

Abstract
The signal pathways that control effector function in human natural killer (NK) cells are little known. In this study, we have identified the critical role of the mitogen-activated protein kinase (MAPK) pathway in NK lysis of tumor cells, and this pathway may involve the mobilization of granule components in NK cells upon interaction with sensitive tumor target cells. Evidence was provided by biological, biochemical, and gene transfection methods. NK cell binding to tumor cells for 5 min was sufficient to maximally activate MAPK/extracellular signal–regulatory kinase 2 (ERK2), demonstrated by its tyrosine phosphorylation and by its ability to function as an efficient kinase for myelin basic protein. MAPK activation was achieved in NK cells only after contact with NK-sensitive but not NK-resistant target cells. In immunocytochemical studies, cytoplasmic perforin and granzyme B were both maximally redirected towards the tumor contact zone within 5 min of NK cell contact with tumor cells. A specific MAPK pathway inhibitor, PD098059, could block not only MAPK activation but also redistribution of perforin/granzyme B in NK cells, which occur upon target ligation. PD098059 also interfered with NK lysis of tumor cells in a 5-h 51Cr-release assay, but had no ability to block NK cell proliferation. Transient transfection studies with wild-type and dominant-negative MAPK/ERK2 genes confirmed the importance of MAPK in NK cell lysis. These results document a pivotal role of MAPK in NK effector function, possibly by its control of movement of lytic granules, and clearly define MAPK involvement in a functional pathway unlinked to cell growth or differentiation.
Keywords: natural killer cell, mitogen-activated protein kinase, perforin, granzyme B, signal transduction
Cytotoxicity is a key strategy used by the immune system to eliminate abnormal cells, be they virus- or parasite-infected or transformed (1–3). NK cells play an important role in this process by their ability to recognize MHC class I–negative target cells, which escape recognition by specific cytotoxic T cells. The mechanisms controlling NK cell cytotoxicity are gradually being elucidated but they remain fragmentary. In addition to direct lysis of target cells, NK cells also display CD16, which allows for their participation in antibody-dependent cell cytotoxicity of Ig-coated tumor cells. Intracellular events linked to FcR triggering in NK cells have been studied more extensively, and activation of protein tyrosine kinases (PTKs)1 appears to be the initial event (4–6). Rapid phosphorylation of p56lck, TCR-ζ, Zap70, and Syk70 have all been reported. This initial event triggered by FcR cross-linking also leads to phosphatidylinositol (PI)-3 kinase and phospholipase C (PLC)–γ activation that, in turn, sets in motion the inositol-1,4,5-triphosphate cascade and mobilization of intracellular calcium (7, 8). However, the distal events that lead ultimately to target lysis are not yet known.
For direct lysis of target cells, several investigators have contributed to the current concept that this action is the result of a balance of stimulatory and inhibitory receptors on NK cells (9). The killer cell–inhibitory receptors and CD94/ NKG2 heterodimers in man and the Ly49 family of receptors in mice negatively control the lytic event (10–13). Upon ligation with MHC class I molecules on the target cells, these inhibitory receptors suppress the lytic process through their association with protein tyrosine phosphatases, PTP-1C and -1D, which may dephosphorylate critical tyrosine kinases (14–17). Some information on the stimulatory receptors and their ligands is currently available (18), but little is known of the internal signal events except that a calcium and cAMP–dependent pathway leading to inositol triphosphate generation is activated upon NK cell binding to its sensitive target (19–22). What other signals might follow are not yet determined.
Thus, previous studies have concentrated on signal events proximal to CD16 or NK receptors. Our goal in this study was to identify downstream events in NK cells that lead to target cell lysis. It is well known that both perforin and serine proteases such as granzyme B are released from NK cells upon target cell contact (23, 24). The cooperation of these two molecules results in lysis and apoptosis of the target cell (25–27). Perforin polymerization and insertion into the target membrane are reported to cause osmotic leakage and to provide target cell entry of granzyme B, which triggers the caspase system to lead to DNA fragmentation (28, 29). Our findings demonstrate that NK lysis of tumor target cells is critically regulated by functional mitogen-activated protein kinase (MAPK)/extracellular signal–regulated kinase (ERK), identified to be MAPK/ERK2. This family of signal molecules was first identified as a serine/threonine kinase belonging to the signal cascade downstream of growth factor receptors, and has been studied largely for its role in cell growth and differentiation (30–36). We report here that the control of NK lysis by ERK2 may be at the level of perforin and granzyme B mobilization, and that ERK2 is involved in the redistribution of these granule components to the contact zone between NK cells and target cells.
Materials and Methods
Cells.
A human NK leukemia cell line, YT (a gift from Dr. Eckhard Podack, University of Miami, Miami, FL), has been used by us previously to investigate cytolytic function (37). Its ability to serve as an NK effector cell against a human B lymphoma cell line, Raji (American Type Culture Collection, Rockville, MD), was exploited in this study. Another NK-sensitive cell line, K562, used for measuring function in fresh NK cells, and two NK-resistant tumor cell lines, Jurkat and HL60, were all obtained from ATCC. All cells were cultured in RPMI 1640 containing 10% FCS with 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 mM Hepes buffer (GIBCO BRL, Gaithersburg, MD), hereafter referred to as “medium.”
Isolation of LGL from Peripheral Blood.
PBMCs were first obtained by Ficoll-Hypaque density centrifugation of leukocyte buffy coats obtained from normal volunteers at the Southwest Florida Blood Bank (Tampa, FL). The mononuclear cells were washed with PBS twice and allowed to adhere to plastic for 1 h at 37°C in medium. The recovered nonadherent cells were further depleted of adherent cells by incubation on nylon wool columns for 30 min at 37°C. The cells passing through the columns were then placed on a four-step discontinuous gradient with a range of Percoll from 40 to 47.5%, as described previously (3). The cells recovered from 42.5–45.0% Percoll were assessed visually for LGL morphology on Giemsa-stained cytocentrifuged slides to contain routinely 75–90% LGL, and were used to test for NK function.
Chemical Reagents and Antibodies.
The MAPK kinase (MEK)1 inhibitor PD098059 (New England Biolabs Inc., Beverly, MA) was dissolved in DMSO and stored in aliquots at −20°C (38, 39). The stock was diluted to the desired working concentration in RPMI 1640 immediately before use. mAbs against panMAPK/ ERK, ERK1, ERK2, and ERK3 were purchased from Transduction Laboratories (Lexington, KY). Rabbit antiactive MAPK (AMAPK) generated against the phosphorylated TEY epitope of MAPK was purchased from New England Biolabs Inc. Monoclonal antiphosphotyrosine, 4G10, rabbit antilyn, and rabbit anti-MAPK were purchased from Upstate Biotechnology Inc. (Lake Placid, NY). mAb against granzyme B (2C5) was generated as described previously (40). mAb against perforin was purchased from Endogen, Inc. (Woburn, MA).
Cytotoxicity Assay.
A 51Cr-release assay was performed as described previously, using Raji tumor cells as targets for YT effector cells, and K562 tumor cells for fresh LGL (3, 37). Briefly, target tumor cells were labeled with 200 μCi of Na [51Cr]chromate (Amersham Corp., Arlington Heights, IL) in 0.2 ml of medium at 37°C in a 5% CO2 atmosphere for 1 h. The cells were then washed three times and added to effector cells at 5 × 103 cells/ well in 96-well round-bottomed microplates, resulting in E/T ratios ranging from 50:1 to 3:1 in a final volume of 0.2 ml in each well. After 5 h incubation at 37°C, 100 μl of culture supernatants was harvested and counted in a γ counter. The percent specific 51Cr release was determined as described previously (3, 37) according to the equation [(experimental cpm − spontaneous cpm)/total cpm incorporated] × 100. All determinations were done in triplicate, and the SEM of all assays was calculated and was typically ∼5% of the mean or less.
In assays using the MEK inhibitor, YT cells or fresh LGL at 2.5 × 106 cells/ml were incubated for 1 h at 37°C with serum-free RPMI 1640, serial dilutions of PD098059, or an equal amount of DMSO used to dilute the highest concentration of PD098059, in a total volume of 5 ml in a 25-cm2 tissue culture flask. The cells were then washed twice with medium and plated into triplicate wells of a 96-well microplate at various dilutions before 51Cr-labeled Raji tumor cells were added.
Thymidine Uptake Assays.
YT cells treated with RPMI 1640, PD098059, or DMSO as described above for 1 h at 37°C were washed twice with medium. The YT cells were then cultured in triplicate wells in a 96-well flat-bottomed plate (5 × 104 cells/100 μl/well) for 24 h at 37°C and pulsed with [3H]thymidine (1 μCi/ well; Amersham Corp.) for another 6 h of culture before harvest. The incorporated radioactivity was measured using an automated liquid scintillation counter.
Fixation of Target Cells.
Raji, Jurkat, or HL60 tumor cells were washed with PBS once and incubated with 1% paraformaldehyde (methanol-free) in PBS, pH 7.4, on ice for 30 min. The cells were then washed four times with PBS to remove all paraformaldehyde.
Immunoprecipitation and Western Blotting.
YT cells were cultured in serum-free medium for 4 h at 37°C before use in order to reduce the background phosphorylation. The rested YT cells (107/ml) were then treated with either serum-free RPMI 1640, DMSO, or PD098059 for 1 h at 37°C, washed, and mixed with an equal number of paraformaldehyde-fixed Raji target cells. The cells were pelleted rapidly at 1,000 rpm in a microcentrifuge at 4°C followed by incubation for 0–15 min at 37°C. Then the cells were solubilized by incubation at 4°C for 30 min in 1% NP-40, 10 mM Tris, 140 mM NaCl, 0.1 mM PMSF, 10 mM iodoacetamide, 50 mM Na fluoride, 1 mM EDTA, 0.4 mM Na orthovanadate, 10 μg/ml leupeptin, 10 μg/pepstatin, and 10 μg/ml aprotinin. Cell lysates were centrifuged at 12,000 g for 15 min to remove nuclei and cell debris. For immunoprecipitation, lysates were precleared of nonspecific binding proteins via incubation with normal rabbit serum or mouse IgG for 1 h at 4°C, followed by formalin-fixed Staphylococcus aureus (Pansorbin; Calbiochem Corp., La Jolla, CA) or protein A–Sepharose beads (GIBCO BRL) for 1 h at 4°C, respectively. Lysates containing 1.0 mg protein/ml were then incubated with 5–10 μg of the indicated antibody for 2 h. Immune complexes were collected with protein A–Sepharose beads and washed three times with washing buffer (0.1% NP-40, 10 mM Tris, 140 mM NaCl, 0.1 mM PMSF, 10 mM iodoacetamide, 50 mM Na fluoride, 1 mM EDTA, 0.4 mM Na orthovanadate). Samples were then boiled for 5 min in loading buffer and separated by 10% SDS-PAGE, followed by Western blot analysis with the desired antibody. The proteins were detected by the enhanced chemiluminescence detection system (ECL; Amersham Corp.).
In Vitro Kinase Assays.
Cell lysates prepared as described above were precleared and immunoprecipitated with 5 μg polyclonal anti-panMAPK/ERK (Upstate Biotechnology Inc.). The immunoprecipitated proteins were collected using protein A–Sepharose beads, washed, and incubated in a volume of 40 μl with 10 μCi of [γ-32P]ATP in 30 mM Tris (pH 8.0), 20 mM MgCl2, 2 mM MnCl2, and 10 μM ATP for 30 min at 30°C with 5 μg of myelin basic protein (MBP; Upstate Biotechnology Inc.) as a substrate for MAPK (34). The reaction was terminated with sample buffer by quick centrifugation, and the mixture was electrophoresed on a 15% SDS-polyacrylamide gel rather than the 10% gel used above, in order to detect the small molecular mass of MBP, 18 kD. After drying the gel, 32P-labeled phosphoproteins were detected by autoradiography.
Transient Transfection.
The CMV promoter–based expression vector pLNCAL, encoding wild-type or mutant dominant-negative human ERK2, was used as described previously (41, 42). The plasmids contained either a catalytically inactive, kinase-deficient (KD) gene, where the critical lysine at position 52 was substituted for arginine (K52R), or the ERK2 gene that has the Thr and Tyr residues within the TEY motif substituted with alanine and phenylalanine (TAYF) or replaced by glutamic acid (TEYE). YT cells were transfected using the PerFect Lipids transfection kit (Invitrogen Corp., San Diego, CA), and transfection efficiency in YT cells was first optimized using the control plasmid pcDNA3.1/ His/lacZ (GAL) included in the kit. Using β-galactosidase expression from the control GAL plasmid in YT cells, it was determined that 4 μg/ml of plasmid DNA provided 35–40% transfection with the least toxicity. Using the same conditions, YT cells were transfected with 4 μg/ml of the plasmid containing each of the various MAPK constructs. 24 h later, the transfected YT cells were checked for viability by trypan blue exclusion, washed, and adjusted to the appropriate concentrations of viable cells before testing for cytotoxicity against Raji tumor cells in a 5-h 51Cr-release assay. Viability of the transfected cells via this liposome method was routinely >90%. An equal aliquot of YT cells, left untransfected but incubated and treated in an identical manner, was included as a control.
Immunostaining.
YT cells pretreated with 100 μM PD098059 or DMSO for 1 h at 37°C and washed were added to Raji cells at a 1:1 ratio in a total volume of 100 μl, spun rapidly at 1,000 rpm for 1 min in a cold microcentrifuge, and incubated for 0–5 min at 37°C. The cells were then centrifuged onto a microscope slide and fixed at −20°C with methanol/acetone (3:1) for 20 min (43). The slides were air-dried and rehydrated for 2 h in several changes of PBS. Polyclonal rabbit antilyn kinase or monoclonal anti–human perforin or granzyme B was diluted 1:200 with 0.1% NP-40 in 1% BSA in PBS and applied to the slide for 1 h at room temperature. After several washes with PBS for 2 h, the slides were incubated for 25 min at room temperature with goat anti– rabbit IgG tetramethyl rhodamine isothiocyanate (TRITC)-labeled antibody (Sigma Chemical Co., St. Louis, MO) diluted 1:80, or goat anti–mouse Ig FITC-labeled antibody (Sigma Chemical Co.) diluted 1:100 in 0.1% NP-40 in PBS containing 1% BSA. After several washes in PBS, the slides were covered with coverslips in mounting media of antifade/4,6-diamino-2-phenylindole (DAPI). Immunofluorescence was observed with an Orthoplan 2 microscope (Ernst Leitz GmbH, Wetzlar, Germany), and images were captured by a charged coupled device (CCD) camera with the Smart Capture program (Vysis Inc., Downers Grove, IL). On each slide, 100 YT–Raji conjugates were evaluated for perforin or granzyme B mobilization.
Several controls were performed, i.e., YT cells alone or Raji tumor cells alone stained with no primary antibody but with FITC-labeled goat anti–mouse Ig or with TRITC-labeled goat anti–rabbit IgG, to check for nonspecific binding of the secondary antibodies. No nonspecific binding of these antibodies was detected in YT cells or Raji tumor cells in any experiment performed, and these results were omitted from Figs. 8 and 9 for clarity.
Figure 8.
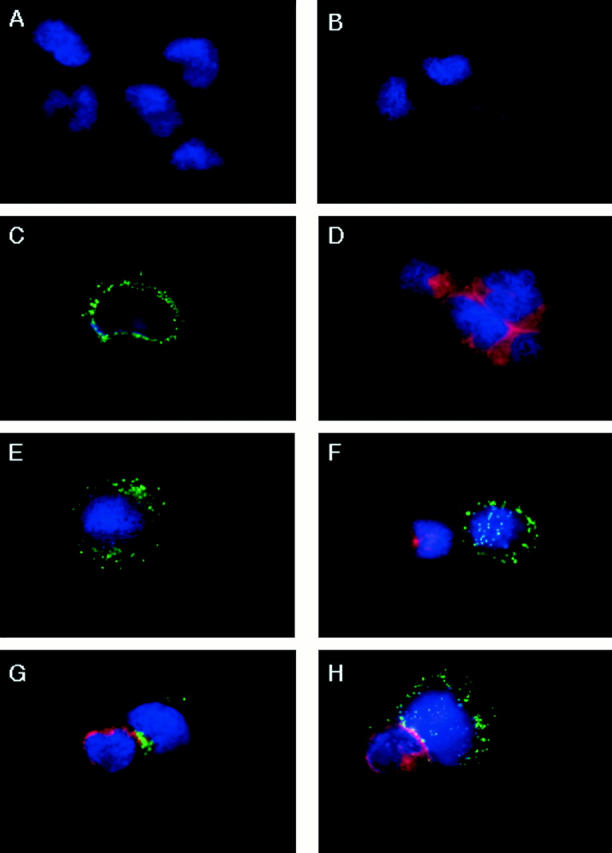
Inhibition of perforin redistribution in NK cells by PD098059. YT cells pretreated with DMSO or 100 μM of PD098059 for 1 h at 37°C were incubated with an equivalent number of Raji tumor cells for 0–5 min at 37°C. The cells were then cytospinned onto microscope slides and stained with anti–perforin-FITC and/or anti–lyn-TRITC. (A) YT cells stained with anti–lyn-TRITC, (B) Raji tumor cells stained with anti–perforin-FITC, (C) YT cells stained with anti–perforin-FITC, (D) Raji cells stained with anti–lyn-TRITC, (E) DMSO-pretreated YT cells stained with anti–perforin-FITC, (F) a DMSO-pretreated YT cell that had bound a Raji tumor cell at 0 min, stained with anti–perforin-FITC/anti–lyn-TRITC, (G) a DMSO-pretreated YT cell that had bound a Raji tumor cell for 5 min at 37°C, stained with anti–perforin-FITC/anti–lyn-TRITC, and (H) a PD098059-pretreated YT cell that had bound a Raji tumor cell for 5 min at 37°C, stained with anti–perforin-FITC/anti–lyn-TRITC.
Figure 9.
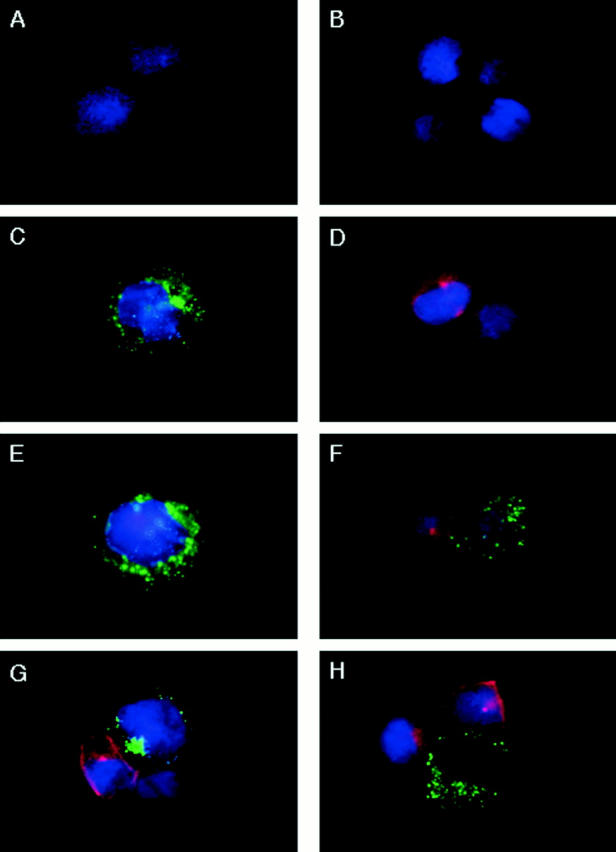
Inhibition of granzyme B redistribution in NK cells by PD098059. YT cells pretreated with DMSO or 100 μM of PD098059 for 1 h at 37°C were incubated with an equivalent number of Raji tumor cells for 0–5 min at 37°C. The cells were then cytospinned onto microscope slides and stained with anti–granzyme B-FITC and/or anti–lyn-TRITC. (A) YT cells stained with anti–lyn-TRITC, (B) Raji tumor cells stained with anti– granzyme B-FITC, (C) YT cells stained with anti–granzyme B-FITC, (D) Raji cells stained with anti–lyn-TRITC, (E) DMSO-pretreated YT cells stained with anti–granzyme B-FITC, (F) a DMSO-pretreated YT cell that had bound a Raji tumor cell for 0 min, stained with anti–granzyme B-FITC/ anti–lyn-TRITC, (G) a DMSO-pretreated YT cell that had bound a Raji tumor cell for 5 min at 37°C, stained with anti–granzyme B-FITC/anti–lyn-TRITC, and (H) a PD098059-pretreated YT cell that had bound a Raji tumor cell for 5 min at 37°C, stained with anti–granzyme B-FITC/anti–lyn-TRITC.
Results
Suppression of NK Lysis of Tumor Cells by Inhibition of MAPK.
In our first attempt to examine MAPK involvement in NK effector function, we tested the effect of the specific MAPK pathway inhibitor PD098059 on lysis of Raji tumor cells by the NK cell line, YT. PD098059 was identified initially to inhibit the unphosphorylated form of MEK1 and a constitutively active mutant of MEK1, and subsequently to inhibit the activation of MEK by c-Raf or MEK kinase 1 but not the activation of MAPK kinase 4 by MEK kinase 1 (38, 39). YT cells were pretreated with a range of concentrations of PD098059 from 10 to 200 μM for 1 h at 37°C before incubation with 51Cr-labeled Raji tumor cells for 5 h at 37°C to test for lysis. Representative data from one of six experiments are shown in Fig. 1 A. A dose-dependent inhibition by PD098059 was evident at all the E/T ratios tested. Marked inhibition was seen with 50 μM of PD098059, with almost complete inhibition achieved with 200 μM. All cells under each treatment remained 100% viable by assessment of trypan blue exclusion, indicating that the inhibitor was not nonspecifically toxic. DMSO, used as a vehicle for PD098059 solubilization, also had no effect. Thus, the NK effector function can be interfered with by a MAPK pathway inhibitor. To confirm that fresh NK cells require this same pathway for lysis of tumor cells, we also tested the effect of PD098059 on highly enriched Percoll-fractionated LGL from normal peripheral blood (Fig. 1 B). Similar inhibition of lytic function was seen with LGL treated with the same dose range of PD098059. This result was reproducible with LGL from two other donors (data not shown).
Figure 1.


Inhibition of NK lysis by PD098059. (A) YT cells untreated or treated 1 h at 37°C with 10–200 μM of the MEK1 inhibitor PD098059 or an amount of DMSO used to dilute the highest concentration of PD098059 were tested in triplicate wells for lysis of 51Cr-labeled Raji tumor cells at the E/T ratios indicated. (B) Highly enriched human LGL, freshly isolated by Percoll gradient centrifugation of nonadherent PBMCs, were preincubated with PD098059 at the indicated concentrations for l h at 37°C before testing for lysis against 51Cr-labeled K562 tumor cells. The SEM for each mean percent cytotoxicity of Raji or K562 tumor cells was <5% of the mean, and was not included.
Inhibition of MAPK Does Not Interfere with NK Cell Proliferation.
YT cells form a continuous cell line that can be maintained in culture. Therefore, we next examined whether MAPK might also control the proliferative cell cycle in YT cells. YT cells were treated with a range of concentrations of PD098059 for 1 h at 37°C and then cultured in medium for 24 h, with [3H]thymidine added for another 6 h. In a representative experiment of four (Fig. 2), it can be seen that none of the PD098059 concentrations interfered with normal growth of YT cells. Even 200 μM of the inhibitor did not have a noticeable effect on YT cell proliferation; neither did DMSO. Thus, the results from the first two sets of experiments suggest that the MAPK pathway controls the effector function rather than the proliferative response in YT cells.
Figure 2.

Effect of PD098059 on YT cell proliferation. YT cells untreated or treated with 20–200 μM of PD098059 or an amount of DMSO used to dilute the highest concentration of PD098059 were cultured in triplicate wells for 24 h at 37°C before another 6 h pulsing with [3H]thymidine. The mean and SEM of triplicate wells are shown.
Induction of Tyrosine Phosphorylation in MAPK within the NK Cells by Interaction with Target Cells.
The ability of a highly specific MAPK pathway inhibitor to block NK function suggests that MAPK must be activated and functional in NK cells during target lysis. Thus, we asked whether MAPK was constitutively active in NK cells or required target cell ligation to activate MAPK. We approached this by biochemical means to determine that MAPK was activated in NK cells through E/T interaction. It is well-established that MAPK upon activation becomes tyrosine phosphorylated (30). Thus, immunoprecipitation of PTK substrates from YT cell lysates with monoclonal antiphosphotyrosine antibody, 4G10, and Western blotting of the immunoprecipitate with the same antibody can first define whether a tyrosine-phosphorylated band of the predicted molecular weight of one of the MAPK is detected upon interaction with tumor cells (Fig. 3 A). To follow this format, YT cells were added to paraformaldehyde-fixed Raji tumor cells and lysed immediately (0 min; Fig. 3, lane 3), or the mixture was incubated at 37°C for 2–15 min (lanes 4–6) before lysis. In another group (lane 7), YT cells were pretreated with 100 μM PD098059 for 1 h at 37°C before incubating with Raji cells for 5 min at 37°C. Using a 10% SDS-polyacrylamide gel for electrophoresis, a tyrosine-phosphorylated band at ∼42/44 kD that correlated to the p42/44 MAPK was detected as early as 2 min after NK cell interaction with target cells. This band peaked at 5 min and was still detectable at 15 min. Similar results were obtained in two other experiments (data not shown). However, in PD098059-treated NK cells, incubation with target cells for 5 min did not induce this phosphorylated band. As a control, immunoprecipitation with an isotype-matched IgG was conducted (Fig. 3, lane 8), and no band was seen in the immunoprecipitate when probed with antiphosphotyrosine. Lysates of YT alone or Raji alone also showed no bands that could be detected with antiphosphotyrosine (lanes 1 and 2). Subsequent experiments used 1-h pretreatment of NK cells with 100 μM of PD098059.
Figure 3.


Detection of tyrosine-phosphorylated p42/p44 MAPK protein in Raji-activated YT effector cells. (A) YT cells were cultured alone or with Raji cells at a 1:1 ratio for 0–15 min at 37°C. YT cells were also pretreated with 100 μM of PD098059 for 1 h at 37°C before incubation for 5 min at 37°C with Raji tumor cells. The cells were then lysed and immunoprecipitated (IP) with monoclonal antiphosphotyrosine, 4G10. Immunoprecipitation of YT cells, which had been preincubated with Raji tumor cells for 5 min at 37°C, with isotype-matched IgG was also performed as a control. Raji cells alone were included to check for background phosphorylation. The immunoprecipitates were then probed with 4G10 by Western blotting (WB). (B) YT cells were untreated or pretreated for 1 h at 37°C with 100 μM of PD098059 or an equivalent amount of DMSO used to dilute PD098059. The cells were then mixed with Raji tumor target cells for 0–5 min at 37°C and lysed. The lysates were immunoprecipitated with antiphosphotyrosine, 4G10, or control isotype-matched IgG and then probed with anti-panERK.
To confirm that the detected band at 42/44 kD was MAPK, another experiment was conducted in which YT cells, before and after treatment with DMSO or 100 μM of PD098059 for 5 min at 37°C, were incubated with Raji cells and then lysed. The lysates were immunoprecipitated with antiphosphotyrosine, 4G10, and probed with anti-panERK, which detects all three known forms (31–33) of MAPK/ERK (Fig. 3 B). Significantly, upon 5 min interaction with tumor cells, only one band at 42/44 kD was detected in NK cells (lane 2) compared with 0 min incubation with target cells (lane 1). PD098059 abrogated the detection of this MAPK in the NK cells (lane 4), whereas DMSO had no effect (lane 3). Immunoprecipitation with a control IgG produced no band (lane 5). These data were reproducible in two other experiments. Thus, it appears that YT–Raji interactions provoke a specific MAPK pathway associated with the p42/44 kD form.
Detection of Active MAPK in NK Cells after Activation with Target Cells.
A signature event that identifies MAPK activation is the phosphorylation of Thr and Tyr in the TEY motif of the protein (30). MAPK activation in NK cells by target cells can thus be confirmed by detection of the active form of MAPK with a specific anti-AMAPK (α-AMAPK) antibody directed towards the phosphorylated TEY epitope of the molecule (30). This approach was next pursued by probing YT whole cell lysates with anti-AMAPK after the YT cells, which had been untreated or treated with 100 μM of PD098059, had interacted with Raji cells for 0–15 min (Fig. 4 A). Compared with 0 min incubation (lane 1, top), AMAPK in NK cells was only detected at 5 min incubation with tumor cells, and it remained high at 15 min (lanes 2 and 3). However, PD098059-pretreated NK cells showed a marked reduction in active MAPK upon 15 min activation with tumor cells (lane 4). To ensure that the differences seen were not due to unequal loading of the samples, the filter was stripped and reblotted with anti-panERK (bottom). Similar levels of p42/44 ERK were detected in all four lanes, indicating that its active phosphorylated form was specifically triggered in NK cells only by target ligation. To examine if MAPK activation is specifically triggered in NK cells only by the appropriate target cells, we also incubated YT cells with two different NK-resistant tumor cells, Jurkat and HL60, and tested the cell lysates for the presence of active MAPK (Fig. 4 B). Neither Jurkat nor HL60 tumor cells can be lysed by YT cells (data not shown). YT cells preincubated with either Jurkat or HL60 tumor cells at 37°C for 5 min showed no active MAPK, whereas YT cells could be triggered by Raji tumor cells in the same experiment to produce active MAPK (top). Stripping and reblotting with panERK ensured that the MAPK proteins were expressed equally before and after 5 min incubation in the three cell mixtures (bottom). Thus, MAPK activation occurs in NK cells only upon contact with sensitive target cells.
Figure 4.



Detection of the activated form of MAPK in Raji-triggered YT effector cells. (A) YT cells untreated or treated with 100 μM of PD098059 for 1 h at 37°C were mixed with equal numbers of Raji tumor cells for 0–15 min at 37°C. Whole cell lysates were then prepared and analyzed by Western blotting (WB) with anti-AMAPK (α-AMAPK) that was generated against the phosphorylated TEY epitope of MAPK (top). The blots were then stripped and reprobed with anti-panERK to show equal loading of all the lanes (bottom). (B) YT cells were mixed with equal numbers of either Jurkat or HL60 tumor cells, both of which are NK-resistant, or with the NK-sensitive Raji tumor cells, for 0 or 5 min at 37°C. Whole cell lysates were prepared and analyzed by Western blotting with anti-AMAPK (top). The blots were then stripped and reprobed with anti-panERK (bottom). (C) YT cells were pretreated with medium (MED), DMSO, or 10–200 μM of PD098059 for 1 h at 37°C before addition of Raji tumor cells for 0 or 5 min at 37°C. Cell lysates were then prepared and probed with anti-AMAPK (top) and consecutively restripped and reprobed with anti-ERK2 (middle) and anti-ERK1 (bottom).
The routine concentration used in this study was 100 μM of PD098059. To examine if this concentration was sufficient to block MAPK activation completely, YT cells were pretreated with 10–200 μM of PD098059 for 1 h at 37°C before incubation with Raji tumor cells for 5 min. Cell lysates were then prepared and probed for the presence of active MAPK by Western blotting. Fig. 4 C shows a dose-dependent inhibition, with 10 μM able to block MAPK activation in YT cells caused by Raji tumor cells, and 50 μM/100 μM able to take the active MAPK level down to a barely detectable level (top). Stripping and reblotting with anti-ERK1 and anti-ERK2, respectively, indicated that both proteins were abundant and equally present in all the lanes (middle and bottom).
Assessment of Kinase Function of MAPK in Target Cell-activated NK Cells.
To ensure that the MAPK in NK cells after target ligation was functional, an in vitro kinase assay was used with MBP as a substrate (34), which is commonly used for this purpose (Fig. 5, top). YT cells with or without DMSO or PD098059 pretreatment were incubated with Raji tumor cells for 0 or 5 min and then lysed for immunoprecipitation with anti-panERK. The ERK immunoprecipitates were tested for their ability to phosphorylate the MAPK substrate, MBP, and were run on a 15% SDS-polyacrylamide gel for easy detection of the small molecular mass 18-kD MBP substrate. Without incubation, YT cell lysates showed no ability to phosphorylate MBP (lane 1), but upon 5 min incubation with target cells, a sharp phosphorylated band at the expected molecular weight of MBP was detected in NK cells treated either with medium or DMSO (lanes 2 and 3). The phosphorylated MBP band was not detected in PD098059-treated NK cells even after 5 min incubation with target cells (lane 4). As expected, the control normal rabbit serum immunoprecipitate from YT cells, incubated 5 min with target cells, did not show phosphorylation of MBP (lane 5). Blotting the same filter with anti-panERK (bottom), equal amounts of both p42 and p44 forms of ERK were shown to be immunoprecipitated by anti-panERK in all the lanes. In contrast to the earlier 10% gels, this experiment using a 15% gel allowed for the detection of the closely migrating but separate p42 and p44 ERKs. These results suggest that MAPK becomes not only tyrosine phosphorylated but also functionally activated by being able to act as a kinase on its substrate, MBP.
Figure 5.

Analysis of kinase function in p42/44 MAPK from Raji-activated YT effector cells. YT cells untreated or treated for 1 h at 37°C with 100 μM of PD098059 or an equal concentration of the diluent, DMSO, were mixed with Raji tumor cells at a 1:1 ratio for 0–5 min at 37°C. The cells were then lysed and immunoprecipitated (IP) with anti-panERK. The immunoprecipitates were incubated with [α- 32P]ATP and the 18-kD MBP as a substrate for the in vitro kinase assay in a 15% SDS gel (top). The filter was then probed with anti-panERK to show equal loading in all the lanes (bottom). WB, Western blotting. MED, Medium.
Identification of ERK2 as the Active MAPK in Target-ligated NK Cells.
Because ERK1/p44, ERK2/p42, and ERK3/ p97 exist in human cells (31–33), our findings tended to narrow the responsible ERK to either ERKl or ERK2. To probe for the specificity of the MAPK that was activated in NK cells, YT cells with or without DMSO or PD098059 pretreatment were incubated with Raji tumor cells for 0 or 5 min, their lysates were immunoprecipitated with antiphosphotyrosine, and the immunoprecipitates were probed with anti-ERK2 (Fig. 6 A). ERK2 was readily detectable among the tyrosine-phosphorylated immunoprecipitates from DMSO-treated (lane 3) or untreated (lane 2) NK cells activated 5 min with tumor cells. PD098059 pretreatment abolished completely the detection of tyrosine-phosphorylated ERK2 (lane 4). Probing with antibodies against ERK1 and ERK3 did not reveal any band (data not shown).
Figure 6.


Identification of ERK2 as the activated MAPK isoform in Raji-triggered YT effector cells. (A) YT cells untreated or pretreated 1 h at 37°C with 100 μM of PD098059 or an equivalent amount of the diluent, DMSO, were mixed with Raji tumor cells at a 1:1 ratio for 0–5 min at 37°C. The cells were then lysed and immunoprecipitated (IP) with antiphosphotyrosine, 4G10, followed by Western blot analysis (WB) with anti-ERK2. MED, Medium. (B) YT cells similarly treated as in A were immunoprecipitated with anti-AMAPK (α-AMAPK) and probed with the same antibody to locate the activated phosphorylated form of MAPK. The filter was then stripped and reprobed with anti-ERK2 to identify the activated MAPK isoform as ERK2. NRS, Normal rabbit serum.
To further confirm that ERK2 was activated upon target ligation of NK cells, NK cells similarly treated as above were lysed and immunoprecipitated with anti-AMAPK, which specifically recognized the phosphorylated TEY epitope of MAPK (Fig. 6 B). The immunoprecipitates were first probed with anti-AMAPK to define accurately when this form becomes detectable (top). The AMAPK was only detectable upon 5 min interaction of NK cells with the tumor cells (lane 2); PD098059 (lane 4) but not DMSO (lane 3) blocked this detection. Because anti-AMAPK was a polyclonal rabbit antibody, normal rabbit serum was used as a control to immunoprecipitate lysates from NK cells activated 5 min with target cells. The control serum did not produce a detectable band (lane 5). Upon stripping and reprobing with anti-ERK2, the same band was again detected only in untreated or DMSO-pretreated NK cells activated 5 min with tumor cells and not in the PD098059-treated NK cells (bottom). Probing with antibodies against ERK1 and ERK3 did not produce any detectable band (data not shown). The results taken together demonstrate that MAPK/ERK2 activation was consistently triggered in NK cells upon target ligation.
Downregulation of NK Function by Transient Transfection with Dominant-Negative MAPK.
The activation of MAPK by NK ligation with target cells and the ability of PD098059 to inhibit NK lysis of the same tumor targets are highly suggestive of the linkage between MAPK activation and NK effector function. To further demonstrate this linkage, we used transient transfection assays (41, 42) to determine whether expression of dominant-negative p42 MAPK/ ERK2 could inhibit NK lysis of tumor cells. YT cells were transfected with plasmids encoding either wild-type ERK2 or ERK2 in which the Thr and Tyr residues within the critical TEY motif that must be phosphorylated for activity were substituted with glutamic acid (TEYE) or alanine and phenylanine (TAYF). A plasmid containing the KD form of ERK2 was also transfected into YT cells. After 24 h of transfection, YT cells were checked for viability and washed, and the viable cells were adjusted to the appropriate concentrations for testing in a 5-h 51Cr-release assay against Raji tumor cells. Comparison was made with an aliquot of parental YT cells that had been handled similarly but without any transfection. Fig. 7 shows a representative experiment of three with similar results. YT cells transfected with either the KD, TEYE, or TAYF form of MAPK/ERK2 were markedly deficient in lysis of tumor cells at all the E/T ratios tested, and all three mutant forms showed significant inhibition of lysis compared with the parental nontransfected YT cells at all the E/T ratios tested. Transfection with wild-type MAPK/ERK2 did not significantly alter NK lysis of tumor cells. Therefore, functional MAPK/ERK2 appears to be required for optimal NK function against tumor cells.
Figure 7.

Inhibition of NK function by transient transfection with dominant-negative MAPK/ERK2 in YT effector cells. Equal aliquots of YT cells were left untransfected or transiently transfected with KD MAPK/ERK2 in which K52R substitution was constructed, or with mutant ERK2 where Thr and Tyr residues in the TEY motif were replaced with glutamic acid (TEYE) or with alanine and phenylalanine (TAYF). The wild-type ERK2 plasmid (WT) was also used to transfect YT cells. After 24 h at 37°C, untransfected and transfected YT cells were assessed for viability and adjusted to the appropriate concentrations of viable cells before testing for lysis of 51Cr-labeled Raji tumor cells at the indicated E/T ratios. The SEM of each mean percent cytotoxicity was <5%, and was not shown.
MAPK Regulation of Perforin Mobilization and Redistribution in NK Cells.
Upon contact with sensitive target cells, NK cells are known to immediately release perforin and granzyme B (20, 23, 24). These molecules work in a coordinated manner, with perforin polymerizing into the target cell membrane at the point of contact and allowing the passage of lytic enzymes, specifically granzyme B, which can initiate the apoptotic process in the affected target cells (25–29). However, little information is available on the intracellular signal mechanisms in NK cells that trigger their polarization and release. In antibody-dependent cell cytotoxicity, FcR triggering in NK cells has been documented to require PI3 kinase activation, which mediates granule release (7). Besides PI3 kinase, several molecules, PLC-γ, cAMP, and calcium influx have been associated with granule exocytosis (7, 8), but further downstream events have not been explored. Thus, we asked whether MAPK controls perforin mobilization and redistribution in NK cells. The approach was to examine the pattern of distribution of cytoplasmic perforin in NK cells before and after conjugation with target cells, and to evaluate the effect of PD098059. Fig. 8 shows a representative YT–RAJI conjugate before and after each treatment. To be able to distinguish the effector cells from the tumor cells, it is necessary to identify a selective marker for each cell type. In screening several intracellular antigens that exist in Raji cells but not in YT cells, we determined that the src kinase, lyn, was a selective marker for Raji cells. YT cells (A) did not stain with TRITC-labeled antilyn, whereas Raji cells did (D). On the other hand, only YT cells (C) stained with FITC-labeled antiperforin; Raji cells did not (B). All cells stained blue with the nucleus marker. In resting YT cells, perforin was observed to be evenly distributed throughout the cytoplasm in punctated forms (C). DMSO pretreatment of NK cells did not adversely affect the pattern of FITC-labeled perforin distribution (E). Upon addition of Raji tumor cells without any incubation (F), the DMSO-pretreated NK cell that had bound to TRITC-labeled Raji again showed even distribution of FITC-labeled perforin in their cytoplasm, much like the pattern in NK cells without target cells (C and E). Upon 5 min incubation at 37°C with tumor cells (G), the DMSO-pretreated NK cell that had conjugated with a target cell showed complete mobilization of intracellular perforin towards the point of contact with the target cell. Further incubation up to 30 min did not change this pattern (data not shown), indicating that 5 min of target cell ligation was sufficient for optimal redistribution of perforin to the contact point. In contrast, the PD098059-pretreated NK cell that had formed a conjugate with Raji tumor cells at 37°C for 5 min showed no redistribution of FITC-labeled perforin, and retained even staining with FITC-labeled perforin throughout the cytoplasm (H). Enumeration of the YT–Raji conjugates indicated that, upon 5 min incubation at 37°C, 27% of the conjugates from DMSO-treated YT cells had mobilized perforin, compared with 4% of the PD098059-treated YT cell conjugates, which is similar to the 5% of conjugates that showed mobilization in the control YT–Raji culture at 0 min. Similar results were obtained in two other experiments (data not shown). Therefore, functional MAPK appears to be required to mobilize and redistribute perforin from NK cells towards the contact point with target cells.
MAPK Regulation of Granzyme B Mobilization and Redistribution in NK Cells.
Because granzyme B appears to be an integral component of the NK lytic process, the next question was whether MAPK could also control this process. Thus, the pattern of distribution of granzyme B in NK cells before and after target ligation, with or without PD098059 treatment, was evaluated, and a representative NK–target conjugate is shown for each group (Fig. 9). The patterns were identical to those seen with perforin. FITC-labeled granzyme B was not detected in Raji cells (B) but was found to be evenly distributed within the cytoplasm of resting YT cells (C). Raji cells were again labeled with TRITC-antilyn (D) to distinguish them from YT cells that are lyn-negative (A). DMSO did not adversely affect the distribution of granzyme B in NK cells (E). The DMSO-pretreated NK cell that had bound Raji cells without any preincubation (F) showed no polarization of granzyme B towards the target cell. However, upon 5 min incubation with Raji cells, the DMSO-pretreated NK cell that had bound a target cell now showed complete redirection of FITC-labeled granzyme B to the tumor contact site (G). As with perforin, 5 min was sufficient for complete granzyme B redirection, and further incubation up to 30 min did not change the pattern (data not shown). In contrast, the PD098059-pretreated NK cell did not respond to target cell ligation by granzyme B polarization (H) and showed an even distribution of this FITC-labeled enzyme throughout the cytoplasm. Enumeration of YT–Raji conjugates indicated that upon 5 min incubation at 37°C, 28% of the DMSO-treated YT cell conjugates had mobilized granzyme B, compared with 3% of the PD098059-treated YT cell conjugates, which is similar to the 5% of YT–Raji conjugates that showed mobilization at 0 min incubation. Two other experiments produced similar results (data not shown). Thus, the data depict that MAPK may also regulate granzyme B redistribution to the tumor cell site.
Discussion
Our analysis of downstream intracellular signals that occur in NK cells upon target cell ligation to trigger target lysis has unequivocally identified the MAPK pathway as a critical component. A specific MAPK, ERK2, is required for NK effector function, and its primary function may be to regulate the mobilization and redistribution of cytoplasmic perforin and granzyme B towards the contact zone with target cells. To our knowledge, this is the first direct evidence of MAPK involvement in a lymphocytic function that is not associated with gene transcription. MAPK activation has been investigated in several systems, in both immune-related and nonrelated cells, but the focus has been on its involvement in either gene induction or cell cycle control. For example, MAPK has been reported to phosphorylate and regulate c-fos, c-myc, myb, Elk-1, Tal-1, Ets-2, and STAT (signal transducer and activator of transcription) proteins (42, 44–49). However, it is also becoming evident that MAPK can act as a kinase for signal molecules that are not direct transcription factors. For example, activation of the superoxide generation system that occurs within minutes in human neutrophils stimulated with FMLP may involve MAPK-mediated serine/threonine phosphorylation of several components of the NADPH oxidase, including p47phox (50). In one report, MAPK/ERK2 was demonstrated to directly phosphorylate a synthetic peptide of p47phox (51). Purified cytosolic phospholipase A2 has also been shown to be directly phosphorylated and activated by ERK2 (52). It was demonstrated recently that nicotine-induced secretion from bovine adrenal chromaffin cells also required MAPK activation (53). In our system, the events controlled by ERK2 were also extremely rapid: they were triggered almost immediately, within 5 min, in NK cells upon target cell binding. From the immunochemistry studies in this paper, we found that both perforin and granzyme B were expressed abundantly and were located in a punctated distribution throughout the cytoplasm of resting NK cells. Microscopic analysis of single NK–target conjugates indicated that interaction with a sensitive tumor target triggered the movement of both perforin and granzyme B towards the point of NK contact with the tumor target, and in 5 min of interaction, redistribution within the NK cell cytoplasm of the two lytic molecules was complete such that only a small area adjacent to the target cell contact zone contained perforin and granzyme B. Use of a specific MAPK pathway inhibitor, PD098059 (38, 39), indicated that pretreatment of NK cells with this reagent could block completely the redirection of both perforin and granzyme B towards the target cell in the NK–target conjugate. To date, studies that have focused on mechanisms of NK lysis have assessed primarily perforin release and granule exocytosis into the supernatant. The extracellular release of these lytic components has been shown to require cAMP, PI3 kinase, PLC-γ, and calcium mobilization (20–22). Our work here focused not on extracellular release but on intracellular redirection of perforin and granzyme B. Thus, this study provides important information on the reorganization of the intracellular granules that harbor both perforin and granzyme B, and suggests that MAPK controls this process.
The redirection of perforin and granzyme B towards the contact zone with target cells apparently mediates target lysis. This is confirmed by the ability of the MAPK pathway inhibitor, PD098059, to also block NK lysis of the tumor cells in a 5-h 51Cr-release assay. In addition to functional interference by MAPK inhibition, we also showed by biochemical means that MAPK becomes rapidly tyrosine phosphorylated and activated in NK cells within 5 min of interaction with tumor targets. The time frame of maximal activation of MAPK, i.e., 5 min, coincided with the time frame of maximal redirection of perforin and granzyme B to the tumor contact site. Immunoprecipitation with antiphosphotyrosine followed by Western blotting with anti-panMAPK as well as with anti-ERK–specific antibodies indicated that only ERK2 and not ERK1 or ERK3 was activated by NK to target ligation. The active form of ERK2 could also be documented using a specific antibody directed against the phosphorylated TEY epitope of MAPK. Additional evidence of functional activation of ERK2 was provided by the ability of the MAPK isolated from NK cells that have interacted 5 min with tumor target cells, but not from NK cells mixed with target cells without prior incubation, to act efficiently as a kinase to phosphorylate MBP. In all of these experiments, PD098059 pretreatment of NK cells abolished the phosphorylation and activation of MAPK. The linkage between MAPK/ERK2 and NK function was also demonstrated via transient transfection studies with dominant-negative MAPK/ERK2. YT cells transiently transfected with either the KD, TEYE, or TAYF form of ERK2 displayed marked reduction in their ability to lyse tumor target cells in a 5-h 51Cr-release assay, whereas wild-type ERK2 transfection had no negative effect. Finally, specificity of MAPK activation was demonstrated by the ability of only NK-sensitive Raji but not NK-resistant Jurkat or HL60 tumor target cells to trigger phosphorylation of MAPK in YT cells. As an important correlate, we also demonstrated that MAPK activation was necessary for fresh NK cell lysis of tumor target cells, by the ability of PD098059 to block LGL lysis of K562 tumor cells.
Taken together, the results identify a key downstream signal molecule, ERK2, that controls NK effector function. The control appears to be at the level of mobilization of lytic components, perforin and granzyme B, upon target cell contact. Thus, our findings add yet another function to MAPK, and identify lytic regulation by MAPK as a key point of control in NK cells in our system.
Acknowledgments
This work was supported by National Institutes of Health grant CA-63724, and by the Pathology Core and Molecular Imaging Core facilities of the H. Lee Moffitt Cancer Center.
Abbreviations used in this paper
- AMAPK
active MAPK
- ERK
extracellular signal–regulatory kinase
- KD
kinase deficient
- MAPK
mitogen-activated protein kinase
- MBP
myelin basic protein
- MEK
MAPK kinase
- PI
phosphatidylinositol
- PLC
phospholipase C
- TRITC
tetramethyl rhodamine isothiocyanate
References
- 1.Trinchieri G. Biology of natural killer cells. Adv Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Welsh RM. Regulation of virus infections by natural killer cells. A review. Nat Immun Cell Growth Regul. 1986;5:169–199. [PubMed] [Google Scholar]
- 3.Blanchard DK, Hall RE, Djeu JY. Role of CD18 in lymphokine-activated killer (LAK) cell mediated lysis of human monocytes: comparison with other LAK targets. Int J Cancer. 1990;45:312–318. doi: 10.1002/ijc.2910450218. [DOI] [PubMed] [Google Scholar]
- 4.Azzoni L, Kamoun M, Salcedo TW, Kanakaraj P, Perussia B. Stimulation of Fc gamma RIIIA results in phospholipase C-gamma 1 tyrosine phosphorylation and p56lck activation. J Exp Med. 1992;176:1745–1750. doi: 10.1084/jem.176.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Shea JJ, Weissman AM, Kennedy ICS, Ortaldo JR. Engagement of the natural killer cell IgG Fc receptor results in tyrosine phosphorylation of the zeta chain. Proc Natl Acad Sci USA. 1991;88:350–354. doi: 10.1073/pnas.88.2.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ting AT, Dick CJ, Schoon RA, Karnitz LM, Abraham RT, Leibson PJ. Interaction between lck and syk family tyrosine kinases in Fcγ receptor-initiated activation of natural killer cells. J Biol Chem. 1995;270:16415–16421. doi: 10.1074/jbc.270.27.16415. [DOI] [PubMed] [Google Scholar]
- 7.Bonnema JD, Karnitz LM, Schoon RA, Abraham RT, Leibson PJ. Fc receptor stimulation of phosphatidylinositol 3-kinase in natural killer cells is associated with protein kinase C-independent granule release and cell-mediated cytotoxicity. J Exp Med. 1994;180:1427–1435. doi: 10.1084/jem.180.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cassatella MA, Anegon I, Cuturi MC, Griskey P, Trinchieri G, Perussia B. FcγR(CD16) interaction with ligand induces Ca2+ mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+in FcγR(CD16)-induced transcription and expression of lymphokine genes. J Exp Med. 1989;169:549–567. doi: 10.1084/jem.169.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanier LL, Phillips JH. Inhibitory MHC class I receptors on NK cells and T cells. Immunol Today. 1996;17:86–91. doi: 10.1016/0167-5699(96)80585-8. [DOI] [PubMed] [Google Scholar]
- 10.Moretta A, Vitale M, Bottino C, Orgeno AM, Morelli L, Augugliaro R, Barbaresi M, Ciccone E, Moretta L. P58 molecules as putative receptors for MHC class I molecules in human natural (NK) cells. Anti-p58 antibodies reconstitute lysis of MHC class I-protected cells in NK clones displaying different specificities. J Exp Med. 1993;178:597–614. doi: 10.1084/jem.178.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Andrea A, Hang C, Franz-Bacon K, McClanahan T, Phillips JH, Lanier LL. Molecular cloning of NKB1. A natural killer receptor for HLA-B allotypes. J Immunol. 1995;155:2306–2310. [PubMed] [Google Scholar]
- 12.Carreto M, Cantoni C, Bellon T, Bottino C, Biassoni R, Rodriguez A, Perez-Villar JJ, Moretta L, Moretta A, Lopez-Botet M. The CD94 and NKG2-A C type lectin covalently assemble to form a natural killer cell inhibitory receptor for HLA class I molecules. Eur J Immunol. 1997;27:563–567. doi: 10.1002/eji.1830270230. [DOI] [PubMed] [Google Scholar]
- 13.Karlhofer FM, Ribaudo RK, Yokoyama WM. MHC class I alloantigen specificity of Ly49+ IL-2 activated natural killer cells. Nature. 1992;358:66–70. doi: 10.1038/358066a0. [DOI] [PubMed] [Google Scholar]
- 14.Campbell KS, Dessing M, Lopez-Botet M, Cella M, Colonna M. Tyrosine phosphorylation of a human natural killer inhibitory receptor recruits protein tyrosine phosphatase 1C. J Exp Med. 1996;184:93–100. doi: 10.1084/jem.184.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olcese L, Lang P, Vely F, Cambiaggi A, Marguet D, Blery M, Hippen KL, Biassoni R, Moretta A, Moretta L, Cambier JC, Vivier E. Human and mouse natural killer cell inhibitory receptors recruit PTP1C and PTP1D protein tyrosine phosphatases. J Immunol. 1996;156:4531–4534. [PubMed] [Google Scholar]
- 16.Fry AM, Lanier LL, Weiss A. Phosphotyrosines in the killer cell inhibitory receptor motif of NKB1 are required for negative signaling and for association with protein tyrosine phosphatase 1C. J Exp Med. 1996;184:295–300. doi: 10.1084/jem.184.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura MC, Niemi EC, Fisher MJ, Shultz LD, Seaman WE, Ryan JC. Mouse Ly-49A interrupts early signal events in natural killer cell cytotoxicity and functionally associates with the SHP-1 tyrosine phosphatase. J Exp Med. 1996;184:673–684. doi: 10.1084/jem.185.4.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biassoni R, Cantoni C, Falco M, Verdiani S, Bottino C, Vitale M, Conte R, Poggi A, Moretta A, Moretta L. The human leukocyte antigen (HLA)-C–specific “activatory” or “inhibitory” natural killer cell receptors display highly homologous extracellular domains but differ in their transmembrane and intracytoplasmic portion. J Exp Med. 1996;183:645–650. doi: 10.1084/jem.183.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henkart P. Mechanism of lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 1985;3:31–58. doi: 10.1146/annurev.iy.03.040185.000335. [DOI] [PubMed] [Google Scholar]
- 20.Seaman WE, Eriksson E, Dobrow R, Imboden JB. Inositol triphosphate is generated by a rat natural killer cell tumor in response to target cells or to cross-linked monoclonal antibody OX-34: possible signaling role for the OX-34 determinant during activation by target cells. Proc Natl Acad Sci USA. 1987;84:4239–4243. doi: 10.1073/pnas.84.12.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Windebank KP, Abraham RT, Powis G, Olsen RA, Barna TJ, Leibson PJ. Signal transduction during human natural killer cell activation: inositol phosphate generation and regulation by cyclic AMP. J Immunol. 1988;141:3951–3957. [PubMed] [Google Scholar]
- 22.Richards AL, Djeu JY. Calcium-dependent natural killer and calcium-independent natural cytotoxic activities in an IL2-dependent killer cell line. J Immunol. 1990;145:3144–3150. [PubMed] [Google Scholar]
- 23.Henkart PA, Millard PJ, Reynolds CW, Henkart MP. Cytolytic activity of purified cytoplasmic granules from cytotoxic rat large granular lymphocyte tumors. J Exp Med. 1984;160:75–93. doi: 10.1084/jem.160.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young JDE, Hengartner H, Podack ER, Cohn ZA. Purification and characterization of a cytolytic pore-forming protein from granules of cloned lymphocytes with natural killer cell activity. Cell. 1986;44:849–859. doi: 10.1016/0092-8674(86)90007-3. [DOI] [PubMed] [Google Scholar]
- 25.Chen G, Shi L, Litchfield DW, Greenberg AH. Rescue from granzyme B-induced apoptosis by Wee1 kinase. J Exp Med. 1995;181:2295–2300. doi: 10.1084/jem.181.6.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapani JA, Browne KA, Smyth MJ, Jans DA. Localization of granzyme B in the nucleus. A putative role in the mechanism of cytotoxic lymphocyte-mediated apoptosis. J Biol Chem. 1996;271:4127–4133. doi: 10.1074/jbc.271.8.4127. [DOI] [PubMed] [Google Scholar]
- 27.Shi L, Mai S, Israels S, Browne K, Trapani JA, Greenberg AH. Granzyme B (GraB) autonomously crosses the cell membrane and perforin initiates apoptosis and GraB nuclear localization. J Exp Med. 1997;185:855–866. doi: 10.1084/jem.185.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T cell-derived granzyme B. Nature. 1995;377:446–448. doi: 10.1038/377446a0. [DOI] [PubMed] [Google Scholar]
- 29.Duan JJ, Orth K, Chinnaiyan AM, Poirrer GG, Forelich CJ, He WW, Dixit VM. ICE-LAP6, a novel member of the ICE/Ced-3 gene family is activated by the cytotoxic T cell protease, granzyme B. J Biol Chem. 1996;271:16720–16724. doi: 10.1074/jbc.271.28.16720. [DOI] [PubMed] [Google Scholar]
- 30.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 31.Boulton TG, Yancopoulos GD, Gregory JS, Slaughter C, Moomaw C, Hsu J, Cobb M. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science. 1990;249:64–67. doi: 10.1126/science.2164259. [DOI] [PubMed] [Google Scholar]
- 32.Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejeska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb M. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- 33.Zhu AX, Zhao Y, Moller DE, Flier JS. Cloning and characterization of p97MAPK, a novel human homolog of rat ERK3. Mol Cell Biol. 1994;14:8202–8211. doi: 10.1128/mcb.14.12.8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pages G, Lenormand P, L'Allemain G, Chambard JC, Meloche S, Pouyssegur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci USA. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of map kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 36.Samuels ML, Weber MJ, Bishop JM, McMahon M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human Raf-1 protein kinase. Mol Cell Biol. 1993;13:6241–6252. doi: 10.1128/mcb.13.10.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu JH, Wei S, Blanchard DK, Djeu JY. Restoration of lytic function in a human natural killer cell line by gene transfection. Cell Immunol. 1994;156:24–35. doi: 10.1006/cimm.1994.1150. [DOI] [PubMed] [Google Scholar]
- 38.Dudley DT, Pand L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 40.Trapani JA, Krowne KA, Dawson M, Smyth MJ. Immunopurification of functional Asp-ase (natural killer cell granzyme B) using a monoclonal antibody. Biochem Biophys Res Commun. 1993;195:910–920. doi: 10.1006/bbrc.1993.2131. [DOI] [PubMed] [Google Scholar]
- 41.Her JH, Lakhani S, Zu K, Vila J, Dent P, Sturgill TW, Weber MJ. Dual phosphorylation and autophosphorylation in mitogen-activated protein (MAP) kinase activation. Biochem J. 1993;296:25–31. doi: 10.1042/bj2960025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.David M, Petricoin E, Benjamin C, Pine R, Weber MJ, Larner AC. Requirement for MAP kinase (ERK2) activity in interferon α- and interferon β-stimulated gene expression through STAT protein. Science. 1995;269:1721–1723. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- 43.Valkov NI, Gump JL, Sullivan DM. Quantitative immunofluorescence and immunoelectron microscopy of the topoisomerase IIα associated with nuclear matrices from wild-type and drug-resistant chinese hamster ovary cell lines. J Cell Biochem. 1997;67:112–130. doi: 10.1002/(sici)1097-4644(19971001)67:1<112::aid-jcb12>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 44.Chen RH, Abate C, Blenis J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci USA. 1993;90:10952–10956. doi: 10.1073/pnas.90.23.10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aziz N, Miglarese MR, Hendrickson RC, Shabanowitz J, Sturgill TW, Hunt DF, Bender TP. Modulation of c-Myb-induced transcription activation by a phosphorylation site near the negative regulatory domain. Proc Natl Acad Sci USA. 1995;92:6429–6433. doi: 10.1073/pnas.92.14.6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH, Shaw PE. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO (Eur Mol Biol Organ) J. 1995;14:951–962. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wadman IA, Hsu HL, Cobb MH, Baer R. The MAP kinase phosphorylation site of TAL1 occurs within a transcriptional activation domain. Oncogene. 1994;9:3713–3716. [PubMed] [Google Scholar]
- 48.Yang BS, Hauser CA, Henkel G, Colman MS, Van Beveren C, Stacey KJ, Hume DA, Maki RA, Ostorwoski MC. Ras-mediated phosphorylation of a conserved threonine residue enhances the transactivation activities of c-Ets1 and c-Ets2. Mol Cell Biol. 1996;16:538–547. doi: 10.1128/mcb.16.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wen Z, Zhong Z, Darnell JJ. Maximal activation of transcription of Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 50.Dusi S, Donini D, Rossi F. Tyrosine phosphorylation and activation of NADPH oxidase in human neutrophils: a possible role for MAP kinases and for a 75 kDA protein. Biochem J. 1994;304:243–250. doi: 10.1042/bj3040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grinstein S, Furuya W, Butler JR, Tseng J. Receptor-mediated activation of multiple serine/threonine kinases in human leukocytes. J Biol Chem. 1993;268:20223–20231. [PubMed] [Google Scholar]
- 52.Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- 53.Cox ME, Parsons SJ. Roles for protein kinase C and mitogen-activated protein kinase in nicotine-induced secretion from bovine adrenal chromaffin cells. J Neurochem. 1997;69:1119–1130. doi: 10.1046/j.1471-4159.1997.69031119.x. [DOI] [PubMed] [Google Scholar]