell. 1992;70:1035–1048. doi: 10.1016/0092-8674(92)90252-8. [DOI] [PubMed] [Google Scholar]
- 17.Silver ML, Guo H-C, Strominger JL, Wiley DC. Atomic structure of a human MHC molecule presenting an influenza virus peptide. Nature. 1992;360:367–369. doi: 10.1038/360367a0. [DOI] [PubMed] [Google Scholar]
- 18.Fremont DH, Matsumura M, Stura EA, Peterson PA, Wilson IA. Crystal structures of two viral peptides in complex with murine MHC class I H-2Kb. Science. 1992;257:919–927. doi: 10.1126/science.1323877. [DOI] [PubMed] [Google Scholar]
- 19.Zhang W, Young AC, Imarai M, Nathanson SG, Sacchettini JC. Crystal structure of the major histocompatibility complex class I H-2kbmolecule containing a single viral peptide: implications for peptide binding and T-cell recognition. Proc Natl Acad Sci USA. 1992;89:8403–8407. doi: 10.1073/pnas.89.17.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falk K, Roetzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 21.Rammensee H-G. Chemistry of peptides associated with MHC class I and class II molecules. Curr Opin Immunol. 1995;7:85–96. doi: 10.1016/0952-7915(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 22.Rammensee H-G, Friede T, Stefanoviic S. MHC ligand motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 23.Gairin JE, Mazarguil H, Hudrisier D, Oldstone MBA. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Dbmajor histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J Virol. 1995;69:2297–2305. doi: 10.1128/jvi.69.4.2297-2305.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klavinskis LS, Whitton JL, Joly E, Oldstone MBA. Vaccination and protection from a lethal viral infection: identification, incorporation, and use of a cytotoxic T lymphocyte glycoprotein epitope. Virology. 1990;178:393–400. doi: 10.1016/0042-6822(90)90336-p. [DOI] [PubMed] [Google Scholar]
- 25.Pircher HP, Moskophidis D, Rohrer U, Bürki K, Hengartner H, Zinkernagel RM. Viral escape by selection of cytotoxic T cell–resistant virus variants in vivo. Nature. 1990;346:629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 26.Oldstone M, Whitton JL, Lewicki H, Tishon A. Fine dissection of a nine amino acid glycoprotein epitope, a major determinant recognized by lymphocytic choriomeningitis virus–specific class I–restricted H-2Db cytotoxic T lymphocytes. J Exp Med. 1988;168:559–570. doi: 10.1084/jem.168.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hany M, Oehen S, Schulz M, Hengartner H, Mackett M, Bishop D, Zinkernagel RM. Anti-viral protection and prevention of lymphocytic choriomeningitis or of the local footpad swelling reaction in mice by immunisation with vaccinia-recombinant virus expressing LCMV-WE nucleoprotein or glycoprotein. Eur J Immunol. 1989;19:417–424. doi: 10.1002/eji.1830190302. [DOI] [PubMed] [Google Scholar]
- 28.Whitton JL, Southern PJ, Oldstone MBA. Analysis of the cytotoxic T lymphocyte responses to glycoprotein and nucleoprotein components of lymphocytic choriomeningitis virus. Virology. 1988;162:321–327. doi: 10.1016/0042-6822(88)90471-0. [DOI] [PubMed] [Google Scholar]
- 29.Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 30.Lehmann-Grube F. Lymphocytic choriomeningitis virus. Virol Monogr. 1971;10:1–173. [Google Scholar]
- 31.Romanowski V, Matsuura Y, Bishop D. Complete sequence of the S RNA of lymphocytic choriomeningitis virus (WE strain) compared to that of pichinde arenavirus. Virus Res. 1985;3:101–114. doi: 10.1016/0168-1702(85)90001-2. [DOI] [PubMed] [Google Scholar]
- 32.Salter RD, Cresswell P. Impaired assembly and transport of HLA-A and -B antigens in a mutant T×B cell hybrid. EMBO (Eur Mol Biol Organ) J. 1986;5:943–949. doi: 10.1002/j.1460-2075.1986.tb04307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohlen C, Bastin J, Ljunggren HG, Imreh S, Klein G, Townsend AR, Kärre K. Restoration of H-2b expression and processing of endogenous antigen in the MHC class I pathway by fusion of a lymphoma mutant to L cells of the H-2k haplotype. Eur J Immunol. 1990;20:1873–1876. doi: 10.1002/eji.1830200837. [DOI] [PubMed] [Google Scholar]
- 34.Baenziger J, Hengartner H, Zinkernagel RM, Cole GA. Induction or prevention of immunopathological disease by cloned cytotoxic T cell lines specific for lymphocytic choriomeningitis virus. Eur J Immunol. 1986;16:387–393. doi: 10.1002/eji.1830160413. [DOI] [PubMed] [Google Scholar]
- 35.Zinkernagel RM, Leist TP, Hengartner H, Althage A. Susceptibility to lymphocytic choriomeningitis virus isolates correlates directly with early and high cytotoxic T cell activity, as well as with footpad swelling reaction, and all three are regulated by H-2D. J Exp Med. 1985;162:2125–2141. doi: 10.1084/jem.162.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruns M, Cihak J, Müller G, Lehmann-Grube F. Lymphocytic choriomeningitis virus. VI. Isolation of a glycoprotein mediating neutralization. Virology. 1983;130:247–251. doi: 10.1016/0042-6822(83)90135-6. [DOI] [PubMed] [Google Scholar]
- 37.Lehmann-Grube F, Assmann U, Loliger C, Moskophidis D, Lohler J. Mechanism of recovery from acute virus infection. I. Role of T lymphocytes in the clearance of lymphocytic choriomeningitis virus from spleens of mice. J Immunol. 1985;134:608–615. [PubMed] [Google Scholar]
- 38.Rötzschke O, Falk K, Deres K, Schild H, Norda M, Metzger J, Jung G, Rammensee HG. Isolation and analysis of naturally processed viral peptides as recognized by cytotoxic T cells. Nature. 1990;348:252–254. doi: 10.1038/348252a0. [DOI] [PubMed] [Google Scholar]
- 39.Falk K, Roetzschke O, Deres K, Metzger J, Jung G, Rammensee HG. Identification of naturally processed viral nonapeptides allows their quantification in infected cells and suggests an allele-specific T cell epitope forecast. J Exp Med. 1991;174:425–434. doi: 10.1084/jem.174.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bishop, D.H.L. 1990. Arenaviruses and their replication. In Virology. 2nd ed. B.N. Fields, D.M. Knipe, R.M. Chanock, M.S. Hirsch, J.L. Melnick, T.P. Monath, and B. Roizman, editors. Raven Press, Ltd., New York. 1231–1267.
- 41.Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci USA. 1996;93:4102–4107. doi: 10.1073/pnas.93.9.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohashi PS, Zinkernagel RM, Leuscher I, Hengartner H, Pircher HP. Enhanced positive selection of a transgenic TCR by a restriction element that does not permit negative selection. Int Immunol. 1993;5:131–138. doi: 10.1093/intimm/5.2.131. [DOI] [PubMed] [Google Scholar]
- 43.Hudrisier D, Oldstone MBA, Gairin JE. The signal sequence of lymphocytic choriomeningitis virus contains an immunodominant epitope that is restricted by both H-2Db and H-2Kbmolecules. Virology. 1997;234:62–73. doi: 10.1006/viro.1997.8627. [DOI] [PubMed] [Google Scholar]
- 44.Aebischer T, Oehen S, Hengartner H. Preferential usage of V alpha 4 and V beta 10 T-cell receptor genes by LCMV-glycoprotein specific H-2Dbrestricted cytotoxic T-cells. Eur J Immunol. 1990;20:523–531. doi: 10.1002/eji.1830200310. [DOI] [PubMed] [Google Scholar]
- 45.Levitsky V, Zhang Q-J, Levitskaya J, Masucci MG. The life span of major histocompatibility complex– peptide complexes influences the efficiency of presentation and immunogenicity of two class I–restricted cytotoxic T lymphocyte epitopes in the Epstein-Barr virus nuclear antigen 4. J Exp Med. 1996;183:915–926. doi: 10.1084/jem.183.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsomides TJ, Aldovini A, Johnson RP, Walker BD, Young RA, Eisen HN. Naturally processed viral peptides recognized by cytotoxic T lymphocytes on cells chronically infected by human immunodeficiency virus type 1. J Exp Med. 1994;180:1283–1293. doi: 10.1084/jem.180.4.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pamer EG. Direct sequence identification and kinetic analysis of an MHC class I–restricted Listeria monocytogenesCTL epitope. J Immunol. 1994;152:686–694. [PubMed] [Google Scholar]
- 48.Vijh S, Pamer EG. Immunodominant and subdominant CTL responses to Listeria monocytogenesinfection. J Immunol. 1997;158:3366–3371. [PubMed] [Google Scholar]
- 49.Harding CV, Unanue ER. Quantitation of antigen-presenting cell MHC class II/peptide complexes necessary for T-cell stimulation. Nature. 1990;346:574–576. doi: 10.1038/346574a0. [DOI] [PubMed] [Google Scholar]
- 50.Demotz S, Grey HM, Sette A. The minimal number of class II MHC–antigen complexes needed for T cell activation. Science. 1990;248:1028–1030. doi: 10.1126/science.2118680. [DOI] [PubMed] [Google Scholar]
- 51.Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence that a single peptide–MHC complex on a target cell can elicit a cytolytic T cell response. Immunity. 1996;4:565–571. doi: 10.1016/s1074-7613(00)80483-5. [DOI] [PubMed] [Google Scholar]
- 52.Zimmermann C, Brduscha-Reim K, Blaser C, Zinkernagel RM, Pircher H. Visualization, characterization, and turnover of CD8+memory T cells in virus-infected hosts. J Exp Med. 1996;183:1367–1375. doi: 10.1084/jem.183.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pihlgren M, Dubois PM, Tomkowiak M, Sjörgen T, Marvel J. Resting memory CD8+T cells are hyperreactive to antigenic challenge in vitro. J Exp Med. 1996;184:2141–2151. doi: 10.1084/jem.184.6.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malarkannan S, Afkarian M, Shastri N. A rare cryptic translation product is presented by Kbmajor histocompatibility complex class I molecule to alloreactive T cells. J Exp Med. 1995;182:1739–1750. doi: 10.1084/jem.182.6.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide–MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 56.Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A. Sustained signaling leading to T cell activation results from prolonged T cell receptor occupancy. Role of T cell actin cytoskeleton. J Exp Med. 1995;181:577–584. doi: 10.1084/jem.181.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]