

Abstract
It is currently believed that latently infected, resting B lymphocytes are central to gammaherpesvirus persistence, whereas mucosal epithelial cells are considered nonessential. We have readdressed the question of nonlymphoid persistence using murine gammaherpesvirus 68 (MHV-68). To dissect lymphoid from nonlymphoid persistence, we used μMT transgenic mice that are defective in B cells. MHV-68 DNA persisted in the lungs of intact and B cell–deficient mice. Both episomal and linear forms of the virus genome were present in lungs, implying the presence of both latency and productive replication. In situ hybridization for virus tRNA transcripts revealed latent MHV-68 in pulmonary epithelial cells. Infectious virus was recovered from the lungs of μMT mice after T cell depletion, showing that the persisting virus DNA was reactivatable. Finally, using adoptive transfer of B cells into B cell–deficient mice, it was shown that virus persisting in lungs seeded splenic B cells, and virus resident in the spleen seeded the lungs. These results show that mucosal epithelia can act as a nonlymphoid reservoir for gammaherpesvirus persistence, and that there is a two-way movement of virus between lymphoid and nonlymphoid compartments during persistence.
Keywords: gammaherpesvirus, latency, Epstein-Barr virus, epithelia, lungs
Gammaherpesviruses have clinical relevance in both the medical and veterinary fields. In humans, EBV is associated with infectious mononucleosis, B lymphomas, and nasopharyngeal carcinoma (1). Likewise, the Kaposi's sarcoma–associated herpesvirus (KSHV)1 also called human herpesvirus type 8, is associated with B lymphomas as well as the lesion from whence its name is derived (2). In the veterinary field, there is a group of closely related viruses, including ovine herpesvirus type 2 and alcelaphine herpesvirus type 1, which are associated with a fatal lymphoproliferative syndrome in domestic ruminants termed malignant catarrhal fever (3).
All gammaherpesviruses are characterized by their ability to become latent in lymphoid cells. These viruses also infect epithelial cells, especially in the respiratory tract, from which they spread from host to host (1). To date, the mode of gammaherpesvirus persistence in the host has been addressed only with EBV. Only productive, not latent, infection has been described at mucosal sites in epithelia, and as such, these sites are considered nonessential to virus persistence (4–6). Indeed, based on evidence currently available, a pool of latently infected, resting B lymphocytes are the central and essential feature of EBV persistence, and pharyngeal epithelial cells are occasionally reinfected from these cells (6–10). However, investigation of the pathogenesis of most gammaherpesviruses, in particular those infecting humans, is difficult. Therefore, we are readdressing the question of alterative sites of persistence and latency using murine gammaherpesviruses 68 (MHV-68).
MHV-68 will infect laboratory mice via intranasal inoculation, after which it establishes a productive infection in alveolar epithelial cells (11). The virus then spreads to the spleen, where a latent infection is established in B lymphocytes (12). Acute virus replication in the lung resolves after 7–10 d postinfection (p.i.), beyond which time no evidence of infection can be found in this organ by biological methods, i.e., direct plaque assay or reactivation assays. In contrast, latent virus can be reactivated from splenic B cells throughout the lifetime of the animal. It was reasoned that, like EBV, the B lymphoid compartment was the major site of MHV-68 persistence and latency (12). However, our recent work with μMT mice, which are transgenically deficient in mature B cells, has cast doubt on this hypothesis (13). Infection of the lungs of these mice by MHV-68 followed a normal course, except that clearance of infectious virus was delayed from 10–17 d p.i. We could find no evidence of splenic or hematogenous infection or infection of other organs either by reactivation assay or by using a sensitive nested PCR assay. In contrast, using the same PCR assay, we were able to show long-term persistence of MHV-68 DNA in lungs. This showed that although the B cell is clearly a major site of latency and persistence, it is not essential for the persistence of the virus, and that this can occur at mucosal sites, in this case the lungs. The aim of this work was to investigate further the long-term persistence of MHV-68 in the lungs of infected mice, and to investigate the trafficking of virus during persistence between B cells in the spleen and epithelial cells in the lung.
Materials and Methods
Cell Lines and Virus.
MHV-68 strain g2.4 was propagated and quantified by plaque assay using BHK-21 cells as described previously (11). The B lymphocyte tumor cell lines S11 (MHV-68–positive) and S31 (MHV-68–negative) were derived in this laboratory, and were grown as described by Usherwood et al. (14).
Mice.
The mice used in this study were μMT/μMT (15) on a C57/BL6 background. They have a targeted lesion in the μ immunoglobulin chain resulting in the failure to express IgM; consequently, B cell development is arrested at the pre–B cell stage. A colony was established from these mice in the Department of Veterinary Pathology, University of Edinburgh. C57/BL6 mice were obtained from Bantin and Kingman Ltd. (Hull, UK). Male and female mice were infected at 4–11 wk of age intranasally with 4 × 105 PFU MHV-68 under halothane anesthesia.
DNA PCR Analysis.
MHV-68 DNA was extracted from cells and tissue using Qiamp tissue kits (QIAGEN Inc., Chatsworth, CA). Accurate DNA quantitation was performed using a DNA fluorometer (DyNA Quant 200; Pharmacia Biotech AB, Uppsala, Sweden). Nested PCR amplification was performed on 100 ng of high molecular weight DNA using primers specific for the gp150 gene as described previously (13). The primer pair PAG1 plus PAG2 (external) was used for an initial 40 cycles, and the primer pair PAG11 and PAG12 was used for a subsequent 25 cycles on 10 μl of product from the first amplification. Products were analyzed on 2% agarose gels containing ethidium bromide.
Using known amounts of cloned template, it was determined that the process could detect one copy of virus genome in 100 ng high molecular weight DNA.
Quantitative Competitive PCR.
This was carried out essentially as described by Gilliland et al. (16) and Siebert and Larrick (17). The quantitative competitive (QC) PCR assay was based on the gp150-specific assay described above. The template for the gp150-specific PCR using primers PAG1 and PAG2 does not contain a site for the restriction enzyme BamHI. A mutant competitor template containing an internal BamHI restriction endonuclease site for use in QC-PCR was made. Two separate PCRs were performed using DNA containing the gp150 gene as template. The first used the external primer PAG1 (sense; see above) along with the primer MUT1, 5′-CAG GAA GAG GAT CCA ACT ATG ACT C (antisense), which is specific for a sequence ∼200 bp downstream in the gp150 gene but with the sequence mutated into a BamHI site. The second used the primer MUT2, which is the reverse complement of MUT1, and the external primer PAG2 (antisense). Two reaction products of ∼200 and 300 bp in length were generated from these PCRs, representing the upstream and downstream portions of the gp150 template. The 3′ 25 bp of the product of PAG1 and MUT1 overlapped with the 5′ 25 bp of the product of MUT2 and PAG2. Thus, when portions (10 μl) of the products were mixed together with primers PAG1 and PAG2 and amplified by PCR, this resulted in the generation of a 501-bp mutant gp150 template containing an internal BamHI site. When cut with BamHI, this template formed fragments of ∼200 and 300 bp in size. This mutant competitor template was inserted using standard molecular cloning techniques into the vector pKS (−) (Stratagene Inc., La Jolla, CA).
Accurate quantification of the copy number of MHV-68 DNA present in samples was performed on 100 ng high molecular weight DNA. A series of PCRs was performed on each sample using primers PAG1 and PAG2, each containing the same amount of high molecular weight DNA but spiked with a dilution series of defined quantities of mutant competitor template. Nested PCR was then performed using primers PAG11 and PAG12 as before, except that PAG12 had been labeled with IRD-41. Reaction products were cut with BamHI, and the amounts of labeled wild-type (501 bp) and competitor (300 bp) product were assessed using a sequencing machine (LI-COR Inc., Lincoln, NE) which detects IRD-41–labeled DNA. A standard curve was constructed using cloned wild-type DNA, and relating log([competitor product]/[wild-type product]) to the log [competitor]. This was found to be a straight line with a slope of one. The exact amount of DNA in each sample was then determined by replacing cloned wild-type DNA with high molecular weight test DNA, and calculating the point at which [competitor product] is equal to [wild-type product] or log([competitor product]/[wild-type product]) is equal to zero.
In Situ–Lysis Gardella Gels and PCR.
Gardella gel electrophoresis with PCR analysis was performed in a horizontal format as described previously (7) with the following modification. Gel slices were melted at 65°C, and 5 μl was analyzed directly by PCR for MHV-68. A single round (nonnested) PCR for MHV-68 gp150 was used with primers PAG1 and PAG2 (see above) for 30–40 cycles.
Dissociation of Lungs to a Single Cell Suspension.
Before removal, lungs were perfused through the right ventricle of the heart with 30 ml PBS. Lungs were removed and placed in ice-cold medium. Larger airways were removed along with lung-associated lymphoid and connective tissue. The remaining tissue was then cut into pieces of ∼1–2 mm3. Diced lung was then incubated with a solution of 150 U/ml collagenase/dispase (Sigma Chemical Co., St. Louis, MO) and 50 U/ml DNase I (Sigma Chemical Co.) in RPMI 1640 medium containing 10% FCS for 2 h at 37°C and then dissociated by passing through a 23-gauge needle. The suspension was then washed twice with 10 ml medium and evaluated by trypan blue staining. Between 2 and 5 × 107 viable cells were obtained per mouse.
In Vivo Depletion of T Cells.
T cell subsets were depleted using the rat mAbs YTS 191.1 (anti-CD4) and YTS 169.4 (anti-CD8). Mice were injected intravenously in the tail with 0.1 ml of antibody at 10 mg/ml and again 2 d later. Subsequently, mice were injected with the same dose intraperitoneally at 7 and 11 d after the original injection.
Cytofluorometry.
Cells were stained with mAbs and analyzed using a FACScan® (Becton Dickinson, San Jose, CA) as described previously (13). Anti-CD4 and anti-CD8 antibodies were from the hybridomas YTS 191.1 and YTS 169.4, and anti-B220 (CD45R) was purchased from PharMingen (San Diego, CA).
Adoptive Transfer of T-depleted Splenocytes.
Splenocytes were first removed from spleens by teasing, and then red cells were lysed by water. Cells were then incubated with anti-CD4 (YTS 191.1) and anti-CD8 (YTS 169.4) both at 100 μg/ml along with a 1:4 dilution of rabbit complement (Lowtox-H; Cedarlane Labs. Ltd., Hornby, Ontario, Canada). After incubation for 1 h at 37°C, the cells were washed, and 5 × 107 cells in 200 μl PBS were given intraperitoneally to each mouse.
In Situ Hybridization.
Radioactive and nonradioactive in situ hybridizations on formalin-fixed, paraffin-embedded sections were carried out essentially as described previously (18). Sense and antisense labeled transcripts to the MHV-68 tRNA molecules 1–4 were made from the plasmid pEH 1.4 (a gift of Dr. S. Efstathiou, University of Cambridge, Cambridge, UK [19]) using either T3 or T7 polymerases, respectively. Likewise, labeled transcripts to the MHV-68 thymidine kinase gene were made using the gene contained in pKS (−) (Stratagene Inc.) (20). Detection of tRNAs and cytokeratin on the same section was performed as follows. Sections were pretreated for in situ hybridization, which included microwave treatment in citrate buffer as described by Sibony et al. (21). Hybridization was performed with a digoxigenin-labeled tRNA probe, and slides were developed using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) substrate (Sigma Chemical Co.) as described previously (18). Sections were then reacted with anticytokeratin/HRP (MNF116-EPOS, U7022; Dakopatts A/S, Copenhagen, Denmark) according to the manufacturer's instructions, and ultimately developed using 3,3′-diaminobenzidine tetrahydrochloride (DAB) substrate (Sigma Chemical Co.).
Results
MHV-68 Persists in the Lungs of Intact Mice.
Our previous results using PCR analysis had shown that MHV-68 could persist in the lungs of B cell–deficient μMT mice after infection by the intranasal route in the absence of infection in the spleen or other organs (13). To confirm that persistence in lungs was not merely an anomaly of μMT mice, we analyzed intact mice that had been infected 12 mo previously with MHV-68. We wanted to distinguish positive signals derived from MHV-68 in B lymphocytes merely trafficking through the lungs in the blood supply from those derived from lung tissue. Therefore, we perfused lungs extensively with PBS before removal and extracted high molecular weight DNA from the blood, bone marrow, and liver in addition to the lungs and spleen of five mice. The DNA was then analyzed for the presence of MHV-68 genome by nested PCR (13). This assay was specific for the MHV-68 pg150 gene and was sensitive to 1 genome copy per 100 ng DNA. The results are shown in Fig. 1. As expected, MHV-68 DNA was found in the lungs and spleen of all five mice. However, none of the mice had MHV-68 in their liver or kidney. Two mice (2 and 3) had evidence of MHV-68 DNA in their blood and bone marrow. Therefore, in the case of mice 2 and 3, the positive signal in lung tissue could have been due to MHV-68 trafficking in the blood. This is unlikely to be the case in the other three mice, since they had no detectable MHV-68 DNA either in their blood or in the liver and kidney. This data shows that MHV-68 persists in the lungs of intact mice as well as the spleen, and shows that our original observations with μMT mice were not due to an artifact of a B cell–deficient system.
Figure 1.

Detection of MHV-68 in C57/BL6 mice by PCR. DNA was extracted from organs of five C57/BL6 mice which had been infected intranasally with MHV-68 for 12 mo and analyzed for the presence of MHV-68 DNA by nested PCR specific for the gp150 gene. Products were analyzed on 2% agarose gels containing ethidium bromide and visualized using a UV transilluminator. Images are shown with the colors reversed for clarity. Molecular weight determinations were made relative to a 1-kb DNA ladder (Mr), and the size of the pertinent bands in bp are shown at the left. In all panels, DNA from the MHV-68–positive tumor cell line S11 was used as a positive control (+VE), and produced the expected product of 368 bp. DNA from the MHV-68–negative tumor cell line S31 (−VE) was used along with deionized water (H2O) as a negative control. (A) The results of analysis of lungs (LUNG 1–5), spleens (SPL 1–5), and bone marrows (BM 1–5) of all five mice. (B) Analysis of blood (BLOOD 1–5) of all five mice. (C) Analysis of livers (LIV 1–5) and kidneys (KID 1–5) of all five mice.
To ascertain whether there was any relationship between the levels of MHV-68 DNA in the lung, spleen, and bone marrow, we measured the exact amount of virus DNA in each organ by QC-PCR. This assay was developed as described in Materials and Methods, and was sensitive to 1 copy of genome per 100 ng DNA. The results of performing this assay on the same five DNA samples as used in Fig. 1 are shown in Fig. 2. The level of MHV-68 DNA present in the lungs showed the greatest variability, from 1 to 1,000 copies/100 ng, whereas the levels in the spleen were more consistent (12–300 copies/100 ng). The level of virus DNA in the bone marrow, where detected, was much lower (1–10 copies/100 ng). There was no direct relationship between levels of virus DNA in the lungs and spleen. In two mice, there was a higher level in spleen than lung, the converse was true in another two mice, and one mouse had equal levels in both. In contrast, the level of virus DNA in the spleen bore a direct relationship to the level in the bone marrow. This suggests that in intact C57/BL6 mice, both the lungs and the spleen may be independent sites of MHV-68 persistence. The presence of and levels of virus in the bone marrow appear to be directly related to the amount of virus in the spleen, suggesting that the bone marrow may not be an independent site of virus persistence but is dependent on trafficking cells in the blood and/or seeding from the spleen. This may also suggest a more central role for splenic persistence in intact mice with regard to seeding of other organs.
Figure 2.

Determination of MHV-68 DNA load in organs by QC-PCR. The precise number of copies of MHV-68 DNA present in the lung, spleen, and bone marrow (BM) in the same samples as in Fig. 1 was determined by QC-PCR. The results are presented on a scatter chart as the number of copies per 100 ng high molecular weight DNA for each organ in each mouse. Mouse 1 (♦); mouse 2 (▪); mouse 3 (▴); mouse 4 (X); and mouse 5 (•).
Both Episomal and Linear Forms of MHV-68 DNA Are Present during Persistence in Lungs.
The state of MHV-68 in lungs during persistence was determined by molecular means using Gardella gel analysis. This technique is able to distinguish linear from episomal genomes. Using this technique, it has been shown that latently infected cells contain genomes that are in a covalently closed, circular (episomal) conformation. In contrast, although many forms of the genome may be present in productively replicating cells, Gardella gel analysis of such cells reveals only genomes that are in a linear conformation (7, 14, 22). In control experiments, we performed Southern analysis of a Gardella gel as described previously (14). We used the S11 B lymphocyte line, which is known to contain both latent and productive forms of MHV-68. Approximately 5% of these cells are known to be undergoing productive replication at any one time while the remainder are latent (14). We also used acutely infected mouse epithelial cells (C127). The results are shown in Fig. 3. A clearly shows the resolution of linear and circular forms of the MHV-68 genome in S11 cells, whereas B shows that only the linear form of the genome was present in acutely infected epithelial cells.
Figure 3.

Analysis of MHV-68 DNA conformation in control cells by Gardella gel analysis. DNA in intact cells was fractionated using a Gardella (in situ–lysis) gel. A and B show Southern blot analysis of a Gardella gel. The gel is shown sideways for comparison with C and D, and the top of the gel is indicated (TOP). A shows the results of using the MHV-68–positive B cell line S11, which is known to contain both episomal and linear DNA. B shows the results of using C127 (murine epithelial cells), which had been acutely infected 24 h previously with 10 PFU/cell MHV-68. C and D show the results of slicing the Gardella gel and analyzing each slice for the presence of MHV-68 DNA by PCR. Therefore, each panel represents one lane of a Gardella gel, and the top is shown next to the first of the sequential slices. PCR products were analyzed on 2% agarose gels containing ethidium bromide and visualized using a UV transilluminator. Images are shown with the colors reversed for clarity. DNA from the MHV-68–positive tumor cell line S11 was used as a positive control (+), and produced the expected product of 368 bp. DNA from the MHV-68–negative tumor cell line S31 (−) was used as a negative control. C shows the results of using S11 cells as for A, and D shows the results of using S11 cells after treatment with the virus DNA replication inhibitor 4′-S-EtdU for 14 d. The positions of circular and linear MHV-68 DNA are shown (top).
To obtain the required sensitivity to be able to detect MHV-68 during persistence in lungs, we needed to use a PCR-based modification of the Gardella gel technique, which has been used successfully to detect latent EBV and KSHV genomes in vivo (7, 22). Instead of analyzing the Gardella gel by Southern blotting, after electrophoresis individual lanes of the gel were excised and cut sequentially into slices. Each slice was then analyzed for the presence of MHV-68 DNA by PCR. Therefore, each lane of the PCR gel represents one sequential slice of the Gardella gel lane. Again in control experiments, we used S11 cells. This line was used either untreated or after treatment for 14 d with the antiviral drug 2′-deoxy-5-ethyl-β-4′-thiouridine (4′-S-EtdU) (23). This drug is thought to act like Acyclovir, is phosphorylated by the viral thymidine kinase, and causes inhibition of DNA synthesis by chain termination. By this means, it is known to inhibit productive MHV-68 replication but does not affect latency. The results are shown below those of conventional Southern blots in Fig. 3. C shows analysis of untreated S11 cells. As in A, the resolution of both episomal and linear forms of the genome in this panel can be seen clearly. After treatment of S11 cells with 4′-S-EtdU (D), the linear form of the genome was absent, representing the predicted blocking of DNA replication and clearance of linear DNA from the cells by the drug. Therefore, the Gardella-PCR system was able to distinguish episomal from linear genomes in control cells.
We next analyzed cells derived from mouse lungs. We used primarily μMT mice in these experiments, since lungs are a site of persistence in these mice, and there is no detectable hematogenous source of virus. Intact C57/BL6 mice were used as comparison. Lungs were taken from mice during the persistent phase of infection: 2 mo p.i. in the case of μMT mice, and 7 mo p.i. in the case of C57/ BL6 mice. Selected mice were treated with the herpesvirus DNA replication inhibitor 4′-S-EtdU. Treated mice received 1 mg/mouse/d of drug in the drinking water continuously for 28 d before sampling. This concentration of drug is known to completely inhibit productive MHV-68 DNA synthesis and replication in vivo. Lungs of both μMT and C57/BL6 mice were perfused through the right ventricle to minimize contamination by lymphocytes in the blood supply, and then disaggregated to a single cell suspension. Cells derived from lungs were then loaded into the wells of a Gardella gel that was analyzed by PCR as above. The experiment was repeated twice with comparable results. A representative selection of the results is shown in Fig. 4. In five out of six cases, the lungs of μMT mice were found to contain both episomal and linear genomes (A), but in one case, we found evidence of only episomal genomes (B). In addition, all (four out of four) intact C57/ BL6 mice contained both linear and episomal forms of the genome (C). After treatment of μMT mice for 28 d with 4′-S-EtdU, we found that in all cases (four out of four), only the episomal form of the genome was present (D).
Figure 4.

Analysis of MHV-68 DNA conformation in lung cells by Gardella gel–PCR analysis. Intact cells were loaded into the wells of a Gardella (in situ–lysis) gel. After electrophoresis, individual lanes were cut sequentially into slices. Slices from each lane were then analyzed by PCR for the presence of MHV-68 DNA. PCR products were analyzed on 2% agarose gels containing ethidium bromide and visualized using a UV transilluminator. Images are shown with the colors reversed for clarity. Therefore, each panel represents one lane of a Gardella gel, and the top is shown next to the first of the sequential slices. The slices representing the positions on the Gardella gel of the circular and the linear forms of the genome are shown (top). In all panels, DNA from the MHV-68–positive tumor cell line S11 was used as a positive control (+), and produced the expected product of 368 bp. DNA from the MHV-68–negative tumor cell line S31 (−) was used along with deionized water (H2O) as a negative control. A, B, and D show results using cells from μMT mouse lungs 2 mo p.i., whereas C shows results using cells from C57/BL6 mice lungs 7 mo p.i. and are labeled accordingly. D shows the results using μMT mouse lungs at 2 mo p.i., but after treatment for the last 28 d of infection with the inhibitor of virus DNA replication 4′-S-EtdU.
Thus, both episomal and linear forms of the MHV-68 genome were present in the majority of mouse lungs, suggesting that both latently and productively infected cells may be normally present. However, episomal DNA was present alone in one μMT mouse, and after 4′-S-EtdU treatment (which inhibits virus DNA replication) in others. This suggests that the persistence of MHV-68 in a latent state in the lung may not depend on the presence of productively infected cells.
Infectious MHV-68 Can Be Reactivated in μMT Mice.
We surmised that if MHV-68 DNA in the lungs of mice represented intact virus, it should be possible to detect it by biological means. All of our previous attempts to induce the production of infectious virus from the lungs of mice during persistent infection had failed. These included immunosuppression by removal of T cells, including CD4+ or CD8+ T cells alone as well as both CD4+ and CD8+ T cells, immunosuppression with cyclosporin A, and treatment with dexamethasone. Since μMT mice have no B cells, they are unable to produce antiviral antibody. Therefore, we attempted to obtain infectious virus from μMT mice as well as intact C57/BL6 mice during the persistent phase of infection, i.e., 54 d p.i. The experiment was repeated twice with comparable results. T cell subsets were depleted using either anti-CD4 (YTS 191.1), anti-CD8 (YTS 169.4), or anti-CD4 and -CD8 in combination. Control mice received PBS alone. FACS® analysis of lymphocytes after depletion revealed a >99% depletion of the relevant T cell subset. After depletion for 14 d, the mice were sampled, and the virus titers present in the lungs were determined. The results are shown in Table 1. As in previous experiments, MHV-68 could not be reactivated from intact mice. However, infectious virus was recovered from μMT mice after depletion of either CD8+ T cells or CD4+ plus CD8+ cells. The amount of virus recovered was small in the case of CD8+ T cell depletion, but was much greater, approaching levels seen during acute infection, in CD4+ plus CD8+–depleted animals.
Table 1.
Induction of Infectious MHV-68 from the Lungs of Persistently Infected Mice by Immunosuppression
| Treatment | Virus titer in C57/BL6 lungs* | Virus titer in μMT lungs* | ||
|---|---|---|---|---|
| PFU/lung | PFU/lung | |||
| PBS | <10 | <10 | ||
| anti-CD4 | <10 | <10 | ||
| anti-CD8 | <10 | 15 | ||
| anti-CD4 + CD8 | <10 | 6 × 105 |
Titers are expressed as the mean of three individual mice.
We have found no other site of persisting MHV-68 than the lungs in μMT mice infected by the intranasal route. Therefore, the above results suggest that the persisting virus detected by molecular methods in lungs may not be defective, and is capable of replicating to high levels in the absence of both antiviral antibody and T cells.
Latent MHV-68 Persists in Lung Epithelial Cells.
To determine the cell type harboring MHV-68 in the lung during persistence, we performed in situ hybridization on tissue sections taken from either μMT (n = 3) or C57/BL6 (n = 3) mice during the persistent phase at 54 d p.i. Control hybridizations were performed on sections from mock-infected μMT and C57/BL6 mice. Sections were hybridized initially with a 35S-labeled riboprobe complementary (antisense) to the MHV-68 tRNA-like genes that are expressed during MHV-68 latency (19). As controls, adjacent serial sections were hybridized with a sense tRNA probe and a probe that hybridized to mRNAs encoding the MHV-68 thymidine kinase and gH proteins which are expressed during the productive phase of MHV-68 infection (24). Representative examples of the results are shown in Fig. 5.
Figure 5.
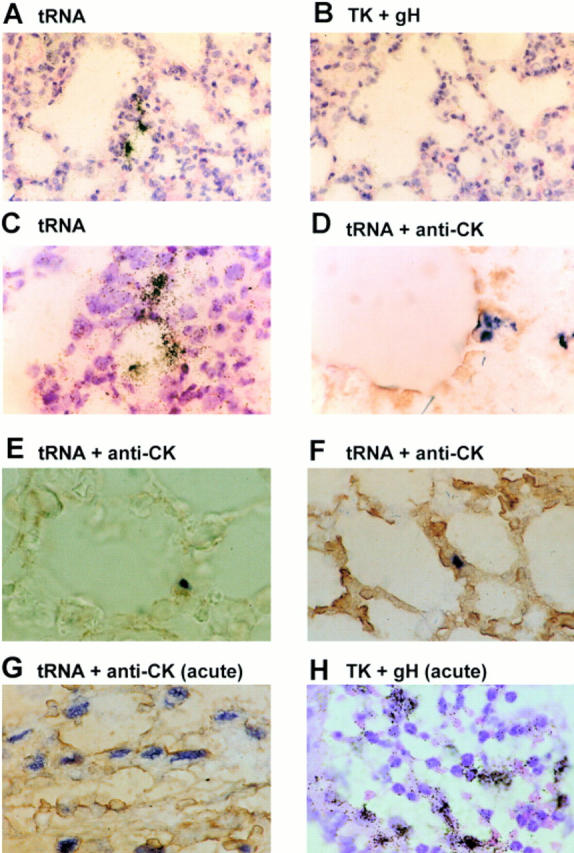
Determination of cell type harboring latent MHV-68 in the lungs by in situ hybridization. Sections of 5 μM thickness were cut from formalin-fixed paraffin-embedded lung tissues from mice during virus persistence at 54 d p.i. (A–F). Control sections were derived from mice at 5 d p.i. (G and H, acute). Sections shown in A–C and H were hybridized with 35S-labeled riboprobes and developed using photographic emulsion. The sections shown in D–G were hybridized with digoxigenin-labeled riboprobes and developed using a combination of alkaline phosphatase–labeled antidigoxigenin followed by NBT/BCIP (blue/black). Probes used were specific for either the latency-associated tRNAs (A and C–G, tRNA) or the productive cycle–associated thymidine kinase (TK) and gH genes (B and H, TK + gH). D–G were additionally reacted with anticytokeratin antibody and developed using DAB (brown, anti-CK). A and B are adjacent serial sections from one μMT mouse biopsy. All other panels are derived from different μMT mice. A–C and H were counterstained with hematoxylin and eosin. A and B, ×230; C–H, ×580.
No reaction was seen using the control sense tRNA probe or antisense tRNA probe on sections from mock-infected mice (not shown). Foci of positive cells were observed in sections from all six infected mice, either μMT or C57/BL6, using the antisense tRNA probe (Fig. 5, A and C). These foci were small, and there were between one and five (mean two) per section. There was no reaction using the thymidine kinase plus gH probe on adjacent serial sections (B); however, control hybridizations showed that the thymidine kinase plus gH probe was capable of detecting productively infected cells present during acute infection (H). Therefore, it seems likely that the tRNA-expressing foci observed were cells latently infected with MHV-68. Further, the position of the foci within the biopsy suggested that these latently infected cells were not centered on areas of inflammation or blood vessels, but were bordering airways and were possibly epithelial cells.
To identify precisely the cell type involved, we performed a second set of in situ hybridizations using a digoxigenin-labeled antisense tRNA probe. The same sections were reacted with an mAb specific for cytokeratin. Positive control sections taken from the acute phase of infection were used for comparison. During the productive phase of infection, MHV-68 tRNAs (dark blue) were expressed in the nuclei of a large number of cells, which also expressed cytokeratin (brown) and were thus epithelial (G). As can be seen in Fig. 5 D–F, during latency only small foci of cells positive for tRNA sequences were present; however, these cells were also positive for cytokeratin. Thus, the lung cells that expressed MHV-68 tRNAs during the persistent phase of infection were epithelial in nature.
Movement of MHV-68 between Lung and Spleen during Virus Latency.
Our data showed that lungs and spleen were sites of MHV-68 latency and persistence. We wished to know whether these sites were truly independent, or whether virus resident in the spleen was able to traffic to the lung and virus resident in the lung was able to spread to the spleen.
To see whether MHV-68 persisting in spleens of mice could spread to the lungs, we took T-depleted cells from the spleens of C57/BL6 mice that had been either mock-infected or infected with MHV-68. We then infused these cells into uninfected μMT mice. The mice were tested pretransfer and found to have no B220+ cells in the blood. When tested 4 d after transfer, they were found to have between 1 and 18% B220+ cells, indicating that adoptively transferred cells were persisting. After 11 d, the spleens, lungs, and blood of these mice were screened for the presence of MHV-68 by PCR. Before removal, the lungs were perfused with PBS to remove contaminating lymphocytes present in the blood supply. The results are shown in Fig. 6 A. None of the mice that received B cells from uninfected mice (Mock) had evidence of MHV-68 infection. Two out of five mice that received cells from infected mice had evidence of infection in both the spleen and the lungs but not the blood. Thus, in the mice where transfer had been successful in establishing infected B cells in the spleen, there was also evidence of infection in the lung. This implies that virus resident in splenic B cells is able to spread and establish infection in the lungs.
Figure 6.


Detection of MHV-68 in organs of μMT mice after adoptive transfer of B lymphocytes. MHV-68 genomes were detected by nested PCR for gp150. Products were analyzed on 2% agarose gels containing ethidium bromide and visualized using a UV transilluminator. Images are shown with the colors reversed for clarity. Molecular weight determinations were made relative to a 1-kb DNA ladder (Mr), and the size of the pertinent bands in bp are shown at the left. In all panels, DNA from the MHV-68–positive tumor cell line S11 was used as a positive control (+VE), and produced the expected product of 368 bp. DNA from the MHV-68–negative tumor cell line S31 (−VE) was used along with deionized water (H2O) as a negative control. (A) T-depleted splenocytes from infected (INF 1–4) or mock-infected (MOCK 1–3) C57/BL6 mice were adoptively transferred into uninfected μMT mice. After 11 d, DNA was extracted and analyzed for MHV-68 by nested PCR. Results from analysis of the spleens, lungs, and blood are indicated above the relevant tracks. (B) Three persistently infected μMT mice (1–3) were analyzed for MHV-68 by nested PCR. Results from either lungs or spleen are shown above the relevant tracks. (C) T cell–depleted splenocytes from uninfected C57/BL6 mice were adoptively transferred into either mock-infected (MOCK 1 and 2) or persistently infected (INF 1–3) μMT mice. After 14 d, DNA was extracted and analyzed for MHV-68 by nested PCR. Results from analysis of the spleens and lungs are indicated above the relevant tracks.
To see whether persisting MHV-68 resident in the lungs of mice could spread to the spleen, we exploited the fact that MHV-68 persists in the lungs of μMT mice in the absence of any evidence of splenic infection. Therefore, we took μMT mice that had been mock-infected or infected with MHV-68 for 80 d. We then infused T-depleted splenocytes from C57/BL6 mice into these mice. The mice were tested pretransfer and found to have no B220+ cells in the blood stream. At 2 d after transfer, they had between 1 and 18% B220+ cells in the blood stream, indicating that adoptively transferred cells were persisting. After 14 d, the mice were sampled, and the lungs and spleen were tested for the presence of MHV-68 by PCR. Again, before removal, the lungs were perfused to remove contaminating B cells in the blood volume. The results are shown in Fig. 6, B and C. B shows in a control experiment that virus was present in the lung but not the spleen of persistently infected μMT mice. This supports our previous observations (13). C shows the results of transfer experiments into similar mice. No virus was evident in either the lungs or spleen of two mock-infected mice. However, MHV-68 infection was seen in both the lungs and the spleen of infected mice. Therefore, this shows that in the presence of B cells, MHV-68 persisting in the lungs can establish infection in the spleen.
Discussion
The above data showed that MHV-68 could persist in the lungs of both intact and B cell–deficient mice. Both episomal and linear forms of the genome were detected by Gardella gel analysis in lungs, implying the presence of latency and productive replication, and we have shown that persisting virus can be recovered in infectious form after immunosuppression. Latent MHV-68 was seen in pulmonary epithelial cells during persistent infection, but productively infected cells were not found. Adoptive transfer experiments suggested that virus persisting in the lungs could seed the spleen, and conversely, virus resident in the spleen could seed the lungs.
We have used the μMT B cell–deficient mouse since we and others have shown that MHV-68 persists in the lungs but not in the spleen or any other organ yet detected after intranasal infection of these mice (13, 25). Other workers have shown that the virus is able to infect and persist in the spleen of μMT mice, but only after intraperitoneal inoculation of a larger dose of virus, which enables a more disseminated, systemic infection to be established (26). This implies that B lymphocytes are required for MHV-68 to traffic and seed the spleen after respiratory challenge, and that in the absence of B cells, alternative cell types within the spleen are capable of harboring persisting virus. Our data using intranasal infection do not conclusively exclude the possibility of a more complicated host–pathogen relationship, such as the one that exists with CMV, where multiple sites have an important input into persistence. However, in μMT mice, we have never detected virus DNA in any organ other than the lung during persistence. This and the absence of virus DNA in the blood of μMT mice (Fig. 6 A) strongly suggest that the lung may be the primary source of persistence in the current experiments. Therefore, we believe that intranasal infection of μMT mice provides a powerful means of studying persistence of a gammaherpesvirus at a mucosal site. Data obtained with the μMT strain are likely representative, because the patterns of persistence in the lungs of these mice are mirrored in all aspects by intact C57/BL6 mice.
Molecular analysis suggested that the mode of MHV-68 persistence was a mixture of latency and productive replication in both μMT and intact mice. Evidence of virus persistence in the lungs has never been obtained using biological criteria such as direct plaque or reactivation assays. However, our data (Table 1) showed that infectious virus could be recovered in the lungs of μMT mice after T cell depletion. We have not found virus in any other organ in this system, although this does not exclude the presence of small amounts of latent virus in some hitherto unrecognized site. Also, one issue that cannot be resolved conclusively here is whether the infectious virus detected was due to the amplification of small amounts of persistent active replication present in the μMT mice because of immunodeficiency or reactivation of latency. Even so, our data strongly suggest that the infectious virus found in the lungs originates there, and that the virus found by molecular means is not defective. Previous failure to detect MHV-68 in lungs by biological methods is unlikely to be due to the level of virus being below the level of detection, since the lungs contained a comparable amount of virus DNA to the spleens (see Fig. 2). A more likely explanation is that epithelial cells in which the virus resides may be less able to support in vitro reactivation than B lymphocytes in the spleen.
The precise mechanisms governing the control of reactivation of gammaherpesviruses in the host are not known. Certainly, T cells play a critical role, since humans who are either iatrogenically depleted of T cells or deficient in T cell function because of disease can succumb to EBV-driven lymphoproliferative disease (27). In addition, AIDS patients often develop Kaposi's sarcoma which is KSHV-associated (2). However, our results have shown that depletion of T cells alone did not result in the reactivation of MHV-68. Reactivation was achieved in μMT mice in the absence of both antibody and T cells. Although additional experiments are required to confirm this, these results strongly suggest a role for antibody in the control of gammaherpesviruses after reactivation. Thus, the presence of antiviral antibody in intact mice presumably prevented the spread of reactivated virus even in the face of severe immunosuppression. This may explain in part why EBV-driven lymphoproliferative disease often only develops after many months of T cell depletion (27). A long time may be required in the absence of T cells for B cell memory and antibody to decline to a level that is sufficient for virus reactivation to occur. Such a role for antibody is not unique. Elegant work, also using the μMT strain of mice, has shown that antiviral antibodies are important for the control of recurrent murine CMV (28).
Detection of the productive form of MHV-68 in lungs during persistence is perhaps not surprising, since this is the target organ for productive replication during acute infection, and productive replication at a mucosal site also provides a means for the virus to spread from host to host. Also, a similar pattern of low-level, sporadic productive replication at mucosal sites is seen during persistence of EBV (1). However, the numbers of cells undergoing productive MHV-68 replication in the lung at any one time must be small, since although we detected them by Gardella gel analysis, they were not seen by in situ hybridization or immunohistochemistry.
Detection of murine gammaherpesvirus in epithelial cells at a mucosal surface in an episomal conformation and in the absence of lytic cycle gene expression suggests that at least some of the epithelial cells at this site were latently infected. As such, this is a novel and important finding. The impact of this is increased by the observations that in μMT mice, lungs are the only known organ of persistence, and treatment with a herpesvirus DNA replication inhibitor for 28 d did not affect persistence at this site. This strongly suggests that the persistence of a latent MHV-68 infection in the lungs did not absolutely require constant reseeding from other sites such as the lymphoid compartment or local productive replication. However, our results do not preclude that reseeding of this site from other organs does not occur during persistence in intact mice. This may have implications for our view of the pathogenesis of other gammaherpesviruses. Persistence at mucosa has been addressed only with one other gammaherpesvirus, EBV. Epithelial cells containing productively replicating EBV and derived from an unknown source can be found in saliva (5). However, latently infected cells have never been seen in mucosal biopsies, and long-term treatment of patients with Acyclovir results in the loss of EBV from the mucosa but not the blood (6, 10). Therefore, it has been suggested that mucosa are not essential for EBV persistence in a given host, and that constant reseeding from the lymphoid compartment is required to maintain a virus presence at these sites. Experiments in humans and large animals are hard because of ethical considerations and the difficulty in obtaining biopsy material. In addition, our data suggest that the amount of gammaherpesvirus at mucosal sites is likely to be low. Therefore, it is possible that virus persisting at mucosal surfaces in other systems may have been missed. An alternative explanation is that the ability to persist at a mucosal site or not might represent a fundamental biological difference between different members of this subfamily. However, this question cannot be resolved until the persistence of EBV, KSHV, and other gammaherpesviruses is reevaluated to include alternative sites of mucosal persistence, such as the lung. This may turn out to be particularly pertinent because of the association of persistent gammaherpesvirus infection with diseases of mucosa. These include the association of EBV with nasopharyngeal carcinoma (1), oral hairy leukoplakia (4), and idiopathic pulmonary fibrosis (29) and the association of KSHV with sarcoidosis (30).
Work with EBV has suggested that latency in B cells is central to the maintenance of persistence, and that virus can traffic from these cells and seed mucosa. Certainly, our results shown in Fig. 2 suggest that virus resident in the lymphoid compartment plays a central role in the persistence of MHV-68 in intact mice. Our data using uninfected μMT mice which had infected splenocytes transferred into them (Fig. 6 A) suggest that, like EBV, virus may be able to traffic from B cell to mucosa during MHV-68 persistence. However, transfer of naive B cells into infected μMT mice where mucosa may be the only site of persistence suggested that in addition, MHV-68 are able to traffic to seed B cells. Therefore, these results suggest that epithelial cells in mucosa may be as important as B cells during MHV-68 persistence, and that there may be a dynamic, two-way movement of virus between the lymphoid and nonlymphoid reservoirs. This may form part of the survival strategy of the virus. Thus, persistence and replication may be required at a mucosal site in order to ensure transmission to new hosts. An additional reservoir of latent virus in lymphoid tissue with a limited flow of virus between the two sites may ensure that the virus is never cleared from either site.
Work is now under way to ascertain the nature of the latent state in the lung of infected mice. A key issue is whether the pattern of latent gene expression seen in the B cell is reflected in lung epithelial cells. The consequences of long-term infection of the lung with MHV-68 provides a unique opportunity to explore links between gammaherpesvirus infection and lung diseases of man and animals of unknown etiology.
Acknowledgments
The authors wish to thank Dr. S. Efstathiou for the generous gift of pEH1.4, and Drs. E. Littler and P. Collins, Glaxo Wellcome PLC (Middlesex, UK) for the generous gift of 4′-S-EtdU.
This work was funded by grants obtained from the United Kingdom Medical Research Council and Biotechnology and Biological Sciences Research Council. J. Stewart is a Royal Society University Research Fellow.
Abbreviations used in this paper
- 4′-S-EtdU
2′-deoxy-5-ethyl-β-4′-thiouridine
- KSHV
Kaposi's sarcoma herpesvirus
- MHV-68
murine gammaherpesvirus 68
- p.i.
postinfection
- QC
quantitative competitive
Footnotes
E.J. Usherwood's current address is Department of Immunology, St. Jude Children's Research Hospital, 332 North Lauderdale, Memphis, TN 38103.
References
- 1.Rickinson, A.B., and E. Kieff. 1996. Epstein-Barr virus. In Fields Virology. B.N. Fields, D.M. Knipe, and P.M. Howley, editors. Lippincott-Raven Publishers, New York. 2397– 2446.
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 3.Roizmann B, Desrosiers RC, Fleckenstein B, Lopez C, Minson AC, Studdert MJ. The family Herpesviridae: an update. The Herpesvirus Study Group of the International Committee on Taxonomy of Viruses. Arch Virol. 1992;123:425–449. doi: 10.1007/BF01317276. [DOI] [PubMed] [Google Scholar]
- 4.Greenspan JS, Greenspan D, Lennette ET, Abrams DI, Conant MA, Peterson V, Freese UK. Replication of Epstein-Barr virus within the epithelial cell of oral “hairy” leukoplakia, an AIDS-associated lesion. N Engl J Med. 1985;313:1564–1571. doi: 10.1056/NEJM198512193132502. [DOI] [PubMed] [Google Scholar]
- 5.Sixbey JW, Nedrud JG, Raab-Traub N, Hanes RA, Pagano JS. Epstein-Barr virus replication in oropharyngeal epithelial cells. N Engl J Med. 1984;310:1225–1230. doi: 10.1056/NEJM198405103101905. [DOI] [PubMed] [Google Scholar]
- 6.Niedobitek G, Young L, Lau R, Brooks L, Greenspan D, Greenspan JS, Rickinson AB. Epstein-Barr virus infection in oral hairy leukoplakia: virus replication in the absence of a detectable latent phase. J Gen Virol. 1991;72:3035–3046. doi: 10.1099/0022-1317-72-12-3035. [DOI] [PubMed] [Google Scholar]
- 7.Decker LL, Klaman LD, Thorley-Lawson DA. Detection of the latent form of Epstein-Barr virus DNA in the peripheral blood of healthy individuals. J Virol. 1996;70:3286–3289. doi: 10.1128/jvi.70.5.3286-3289.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gratama JW, Oosterveer MAP, Zwaan FE, Lepooutre J, Klein G, Ernberg I. Eradication of Epstein-Barr virus by allogeneic bone-marrow transplantation: implications for sites of viral latency. Proc Natl Acad Sci USA. 1988;85:8693–8696. doi: 10.1073/pnas.85.22.8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyashita EM, Yang B, Lam KMC, Crawford DH, Thorley-Lawson DA. A novel form of Epstein-Barr virus latency in normal B cells in vivo. Cell. 1995;80:593–601. doi: 10.1016/0092-8674(95)90513-8. [DOI] [PubMed] [Google Scholar]
- 10.Yao QY, Ogan P, Rowe M, Wood M, Rickinson AB. Epstein-Barr virus-infected B cells persist in the circulation of acyclovir-treated carriers. Int J Cancer. 1989;43:67–71. doi: 10.1002/ijc.2910430115. [DOI] [PubMed] [Google Scholar]
- 11.Sunil-Chandra NP, Efstathiou S, Arno J, Nash AA. Virological and pathological features of mice infected with murine gammaherpesvirus 68. J Gen Virol. 1992;73:2347–2356. doi: 10.1099/0022-1317-73-9-2347. [DOI] [PubMed] [Google Scholar]
- 12.Sunil-Chandra NP, Efstathiou S, Nash AA. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. . J Gen Virol. 1992;73:3275–3279. doi: 10.1099/0022-1317-73-12-3275. [DOI] [PubMed] [Google Scholar]
- 13.Usherwood EJ, Stewart JP, Robertson K, Allen DJ, Nash AA. Absence of splenic latency in murine gammaherpesvirus 68-infected B cell-deficient mice. J Gen Virol. 1996;7:2819–2825. doi: 10.1099/0022-1317-77-11-2819. [DOI] [PubMed] [Google Scholar]
- 14.Usherwood EJ, Stewart JP, Nash AA. Characterization of tumor cell lines derived from murine gammaherpesvirus 68-infected mice. J Virol. 1996;70:6516–6518. doi: 10.1128/jvi.70.9.6516-6518.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 16.Gilliland G, Perrin GG, Blanchard K, Bunn FF. Analysis of cytokine mRNA and DNA: detection and quantitation by competitive polymerase chain reaction. Proc Natl Acad Sci USA. 1990;87:2725–2729. doi: 10.1073/pnas.87.7.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siebert PD, Larrick JW. Competitive PCR. Nature. 1990;359:557–558. doi: 10.1038/359557a0. [DOI] [PubMed] [Google Scholar]
- 18.Fazakerley JF, Pathak S, Scallan M, Amor S, Dyson H. Replication of the A7(74) strain of Semliki Forest virus is restricted in neurons. Virology. 1993;195:627–637. doi: 10.1006/viro.1993.1414. [DOI] [PubMed] [Google Scholar]
- 19.Bowden RJ, Simas JP, Davis A, Efstathiou S. Murine gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J Gen Virol. 1997;78:1675–1687. doi: 10.1099/0022-1317-78-7-1675. [DOI] [PubMed] [Google Scholar]
- 20.Pepper SD, Stewart JP, Arrand JR, Mackett M. Murine gammaherpesvirus-68 encodes homologues of thymidine kinase and glycoprotein H: sequence, expression, and characterization of pyrimidine kinase activity. Virology. 1996;219:475–479. doi: 10.1006/viro.1996.0274. [DOI] [PubMed] [Google Scholar]
- 21.Sibony M, Commo F, Callard P, Gasc J-M. Enhancement of mRNA in situhybridization signal by microwave heating. Lab Invest. 1995;73:586–591. [PubMed] [Google Scholar]
- 22.Decker LL, Shankar P, Khan G, Freeman RB, Dezube BJ, Lieberman J, Thorley-Lawson DA. The Kaposi sarcoma–associated herpesvirus (KSHV) is present as an intact latent genome in KS tissue but replicates in the peripheral blood mononuclear cells of KS patients. J Exp Med. 1996;184:283–288. doi: 10.1084/jem.184.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahim SG, Trivedi N, Bogunovic-Batchelor MV, Hardy GW, Mills G, Selway JWT, Snowden W, Littler E, Coe PL, Basnak I, et al. Synthesis and anti-herpes virus activity of 2′-deoxy-4′-thiopyrimidine nucleosides. J Med Chem. 1996;39:789–795. doi: 10.1021/jm950029r. [DOI] [PubMed] [Google Scholar]
- 24.Mackett M, Stewart JP, Pepper S, Chee M, Efstanthiou S, Nash AA, Arrand JR. Genetic content and preliminary transcriptional analysis of a representative region of murine gammaherpesvirus 68. J Gen Viol. 1997;78:1425–1433. doi: 10.1099/0022-1317-78-6-1425. [DOI] [PubMed] [Google Scholar]
- 25.Kulkarni AB, Holmes KL, Fredrickson TN, Hartley JW, Morse HC., III Characteristics of a murine gammaherpesvirus infection in immunocompromised mice. In Vivo. 1997;11:281–292. [PubMed] [Google Scholar]
- 26.Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW. Mature B cells are required for acute splenic infection but not for establishment of latency by murine gammaherpesvirus 68. J Virol. 1996;70:6775–6780. doi: 10.1128/jvi.70.10.6775-6780.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanto DW, Frizzera G, Gajl-Peczalska PK, Simmons RL. Epstein-Barr virus, immunodeficiency, and B cell lymphoproliferation. Transplantation (Baltimore) 1985;39:461–472. doi: 10.1097/00007890-198505000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Jonjic S, Pavic I, Polic B, Crnkovic I, Lucin P, Koszinowski UH. Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J Exp Med. 1994;179:1713–1717. doi: 10.1084/jem.179.5.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Egan JJ, Stewart JP, Hasleton PS, Arrand JR, Carroll K, Woodcock A. Epstein-Barr virus replication within pulmonary epithelial cells in cryptogenic fibrosing alveolitis. Thorax. 1995;50:1234–1239. doi: 10.1136/thx.50.12.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Alberti L, Piattelli A, Artese L, Favia G, Patel S, Saunders N, Porter SR, Scully CM, Ngui S-L, Teo C-G. Human herpesvirus 8 variants in sarcoid tissues. Lancet. 1997;350:1655–1661. doi: 10.1016/s0140-6736(97)10102-7. [DOI] [PubMed] [Google Scholar]