

Abstract
The serine proteinase inhibitor (serpin) plasminogen activator inhibitor type 2 (PAI-2) is well characterized as an inhibitor of extracellular urokinase-type plasminogen activator. Here we show that intracellular, but not extracellular, PAI-2 protected cells from the rapid cytopathic effects of alphavirus infection. This protection did not appear to be related to an effect on apoptosis but was associated with a PAI-2–mediated induction of constitutive low-level interferon (IFN)-α/β production and IFN-stimulated gene factor 3 (ISGF3) activation, which primed the cells for rapid induction of antiviral genes. This primed phenotype was associated with a rapid development of resistance to infection by the PAI-2 transfected cells and the establishment of a persistent productive infection. PAI-2 was also induced in macrophages in response to viral RNA suggesting that PAI-2 is a virus response gene. These observations, together with the recently demonstrated PAI-2–mediated inhibition of tumor necrosis factor-α induced apoptosis, (a) illustrate that PAI-2 has an additional and distinct function as an intracellular regulator of signal transduction pathway(s) and (b) demonstrate a novel activity for a eukaryotic serpin.
Keywords: plasminogen activator inhibitor, serpin, interferon, alphavirus, Ross River virus
The serine proteinase inhibitors, or serpins,1 are a large family of single chain molecules that are structurally related to α1-antitrypsin. They participate in the regulation of proteinase-activated physiological and pathological processes, including blood coagulation, fibrinolysis, extracellular matrix remodelling, prohormone activation, and tumor metastasis. Ovalbumin is the parent prototype of a distinctive and growing subfamily of serpins, known as ov-serpins (1). Ov-serpins lack typical, cleavable hydrophobic signal sequences, which results in inefficient (and often undetectable) translocation and secretion, and thus a predominantly cytoplasmic localization of these proteins. Members of this subfamily include the viral serpin crmA, plasminogen activator inhibitor type 2 (PAI-2), elastase inhibitor, squamous cell carcinoma antigen, the tumor suppressor maspin, placental thrombin inhibitor (PI)-6, and the recently described PI-8 and PI-9 (1–6). The best characterized of these serpins is the intracellular viral serpin crmA, which blocks apoptosis by inhibition of IL-1β–converting enzyme (ICE) and other related caspases (7). The vaccinia virus–encoded serpin, B13R (SPI-2), is also an inhibitor of ICE, and blocks both TNF-α– and FAS-mediated apoptosis (8). Intracellular functions for some mammalian ov-serpins have been recently postulated (3, 5), suggesting that this family may also participate in a range of diversified intracellular activities.
PAI-2 was originally characterized as an inhibitor of the extracellular serine proteinase, urokinase-type plasminogen activator (uPA) (9). uPA generates plasmin from its precursor plasminogen and regulates extracellular proteolysis involved in tissue remodelling, inflammatory disease, and tumor cell invasion/metastasis. However, the predominant proportion of newly synthesized PAI-2 remains intracellular, with only a fraction of PAI-2 under certain circumstances being secreted as a glycosylated product (10). Recently, an intracellular role for PAI-2 has been postulated based on the ability of cytoplasmic expression of PAI-2 to protect cells from TNF-α–mediated apoptosis (11, 12). PAI-2 is rapidly induced in monocytes/macrophages in response to TNF-α and LPS, and Dickinson et al. (12) have suggested that the physiological role of intracellular PAI-2 in inflammatory macrophages may be to protect these cells from the cytotoxic effects of their own TNF-α. A role for PAI-2 in the inhibition of apoptosis has been supported by the observation that PAI-2 can inhibit Mycoplasma avium– induced apoptosis of macrophages (13).
In this study, we show that HeLa cells expressing PAI-2 are protected from the cytopathic effect (CPE) of the alphaviruses, Ross River virus (RRV) and Sindbis virus. Alphaviruses are single-stranded positive-sense RNA viruses that induce a rapid, lytic infection in most vertebrate cells (14). RRV infection did not induce apoptosis in HeLa cells, indicating that protection against CPE in PAI-2–expressing cells was unrelated to a PAI-2–mediated inhibition of apoptosis. Instead, protection was associated with a PAI-2–mediated induction of constitutive low-level autocrine IFN-α/β production, which primed the cells for rapid, IFN-α/β–independent induction of antiviral resistance. Thus, after virus infection, PAI-2–transfected cells induced antiviral genes (without further IFN-α/β), which was associated with a rapid inhibition of viral replication. In contrast, virus infection of control cells did not result in IFN-α/β or antiviral gene induction and was associated with rapid viral replication and cell death.
Intracellular PAI-2 expression thus produced at least two potentially related phenotypes, resistance to TNF-α (11, 12), and induction of constitutive autocrine IFN-α/β priming. These phenotypes are entirely distinct from the well-characterized effects of extracellular PAI-2 and suggest that PAI-2 also has an intracellular function as a regulator of signal transduction pathway(s).
Materials and Methods
Cells and Cell Culture.
Stable, cloned HeLa cell lines expressing sense (S1a, S1b) and antisense (A2/7, A2/17) PAI-2 cDNA were generated as previously described (12) by inserting a DNA fragment containing the entire PAI-2 coding sequence and the 3′ untranslated region in both orientations into the expression vector, pRcCMV, under control of the constitutive CMV promoter. As a positive control for an irrelevant gene, the coding sequence for the chloramphenicol acetyl transferase (CAT) gene was inserted into the same vector. Stable transfectants containing these constructs and vector alone (CMV) were selected by resistance to G418 and characterized by Northern and immunoblot analyses (12). The human macrophage cell line, MonoMac6, was obtained from Professor H.W.L. Ziegler-Heitbrock, University of Munich (Munich, Germany; reference 15). All cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 60 μg/ml penicillin G, and 100 μg/ml streptomycin sulfate, and were maintained at 37°C in a 5% CO2 and 95% air atmosphere. The transfected HeLa lines were also maintained with 200 μg/ml G418, which was removed at least 48 h before their use in an experiment. Cells (∼107) were treated with 500 U/ml human IFN-γ (Boehringer Mannheim GmBH, Mannheim, Germany), 500 U/ml IFN-α–2B (Intron A; Schering-Plough, Pty. Ltd., New South Wales, Australia), or 20 μg/ml polyinosinic-polycytidylic acid (poly IC) (Sigma Chemical Co., St. Louis, MO) for the times indicated. All cell lines were checked routinely and determined to be mycoplasma free.
Virus Infections.
The following viruses were used in this study: RRV, T48 strain, stock 108 supplied at 50% tissue culture infectivity dose (TCID50) per ml (16); Sindbis virus, ∼108 PFU/ ml (17), obtained from Professor D.E. James (University of Queensland, Brisbane, Australia); Adenovirus 5, 108 infectious doses per ml (18); Influenza virus (reassortant with external antigens from A/Taiwan/1/86 and internal antigens from A/PR/8/34), 109 egg infectious U/ml, supplied as an egg allantoic fluid– derived stock by A. Hampson (CSL Ltd., Parkville, Australia); Vaccinia virus, thymidine kinase negative WR strain, stock supplied at 109 PFU/ml (19). The multiplicity of infection (MOI) was based on the units given for each virus; thus, MOI 1 (or log MOI 0) for RRV was 1 TCID50/cell, whereas MOI 1 for vaccinia virus refers to 1 PFU/cell.
Cell Survival Assays.
Cell survival was quantitated by both crystal violet staining and tetrazolium dye–based MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assays. Cell lines were plated into 96-well plates at 2 × 104 cells/well in duplicate or triplicate and allowed to adhere overnight. After addition of virus, the plates were incubated at 37°C for 7 d or for the indicated time period before staining with crystal violet or MTT assay. For the crystal violet assay, medium was removed and the cells were stained for 15 min with 0.05% crystal violet in 10% formaldehyde. The plates were then washed with water and dried, and 100 μl of 100% methanol was added to dissolve the dye, then the OD at 595 nm was measured using a Bio-Rad ELISA plate reader (Bio-Rad, Hercules, CA). The MTT assay was performed as previously described (12) with cells plated as above. Cell survival for both assays was expressed as a percentage calculated as OD of experimental wells/OD of control wells.
RRV Virus Titration (TCID50) and Immunofluorescent Antibody Staining.
The TCID50 assay was performed in duplicate using 10-fold serial dilutions of 100 μl of sample supernatants in 96-well flat-bottomed plates. Vero cells (2 × 104 cells in 100 μl) were then added into each well and the plates were incubated at 37°C for 7 d. The plates were stained with 0.05% crystal violet in 10% formaldehyde and the RRV titer was expressed as a TCID50 (16).
For indirect immunofluorescence staining, cells cultured on coverslips were fixed in cold 1:1 (vol/vol) acetone/methanol for 2 min, washed in PBS, blocked with 10% FCS, and stained with a rabbit polyclonal anti-RRV sera (gift from Dr. J. Aaskov, Queensland University of Technology, Brisbane, Australia), followed by an anti–rabbit FITC-labeled F(ab′)2 (Silenus Labs., Hawthorn, Australia).
Immunoblot Analyses.
For immunoblot analyses of whole cell lysates, cells were plated at 0.5–1 × 106 cells/well per 6-well plate and infected with RRV (MOI 1) for 90 min at 37°C followed by washing. At the indicated time points, cell monolayers were washed three times in PBS and harvested by gentle scraping into cold PBS. The cells were pelleted by centrifugation and quickly lysed in cold PBS containing 0.5% Triton X-100, 5 mM EDTA, and 20 μg/ml of the proteinase inhibitor, 4-(2-aminoethyl)benzenesulfonylfluoride (AEBSF). Cell debris was removed by centrifugation at 12,000 g for 10 min, and the protein concentration of each sample was determined by Bio-Rad protein assay (Bio-Rad). The solubilized proteins (60 μg) were separated by SDS-PAGE under nonreducing conditions using a 10% acrylamide gradient gel, and the proteins were electrophoretically blotted onto a nitrocellulose membrane (Bio-Rad) for 16 h at 30 V. Specific antigens were detected by incubation for 1 h with polyclonal rabbit anti-RRV antisera, 1 μg/ml anti–PAI-2 monoclonal antibody (American Diagnostica, Epping, Australia), 1 μg/ml anti- bcl-2 monoclonal antibody (Oncogene Sciences, Uniondale, NY), or 1 μg/ml anti–α-tubulin antibody (Sigma Chemical Co.). Antibody binding was visualized by ECL detection (Amersham International, Little Chalfont, UK) and membranes were exposed to Kodak XK-1 film (Eastman Kodak Co., Rochester, NY).
For immunoblot analysis of nuclear and cytoplasmic lysates, extracts were prepared as previously described (20), except that nuclei were obtained using 0.15% NP-40 in the lysis buffer. Protein extracts were electrophoretically separated on 8% SDS-polyacrylamide gels, blotted to nitrocellulose membranes, and probed with 2 μg/ml anti–interferon-stimulated gene factor 3 (ISGF3)γ polyclonal antibody (C-20) (Santa Cruz Biotech, Santa Cruz, CA) or 2 μg/ml anti–interferon response factor (IRF)-1 polyclonal antibody (C-20) (Santa Cruz Biotechnology).
EMSA Assays.
Protein–DNA complexes were formed in 20-μl reaction mixtures containing 12 mM Hepes, pH 7.9, 30 mM KCl, 0.06 mM EDTA, 0.06 mM EGTA, 1 mM MgCl2, 2.5% Ficoll, 0.5 mM dithiothreitol, 2 μg poly [d(I)–d(C)] (Boehringer Mannheim Australia Pty. Ltd., Castle Hill, Australia), and 20 μg nuclear extract, to which 1 μl of purified 32P-radiolabeled oligonucleotide was added. Complexes formed after 20 min on ice were resolved on 5% polyacrylamide gels at 150 V in TBE (100 mM Tris HCl, 100 mM Borate, and 2 mM EDTA, pH 8.3). Double stranded oligonucleotides were radiolabeled with T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and γ-[32P]ATP (6000 Ci/mmole, Amersham International). The oligonucleotides, containing the interferon-stimulated response element (ISRE) consensus binding motif (21) were as follows: forward, 5′GTTCGGGAAAGGGAAACCGAAACTGAAGCC3′; and reverse, 5′GATCGGCTTCAGTTTCGGTTTCCCTTTCCC3′. Mutant oligonucleotides containing changes in the ISRE consensus sequence were used in gel shift assays to confirm the specificity of the binding interactions (data not shown). Immunoblotting of EMSA gels was performed as described by Demczuk et al. (22) using 2 μg/ml anti-ISGF3γ antibody (Santa Cruz Biotech).
Recombinant PAI-2.
Purified recombinant PAI-2 was provided by Biotech Australia Pty. Ltd. (Sydney, Australia) and had a specific activity of 150,000 IU/mg as assayed for inhibitory activity against uPA using the Spectrolyse® Urokinase assay (American Diagnostica).
IFN-α and IFN-β reverse transcriptase (RT)-PCR.
Cell lines (5 × 106cells/well per 6-well plate) were plated 48 h before addition of RRV (MOI 1) or 5 μg/ml poly IC (Boehringer Mannheim). After 8 h, the medium was removed and the cells lysed with 1 ml Total RNA Isolation Reagent (Advanced Biotechnologies Ltd., Surrey, UK). After extraction, the RNA was reverse transcribed at room temperature for 20 min and then at 40°C for 1 h in a 20 μl reaction containing 1 μg RNA, 1 μg random hexamer primers (Boehringer Mannheim), 1 μl 10 mM dNTP (Promega, Madison, WI) and the following from the Superscript™ II RT kit (GIBCO BRL, Bethesda, MD): 4 μl 5× first strand buffer, 2 μl 0.1 M dithiothreitol, and 1 μl Superscript™ II RNase− RT. Control reactions contained no RT. After hydrolysis of the RNA with sodium hydroxide, the cDNA was purified by QIAquick-spin PCR Purification (QIAGEN, Clifton Hill, Australia) and eluted in 50 μl.
PCR for IFN-α and IFN-β was performed in a 20-μl reaction volume containing 1 μl cDNA and 0.5 μl IFN-α primers (forward: 5′CAGGAGGAGTTTGATGGCAACCAG3′; reverse: 5′GACAACCTCCCAGGCACAAGGGC3′; Stratagene, La Jolla, CA), or 0.5 μl (10 μM) of each of the IFN-β primers (forward: 5′ATGACCAACAAGTGTCTCCTCCAAA3′; reverse: 5′GTCTGAATGTCCAATGGAGGCTTTG3′) or β actin primers (forward: 5′CACAGAGCCTCGCCTTTG3′; reverse: 5′GTACATGCAACGATAGGT3′) (23). Each PCR reaction for IFN-α and β actin contained 0.3 μl (2,500 U/ml) AmpliTaq DNA polymerase, 2 μl 10× AmpliTaq PCR Buffer II (Perkin-Elmer, Norwalk, CT), 1.1 μl 25-mM MgCl2, and 0.4 μl dNTPs. PCR was performed using a GeneAmp PCR system 9600 with 5 min of denaturation at 95°C, followed by 35 cycles at 94°C for 45 s, 64°C for 45 s, and 72°C for 90 s, followed by a 10 min extension. The PCR reaction for IFN-β contained 0.2 μl (2 U/μl) DyNAZyme II DNA polymerase, 2 μl DyNAZyme 10× buffer (Finnzymes Oy, Espoo, Finland), and 1.2 μl dNTPs. PCR was performed with a 2-min denaturation at 95°C, followed by 35 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, followed by a 10 min extension at 72°C. Amplification products were resolved by agarose gel electrophoresis and stained with ethidium bromide.
IFN-α/β Bioassay.
Bioactive IFN-α/β was measured by CPE inhibition bioassay using Sindbis virus infection of Vero cells, a cell line defective for IFN-α/β production. Duplicate 10-fold serial dilutions of test supernatants were cultured with Vero cells (104/well in 96-well plates) for 24 h. Sindbis virus was then added at a MOI 1 and after 4 d the plates were stained with crystal violet. The assay was standardized against recombinant IFN-α2B (Intron A; Schering-Plough Pty. Ltd.) and had a detection sensitivity of 10 IU/ml. A 50% reduction in viral CPE was obtained at 25 IU/ml. RRV containing supernatants were exposed to 960 μW/cm2 UV-C for 2 h to inactivate the RRV (16) before addition to Vero cells.
Supernatant Transfers.
T75 flasks containing S1b and A2/7 cells were maintained in standard medium until the cultures had become overconfluent. The medium was then replaced with 10 ml RPMI 1640 and supplemented with antibiotics and 1% FCS. After 24 h the supernatant was removed, and cell debris was removed by spinning at 2000 g for 20 min and passing the supernatants through a 0.22-μM filter. The supernatants were then concentrated using a Centricon 10 concentrator (Amicon Inc., Beverly, MA). The unconcentrated supernatant and the concentrates from A2/7 and S1b cells were then added separately in quadruplicate to 103 A2/7 cells that had been seeded in wells of a flat 96-well plate the day before. After overnight incubation at 37°C, 200 μl of RPMI 1640, supplemented with antibiotics and 10% FCS, was added and the cells were infected with RRV, MOI = 1. The plates were then stained with crystal violet on day 3. Percentage of cell survival was calculated as mean OD595 of test cells/ mean OD595 of cells which had received medium instead of supernatant and were not infected with RRV.
Treatment of S1b Cells with Anti–IFN-α/β Antibody.
200 S1b cells were seeded per well in a 96-well plate in triplicate and cultured overnight in standard medium, after which the cells were cultured in 100 μl RPMI 1640 (supplemented with antibiotics) containing 1% by volume polyclonal rabbit anti–human IFN-αA (106 neutralizing U/ml) and 4% polyclonal sheep anti–human IFN-β (5 × 104 neutralizing U/ml) (BioSource Int., Camarillo, CA). Control cells were cultured in 1% normal rabbit serum and 4% normal sheep serum. Every 3–4 d the cells were scrapped from the well, disaggregated by repeated pipetting, and reseeded at 200 cells/well. After 18 d, 100 μl of standard medium with 10% FCS was added and the cells were infected with RRV MOI = 1. After 6 d, cell survival was monitored by crystal violet assay and the percentage of cell survival was calculated as a percentage of mean OD595 of test cells/mean OD595 of control cells which had been cultured in 1% normal rabbit serum and 4% normal sheep serum and were not infected with RRV.
Northern Blot Analysis.
For Northern blotting of MonoMac6 cells, 107 cells were infected in 10-ml round-bottomed tubes with RRV (MOI 1) for 1 h at 37°C with shaking every 15 min, followed by three washes before culture at 37°C. Infection of MonoMac6 cells by antibody-dependent enhancement using subneutralizing levels of antibody was performed as previously described (16). MonoMac6 cells were also treated with 100 μg/ml poly IC, 30 ng/ml PMA, or 1 μg/ml LPS. UV-inactivated RRV was prepared as previously described (16). After 4 h the cells were pelleted and RNA was isolated as above. Purified total RNA was electrophoresed on denaturing agarose gels containing 1.1% formaldehyde. The gel was stained with ethidium bromide as a measure of total RNA loaded in each lane. RNA was transferred to Hybond N nylon membranes (Amersham International) by capillary diffusion as described previously (12). The blots were probed with a radiolabeled 2.0-kb HindIII/EcoR1 DNA fragment encoding PAI-2 cDNA (3).
For Northern blotting of S1b and A2/7 cells, 107 cells were infected with RRV (MOI = 1) and cultured for the times indicated before isolation of RNA as described above. Blots were probed with a radiolabeled 300-bp PvuII/EcoRI DNA fragment encoding 2′5′oligoadenylate synthetase (OAS) cDNA (24). Signals were quantitated using a scanning densitometer (Molecular Dynamics, Victoria, Australia) driven by ImageQuant software.
Results
HeLa Cells Expressing PAI-2 Were Protected from the CPE of Certain Viruses.
We have previously shown that HeLa cells stably transfected with PAI-2 were protected from apoptosis induced by TNF-α (12). To explore a possible role for PAI-2 in protection against virus induced apoptosis, PAI-2–expressing HeLa cell lines were infected with a panel of cytopathic viruses which have been reported to kill cells by induction of apoptosis: alphavirus (25); adenovirus (26); influenza (27); and vaccinia (28). HeLa cells stably expressing PAI-2 (S1a, S1b), antisense PAI-2 mRNA (A2/7, A2/17), the negative control expressing CAT, and HeLa cells transfected with plasmid vector alone (CMV) (12) were infected with 10-fold serial dilutions of adenovirus, influenza virus, vaccinia virus, and two alphaviruses, RRV and Sindbis. The percentage of cells surviving the virus-induced CPEs after 7 d was measured by crystal violet assay (Fig. 1). The PAI-2–expressing cells (S1a, S1b) were considerably less sensitive than control cell lines (A2/7, A2/17, CAT, CMV) to the CPE induced by the alphaviruses and adenovirus. The effect was particularly pronounced for RRV, where the PAI-2–expressing lines were resistant to 105 more virus than were the control lines.
Figure 1.

PAI-2–expressing HeLa cell lines are protected from the CPEs of some viruses. Each of the indicated HeLa cell lines were seeded into 96-well plates in duplicate at 2 × 104 cells/well and were infected after 10–24 h with 10-fold serial dilutions of the indicated virus stock. S1a and S1b are PAI-2–expressing cell lines and A2/7, A2/17, CAT, and CMV are controls which do not express PAI-2 (see Materials and Methods). The multiplicity of infection (MOI) is expressed as a log value, thus log MOI of 0 represents 1 virus unit per cell (see Materials and Methods for different units) and log MOI -7 refers to seven 10-fold serial dilutions of log MOI 0. Cell survival was measured after 7 d by crystal violet assay and is expressed as percent survival relative to untreated cells.
PAI-2 Expression Changed the Rapid Lytic Alphavirus Infection into a Persistent Productive Infection.
To explore further the pronounced protective effect of PAI-2 expression against RRV-induced CPE, the survival of infected cells was monitored over 7 d (Fig. 2 A). RRV produced the characteristic rapid lytic infection in A2/7, A2/17, and CAT cell lines, resulting in complete cell death by day 4. In contrast, RRV-infected S1a and S1b cultures showed no detectable change in cell survival compared with uninfected cultures (Fig. 2 A). Analysis of RRV replication and cell infection for RRV infected A2/7 and S1b cultures over an extended period showed that 80% of A2/7 cells were infected on day 2, resulting in a high virus yield (Fig. 2 B). By day 7, a complete loss of virus production was observed, consistent with the widespread CPE and cell death seen in Fig. 2 A. In contrast, 5–10% of S1b cells were initially infected but this fell to a consistent level of <1% after a few days (Fig. 2 B). These data illustrated that the classical rapid lytic alphaviral infection of control cultures was converted to a persistent productive infection in PAI-2–expressing S1b cells. In addition, the development of resistance to infection in S1b cultures illustrated that these cells were not innately resistant to infection but developed antiviral activity after exposure to virus.
Figure 2.

Cell survival and RRV infection in PAI-2–expressing and control HeLa cell lines. (A) Time course of cell survival. The survival of the panel of cell lines infected (dashed lines) or not infected (solid lines) with RRV was assayed daily in replicate plates using the MTT assay. RRV infection was at MOI = 1. (B) Time course of RRV infection. RRV replication (TCID50) and percentage of cells infected (estimated by indirect immunofluorescence on replicate cultures and indicated by % and arrow) was monitored for RRV-infected S1b and A2/7 cell lines. PAI-2–expressing (S1b) and nonexpressing (A2/7) cell lines were seeded into 24-well plates, 106 cells/well in duplicate, and infected at MOI = 1 for 90 min. Free virus was removed by five washes and at the indicated time points, 100 μl was removed for viral titer assay. Medium was changed every 3 d and S1b cultures were split 1 in 3 every 4–5 d after day 6.
PAI-2 Did Not Inhibit RRV Structural Polyprotein Cleavage.
Alphavirus replication involves the synthesis of two polyproteins, the nonstructural polyprotein, which is produced in low abundance and is not readily detected by immunoblotting, and the structural polyprotein, which includes the surface glycoproteins E1 and E2, the E3 protein, and the capsid. Individual proteins are generated from the polyproteins by specific proteolytic cleavages (29). To determine whether PAI-2 might be interfering with these proteolytic events and thereby inhibiting viral replication, protein extracts from RRV-infected S1b and A2/7 cells were analyzed by immunoblotting using a polyclonal antibody generated against RRV viral proteins (Fig. 3). The immunoblots showed no evidence for inhibition of structural polyprotein cleavage by the presence of PAI-2, with no accumulation of the uncleaved precursors, p95 or pE2, evident in S1b cells. Consistent with the higher level of RRV infection in A2/7 cells detected by indirect immunofluorescence (Fig. 2 B), the extracts from A2/7 cells contained more RRV proteins relative to S1b cell extracts at each time point.
Figure 3.

Protein expression in S1b and A2/7 cell lines infected with RRV. PAI-2–expressing (S1b) and nonexpressing (A2/7) cell lines were seeded into 6-well plates (5 × 106 cells/well in duplicate), infected at MOI = 1 for 90 min, and extracellular virus was removed by five washes. At the indicated times, the cells were harvested and protein extracts were electrophoretically separated by SDS-PAGE and immunoblotted. Identical blots were probed with antibodies directed against RRV proteins, PAI-2, and bcl-2 (see Materials and Methods). The RRV proteins are p95 (the capsid-E1-E2-E3 polyprotein); pE2 (pre-E2); E1 and E2 (the structural surface proteins of the virus); the capsid (which is cleaved from the capsid-E1-E2-E3 polyprotein); and E3, which does not become part of the final virion. Equal amounts of protein as determined by Bio-Rad protein assay were loaded in each lane and the blots were reprobed with anti–α-tubulin as a control for protein loading in each lane.
PAI-2 Did Not Protect S1b Cells from RRV-induced CPE by Inhibiting Apoptosis.
Since HeLa cells expressing PAI-2 (S1a, S1b) were resistant to TNF-α–mediated apoptosis (12), the resistance of these cells to alphavirus-induced CPE (Fig. 1) may suggest that PAI-2 expression protected HeLa cells from RRV-induced CPE by inhibiting apoptosis. In a potentially related alphavirus system, Sindbis virus infection of the AT3 prostate carcinoma cell line induces apoptosis, which can be inhibited by expression of bcl-2 (24, 30). However, in contrast to Sindbis virus infection of AT-3 cells, several lines of evidence indicated that PAI-2 did not inhibit RRV-induced apoptosis, as follows. (a) Electron micrographic analysis of RRV-induced CPE in A2/7 and S1b cells showed no evidence of the distinctive ultrastructural morphology characteristic of apoptosis, such as chromatin condensation and margination and cytoplasmic shrinkage (data not shown). (b) PAI-2 expression did not protect from infection with influenza virus, a virus demonstrated to induce apoptosis in HeLa cells (27). (c) Expression of bcl-2 did not correlate with sensitivity of HeLa cells to RRV-induced CPE or with expression of PAI-2 (Fig. 3). (d) Although cleavage of PAI-2 has been reported to be a marker of apoptosis in human myeloleukemic cells (31), S1b cultures showed no evidence of changes in PAI-2 protein expression, or cleavage of PAI-2 after RRV infection (Fig. 3). (e) Pretreatment of A2/7 cells with inhibitors of Sindbis virus induced apoptosis of AT-3 cells, N-acetylcysteine (25 mM) and pyrrolidine dithiocarbamate (150 μg/ml) (30), followed by addition of RRV (as for Fig. 1), did not prevent RRV-induced CPE of A2/7 cells (data not shown).
Extracellular PAI-2 was not responsible for protection against RRV mediated CPE. Our previous data suggested that PAI-2 inhibition of TNF-α–mediated apoptosis was mediated by intracellular PAI-2 (12), which contrasts with the ability of 100 ng/ml (170,000 IU/mg) of PAI-2 added extracellularly, to prevent Mycobacterium avium–induced apoptosis of macrophages (13). In contrast to the latter observations, extracellular PAI-2 did not protect against RRV-mediated CPE, as no protection against RRV-mediated CPE was observed when up to 8,000 ng/ml recombinant PAI-2 was added to A2/7 cells for 24 h before infection with RRV (Fig. 4). Increasing the preincubation time to 48 h had no effect on this result (data not shown).
Figure 4.
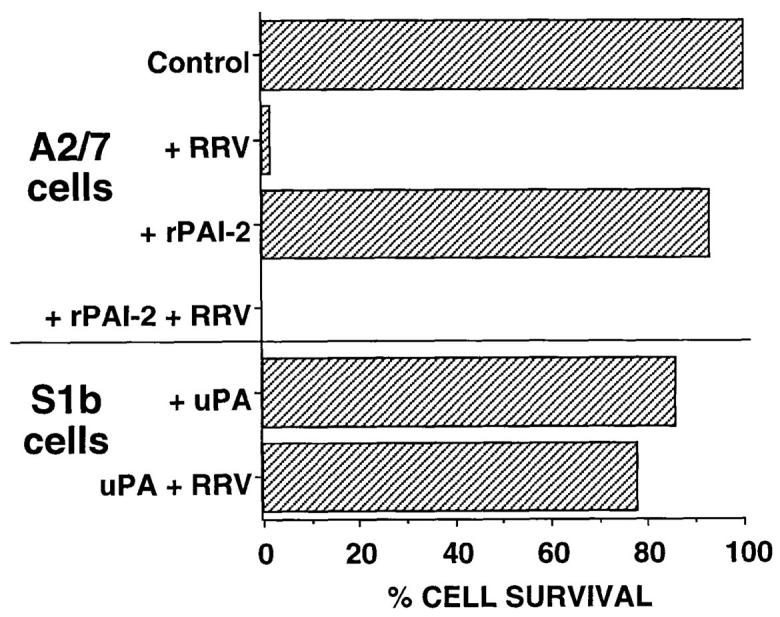
Extracellular PAI-2 was not implicated in protection against CPE. S1b and A2/7 cells were seeded into 96-well plates in duplicate at 2 × 104 cells/ well. RRV (MOI = 1) was added where indicated and cells incubated at 37°C for 7 d before measurement of CPE by crystal violet assay. A2/7 cells were pretreated for 24 h with recombinant PAI-2 (8 μg/ml). S1b cells were incubated with uPA at 500 ng/ml for the 7-d CPE assay. Cell survival is expressed as percentage of survival relative to untreated cells.
PAI-2 was not detectable in concentrated S1b culture supernatants as measured by immunoprecipitation/immunoblot and ELISA analyses (data not shown). In addition, inhibition of possible cell surface PAI-2 activity by addition of uPA, a serine proteinase that forms an essentially irreversible 1:1 complex with PAI-2, also failed to abolish resistance of S1b cells to RRV CPE (Fig. 4). Taken together, these data indicated that neither extracellular nor cell surface PAI-2 were involved in protection against RRV-induced CPE.
PAI-2–expressing HeLa Cells, unlike Control HeLa Cells, Were Primed to Respond to poly IC.
Several lines of evidence implicated IFN-α/β and/or IFN-α/β induced antiviral genes in the protection mediated by PAI-2 against alphavirus-induced CPE: (a) alphaviruses are well known to be highly sensitive to the antiviral activity of IFN-α/β, with as little as 2 IU/ml IFN-α significantly inhibiting alphaviral replication in HeLa cells (32); (b) influenza and vaccinia viruses are known to be able to evade IFN-α/β induced antiviral activity in HeLa cells (32, 33) and PAI-2–expressing cells were not protected from CPE induced by these viruses (Fig. 1); and (c) IFN-α/β–induced genes have a well established role in the maintenance of persistent infections in vitro (34) and such infections could be established in PAI-2–expressing cells (Fig. 2 B). However, HeLa cells do not normally synthesize IFN-α/β even after virus infection or treatment with the synthetic dsRNA analogue, poly IC, unless they have first been primed with IFN (35). How priming operates is not fully understood; however, one of the consequences of IFN priming in HeLa cells is the induction of ISGF3γ (36, 37). ISGF3γ combines with activated ISGF3α to form the active transcription factor complex ISGF3. This complex is reported to be responsible for autocrine amplification of IFN-α/β expression (38) and therefore ISGF3γ induction may be required before virus or poly IC can induce IFN-α/β production in HeLa cells.
As expected for unprimed HeLa cells (35), no constitutive IFN-α/β bioactivity was detected in the supernatants of HeLa, A2/7, or CAT cells, and none was detected after poly IC treatment (Fig. 5 A). No constitutive IFN-α/β bioactivity was detected in S1b culture supernatants, but, a dramatic production of IFN-α/β followed exposure of S1b cells to poly IC (Fig. 5 A). These data clearly illustrated that PAI-2–expressing S1b cells were primed for IFN-α/β production. The implication from this observation was that PAI-2 expression induced autocrine IFN-α/β production below the level detectable by the bioassay (∼10 IU/ml) but at a level sufficient to prime the S1b cells. In HeLa (35) and other cells (39), priming by IFN-α/β treatment is known to confer the ability to synthesize IFN-α/β after poly IC treatment.
Figure 5.




The role of IFN-α/β. (A) Bioassay of IFN-α/β protein activity in the supernatants of S1b, A2/7, CAT, and parental HeLa cell lines. Cells were cultured for 24 h in the presence of medium alone, RRV (MOI = 1), or 20 μg/ml poly IC. Assays were performed in triplicate and error bars represent SD. (B) Assay by RT-PCR for IFN-α, IFN-β, and β actin mRNAs in S1b and A2/7 cells 24 h after treatment with medium alone (control), RRV (MOI = 1), or poly IC (20 μg/ml). As the PCR amplification efficiency is quite different for IFN-α and IFN-β mRNA transcripts, the different intensities for IFN-α and IFN-β amplification products do not necessarily demonstrate different relative levels of these two cytokines. (C) Transfer of supernatants from S1b cell and A2/7 cells onto A2/7 cells followed by infection with RRV. Neat (1×) or 15- or 30-fold concentrated 24-h supernatants from S1b and A2/7 were added to A2/7 cells overnight before infection with RRV (MOI = 1). Control represents addition of medium rather than supernatant followed by RRV infection. Cell survival was determined by crystal violet assay and is expressed as a percentage of A2/7 cells, which were incubated with medium and were not infected with RRV (NO RRV). (Supernatant alone in the absence of RRV had no significant effect on cell survival; data not shown). (D) Long-term incubation of S1b cells with anti–IFN-α and anti–IFN-β antibodies. S1b cells were maintained at low cell density in the presence of anti–IFN-α/β antibodies (anti–IFN-αβ + RRV) or control antibody (control antibody + RRV) for the indicated number of days (gray bars, day 2; hatched bars, day 18; black bars, day 25) followed by infection with RRV; MOI = 1. S1b cells were also incubated with anti–IFN-α/β antibodies without RRV infection (anti–IFN-α/β NO RRV). Cell survival was measured on day 6 using the crystal violet assay and was expressed as a percentage of the survival of cells that had been incubated with control antibody and that had not been infected with RRV.
Perhaps unexpectedly, IFN-α/β bioactivity was not detected in supernatants from PAI-2–expressing S1b cells after RRV infection (Fig. 5 A). Thus the priming phenomena resulted in IFN-α/β production after poly IC treatment but not RRV infection. This data indicated that viral induction of IFN-α/β was not responsible for protection from alphaviral CPE, but that S1b cells were protected as a consequence of being primed. Primed cells have been demonstrated to induce protective (IFN-α/β–inducible) antiviral genes after viral infection, without induction of further IFN-α/β production (see Discussion).
Detection of IFN-α/β mRNA by RT-PCR in PAI-2–expressing Cells.
The previous data (Fig. 5 A) suggested that PAI-2–expressing cells have a primed phenotype. This phenotype is a known consequence of prior exposure to IFN-α/β (35), suggesting that PAI-2–expressing cells may produce autocrine IFN-α/β at levels not detectable by the bioassay. A more sensitive method for detection of cytokines is the use of RT-PCR technology, whereby cytokine mRNA is amplified using specific oligonucleotide primers in RT-PCR assays (23). These RT-PCR assays were performed using standard conditions and the quality and the quantity of the cDNA was assessed by the amplification of β actin in the same samples. However, the RT-PCR results should be considered qualitative. Examination of mRNA isolated from S1b and A2/7 cells showed that S1b cells constitutively expressed both IFN-α and IFN-β mRNAs, whereas in A2/7 cells no IFN-β and only a weak IFN-α signal was detectable (Fig. 5 B, control). (The PCR amplification efficiency differs considerably for IFN-α and IFN-β mRNA transcripts, so different intensities for the respective amplification products should not be interpreted as demonstrating different relative levels of these two cytokines.) The increase in the intensities of the IFN-α and IFN-β mRNAs detected in S1b cells in response to poly IC, but not RRV, infection was consistent with IFN-α/β bioactivity seen in the Vero cell assay (Fig. 5 A).
The detection of more IFN-α/β mRNA in PAI-2–expressing cells strongly supported the contention that S1b cells produced low levels of autocrine IFN-α/β, which was responsible for the primed phenotype demonstrated in Fig. 5 A.
Concentrated Supernatants Transferred Antiviral Activity.
If S1b cells secrete low levels of IFN-α/β, which leads to protection against RRV CPE, protective activity should be transferable in tissue culture supernatants from S1b cells. Although transfer of neat (1×) supernatants from S1b cells to A2/7 cells did not protect the A2/7 cells from RRV CPE, preincubation of A2/7 cells with S1b supernatants concentrated 15- and 30-fold conferred 44 and 100% protection to RRV-infected A2/7 cells, respectively (Fig. 5 C). Similar supernatants from A2/7 cells and control supernatant (medium) failed to provide any significant protection from RRV-mediated CPE (Fig. 5 C). Thus, concentrated supernatants from S1b cells were able to confer antiviral resistance to A2/7 cells further supporting a role for autocrine IFN-α/β.
Treatment of S1b Cells with Anti–IFN-α/β Antibodies.
If S1b cells are protected from RRV–induced CPE by low-level autocrine IFN-α/β priming, then incubation of S1b cells with anti–IFN-α/β antibodies should block autocrine IFN-α/β bioactivity and confer sensitivity to RRV-induced CPE. However, incubation of S1b cells with anti– IFN-α/β antibodies for 48 h failed to confer sensitivity (Fig. 5 D). In addition, short-term (24 h) treatment of S1a and S1b cells with inhibitors of IFN-α/β signal transduction (2-aminopurine and staurosporine) also failed to induce sensitivity to RRV-mediated CPE. Long-term exposure was found to be toxic and inhibited viral replication (data not shown). However, the primed phenotype is known to persist for up to 18 d in the absence of IFN-α/β (40). Therefore, autocrine IFN-α/β activity was blocked by incubation of S1b cells in the presence of anti–IFN-α/β antibodies for 18, 21, and 25 d. A low cell density was used to allow access of antibody to the basolateral cell surfaces. After the indicated number of days, the cells were infected with RRV and left for 6 d. S1b cells treated with nonneutralizing, control antibody remained protected against CPE, whereas cells treated for extended periods with anti–IFN-α/β antibodies showed significant CPE (Fig. 5 D).
PAI-2–expressing Cells, but not Control Cells, Showed Constitutive and Inducible Activation of ISGF3.
If PAI-2–expressing HeLa cells secreted low levels of autocrine IFN-α/β that was biologically active, then the consequence should be the presence of constitutively activated ISGF3 in S1b cells. ISGF3, the complex of ISGF3γ and ISGF3α, is the principle transcription factor activated by IFN-α/β (21). In addition, PAI-2–expressing cells should also contain ISGF3γ, a protein known to be induced in HeLa cells after priming with IFN-α/β or -γ (36, 37). Control HeLa cells, in contrast, lacking significant ISGF3γ, should be unable to form ISGF3 after IFN-α/β or poly IC treatment (36–38).
The predicted presence or absence of active ISGF3 was determined by EMSA using a radiolabeled oligonucleotide containing the ISRE to which activated ISGF3 binds (37, 38). EMSA experiments using nuclear extracts showed the presence of a weak ISGF3 band in S1b cells, which became more intense after poly IC exposure (Fig. 6 A). The identity of ISGF3 was confirmed by immunoreactivity of the complex with anti-ISGF3γ antibodies in combination EMSA/immunoblot experiments (data not shown). ISGF3, as expected, was undetectable in A2/7 nuclear extracts from control and poly IC treated cells (Fig. 6 A). These experiments demonstrated that PAI-2 expression resulted in (a) constitutive low-level ISGF3 activation and (b) induction of ISGF3γ, which permitted enhanced ISGF3 activation after poly IC treatment. Both of these effects are recognized consequences of IFN-α/β stimulation (21, 36, 37), strongly suggesting that the low level of autocrine IFN-α/β was biologically active in S1b cells. The activation of ISGF3 in S1b cells (but not A2/7 cells) after poly IC treatment (Fig. 6 A), is consistent with the induction of IFN-α/β by poly IC seen in Fig. 5 A (38) and provides further evidence that PAI-2–expressing cells have been primed by autocrine IFN-α/β exposure.
Figure 6.
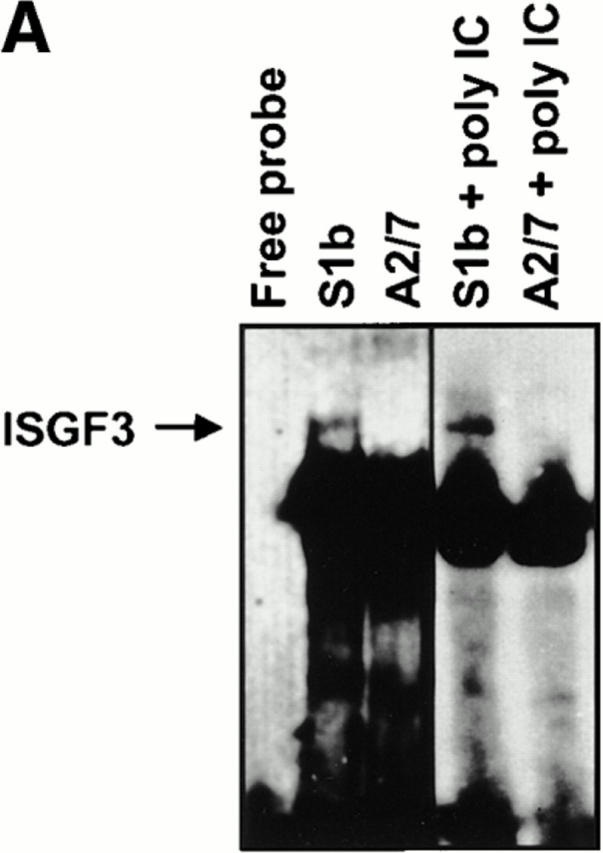

(A) EMSA assay for ISGF3. Nuclear extracts isolated from S1b and A2/7 cells, either untreated or treated with 20 μg/ml poly IC as indicated, were analyzed by EMSA using a radiolabeled ISRE oligonucleotide probe. The ISGF3 complex detected in S1b cells is indicated by the arrow. Free probe is shown at the bottom of the gel. The identity of ISGF3 was confirmed by immunoreactivity of this band with anti-ISGF3γ antibody (data not shown). (B) ISGF3γ and IRF-1 expression in S1b and A2/7 cells. Nuclear and cytoplasmic extracts from S1b and A2/7 cells were analyzed by immunoblotting using antibodies against ISGF3γ and IRF-1. Where indicated, cells were pretreated for 16 h with 500 U/ ml IFN-γ. Equal amounts of protein as determined by Bio-Rad protein assay were loaded in each lane.
PAI-2–expressing Cells, but not Control Cells, Showed Constitutive Expression and Nuclear Translocation of ISGF3γ.
A series of Western blotting experiments using anti-ISGF3γ antibodies were undertaken to provide further evidence that PAI-2 expression resulted in constitutive induction of ISGF3γ synthesis, which is normally low or absent in HeLa cells (36, 37), and in constitutive IFN-α/β–mediated activation of ISGF3α, which results in the nuclear translocation of the ISGF3 complex (21, 36, 37, 40). As expected, control A2/7 cells (No treatment, Fig. 6 B), like the parent HeLa cells (36, 37), synthesized low to undetectable levels of ISGF3γ and no ISGF3γ could be detected in nuclear extracts. In contrast, S1b cells showed a weak but clear ISGF3γ band in the nuclear extract of resting cells (No treatment, Fig. 6 B). As a positive control for these experiments, the cells were treated with IFN-γ, a known inducer of ISGF3γ (36, 37). As expected IFN-γ induced ISGF3γ in both cell lines (IFN-γ, Fig. 6 B). Thus as might be expected (36, 37) autocrine IFN-α/β induced expression of ISGF3γ in S1b cells.
The levels of nuclear IRF-1, another transcriptional regulator of IFN and IFN-regulated genes (21), were also analyzed in the same extracts by immunoblot analysis. In the absence of IFN-γ treatment, the IRF-1 levels in S1b and A2/7 extracts were found to be weak and relatively equivalent (Fig. 6 B). However, after IFN-γ treatment, less nuclear IRF-1 was induced in S1b cells, an observation which may explain the comparative resistance of these cells to IFN-γ–mediated cytostasis (12).
PAI-2–expressing Cells Were Primed for Rapid Induction of Antiviral Genes.
Although PAI-2–mediated expression of low-level autocrine IFN-α/β appeared to prime S1b cells for induction of IFN-α/β after poly IC treatment, this priming did not result in the induction of IFN-α/β after RRV infection (Fig. 5 A). To determine whether low-level autocrine IFN-α/β production in S1b cells (a) induced constitutive expression of antiviral genes or (b) primed cells for activation of antiviral genes after RRV infection, the synthesis of the IFN-α/β inducible antiviral gene, 2′-5′-OAS (24) was monitored by Northern blot analysis (Fig. 7 A). The antiviral activity of OAS is well characterized and is mediated by the synthesis of oligoadenylate polymers, which activate ribonuclease L, which in turn degrades viral and cellular RNA (21).
Figure 7.


Induction of 2′-5′-OAS mRNA in S1b and A2/7 cells after RRV infection. (A) S1b and A2/7 cells (107 cells) were infected with RRV (MOI = 1) for the times indicated. Total RNA was isolated and analyzed by Northern blot using a 32P-radiolabeled OAS cDNA probe. OAS mRNA is present as at least three species of 3.6, 1.85, and 1.65 kb as previously described (23). The lower panel represents the level of 18S rRNA present in each lane as a control for loading. (B) Plot of the intensity of the OAS signal in each lane divided by the intensity of the 18S rRNA in the same sample.
There was no significant difference in the constitutive levels of OAS mRNA in S1b cells compared with A2/7 cells, indicating that low-level autocrine IFN-α/β production in S1b cells was insufficient to upregulate OAS mRNA in these cells. However, after RRV infection, S1b cells synthesized significant levels of OAS mRNA within 4 h of RRV infection, whereas no OAS induction was apparent in A2/7 cells (Fig. 7 B). As RRV infection shuts down host protein synthesis within 5–8 h, rapid induction of OAS is likely to be required to protect the cells against RRV-induced CPE. The drop in the OAS mRNA in S1b cells at 16 h is consistent with mRNA degradation by activated ribonuclease L.
These experiments illustrated that, in contrast to control cells, PAI-2–expressing cells were primed for a rapid induction of the antiviral gene OAS (in the absence of significant further IFN-α/β production; see Fig. 5, A and B). This rapid induction of OAS, and perhaps other antiviral genes, is likely to be responsible for the inhibition of viral replication (Figs. 2 B and 3) (21) and is likely to be instrumental in protecting these cells against CPE (Figs. 1 and 2 A). PAI-2–mediated priming thus conferred on S1b cells the capacity to produce IFN-α/β after poly IC treatment and to synthesize OAS after alphavirus infection.
PAI-2 as a Virus Response Gene.
PAI-2 is strongly and rapidly induced by monocytes/macrophages in response to activation and differentiation agents, e.g., LPS (41) and PMA (42, 43). The data presented here shows that expression of PAI-2 can influence viral CPE and IFN-α/β signaling, suggesting that PAI-2 might be involved in the response of the monocyte/macrophage to virus infection. To determine whether macrophages synthesize PAI-2 in response to viral infection, PAI-2 mRNA levels were analyzed in the human macrophage cell line, MonoMac6, after exposure to neutralized/inactivated RRV, infectious RRV, RRV in the presence of subneutralizing levels of antisera, and poly IC (Fig. 8). MonoMac6 cells treated with RRV alone and RRV plus a 1:10 dilution of antisera (Ab−1), which neutralizes RRV, do not become infected with RRV. Our previous studies have shown that MonoMac6 cells may be infected with RRV only in the presence of subneutralizing levels of anti-RRV antibody through an antibody-dependent enhancement mechanism (16). Thus, MonoMac6 cells treated with RRV in the presence of subneutralizing dilutions of antibody 1:100 (Ab−2), 1:1,000 (Ab−3), and 1:10,000 (Ab−4) become infected with ∼15, 5, and 1% of the cells staining positive by indirect immunofluorescence for RRV after 24 h, respectively (reference 16 and data not shown). PAI-2 mRNA was induced in all virus- and poly IC–treated samples irrespective of whether the cells were infected. In addition, PAI-2 mRNA was induced to levels comparable with those seen after exposure of MonoMac6 cells to known PAI-2 inducers LPS and PMA (Fig. 8). These experiments demonstrated that PAI-2 was induced by viral RNA and illustrated that PAI-2 could be classed as a virus response gene.
Figure 8.

Induction of PAI-2 mRNA in human macrophage MonoMac6 cells by viral RNA. Total RNA was isolated from ∼107 MonoMac6 cells after exposure for 4 h to the following: lane 1, untreated; lane 2, ultraviolet irradiated, inactivated RRV plus Ab−1; lane 3, RRV infection; lane 4, RRV plus Ab−1; lane 5, RRV plus Ab−2; lane 6, RRV plus Ab−3; lane 7, RRV plus Ab−4; lane 8, 1 μg/ml LPS; lane 9, 40ng/ml PMA; lane 10, 20 μg/ml poly IC. RRV infection was performed at MOI = 1. RNA was analyzed by Northern blot using a 32P-labeled probe derived from PAI-2 cDNA. The lower panel represents the level of 18S rRNA present in each lane as a control for loading.
Discussion
This study demonstrated that intracellular (Fig. 4) PAI-2–protected cells from cytopathic alphavirus and adenovirus infections, but not influenza or vaccinia virus infections (Figs. 1 and 2). The protection against CPE was associated with a PAI-2–mediated induction of constitutive bioactive autocrine IFN-α/β production (Figs. 5 and 6), which primed cells for rapid induction of antiviral genes (Fig. 7). Thus, after virus infection of PAI-2–transfected HeLa cells, antiviral genes were rapidly induced (Fig. 7) and this was associated with a rapid inhibition of viral replication (Figs. 2 B and 3), development of antiviral resistance (Fig. 2 B), and protection against CPE (Figs. 1 and 2 A). In contrast, virus infection of control cells did not result in IFN-α/β or antiviral gene induction and was associated with rapid viral replication and cell death. This represents the first report of an association between a mammalian serpin and the regulation of IFN-α/β or IFN-α/β–inducible genes and provides further evidence for cross-talk between the TNF-α and IFN-α/β signaling pathways (44, 45). In addition, PAI-2 was shown to be a virus response gene in macrophages (Fig. 8), further supporting the latter contention. Protection from TNF-α–induced apoptosis and virus-induced CPE are characteristics that cannot readily be ascribed to, or associated with, extracellular PAI-2 activity (Fig. 4), strongly indicating that the function of intracellular PAI-2 is entirely distinct from its extracellular role as a uPA inhibitor.
The reported role for PAI-2 in conferring protection against apoptosis mediated by TNF-α (11, 12) and Mycobacterium avium infection (13) and the reported induction of apoptosis in neural cells by Sindbis infection (25, 30) suggested that the PAI-2–induced protection seen in Fig. 1 was mediated by a mechanism involving the inhibition of apoptosis. However, a series of experiments did not support a role for apoptosis inhibition in the protection against virus-induced CPE (see Results). In addition, PAI-2 did not inhibit several caspases involved in the execution of apoptosis (reference 12 and our unpublished data). Instead, our data suggested that PAI-2–mediated protection occurred through a mechanism involving induction of low-level, autocrine production of IFN-α/β.
A series of data support the contention that PAI-2 expression induced low-level autocrine IFN-α/β activity, which primed cells for induction of IFN-α/β and/or antiviral genes. In summary, the evidence showed that (a) PAI-2–expressing cells, but not control cells, were primed for IFN-α/β production after poly IC treatment (Fig. 5 A); (b) S1b cells contained elevated mRNA for IFN-α/β (Fig. 5 B); (c) concentrated supernatants from S1b cells, but not A2/7 cells, conferred protection to A2/7 cells (Fig. 5 C); (d) long-term incubation of S1b cells with anti–IFN-α/β antibodies resulted in loss of protection against RRV CPE (Fig. 5 D); (e) S1b cells, but not A2/7 cells, showed constitutive and inducible activation of ISGF3 (Fig. 6 A), constitutive expression and nuclear localization of ISGF3γ (Fig. 6 B); and (f) S1b cells, but not A2/7 cells, showed rapid induction of the IFN-α/β–inducible antiviral gene OAS after alphaviral infection (Fig. 7). There is considerable precedence for IFN-α/β exposure to prime cells for a rapid antiviral response (35, 39), and for primed induction of IFN-α/β– inducible antiviral genes by virus to occur in the absence of further IFN-α/β production (see below). Antiviral genes like OAS are also known to be highly effective in inhibiting alphaviral replication in HeLa cells (32) and other virus infections generally (21). Importantly, vaccinia and influenza infections in HeLa cells are known to be resistant to IFN-α/β antiviral activity (32, 33), and PAI-2–expressing cells were not protected against CPE induced by these viruses (Fig. 1). In addition, the ability to establish a persistent infection (Fig. 2 B) is known to be dependent in many in vitro systems on activation of IFN-α/β–inducible antiviral genes (34). The reduction in the number of infected S1b cells seen after initial RRV infection (Fig. 2 B) also illustrated that S1b cultures rapidly developed antiviral resistance in response to RRV rather than being somehow innately resistant to infection by this virus.
Cells expressing PAI-2 produced high levels of IFN-α/β in response to dsRNA (poly IC) and might have been expected to produce IFN-α/β after RRV infection. However, viruses and poly IC can stimulate distinct transcription factor pathways (46) and IFN-α/β induction is known to be regulated by multiple independent positive and negative regulatory elements (21, 35). The exact mechanisms by which priming and the rapid onset of antiviral resistance might occur are not well understood. However, constitutive low-level autocrine IFN-α/β priming has been shown in several cases to be responsible for IFN-α/β–independent, rapid, virus-inducible antiviral resistance (47–49). IFN-α/β priming has been reported to lead to a significant enhancement of virus-inducible, but IFN-α/β–independent, transactivation of IRSFs (which include OAS) through a mechanism involving IRF-3 (47). Other virus inducible, IFN-α/β/ISGF3 independent pathways, which can rapidly induce antiviral resistance genes, are also reported to exist and involve the dsRNA-activated factors (48) and/or IRF-1 and p91 (49, 50).
The postulated causal link between cytoplasmic expression of the serpin, PAI-2, and low-level autocrine IFN-α/β production and the subsequent primed induction of antiviral resistance represents a potentially complex set of interactions between a number of signal transduction pathways. Whether PAI-2 affects the IFN-α/β pathway directly or indirectly or whether this effect is independent of the cell in which PAI-2 is expressed, remains unclear. The molecular mechanisms responsible for the primed condition are also poorly understood, although the primed condition appears to be remarkably stable, taking many days to decay after removal of IFN (40). IFN priming is well recognized in many cells, but priming may have distinct molecular and phenotypic consequences in different cell types and under different conditions (51). Despite these complexities, the two phenotypes of PAI-2–expressing cells, resistance to TNF-α–mediated apoptosis and induction of IFN-α/β priming, suggest that PAI-2 can influence a signaling pathway common to both the TNF-α and IFN-α/β pathways. Addition of IFN-α–2B to HeLa or A2/7 cells did not render them resistant to TNF-α–induced apoptosis (data not shown), indicating that protection of PAI-2–expressing cells against TNF-α–induced apoptosis was not a direct consequence of low-level IFN-α/β production. That cross-talk exists between the TNF-α and IFN-α/β pathways has recently been demonstrated (44, 45) and may involve double-stranded RNA activated protein kinase. One might speculate that the NF-κB signaling complex, which plays an important role in promoting survival after TNF-α activated signal transduction (52), may be the target of PAI-2 action as NF-κB is also the primary regulator of IFN-β synthesis (21). Production of both IFN-α and IFN-β might then occur via positive feedback through activation of ISGF3 (38). The demonstration of PAI-2–mediated influences on transcription factor pathways indicates that the levels of certain transcription factors might be regulated by the activity of specific cytoplasmic proteinases (53), which may in turn be controlled by specific proteinase inhibitors.
The ability of virus and poly IC to induce PAI-2 in the monocyte line, MonoMac6, suggests that PAI-2 induction may be a physiological response to viral RNA. Similar induction of PAI-2 expression has been reported after Dengue virus infection of primary monocytes (54). TNF-α is a primary inducer of PAI-2 (55), however, virus-induced autocrine TNF-α production by MonoMac6 cells is unlikely to cause PAI-2 induction in these cells, since RRV infection of MonoMac6 cells did not result in the release of detectable TNF-α (16). The presence of an IFN-γ activation sequence (GAS)–like element in the upstream promoter region of PAI-2 (56) might suggest that PAI-2 represents an IFN-α/β response gene, perhaps activated by IFN-α/β– activated STAT1–STAT2 heterodimers, which have recently been shown to bind to GAS elements (57).
PAI-2 thus appears to have a physiological intracellular role as a regulator of a cellular signaling pathway(s), which results in at least two phenotypes, resistance to TNF-α–mediated apoptosis, and induction of low-level autocrine IFN-α/β. For activated macrophages in an inflammatory site, such phenotypes would clearly be advantageous, conveying the ability to resist both autocrine TNF-α and to be primed for rapid antiviral responses. The other major site of intracellular PAI-2 expression is in epidermal keratinocytes (58), where synthesis of PAI-2 is constitutive. These cells may also express constitutive IFN-α/β (59) and are a major source of TNF-α after trauma to the skin (60). Constitutive PAI-2 expression in keratinocytes may thus have a similar role to that postulated for macrophages, priming these cells for rapid responses to invading pathogens and protecting them against autocrine TNF-α.
Viral induction of PAI-2 and the PAI-2–mediated establishment of a persistent infection may also point to a potential pathological role for PAI-2. Persistence of organisms in macrophages is implicated in the pathogenesis of several diseases including HIV (61) and certain chronic infectious arthritides, including epidemic polyarthritis, which is caused by RRV (16).
Acknowledgments
We wish to thank Julie Muddiman for technical assistance in this study. We also wish to thank Dr. Peter G. Parsons (Queensland Institute of Medical Research) and Dr. Paul Hertzog (Monash University, Victoria, Australia) for contribution of reagents and valuable discussions. We wish to thank Dr. Clive Bunn, Biotech Australia, Pty. Ltd., for purified recombinant PAI-2; and Schering-Plough, Pty. Ltd. for the supply of IFN– α-2B.
This work was supported financially by the following Australian organizations: The Queensland Cancer Fund, the National Health and Medical Research Council, the Australian Centre for International and Tropical Health and Nutrition, and the Queensland Health Arbovirus Research Fund. J.L. Dickinson was supported in part by the Dora Lush Post-Graduate Biomedical Scholarship from the National Health and Medical Research Council of Australia.
Abbreviations used in this paper
- CAT
chloramphenicol acetyl transferase
- CPE
cytopathic effect
- IRF
interferon response factor
- ISGF3
interferon-stimulated gene factor 3
- ISRE
interferon-stimulated response element
- MOI
multiplicity of infection
- OAS
oligoadenylate synthetase
- PAI-2
plasminogen activator inhibitor
- poly IC
polyinosinic-polycytidylic acid
- RRV
Ross River virus
- serpin
serine proteinase inhibitor
- TCID50
50% tissue culture infectivity dose
- uPA
urokinase-type plasminogen activator
References
- 1.Remold-O'Donnell E. The ovalbumin family of serpin proteins. FEBS Lett. 1993;315:105–108. doi: 10.1016/0014-5793(93)81143-n. [DOI] [PubMed] [Google Scholar]
- 2.Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, Pickup DJ. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1β converting enzyme. Cell. 1992;69:597–604. doi: 10.1016/0092-8674(92)90223-y. [DOI] [PubMed] [Google Scholar]
- 3.Antalis TM, Clark MA, Barnes T, Lehrbach PR, Devine PL, Schevzov G, Goss NH, Stephens RW, Tolstoshev P. Cloning and expression of a cDNA coding for a human monocyte-derived plasminogen activator inhibitor. Proc Natl Acad Sci USA. 1988;85:985–989. doi: 10.1073/pnas.85.4.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou Z, Anisowicz A, Hendrix MJC, Thor A, Neveu M, Sheng S, Rafidi K, Seftor E, Sager R. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526–529. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]
- 5.Scott FL, Coughlin PB, Bird C, Cerruti L, Hayman JA, Bird P. Protease inhibitor 6 cannot be secreted, which suggests it's a new type of cellular serpin. J Biol Chem. 1996;271:1605–1612. doi: 10.1074/jbc.271.3.1605. [DOI] [PubMed] [Google Scholar]
- 6.Sun J, Bird CH, Sutton V, McDonald L, Coughlin PB, DeJong TA, Trapani JA, Bird PI. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J Biol Chem. 1996;271:27802–27809. doi: 10.1074/jbc.271.44.27802. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem. 1997;272:7797–7800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 8.Kettle S, Alcami A, Khanna A, Ehret R, Jassoy C, Smith GL. Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1β–converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1β–induced fever. J Gen Virol. 1997;78:677–685. doi: 10.1099/0022-1317-78-3-677. [DOI] [PubMed] [Google Scholar]
- 9.Kruithof EKO, Baker MS, Bunn CL. Biological and clinical aspects of plasminogen activator inhibitor type 2. Blood. 1995;86:4007–4024. [PubMed] [Google Scholar]
- 10.Von Heijne G, Liljestrom P, Mikus P, Andersson H, Ny T. The efficiency of the uncleaved secretion signal in the plasminogen activator inhibitor type 2 protein can be enhanced by point mutations that increase its hydrophobicity. J Biol Chem. 1991;266:15240–15246. [PubMed] [Google Scholar]
- 11.Kumar S, Baglioni C. Protection from tumour necrosis factor–mediated cytolysis by overexpression of plasminogen activator inhibitor type-2. J Biol Chem. 1991;266:20960–20964. [PubMed] [Google Scholar]
- 12.Dickinson JL, Bates EJ, Ferrante A, Antalis TM. Plasminogen activator inhibitor type 2 inhibits tumor necrosis factor alpha–induced apoptosis. Evidence for an alternate biological function. J Biol Chem. 1995;270:27894–27904. doi: 10.1074/jbc.270.46.27894. [DOI] [PubMed] [Google Scholar]
- 13.Gan H, Newman GW, Remold HG. Plasminogen activator inhibitor type 2 prevents programmed cell death of human macrophages infected with Mycobacterium avium, serovar 4. J Immunol. 1995;155:1304–1315. [PubMed] [Google Scholar]
- 14.Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziegler-Heitbrock HWL, Thiel E, Fuetterer A, Herzog V, Wirtz A, Riethmueller U. Establishment of a human cell line (MonoMac6) with characteristics of mature monocytes. Int J Cancer. 1988;41:456–461. doi: 10.1002/ijc.2910410324. [DOI] [PubMed] [Google Scholar]
- 16.Linn ML, Aaskov J, Suhrbier A. Antibody dependent enhancement and persistence in macrophages of an arbovirus associated with arthritis. J Gen Virol. 1996;77:407–412. doi: 10.1099/0022-1317-77-3-407. [DOI] [PubMed] [Google Scholar]
- 17.Piper RC, Slot JW, Li G, Stahl PD, James DE. Recombinant Sindbis virus as an expression system for cell biology. Methods Cell Biol. 1994;43:55–78. doi: 10.1016/s0091-679x(08)60598-1. [DOI] [PubMed] [Google Scholar]
- 18.Parsons PG, Lean J, Khoo SK, Lark J. Effects of adriamycin and etoposide on the replication of adenovirus 5 in sensitive and resistant human tumour cells. Biochem Pharmacol. 1989;38:31–37. doi: 10.1016/0006-2952(89)90145-7. [DOI] [PubMed] [Google Scholar]
- 19.Boyle DB, Coupar BE, Both GW. Multiple-cloning-site plasmids for the rapid construction of recombinant poxviruses. Gene. 1985;35:169–177. doi: 10.1016/0378-1119(85)90169-6. [DOI] [PubMed] [Google Scholar]
- 20.Antalis TM, Godbolt D. Isolation of intact nuclei from hematopoietic cell types. Nucleic Acids Res. 1991;19:4301. doi: 10.1093/nar/19.15.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalvakolanu DV, Borden EC. An overview of the interferon system: signal transduction and mechanisms of action. Cancer Invest. 1996;14:25–53. doi: 10.3109/07357909609018435. [DOI] [PubMed] [Google Scholar]
- 22.Demczuk S, Harbers M, Vennström B. Identification and analysis of all components of a gel retardation assay by combination with immunoblotting. Proc Natl Acad Sci USA. 1993;90:2574–2578. doi: 10.1073/pnas.90.7.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Somoza N, Vargas F, Roura-Mir C, Vives-Pi M, Fernandez-Figueras MT, Ariza A, Gomis R, Bragado R, Marti M, Jaraquemada D, Pujol-Borrell R. Pancreas in recent onset insulin-dependent diabetes mellitus. Changes in HLA, adhesion molecules and autoantigens, restricted T cell receptor V beta usage, and cytokine profile. J Immunol. 1994;153:1360–1377. [PubMed] [Google Scholar]
- 24.Merlin G, Chebath J, Benech P, Metz R, Revel M. Molecular cloning and sequence of partial cDNA for interferon-induced (2′–5′) oligo(A) synthetase mRNA from human cells. Proc Natl Acad Sci USA. 1983;80:4904–4908. doi: 10.1073/pnas.80.16.4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine B, Huang Q, Isaacs JT, Reed JC, Griffin DE, Hardwick JM. Conversion of lytic to persistent alphavirus infection by the bcl-2cellular oncogene. Nature. 1993;361:739–742. doi: 10.1038/361739a0. [DOI] [PubMed] [Google Scholar]
- 26.Chiou SK, Tseng CC, Rao L, White E. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J Virol. 1994;68:6553–6566. doi: 10.1128/jvi.68.10.6553-6566.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takizawa T, Matsukawa S, Higuchi Y, Nakamura S, Nakanishi Y, Fukuda R. Induction of programmed cell death (apoptosis) by influenza virus infection in tissue culture cells. J Gen Virol. 1993;74:2347–2355. doi: 10.1099/0022-1317-74-11-2347. [DOI] [PubMed] [Google Scholar]
- 28.Ink BS, Gilbert CS, Evan GI. Delay of vaccinia virus–induced apoptosis in nonpermissive Chinese hamster ovary cells by the cowpox virus CHOhr and adenovirus E1B 19K genes. J Virol. 1995;69:661–668. doi: 10.1128/jvi.69.2.661-668.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin JH, Weir RC, Dalgarno L. Replication of standard and defective Ross River virus in BHK cells: patterns of viral RNA and polypeptide synthesis. Arch Virol. 1979;61:87–103. doi: 10.1007/BF01320594. [DOI] [PubMed] [Google Scholar]
- 30.Lin KI, Lee SH, Narayanan R, Baraban JM, Hardwick JM, Ratan RR. Thiol agents and Bcl-2 identify an alphavirus-induced apoptotic pathway that requires activation of the transcription factor NF-κB. J Cell Biol. 1995;131:1149–1161. doi: 10.1083/jcb.131.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen PH, Cressey LI, Gjertsen BT, Madsen P, Mellgren G, Hokland P, Gliemann J, Doskeland SO, Lanotte M, Vintermyr OK. Cleaved intracellular plasminogen activator inhibitor 2 in human myeloleukaemia cells is a marker of apoptosis. Br J Cancer. 1994;70:834–840. doi: 10.1038/bjc.1994.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munoz A, Carrasco L. Action of human lymphoblastoid interferon on HeLa cells infected with RNA-containing animal viruses. J Gen Virol. 1984;65:377–390. doi: 10.1099/0022-1317-65-2-377. [DOI] [PubMed] [Google Scholar]
- 33.Beattie E, Denzler KL, Tartaglia J, Perkus ME, Paoletti E, Jacobs BL. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J Virol. 1995;69:499–505. doi: 10.1128/jvi.69.1.499-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedman RM, Ramseur JM. Mechanisms of persistent infections by cytopathic viruses in tissue culture. Arch Virol. 1979;60:83–103. doi: 10.1007/BF01348025. [DOI] [PubMed] [Google Scholar]
- 35.King P, Goodbourn S. The beta-interferon promoter responds to priming through multiple independent regulatory elements. J Biol Chem. 1994;269:30609–30615. [PubMed] [Google Scholar]
- 36.Bandyopadhyay SK, Sen GC. Role of protein phosphorylation in activation of interferon-stimulated gene factors. J Biol Chem. 1992;267:6389–6395. [PubMed] [Google Scholar]
- 37.Bandyopadhyay SK, Kalvakolanu DVR, Sen GC. Gene induction by interferons: functional complementation between trans-acting factors induced by alpha interferon and gamma interferon. Mol Cell Biol. 1990;10:5055–5063. doi: 10.1128/mcb.10.10.5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoneyama M, Suhara W, Fukuhara Y, Sato M, Ozato K, Fujita T. Autocrine amplification of type I interferon gene expression mediated by interferon stimulated gene factor 3 (ISGF3) J Biochem. 1996;120:160–169. doi: 10.1093/oxfordjournals.jbchem.a021379. [DOI] [PubMed] [Google Scholar]
- 39.Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, Williams BR, Aguet M, Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO (Eur Mol Biol Organ) J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L. Cell to cell transmission of the “priming” effect on the induction of human fibroblast interferon. J Interferon Res. 1985;5:111–119. doi: 10.1089/jir.1985.5.111. [DOI] [PubMed] [Google Scholar]
- 41.Schwartz BS, Bradshaw JD. Regulation of plasminogen activation inhibitor mRNA levels in lipopolysaccharide-stimulated human monocytes. Correlation with production of the protein. J Biol Chem. 1992;267:7089–7094. [PubMed] [Google Scholar]
- 42.Antalis TM, Dickinson JL. Control of plasminogen-activator inhibitor type 2 gene expression in the differentiation of monocytic cells. Eur J Biochem. 1992;205:203–209. doi: 10.1111/j.1432-1033.1992.tb16769.x. [DOI] [PubMed] [Google Scholar]
- 43.Schleuning WD, Metcalf RL, Hession C, Rothenbuehler R, Shaw A, Kruithof EKO. Plasminogen activator inhibitor 2: regulation of gene transcription during phorbol ester–mediated differentiation of U-937 human histiocytic lymphoma cells. Mol Cell Biol. 1987;7:4564–4567. doi: 10.1128/mcb.7.12.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeung MC, Liu J, Lau AS. An essential role for the interferon-inducible, double-stranded RNA-activated protein kinase PKR in the tumor necrosis factor–induced apoptosis in U937 cells. Proc Natl Acad Sci USA. 1996;93:12451–12455. doi: 10.1073/pnas.93.22.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruby J, Bluethmann H, Peschon JJ. Antiviral activity of tumor necrosis factor (TNF) is mediated via p55 and p75 TNF receptors. J Exp Med. 1997;186:1591–1596. doi: 10.1084/jem.186.9.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsuyama T, Kimura T, Kitagawa M, Pfeffer K, Kawakami T, Watanabe N, Kundig TM, Amakawa R, Kishihara K, Wakeham A, et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type-1 IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- 47.Au WC, Moore PA, Lowther W, Juang YT, Pitha PM. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc Natl Acad Sci USA. 1995;92:11657–11661. doi: 10.1073/pnas.92.25.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daly C, Reich NC. Characterisation of specific DNA-binding factors activated by double stranded RNA as positive regulators of interferon α/β–stimulated genes. J Biol Chem. 1995;270:23739–23746. doi: 10.1074/jbc.270.40.23739. [DOI] [PubMed] [Google Scholar]
- 49.Bandyopadhyay SK, Leonard GT, Bandyopadhyay T, Stark GR, Sen GC. Transcriptional induction by double stranded RNA is mediated by interferon-stimulated gene factor 3. J Biol Chem. 1995;270:19624–19629. doi: 10.1074/jbc.270.33.19624. [DOI] [PubMed] [Google Scholar]
- 50.Pine R. Constitutive expression of an ISGF2/IRF1 transgene leads to interferon-independent activation of interferon-inducible genes and resistance to virus infection. J Virol. 1992;66:4470–4478. doi: 10.1128/jvi.66.7.4470-4478.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meldrum DR, Cleveland JC, Jr, Moore EE, Patrick DA, Banerjee A, Harken AH. Adaptive and maladaptive mechanisms of cellular priming. Ann Surg. 1997;226:587–598. doi: 10.1097/00000658-199711000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α–induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 53.Whiteside ST, King P, Goodbourn S. A truncated form of the IRF-2 transcription factor has the properties of a postinduction repressor of interferon-beta gene expression. J Biol Chem. 1994;269:27059–27065. [PubMed] [Google Scholar]
- 54.Krishnamurti C, Wahl LM, Alving BM. Stimulation of plasminogen activator inhibitor activity in human monocytes infected with dengue virus. Am J Trop Med Hyg. 1989;40:102–107. doi: 10.4269/ajtmh.1989.40.102. [DOI] [PubMed] [Google Scholar]
- 55.Metcalf RL, Kruithof EKO, Schleuning W-D. Plasminogen activator inhibitor 1 and 2 are tumor necrosis factor/cachectin-responsive genes. J Exp Med. 1988;168:751–759. doi: 10.1084/jem.168.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Antalis TM, Godbolt D, Donnan KD, Stringer BW. Southwestern blot mapping of potential regulatory proteins binding to the DNA encoding plasminogen activator inhibitor type 2. Gene. 1993;134:201–208. doi: 10.1016/0378-1119(93)90094-j. [DOI] [PubMed] [Google Scholar]
- 57.Li XX, Leung S, Qureshi S, Darnell JE, Stark GR. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon alpha. J Biol Chem. 1996;271:5790–5794. doi: 10.1074/jbc.271.10.5790. [DOI] [PubMed] [Google Scholar]
- 58.Jensen PJ, Wu Q, Janowitz P, Ando Y, Schechter NM. Plasminogen activator inhibitor type 2: an intracellular keratinocyte differentiation product that is incorporated into the cornified envelope. Exp Cell Res. 1995;217:65–71. doi: 10.1006/excr.1995.1064. [DOI] [PubMed] [Google Scholar]
- 59.Yaar M, Palleroni AV, Gilchrest BA. Normal human epidermis contains an interferon-like protein. J Cell Biol. 1986;103:1349–1354. doi: 10.1083/jcb.103.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cumberbatch M, Kimber I. Tumour necrosis factor–alpha is required for accumulation of dendritic cells in draining lymph nodes and for optimal contact sensitization. Immunology. 1995;84:31–35. [PMC free article] [PubMed] [Google Scholar]
- 61.Angelici E, Contini C, Romani R, Epifano O, Serra P, Canipari R. Production of plasminogen activator and plasminogen activator inhibitors by alveolar macrophages in control subjects and AIDS patients. AIDS. 1996;10:283–290. doi: 10.1097/00002030-199603000-00007. [DOI] [PubMed] [Google Scholar]