

Abstract
Chemokines are essential mediators of normal leukocyte trafficking as well as of leukocyte recruitment during inflammation. We describe here a novel non-ELR CXC chemokine identified through sequence analysis of cDNAs derived from cytokine-activated primary human astrocytes. This novel chemokine, referred to as I-TAC (interferon-inducible T cell alpha chemoattractant), is regulated by interferon (IFN) and has potent chemoattractant activity for interleukin (IL)-2–activated T cells, but not for freshly isolated unstimulated T cells, neutrophils, or monocytes. I-TAC interacts selectively with CXCR3, which is the receptor for two other IFN-inducible chemokines, the IFN-γ–inducible 10-kD protein (IP-10) and IFN-γ– induced human monokine (HuMig), but with a significantly higher affinity. In addition, higher potency and efficacy of I-TAC over IP-10 and HuMig is demonstrated by transient mobilization of intracellular calcium as well as chemotactic migration in both activated T cells and transfected cell lines expressing CXCR3. Stimulation of astrocytes with IFN-γ and IL-1 together results in an ∼400,000-fold increase in I-TAC mRNA expression, whereas stimulating monocytes with either of the cytokines alone or in combination results in only a 100-fold increase in the level of I-TAC transcript. Moderate expression is also observed in pancreas, lung, thymus, and spleen. The high level of expression in IFN- and IL-1–stimulated astrocytes suggests that I-TAC could be a major chemoattractant for effector T cells involved in the pathophysiology of neuroinflammatory disorders, although I-TAC may also play a role in the migration of activated T cells during IFN-dominated immune responses.
Keywords: neuroinflammation, astrocytes, calcium, signaling, brain
Chemokines are a superfamily of cytokines important in inflammatory and immune responses due primarily to their chemotactic activities towards subsets of leukocytes (1–4). The superfamily is divided into four groups, CXC, CC, C, and CX3C, based on the number and arrangement of conserved cysteine motifs. The CXC, CC, and CX3C chemokines are distinguished by the presence of one, none, or three amino acids, respectively, between the first two cysteines. The CXC chemokines are primarily active on neutrophils, whereas the CC chemokines are active on monocytes and other leukocytes, including lymphocytes, eosinophils, and basophils. Fractalkine and its murine homolog, neurotactin, are the only known CX3C chemokines, and, unlike other chemokines, are displayed on the cell surface by means of a mucin-like stalk that connects the functional NH2-terminal domain with the COOH-terminal membrane-spanning domain (5, 6). The C chemokine group, distinguished by the absence of the second and fourth cysteines, has only one known member, lymphotactin (7–9). Both fractalkine and lymphotactin show predominant activity toward T cells.
The CXC subfamily is subdivided into ELR1 and non-ELR CXC chemokines based on the presence or absence of this Glu-Leu-Arg tripeptide sequence adjacent to the CXC motif. ELR-containing CXC chemokines include IL-8; NAP (neutrophil-activating protein)-2; ENA (epithelial-derived neutrophil-activating protein)-78; and GRO (growth-related protein)-α, -β, and -γ. The ELR motif is essential for biological activity in that a mutation in any of these amino acids dramatically affects receptor binding and induction of leukocyte migration (10–12). The non-ELR CXC chemokines include the IFN-γ–inducible 10-kD protein (IP-10), the IFN-γ–induced human monokine (HuMig), and stromal cell–derived factor (SDF)-1. IP-10 and HuMig have a unique selectivity for T cells that have been activated by IL-2, whereas SDF-1 has a broader range of activities on resting and activated memory T cells, monocytes, and granulocytes (13–16). IP-10 has been shown to be expressed in delayed-type hypersensitivity reactions in the skin, psoriatic plaques, tuberculoid leprosy, and certain tumors (17–19). Due to these observations, it has been suggested that local high concentrations of IFN upregulate IP-10 and HuMig, which then results in the recruitment of activated/effector T cells, thereby initiating the effector arm of T cell immunity (20).
Chemokines exert their biological activities through G protein–coupled receptors on the surface of target cells. In humans, four CXC chemokine receptors (CXCR1–4) and eight CC chemokine receptors (CCR1–8) have been identified (21–32). The CXC chemokine receptors have been shown to have the following ligand specificities: CXCR1 binds IL-8 (33); CXCR2 binds IL-8 and other ELR-containing CXC chemokines (34); CXCR3 binds IP-10 and HuMig (15); and CXCR4 binds SDF-1α (35, 36). CXCR3 is selectively expressed in activated T cells and not in other leukocyte subpopulations. This underscores the importance of the selective recruitment of activated T cells by IP-10 and HuMig during inflammation (15, 20).
Localized upregulation of chemokines by proinflammatory cytokines like TNF, IL-1, and IFN, and the subsequent infiltration of leukocytes, is extremely important in the development of both acute and chronic inflammatory responses including immediate- and delayed-type hypersensitivity responses. Chemokine expression has been detected in the T cell–mediated disease experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis. Expression of IP-10 and two CC chemokines, MCP (monocyte chemoattractant protein)-1 and RANTES (regulated on activation, normal T cell expressed and secreted), coincides with the onset of clinical EAE (37–39). Presumably, autoreactive T cells enter the central nervous system and secrete proinflammatory cytokines that cause local production of chemokines. These chemokines promote further infiltration of inflammatory cells, resulting in lesion formation and demyelination indicative of multiple sclerosis (40). The astrocyte has been identified as an important source of chemokine expression in EAE (37). Furthermore, astrocytoma cells stimulated with TNF produce IL-8, MCP-1, and IP-10 (41). To identify additional genes that might contribute to neuroinflammatory disease pathology, we sequenced cDNAs derived from proinflammatory cytokine-stimulated primary human astrocytes and performed bioinformatic analysis. In this study, we report a novel non-ELR CXC chemokine identified through this approach, and describe its expression, biological activity, and receptor usage.
Materials and Methods
Reagents.
The recombinant chemokines IP-10 and HuMig were obtained from PeproTech, Inc. (Rocky Hill, NJ). Restriction enzymes and molecular biology reagents were from New England Biolabs (Beverly, MA), GIBCO BRL (Gaithersburg, MD), or Boehringer Mannheim (Indianapolis, IN). BSA and human collagen type IV were obtained from GIBCO BRL. RPMI 1640 medium was from BioWhittaker (Walkersville, MD). RNAs used in reverse transcriptase PCR analysis were either purchased (Clontech, Palo Alto, CA) or generated using Trizol reagent (GIBCO BRL) as specified by the manufacturer. Human primary monocytes were isolated and cultured as described previously (22); human microglial cell line SV-A3 (GFAP−; CD68+) was derived by immortalizing primary human microglia with adenovirus E1b and SV40 large T antigens. Eosinophils were isolated by negative selection using magnetic beads. Neutrophils were isolated as described previously (42).
Identification of I-TAC cDNA Sequence.
A cDNA library was constructed at Incyte Pharmaceuticals, Inc. (Palo Alto, CA) from polyA+ RNA isolated from human fetal astrocytes cultured in DMEM plus 10% FCS for up to 1 mo. A second cDNA library was constructed from polyA+ RNA isolated from similar cells that had been stimulated with TNF-α, IFN-γ, and IL-1β (each at 200 U/ml) for 12 h. Library synthesis was initiated using a 3′ oligo dT primer that contained a NotI site, followed by second strand synthesis, blunting with T4 DNA polymerase, ligation to SalI adapters, and digestion with SalI and NotI. cDNAs were then ligated directionally into pSPORT1 (GIBCO BRL) using SalI (5′) and NotI (3′). Single pass sequence information from the 5′ end of random cDNA clones was generated at Incyte by automated DNA sequencing. Novel sequences were further analyzed using the Basic Local Alignment Search Tool (BLAST, National Center for Biotechnology Information, Bethesda, MD; reference 43) to define similarities to known genes or gene families.
Cloning of Full-length I-TAC cDNA.
Sequence information from the expressed sequence tag cluster NCY580854 (which contained the 5′ end of a novel chemokine) was used to design the following internal primers for 3′ RACE: 5′-GTGTGCTACAGTTGTTCAAGGC-3′ and 5′-GGACGCTGTCTTTGCATAGGC-3′. 3′ RACE products were generated using the 3′ RACE System for Rapid Amplification of cDNA Ends (GIBCO BRL) according to manufacturer's instructions with stimulated astrocyte RNA as template. Amplification products were gel purified and subcloned into pCR2.1 (Invitrogen, Carlsbad, CA) for automated sequence analysis using an Applied Biosystems Sequencer (model 373; Applied Biosystems, Inc., Foster City, CA). Sequence data obtained from these clones was assembled with the original NCY580854 cluster sequences to generate an ∼1,400 bp consensus sequence for I-TAC. The entire coding sequence was then PCR amplified using the following primers, designed to incorporate a Kozak sequence and restriction sites convenient for subcloning into pcDNA3 (Invitrogen): 5′ primer: 5′-CGGGATCCGCCGACCATGAGTGTGAAGGGCATGGCTATAGC-3′; 3′ primer: 5′-CCGCTCGAGCGGTCATTAAAAATTCTTTCTTTCAACTTTTTTGATTATAAGCC-3′. The PCR product was gel isolated, digested with BamHI and XhoI, ligated to pcDNA3, and used to transform Escherichia coli DH5α cells. Correct clones were identified by restriction analysis and the sequence was confirmed by automated sequence analysis as above.
Northern Analysis.
Human Multiple Tissue Northern Blots were purchased from Clontech and hybridized with a 224-bp fragment of I-TAC coding sequence labeled by random priming (Prime-It II; Stratagene, La Jolla, CA) in the presence of [32P]dCTP. Hybridization was carried out in Express-Hyb Solution (Clontech) under stringent conditions as specified by the manufacturer.
Reverse Transcriptase PCR Analysis of I-TAC Expression.
First strand cDNA was generated from total RNA via oligo dT primed reverse transcription. Real time quantitative PCR was performed using a sequence detection system (ABI Prism 7700; Perkin-Elmer Corp., Norwalk, CT) using TaqMan Fluorogenic oligonucleotide probes (Perkin-Elmer Corp.). Relative I-TAC levels were calculated by normalizing to β-actin. Probe and primer sequences are as follows: β-actin forward, AGATCATTGCTCCTCCTGAGC; β-actin reverse, ACGCAACTAAGTCATAGTCCGC; β-actin probe, (6FAM)AGCAGATGTGGATCAGCAAGCAGGA(TAMRA); I-TAC forward, GCTATAGCCTTGGCTGTGATAT; I-TAC reverse, CAGGGCCTATGCAAAGACA; and I-TAC probe, (6FAM)TGTGTGCTACAGTTGTTCAAGGCTTCCC(TAMRA). All reactions were carried out using the TaqMan PCR reagent kit with final reagent concentrations at 10% glycerol, 5.0 mM MgCl2, 200 μM each of dATP, dCTP, and dGTP, 400 μM dUTP, and 1× TaqMan Buffer A. Primer concentrations were 50 nM and probe concentrations were 200 nM. 1.0 μl template cDNA was used for each reaction along with 1.25 U Amplitaq Gold and 0.5 U AmpErase Uracil N-Glycosylase (both from Perkin-Elmer Corp.). Cycling conditions were 2 min at 50°C, 10 min at 94°C, followed by 40 cycles of 15 s at 94°C and 90 s at 60°C. Three replicate reactions were set up for each sample for both β-actin and I-TAC. Relative quantities were calculated by first subtracting the average threshold cycle (Ct) for I-TAC from the β-actin average Ct to generate normalized I-TAC Ct values and then adding the lowest normalized Ct value to each to generate the value for n. Then take 2n to generate a relative quantity for each with the lowest level sample = 1.00.
Chromosomal Localization.
A panel of human/rodent somatic cell-hybrid genomic DNAs (BIOS Laboratories, New Haven, CT) was amplified by PCR using I-TAC–specific primers (forward, 5′-CCTTGGCTGTGATATTGTGTGC-3′ and reverse, 5′-CTGCCACTTTCACTGCTTTTACC-3′). PCR conditions were 30 cycles of denaturation at 94°C for 45 s, annealing at 58°C for 30 s, and extension at 68°C for 5 min using the Robocycler (Stratagene). PCR products were analyzed by agarose gel electrophoresis and discordance analysis was performed as specified by the manufacturer.
Chemical Synthesis of I-TAC.
The predicted mature I-TAC amino acid sequence was synthesized using automated Fmoc-based solid phase peptide chemistry on a Solid Phase Peptide Synthesizer (model 431A) using software version Synthassist 2.0 (both from Applied Biosystems, Inc.) retrofitted with deprotection monitoring. HBTU (benzotriazole-yl tetramethyluronium hexafluorophosphate) activation and single amino acid coupling cycles were used, except after sluggish deprotection steps, in which case the following residue was double coupled. Syntheses were started using 0.25 mmol preloaded Fmoc Lys (Boc) resin (Applied Biosystems, Inc.), 50% of the resin cake was removed midway through the synthesis, and the synthesis was completed. The final synthesis resin was cleaved and deprotected by treatment with a solution of 83% TFA (trifluoroacetic acid), 5% phenol, 5% water, and 2.5% ethandithiol for 1 h at 23°C. The mixture was filtered, the TFA filtrate was diluted into 50 ml of diethylether, and the precipitated crude peptide salt was collected by centrifugation. The peptide was purified by preparative reverse-phase HPLC (20 × 250 mm C18 column; Waters, Milford, MA) using a water/acetonitrile (0.1% TFA) gradient. Fractions were assayed by analytical HPLC (4.5 × 250 mm C18 column; Vydac, Hesparia, CA). Appropriate fractions were pooled (>85% purity) and the resulting solution was adjusted with folding buffer additives (final concentrations: 0.5 mM cysteine, 0.5 mM cystine, 10 mM methionine, 75 mM Hepes, pH 8.0, 0.05–0.5 mg/ml peptide). The folding reaction was essentially complete after 15 h at 4°C as indicated by shift to shorter retention time on analytical HPLC. The oxidized product was purified as above to >98% purity. Concentration was estimated by UV absorbance at 280 nm and confirmed by amino acid analysis (University of Michigan Protein Structure Facility, Ann Arbor, MI). The product was aliquoted, lyophilized, and stored at −80°C. Electrospray (Sciex API100; Perkin-Elmer, Applied Biosystems Division, Foster City, CA) mass of reduced chemokine: 8046.6 daltons (calculated: 8045.8 daltons); mass of folded chemokine: 8042.6 daltons (calculated: 8041.8 daltons).
Isolation and Culture of Human T Lymphocytes.
PBMCs were isolated from heparinized human blood using Accuspin tubes (Sigma Chemical Co., St. Louis, MO). Two different cell culture procedures were used. In the first method, blast cells were prepared by washing the mononuclear cells in RPMI medium, culturing for 3 d in RPMI medium with PHA (2 μg/ml; Murex, Dartford, England), and propagating in 50 U/ml IL-2 (Collaborative Biomedical Products, Bedford, MA). Cells were used 9–21 d after addition of IL-2. In the second cell preparation method, T cells were first separated from the mononuclear fraction using T cell purification columns (R&D Systems, Minneapolis, MN) and then cultured in human IL-2 (400 U/ml) for 7 days in RPMI medium containing 10% FCS.
Generation of Stable CXCR3 Transfectants.
The cDNA encoding CXCR3 was amplified from mRNA isolated from activated T cells with the following primers: forward primer, 5′-GCGAATTCAAGCACCAAAGCAGAGGGG-3′ and reverse primer, 5′-GCTCTAGATGGGCGAAAGGGGAGCCCG-3′. The PCR product obtained was gel purified, digested with EcoRI and XbaI, and cloned into the EcoRI and XbaI sites of pcDNA3 (Invitrogen). The sequence was verified by automated sequence analysis as above. Human embryonic kidney (HEK)293 cells were transfected with the CXCR3 expression construct using lipofectamine as specified by the manufacturer (GIBCO BRL), and selection for G418 resistance (1 mg/ml, wt/vol) was initiated 24 h after transfection. After ∼2 wk, G418-positive colonies were picked and further cloned to obtain clonal cell lines. Clonal cell lines were screened by their ability to mobilize intracellular calcium in response to IP-10. CXCR3 stable transfectants in a mouse pre-B cell line, 300-19, and in Jurkat cells have been described previously (15).
Calcium Mobilization Assay.
T cell blasts and CXCR3 transfectants described above were harvested in flux buffer (1× Hanks, 10 mM Hepes, 1.6mM CaCl2, pH 7.3), loaded with INDO-1-AM (20μg/ml) for 30 min at 37°C, and washed in flux buffer. Cells (0.25 × 106/ml flux buffer) were placed in a continuously stirred cuvette at 37°C in a Photon Technology Inc. (South Brunswick, NJ) fluorimager. Cells were stimulated with various chemokines and the calcium-related fluorescence changes were recorded. The intracellular concentration of Ca2+ was determined as described previously (22).
In Vitro Chemotaxis Assay.
Lymphocyte and PHA blast chemotaxis to various chemokines was measured using a 48-well Boyden's chamber (Neuro Probe, Inc., Cabin John, MD). In brief, agonists were diluted in RPMI medium containing 0.1% BSA and added to the bottom wells of a 48-well chemotaxis chamber. Cells were resuspended in RPMI/BSA and 105 cells were added to the top wells of the chamber. A 5-μm PVP-free filter coated on the bottom with 10 μg/ml of human type IV collagen was used. Chambers were incubated for 60 min in a 5% CO2-humidified incubator at 37°C. After the incubation period, the filters were stained with Dif-Quik and the number of PBMCs migrating to the lower surface was counted in six randomly chosen high powered fields.
I-TAC Binding to CXCR3 Transfectants.
I-TAC was 125I-iodinated using the Bolton and Hunter procedure as described previously (44). Binding assays were performed in triplicate in 200-μl volumes of binding buffer (50 mM Hepes, pH 7.4, 0.5% BSA, 1 mM CaCl2, and 5 mM MgCl2) in the presence of 0.27 nM of radiolabeled I-TAC and various concentrations of unlabeled chemokines at 4°C for 60 min. I-TAC bound to cells was separated from unbound by first diluting the samples in 10 vol of 10 mM Hepes, pH 7.4, and 0.5 M NaCl and then filtering on GF/C filters (presoaked 20 min in 0.3% polyethylenimine) using a vacuum aspirator. Filters were then washed twice with the same buffer, air-dried and counted on a Beckman gamma counter. Scatchard analysis was performed as described previously (22).
Results
Identification of I-TAC.
Through a collaboration with Incyte Pharmaceuticals, Inc., a cDNA library was constructed from RNA isolated from primary human astrocytes stimulated for 12 h with IFN-γ, IL-1β, and TNF-α (200 U/ml each). Approximately 4,000 sequences were generated at Incyte and screened to identify known and novel sequences using the program BLAST (43). The sequence of a novel clone, NCY580854, was most closely related to the non-ELR CXC chemokine, IP-10. This sequence was representative of a cluster of clones present at 0.35% in this stimulated astrocyte library. Full-length sequence of the cDNA corresponding to clone NCY580854 was obtained by 3′-RACE as described in Materials and Methods. The consensus sequence of various overlapping cDNAs corresponding to clone NCY580854 is shown in Fig. 1. The sequence contains an open reading frame of 94 amino acids with a 3′ untranslated region of ∼1,000 bp. (Fig. 1). A rare polyadenylation signal (AATACA; reference 45) is present 10 bases upstream of the polyA+ track (Fig. 1). Part of the sequence of clone NCY580854 is identical to the recently described β-R1 cDNA fragment (46), except that the published β-R1 sequence has an insertion of six bases (TCGAGC) at position 302, a single base deletion at positions 275, 277, and 283, a C→ T change at position 335, a G→ A change at position 353, and an A→ T change at position 410 (Fig. 1). A re-examination of the β-R1 sequence data revealed that the deletions and insertions are not present in that cDNA and the base differences are most likely sequence polymorphisms (Ransohoff, R., personal communication). Therefore, we conclude that NCY580854 and β-R1 are encoded by the same gene. Due to its IFN inducibility and biological activity (see below), we have named this novel chemokine I-TAC (IFN-inducible T cell α chemoattractant). Based on the hydrophobicity profile of the coding sequence of NCY580854 and sequence comparisons with IP-10 and HuMig, we propose that signal peptide cleavage occurs between Gly21 and Phe22 (Fig. 1). The predicted mature polypeptide is 72 amino acids in length, contains 4 conserved cysteines typical of chemokines, and does not contain an ELR motif. Like other chemokines, it is highly basic with an isoelectric point of 10.79. I-TAC has greater similarity (∼40%) to the non-ELR CXC chemokines, IP-10 and HuMig, than to any other known chemokines. The alignment and phylogenetic relationship of I-TAC to other CXC chemokines is shown in Fig. 2, A and B.
Figure 1.

Nucleotide sequence and predicted amino acid sequence of human I-TAC. The deduced amino acid sequence is indicated in single letter codes below the cDNA sequence, with the stop codon indicated by an asterisk. The putative signal peptide is underlined. The polyadenylation signal is of a rare type and is marked with a double underline. The sequence is available from EMBL/GenBank/DDBJ under accession number AF030514.
Figure 2.


Sequence alignment and phylogenetic relationship of I-TAC with other known CXC chemokines. (A) Amino acid alignment of I-TAC with CXC chemokines. The alignment was generated with the program Megalign (DNA Star, Madison, WI) using the Clustal method and then manually aligned to obtain the maximum amino acid similarity. Amino acid residues conserved in all sequences are boxed and shaded. Unshaded boxed regions indicate amino acid residues conserved between I-TAC and at least two other CXC chemokines. (B) Phylogenetic tree showing the relationships between I-TAC and known CXC chemokines. The alignment above was used to generate a phylogenetic tree by the Megalign program and the extent of similarity between sequences is indicated below the phylogenetic tree.
Chromosomal Localization of I-TAC.
The chromosome location of I-TAC was determined by PCR amplification of human/rodent somatic cell-hybrid genomic DNAs with primers designed to flank the predicted position of intron 1 (based on alignment of I-TAC cDNA with the known gene structure of IL-8; reference 47). These primers amplified a genomic product of ∼700 bp, whereas the cDNA product is 105 bp (data not shown). No product was detected in either hamster or mouse genomic DNA controls. The PCR analysis of the somatic cell-hybrid genomic DNA panel using these primers yielded products only in hybrids containing human chromosome 4 (data not shown). Thus, the gene encoding I-TAC maps to chromosome 4 as do other CXC chemokines, with the exception of SDF-1.
Expression of I-TAC mRNA.
To determine the tissue distribution of I-TAC mRNA, we performed Northern blot hybridization using the I-TAC cDNA as probe. The highest levels of I-TAC mRNA were detected in peripheral blood leukocytes, pancreas, and liver, followed by thymus, spleen, and lung (Fig. 3). Lower levels were detected in placenta, prostate, and small intestine. The size of the I-TAC mRNA is ∼1.4 kb, the same size as the full length cDNA sequence in Fig. 1. However, a transcript of ∼4.5 kb was also detected in pancreas, peripheral blood leukocytes, and spleen (Fig. 3). Interestingly, a heterogeneous range of transcripts from ∼1.3 to 6 kb was detected in liver and small intestine, which cannot be explained by RNA degradation, since discreet bands were detected in these lanes using other genes as probes (data not shown). These bands may represent alternatively spliced I-TAC transcripts, or cross-hybridizing mRNAs, although we have not investigated this further.
Figure 3.

Northern blot analysis of the tissue distribution of I-TAC. Multiple Tissue Northern Blot filters (Clontech), containing 2 μg poly(A)+ mRNA/lane, were hybridized with a 32P-labeled human I-TAC cDNA probe. Conditions for hybridization and subsequent washings were as indicated in Materials and Methods. Autoradiography was performed at −70°C with intensifying screen for 24–72 h. Migration of molecular weight markers in kilobases are shown between the blots.
To investigate the regulation of I-TAC mRNA by IFN, we used real time quantitative reverse transcriptase PCR (48). β-actin mRNA was simultaneously amplified and the levels of I-TAC PCR product were normalized to the levels of β-actin PCR product. The lowest level of I-TAC PCR product detected within the samples analyzed was arbitrarily set at one and corresponds to the mRNA detected in cultured unstimulated astrocytes (Table 1). IFN-γ upregulated I-TAC mRNA in all cell types examined. The greatest induction, 14,000-fold, was observed in astrocytes. Furthermore, IL-1 synergized with IFN-γ to give an ∼400,000 fold increase of I-TAC mRNA in astrocytes. This synergy was not observed in monocytes. In monocytes, IFN-β induction of I-TAC mRNA was similar to IFN-γ induction. Finally, IFN induction resulted in a significantly higher I-TAC mRNA level in astrocytes compared with the basal level detected in pancreas, lung, or liver (Table 1). These results demonstrate a tremendous upregulation of I-TAC mRNA in astrocytes stimulated with IFN-γ together with IL-1, and further confirm the previous report that the β-R1 gene encoding I-TAC is regulated by IFN (46).
Table 1.
Real Time Quantitative Reverse Transcriptase PCR of I-TAC mRNA
| Cell type (± stimulation) | Relative level of I-TAC mRNA* | |
|---|---|---|
| Astrocytes | 1.00 | |
| Astrocytes + IL-1 | 3,221 | |
| Astrocytes + IFN-γ | 14,164 | |
| Astrocytes + IL-1 and IFN-γ | 467,087 | |
| Monocytes | 4 | |
| Monocytes + IL-1 | 5 | |
| Monocytes + IFN-γ | 399 | |
| Monocytes + IL-1 and IFN-γ | 222 | |
| Monocytes + IFN-β | 425 | |
| SV-A3 (microglial cell line) | 5 | |
| SV-A3 + IFN-γ | 48,308 | |
| Pancreas | 4,021 | |
| Lung | 637 | |
| Liver | 35 |
Levels of I-TAC mRNA were calculated as described in Materials and Methods.
Chemotactic Activity of I-TAC.
Since I-TAC shares the greatest sequence similarity with IP-10 and HuMig, we predicted that it would have similar biological activities and evoke similar responses in T cells. To test this hypothesis, we chemically synthesized the predicted mature I-TAC polypeptide and used the purified material to perform chemotaxis assays on PHA-stimulated peripheral blood T cells cultured in the presence of IL-2 for 8–15 d. I-TAC induced a potent chemotactic response in activated T cells that peaked at 10 nM and decreased at higher concentrations in a typical bell-shaped chemotactic response curve (Fig. 4). I-TAC was equipotent to IP-10, but the efficacy was much higher, i.e., twice as many cells migrated in response to 10 nM I-TAC than to 10 nM IP-10 (Fig. 4). No response was observed with freshly isolated, untreated T cells, monocytes, or granulocytes (data not shown), indicating that, like IP-10 and HuMig, I-TAC is selective for activated T cells (13).
Figure 4.

Chemotactic response of activated T cells to I-TAC. PHA-stimulated T cells grown in the presence of IL-2 for 10–15 d were used in chemotaxis assays as described in Materials and Methods. Each assay was done in triplicate and the cells migrating in response to the indicated concentration of chemokine were scored in five to eight high powered fields (HPF). Error bars were determined by first calculating the average number of cells in the HPF and then determining the standard deviation within the three different wells.
Intracellular [Ca2+] Measurements.
We then determined if I-TAC caused changes in intracellular calcium levels in activated T cells, as is typical for chemokine-induced signaling in leukocytes. I-TAC caused a rapid and transient increase in intracellular calcium level in a dose-dependent manner with peak activity at 10 nM (Fig. 5 A). I-TAC was considerably more potent (EC50: ∼6 nM) than IP-10 and HuMig (EC50: ∼20 and ∼30 nM, respectively). In addition, the I-TAC response was more robust: up to 800 nM calcium was mobilized by I-TAC compared with 200 and 400 nM by HuMig and IP-10, respectively (Fig. 5 B). No change in intracellular calcium level was detected in neutrophils, eosinophils, or monocytes, consistent with the lack of chemotaxis in these cells (data not shown). These results confirmed the activity and specificity observed in the chemotaxis assay and in addition showed that I-TAC is more potent and efficacious than either IP-10 or HuMig.
Figure 5.

Mobilization of intracellular calcium in activated T cells. (A) Concentration-dependent [Ca2+]i changes induced by I-TAC in IL-2–stimulated T cell blasts. I-TAC was added at 50, 10, 5, and 0.5 nM to INDO-1–loaded cells and [Ca2+]i-dependent fluorescence changes were recorded. The data shown is from one representative experiment of at least four separate experiments using cells from different donors. (B) Dose- response curves of I-TAC, IP-10, and HuMig on IL-2–stimulated T cell blasts. Chemokine was added at 0.25, 0.5, 5, 10, 50, and 500 nM to INDO-1–loaded cells and intracellular [Ca2+] changes were monitored as above. These data are compiled from two separate experiments using cells from different donors.
Cross-Desensitization Analysis.
To examine receptor use by I-TAC, we used calcium mobilization as a read-out and performed cross-desensitization experiments using activated T cells. Although homologous desensitization was seen with all three chemokines (data not shown), an interesting pattern of cross-desensitization was observed (Fig. 6). First, IP-10 at 1,000 nM and HuMig at 500 nM completely cross-desensitized each other, indicating that they interact with one receptor, most likely CXCR3 (15). 50 nM I-TAC desensitized the response to 1,000 nM IP-10 and 500 nM HuMig, indicating that I-TAC most likely interacts with the IP-10/HuMig receptor. However, neither IP-10 at 1,000 nM nor HuMig at 500 nM completely desensitized the response to 50 nM I-TAC (Fig. 6). These results suggest two possibilities. The first is that I-TAC interacts with multiple receptors, at least one of which does not bind IP-10 or HuMig. Alternatively, it is possible that all three chemokines interact with one receptor, but I-TAC interacts with at least one site or conformational form bound by neither IP-10 nor HuMig.
Figure 6.

Cross-desensitization of IL-2–stimulated T cell blasts. Chemokine concentrations used were 1,000 nM for IP-10, 500 nM for HuMig, and 50 nM for I-TAC. INDO-1–loaded cells were exposed sequentially to the indicated chemokines. These data are from one representative experiment of four separate experiments using cells from different donors.
Interaction of I-TAC with CXCR3.
Cross-desensitization experiments suggested that IP-10, HuMig, and I-TAC can all interact with the same receptor. Since an IP-10/HuMig receptor (CXCR3) has been identified, we tested the ability of I-TAC to interact with CXCR3. I-TAC caused a potent chemotactic response in 300-19 transfectants stably expressing CXCR3. The response peaked at 10 nM and decreased at higher concentrations in a typical chemotactic response (Fig. 7). No response to I-TAC was observed in parental 300-19 cells. I-TAC was both more potent (maximum migration at 10 nM for I-TAC versus ∼100 nM for IP-10 and HuMig) and more efficacious (twice as many cells migrated in response to 10 nM I-TAC) than IP-10 or HuMig (Fig. 7). To further examine the interaction with CXCR3, the change in the level of intracellular calcium in 300-19 CXCR3 transfectants was monitored after challenge with various chemokines. I-TAC caused a dose-dependent response that peaked at 10 nM (Fig. 8), while no change in intracellular calcium was observed in parental 300-19 cells (data not shown). In comparison to IP-10 or HuMig, I-TAC mobilized twice as much calcium, ∼2,000 nM compared with ∼1,000 nM for IP-10 and HuMig. The EC50 of I-TAC was ∼1 nM compared with ∼10 nM for IP-10 and ∼50 nM for HuMig (Fig. 8). The results of Ca2+ cross-desensitization experiments using the 300-19 CXCR3 transfectants are shown in Fig. 9. Similar to what was observed on T cells, a relatively low concentration of I-TAC (50 nM) completely desensitized the response to either 1,000 nM IP-10 or 500 nM HuMig, although IP-10 or HuMig at these elevated concentrations caused only a partial desensitization of I-TAC responses (Fig. 9). The failure of IP-10 or HuMig to completely desensitize I-TAC responses in cloned CXCR3 transfectants is consistent with the hypothesis that I-TAC binds multiple sites or conformational forms of this receptor. Regardless, these results indicate that I-TAC is a highly potent and efficacious ligand for CXCR3.
Figure 7.

Chemotaxis of stable CXCR3 transfectants. 300-19 transfectants expressing CXCR3 (clone MLRA-A5) were challenged with I-TAC (squares), HuMig (circles), and IP-10 (diamonds). Chemotaxis was done as previously described (15). The incubation time for chemotaxis was 2 h. I-TAC responses to nontransfected 300-19 cells are also shown (triangles).
Figure 8.
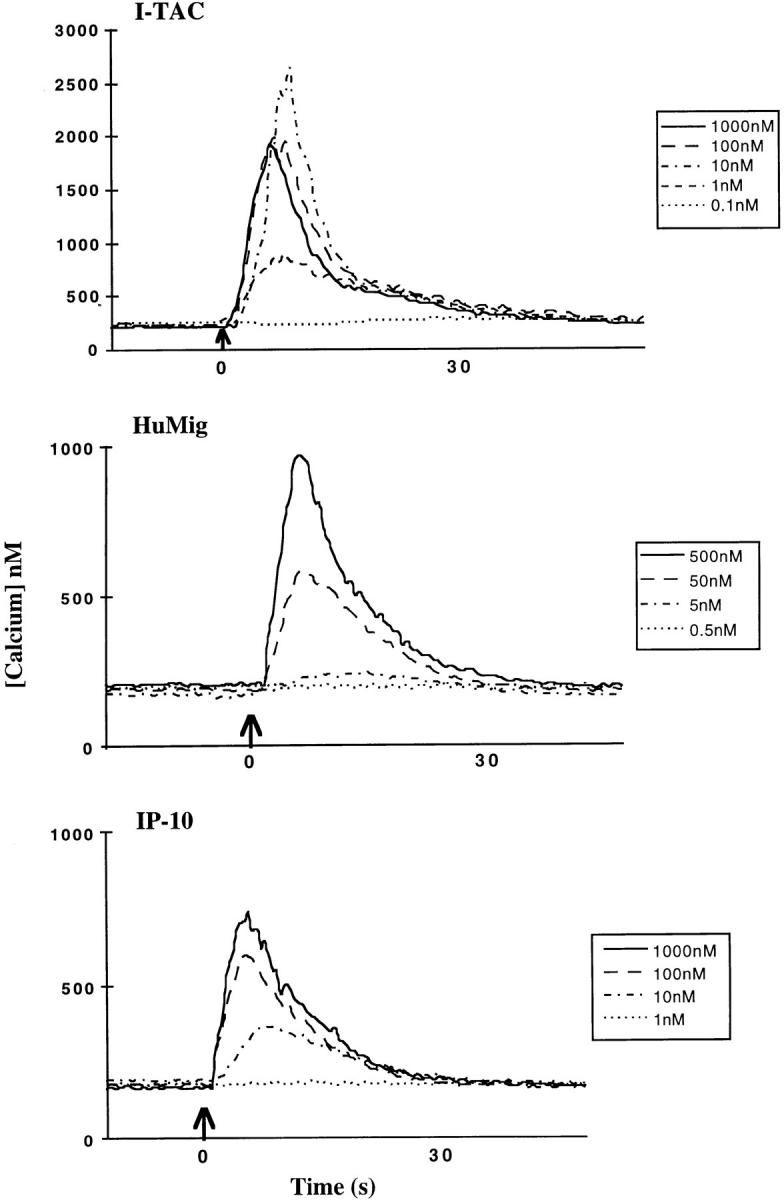
Mobilization of intracellular calcium in stable CXCR3 transfectants. Concentration-dependent [Ca2+]i changes induced by I-TAC, HuMig, or IP-10 on INDO-1–loaded 300-19/CXCR3 transfectants were determined as described above. Doses were as indicated.
Figure 9.

Cross-desensitization of 300-19/CXCR3 transfectants. Chemokine concentrations used were 1,000 nM for IP-10, 500 nM for HuMig, and 50 nM for I-TAC. INDO-1–loaded cells were exposed sequentially to the indicated chemokines, and intracellular [Ca2+] changes were monitored as above.
Binding of I-TAC to CXCR3 Transfectants.
To demonstrate that I-TAC binds to CXCR3, and to measure its affinity compared with IP-10 and HuMig, we performed binding assays. No I-TAC binding was observed over background on the 300-19 transfectants, presumably due to low receptor expression (data not shown). However, radiolabeled I-TAC bound specifically to HEK293 transfectants stably expressing CXCR3 (5976 ± 90 cpm) and not to parental HEK293 cells (1551 ± 48 cpm). The binding to the HEK293 transfectants was displaceable with increasing concentrations of unlabeled I-TAC (Fig. 10). Scatchard analysis revealed two sites, a high affinity site of 0.3 nM and a low affinity site of 36 nM (Fig. 10, n = 3 experiments). When unlabeled IP-10 or HuMig was used to displace radiolabeled I-TAC, the dose-response curve shifted to the right. The IC50 for I-TAC was ∼1 nm, whereas IP-10 and HuMig IC50 values were ∼70 and ∼300 nM, respectively. Direct binding of labeled IP-10 or HuMig to CXCR3 was not detectable above background binding (data not shown). Taken together, these data suggest that IP-10 and HuMig have a low affinity for CXCR3 expressed in HEK293 cells. Since I-TAC has a significantly higher affinity for CXCR3, as well as higher potency and efficacy than IP-10 or HuMig, we conclude that I-TAC is the dominant ligand for this receptor.
Figure 10.

I-TAC binding to HEK293 cells stably expressing CXCR3. CXCR3 transfectants were incubated with radiolabeled I-TAC and various concentrations of unlabeled chemokines. Radiolabeled I-TAC bound to cells was determined as indicated in Materials and Methods. The top panel shows the displacement of radiolabeled I-TAC by unlabeled I-TAC, and the Scatchard analysis used to derive affinities for the two binding sites. The lower panels are the curves showing the displacement of radiolabeled I-TAC by IP-10 and HuMig. The maximum amount of I-TAC bound was 5976 ± 90 cpm. Parental HEK293 cells bound 1551 ± 48 cpm.
Discussion
We have identified a novel non-ELR CXC chemokine, termed I-TAC, through sequencing of cDNAs derived from stimulated astrocytes. The cDNA encoding I-TAC is identical to β-R1, a recently described gene that is differentially regulated by INF-β (46). I-TAC shares greatest sequence similarity with the chemokines IP-10 and HuMig and we provide evidence that I-TAC binds to the IP-10/ HuMig receptor, CXCR3. However, I-TAC has a much higher affinity for CXCR3 than have either IP-10 or HuMig, as demonstrated by displaceable binding. Scatchard analysis revealed two sites for I-TAC, one high affinity and one low affinity. No direct binding was detected on transfectants or activated T cells with radiolabeled IP-10 or HuMig, possibly due to lower affinity for CXCR3. The importance of these interactions in vivo is not clear. IP-10, for example, has been shown to interact with proteoglycans, as may also be possible for HuMig and I-TAC. The influence of these associations on receptor binding and activation is not yet fully understood (49).
I-TAC is more potent and efficacious than either IP-10 or HuMig in its ability to mobilize intracellular calcium and as a chemotactic factor (rank order potency I-TAC > HuMig ≈ IP-10). However, calcium desensitization experiments revealed an interesting phenomenon. In both activated T cells and transfected cell lines expressing CXCR3, I-TAC completely blocked the response to a subsequent challenge with IP-10 or HuMig, consistent with their binding to the same receptor. Unexpectedly, the reverse was not true. Even a high concentration of IP-10 or HuMig could not completely desensitize I-TAC responses. Since these results are seen with the cloned CXCR3, it appears that these three chemokines may interact differently with the receptor. It is conceivable that the receptor may exist in different conformations, or have multiple binding sites, and that I-TAC interacts with conformations or sites which cannot interact with IP-10 and HuMig. Nevertheless, our results show that I-TAC is the dominant ligand for CXCR3 due to higher affinity, potency, and efficacy when compared with IP-10 and HuMig. It is important to note that dominant and weak ligands for CCR2 have also been reported. MCP-1 is considerably more potent than MCP-3 in the mobilization of intracellular calcium, making it the dominant ligand. In addition, saturating levels of MCP-3 do not completely desensitize MCP-1 responses (50).
The existence of dominant and weak ligands may have implications for the roles of multiple chemokines at inflammatory sites. For example, weaker ligands captured on endothelial cells may initiate firm adhesion of leukocytes, then more dominant ligands produced at inflammatory loci direct leukocyte recruitment. Such a “hierarchy” of chemoattractants has been demonstrated in vitro where different chemotactic factors, acting via multiple receptors, “navigate” leukocytes in different directions (51). Our studies would suggest that this phenomenon could also occur by chemokines acting at the same receptor.
In agreement with studies describing the regulation of CXCR3, I-TAC has potent chemotactic activity for IL-2–stimulated T cells and is not active on resting and naive T cells. As such, I-TAC may not play a role in T cell trafficking under normal conditions, but may have its greatest effect during immune response to foreign antigens, where IL-2 has been generated. Antigen recognition by unstimulated naive/memory T cells in secondary lymphoid organs leads to T cell activation, IL-2 production, and clonal expansion of antigen-selective T cells (52, 53). I-TAC released at the site of initial insult may then recruit these effector T cells. This scenario is consistent with the upregulation of I-TAC by acute phase proteins, such as IL-1 and the IFNs, which would be expected to be present at high concentrations at sites of inflammation.
Recently, several chemokines having specificity for T cells have been described, including dendritic cell CC chemokine 1/PARC (pulmonary and activation-regulated chemokine), TARC (thymus and ARC), and MIP (macrophage inflammatory protein)-3a/LARC (liver and ARC) (28, 54–56). I-TAC demonstrated significantly greater potency than any of these three chemokines for IL-2–activated T cells (Gladue, R.P., and T.J. Paradis, unpublished data). The only chemokine more potent than I-TAC was SDF-1α, which interacts with CXCR4 (35, 36, Gladue, R.P., et al., unpublished data). Unlike I-TAC, SDF-1α is active on naive as well as activated T cells, monocytes, and granulocytes. It has an important role in development, as evidenced by the prenatal death that occurs in SDF-1α knock-out mice, most likely due to a defect in the development of the cardiac ventricular septal, as well as defects in B-cell lymphopoiesis and bone-marrow myelopoiesis (57). I-TAC differs from SDF-1α in its unique selectivity for effector T cells and this underscores the importance of I-TAC in T cell–mediated diseases.
The functional relationship of I-TAC to IP-10 and HuMig is not clear. Like IP-10, I-TAC is expressed in normal tissues including thymus, spleen, and pancreas, where it may be involved in the trafficking of activated/effector T cells. In addition, IP-10, HuMig, and I-TAC may also play similar roles in inflammation. IP-10 has been shown to be expressed in delayed-type hypersensitivity reactions in the skin, psoriatic plaques, tuberculoid leprosy, and certain tumors (17–19), where it may be important for the recruitment of effector T cells. It is tempting to speculate that I-TAC might also be expressed in these disease states.
In contrast to the low level of I-TAC expression in normal tissues, I-TAC is dramatically upregulated by IFN, and IL-1 synergizes to further increase expression. This, in addition to I-TAC's selective activity on T cells, suggests a role for I-TAC in T cell–mediated inflammatory diseases. These would most likely be diseases associated with a Th1-type cytokine profile, where abundant IFN-γ and IL-1 would be produced. Examples include autoimmune diseases, delayed-type hypersensitivity responses, certain viral diseases, and transplant rejection (58). However, the marked upregulation of I-TAC in astrocytes and microglial cells upon stimulation by IL-1 and IFN raises the possibility that I-TAC has a central role in the pathogenesis of neuroinflammatory diseases such as meningitis, encephalitis, and multiple sclerosis.
The role of IL-1 in EAE, an animal model for multiple sclerosis, has been demonstrated, in that an IL-1 receptor antagonist has shown beneficial effects (59). Furthermore, acquisition of a Th1-type cytokine profile is required for EAE disease progression, highlighting the importance of IL-2 and IFN-γ (52, 53). Since IL-2 has been shown to upregulate CXCR3 (15), and IFN-γ synergizes with IL-1 to produce I-TAC, it is tempting to speculate that the following sequence of events might cause pathology in multiple sclerosis. Autoreactive T cells that have entered the central nervous system initiate the generation of a localized Th1-type cytokine environment, which leads to the production of I-TAC by astrocytes and microglia. This causes an amplification loop that results in increased infiltration of autoreactive and bystander effector T cells. Eventually, a nonspecific inflammatory response is triggered that may cause lesion formation and demyelination indicative of multiple sclerosis disease pathology.
In conclusion, we have identified and characterized I-TAC, a novel non-ELR CXC chemokine highly expressed in cytokine-stimulated astrocytes. I-TAC has potent chemotactic activity on activated T cells and is a higher affinity ligand for CXCR3 than is IP-10 or HuMig. The unique regulation of I-TAC by IL-1 and IFN-γ, and its high level production by astrocytes and microglia, suggests an important role for this chemokine in central nervous system diseases which involve T cell recruitment.
Acknowledgments
We thank Suzanne P. Williams, Yevette C. Clancy, Melissa T. Cronan, and Lisa S. Hayes for DNA sequencing support; and Michael Fogliano, David P. Dean, and Lynn M. Hames for bioinformatic analysis. We also thank Robin T. Nelson for providing RNAs from leukocytes and hematopoietic cell lines, and Robert B. Nelson for SV-A3 cells. We acknowledge Douglas A. Fisher and John B. Cheng for critical comments on the manuscript. The authors are grateful to Sue Hu and Chun Chao of the Minnesota Medical Research Institute for providing primary human fetal astrocytes.
Abbreviations used in this paper
- EAE
experimental autoimmune encephalomyelitis
- ELR
Glu-Leu-Arg
- HEK
human embryonic kidney
- HuMig
IFN-γ–induced human monokine
- IP-10
IFN-γ–inducible 10-kD protein
- I-TAC
IFN-inducible T cell alpha chemoattractant
- MCP
monocyte chemotactic protein
- SDF
stromal cell–derived factor
References
- 1.Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines—CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 2.Schall TJ, Bacon KB. Chemokines, leukocyte trafficking, and inflammation. Curr Opin Immunol. 1994;6:865–873. doi: 10.1016/0952-7915(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 3.Taub DD, Oppenheim JJ. Chemokines, inflammation and the immune system. Ther Immunol. 1994;1:229–246. [PubMed] [Google Scholar]
- 4.Baggiolini M, Dewald B, Moser B. Human chemokines: an update. Annu Rev Immunol. 1997;15:675–705. doi: 10.1146/annurev.immunol.15.1.675. [DOI] [PubMed] [Google Scholar]
- 5.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 6.Pan Y, Lloyd C, Zhou H, Dolich S, Deeds J, Gonzalo J-A, Vath J, Gosselin M, Ma J, Dussault B, et al. Neurotactin, a membrane-anchored chemokine upregulated in brain inflammation. Nature. 1997;387:611–617. doi: 10.1038/42491. [DOI] [PubMed] [Google Scholar]
- 7.Kelner GS, Kennedy J, Bacon KB, Kleyensteuber S, Largaespada DA, Jenkins NA, Copeland NG, Bazan JF, Moore KW, Schall TJ, et al. Lymphotactin: a cytokine that represents a new class of chemokine. Science. 1994;266:1395–1399. doi: 10.1126/science.7973732. [DOI] [PubMed] [Google Scholar]
- 8.Kennedy J, Kelner GS, Kleyensteuber S, Schall TJ, Weiss MC, Yssel H, Schneider PV, Cocks BG, Bacon KB, Zlotnik A. Molecular cloning and functional characterization of human lymphotactin. J Immunol. 1995;155:203–209. [PubMed] [Google Scholar]
- 9.Yoshida T, Imai T, Kakizaki M, Nishimura M, Yoshie O. Molecular cloning of a novel C or gamma type chemokine, SCM-1. FEBS (Fed Eur Biochem Soc) Lett. 1995;360:155–159. doi: 10.1016/0014-5793(95)00093-o. [DOI] [PubMed] [Google Scholar]
- 10.Clark-Lewis I, Schumacher C, Baggiolini M, Moser B. Structure-function relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of the NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J Biol Chem. 1991;266:18989–18994. [PubMed] [Google Scholar]
- 11.Hebert CA, Vitangcol RV, Baker JB. Scanning mutagenesis of interleukin-8 identifies a cluster of residues required for receptor binding. Proc Natl Acad Sci USA. 1991;90:3574–3577. [PubMed] [Google Scholar]
- 12.Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M. Platelet factor 4 binds to interleukin-8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc Natl Acad Sci USA. 1993;90:3574–3577. doi: 10.1073/pnas.90.8.3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ. Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao F, Rabin RL, Yannelli JR, Koniaris LG, Vanguri P, Farber JM. Human Mig chemokine: biochemical and functional characterization. J Exp Med. 1995;182:1301–1314. doi: 10.1084/jem.182.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, Baggiolini M, Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J Exp Med. 1996;184:1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan G, Luster AD, Hancock G, Cohn ZA. The expression of a γ interferon-induced protein (IP-10) in delayed immune responses in human skin. J Exp Med. 1987;166:1098–1108. doi: 10.1084/jem.166.4.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gottlieb AB, Luster AD, Posnett DN, Carter DM. Detection of a γ interferon-induced protein IP-10 in psoriatic plaques. J Exp Med. 1988;168:941–948. doi: 10.1084/jem.168.3.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarris AH, Esgleyes-Ribot T, Crow M, Broxmeyer HE, Karasavvas N, Pugh W, Grossman D, Deisseroth A, Duvic M. Cytokine loops involving interferon-gamma and IP-10, a cytokine chemotactic for CD4+ lymphocytes: an explanation for the epidermotropism of cutaneous T-cell lymphoma? . Blood. 1995;86:651–658. [PubMed] [Google Scholar]
- 20.Mackay CR. Chemokine receptors and T cell chemotaxis. J Exp Med. 1996;184:799–802. doi: 10.1084/jem.184.3.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao JL, Kuhns DB, Tiffany HL, McDermott D, Li X, Francke U, Murphy PM. Structure and functional expression of the human macrophage inflammatory protein 1 alpha/RANTES receptor. J Exp Med. 1993;177:1421–1427. doi: 10.1084/jem.177.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neote K, DiGregorio D, Mak JY, Horuk R, Schall TJ. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell. 1993;72:415–425. doi: 10.1016/0092-8674(93)90118-a. [DOI] [PubMed] [Google Scholar]
- 23.Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci USA. 1994;91:2752–2756. doi: 10.1073/pnas.91.7.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponath PD, Qin S, Post TW, Wang J, Wu L, Gerard NP, Newman W, Gerard C, Mackay CR. Molecular cloning and characterization of a human eotaxin receptor expressed selectively on eosinophils. J Exp Med. 1996;183:2437–2448. doi: 10.1084/jem.183.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daugherty BL, Siciliano SJ, DeMartino JA, Malkowitz L, Sirotina A, Springer MS. Cloning, expression, and characterization of the human eosinophil eotaxin receptor. J Exp Med. 1996;183:2349–2354. doi: 10.1084/jem.183.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoogewerf A, Black D, Proudfoot AE, Wells TN, Power CA. Molecular cloning of murine CC CKR-4 and high affinity binding of chemokines to murine and human CC CKR-4. Biochem Biophys Res Commun. 1996;218:337–343. doi: 10.1006/bbrc.1996.0059. [DOI] [PubMed] [Google Scholar]
- 27.Samson M, Stordeur P, Labbe O, Soularue P, Vassart G, Parmentier M. Molecular cloning and chromosomal mapping of a novel human gene, ChemR1, expressed in T lymphocytes and polymorphonuclear cells and encoding a putative chemokine receptor. Eur J Immunol. 1996;26:3021–3028. doi: 10.1002/eji.1830261230. [DOI] [PubMed] [Google Scholar]
- 28.Hieshima K, Imai T, Opdenakker G, Van Damme J, Kusuda J, Tei H, Sakaki Y, Takatsuki K, Miura R, Yoshie O, Nomiyama H. Molecular cloning of a novel human CC chemokine liver and activation-regulated chemokine (LARC) expressed in liver. Chemotactic activity for lymphocytes and gene localization on chromosome 2. J Biol Chem. 1997;272:5846–5853. doi: 10.1074/jbc.272.9.5846. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida R, Imai T, Hieshima K, Kusuda J, Baba M, Kitaura M, Nishimura M, Kakizaki M, Nomiyama H, Yoshie O. Molecular cloning of a novel human CC chemokine EBI1-ligand chemokine that is a specific functional ligand for EBI1, CCR7. J Biol Chem. 1997;272:13803–13809. doi: 10.1074/jbc.272.21.13803. [DOI] [PubMed] [Google Scholar]
- 30.Baba M, Imai T, Nishimura M, Kakizaki M, Takagi S, Hieshima K, Nomiyama H, Yoshie O. Identification of CCR6, the specific receptor for a novel lymphocyte-directed CC chemokine LARC. J Biol Chem. 1997;272:14893–14898. doi: 10.1074/jbc.272.23.14893. [DOI] [PubMed] [Google Scholar]
- 31.Tiffany HL, Lautens LL, Gao JL, Pease J, Locati M, Combadiere C, Modi W, Bonner TI, Murphy PM. Identification of CCR8: a human monocyte and thymus receptor for the CC chemokine I-309. J Exp Med. 1997;186:165–170. doi: 10.1084/jem.186.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roos RS, Loetscher M, Legler DF, Clark-Lewis I, Baggiolini M, Moser B. Identification of CCR8, the receptor for the human CC chemokine I-309. J Biol Chem. 1997;272:17251–17254. doi: 10.1074/jbc.272.28.17251. [DOI] [PubMed] [Google Scholar]
- 33.Holmes WE, Lee J, Kuang WJ, Rice GC, Wood WI. Structure and functional expression of a human interleukin-8 receptor. Science. 1991;253:1280–1283. [PubMed] [Google Scholar]
- 34.Murphy PM, Tiffany HL. Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science. 1991;253:1280–1283. [PubMed] [Google Scholar]
- 35.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 36.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line–adapted HIV-1. Nature. 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 37.Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB (Fed Am Soc Exp Biol) J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- 38.Glabinski AR, Tani M, Tuohy VK, Tuthill RJ, Ransohoff RM. Central nervous system chemokine mRNA accumulation follows initial leukocyte entry at the onset of acute murine experimental autoimmune encephalomyelitis. Brain Behav Immun. 1995;9:315–330. doi: 10.1006/brbi.1995.1030. [DOI] [PubMed] [Google Scholar]
- 39.Godiska R, Chantry D, Dietsch GN, Gray PW. Chemokine expression in murine experimental allergic encephalomyelitis. J Neuroimmunol. 1995;58:167–176. doi: 10.1016/0165-5728(95)00008-p. [DOI] [PubMed] [Google Scholar]
- 40.Glabinski AR, Tani M, Aras S, Stoler MH, Tuohy VK, Ransohoff RM. Regulation and function of central nervous system chemokines. Int J Dev Neurosci. 1995;13:153–165. doi: 10.1016/0736-5748(95)00017-b. [DOI] [PubMed] [Google Scholar]
- 41.Ransohoff RM, Aras S, Chaturvedi P. Regulation of chemokine genes in human astrocytoma cells by IFN-gamma and TNF-alpha. FASEB (Fed Am Soc Exp Biol) J. 1994;8:A199. (Abstr.) [Google Scholar]
- 42.McColl SR, Hachicha M, Levasseur S, Neote K, Schall TJ. Uncoupling of early signal transduction events from effector function in human peripheral blood neutrophils in response to recombinant macrophage inflammatory proteins-1 alpha and -1 beta. J Immunol. 1993;150:4550–4560. [PubMed] [Google Scholar]
- 43.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.McFarthing, K.G. 1992. Selection and synthesis of receptor-specific radioligands. In Receptor-Ligand Interactions. E.C. Hulme, editor. Oxford University Press, New York. 1–18.
- 45.Wickens M, Stephenson P. Role of the conserved AAUAAA sequence: four AAUAA point mutations prevent messenger RNA 3′ end formation. Science. 1984;226:1045–1051. doi: 10.1126/science.6208611. [DOI] [PubMed] [Google Scholar]
- 46.Rani MRS, Foster GR, Leung S, Leaman D, Stark GR, Ransohoff RM. Characterization of beta-R1, a gene that is selectively induced by interferon beta (IFN-beta) compared with IFN-alpha. J Biol Chem. 1996;271:22878–22884. doi: 10.1074/jbc.271.37.22878. [DOI] [PubMed] [Google Scholar]
- 47.Mukaida N, Shiroo M, Matsushima K. Genomic structure of the human monocyte-derived neutrophil chemotactic factor IL-8. J Immunol. 1989;143:1366–1371. [PubMed] [Google Scholar]
- 48.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 49.Luster AD, Greenberg SM, Leder P. The IP-10 chemokine binds to a specific cell surface heparan sulfate site shared with platelet factor 4 and inhibits endothelial cell proliferation. J Exp Med. 1995;182:219–231. doi: 10.1084/jem.182.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franci C, Wong LM, Van Damme J, Proost P, Charo IF. Monocyte chemoattractant protein-3, but not monocyte chemoattractant protein-2, is a functional ligand of the human monocyte chemoattractant protein-1 receptor. J Immunol. 1995;154:6511–6517. [PubMed] [Google Scholar]
- 51.Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol. 1997;139:1349–1360. doi: 10.1083/jcb.139.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mosmann TR, Sad S. The expanding universe of T-cell subsets: TH1, TH2 and more. Immunol Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 53.Reiner SL, Seder RA. T helper cell differentiation in immune response. Curr Opin Immunol. 1995;7:360–366. doi: 10.1016/0952-7915(95)80111-1. [DOI] [PubMed] [Google Scholar]
- 54.Adema GJ, Hartgers F, Verstraten R, de Vries E, Marland G, Menon S, Foster J, Xu Y, Nooyen P, McClanahan T, et al. A dendritic-cell–derived C-C chemokine that preferentially attracts naive T cells. Nature. 1997;387:713–717. doi: 10.1038/42716. [DOI] [PubMed] [Google Scholar]
- 55.Hieshima K, Imai T, Baba M, Shoudai K, Ishizuka K, Nakagawa T, Tsuruta J, Takeya M, Sakaki Y, Takatsuki K, et al. A novel human CC chemokine PARC that is most homologous to macrophage-inflammatory protein-1 alpha/LD78 alpha and chemotactic for T lymphocytes, but not for monocytes. J Immunol. 1997;159:1140–1149. [PubMed] [Google Scholar]
- 56.Imai T, Baba M, Nishimura M, Kakizaki M, Takagi S, Yoshie O. The T cell–directed CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J Biol Chem. 1997;272:15036–15042. doi: 10.1074/jbc.272.23.15036. [DOI] [PubMed] [Google Scholar]
- 57.Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, Yoshida N, Kikutani H, Kishimoto T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–638. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- 58.Romagnani S. The Th1/Th2 paradigm. Immunol Today. 1997;18:263–266. doi: 10.1016/s0167-5699(97)80019-9. [DOI] [PubMed] [Google Scholar]
- 59.Martin D, Near SL. Protective effect of the interleukin-1 receptor antagonist (IL-ra) on experimental allergic encephalomyelitis in rats. J Neuroimmunol. 1995;61:241–251. doi: 10.1016/0165-5728(95)00108-e. [DOI] [PubMed] [Google Scholar]