

Abstract
The oncogenic BCR/ABL protein protects hematopoietic cells from apoptosis induced by growth factor deprivation, but the mechanisms are only partially understood. A BCR/ABL mutant lacking amino acids 176–426 in the BCR domain (p185ΔBCR) failed to protect interleukin 3–deprived 32Dcl3 myeloid precursor cells from apoptosis, although it possessed tyrosine kinase activity and was capable of activating the Ras-Raf-MAP kinase pathway. Compared to p185 wild-type transfectants, p185ΔBCR-transfected cells showed markedly reduced levels of Bcl-2 and expressed the hypophosphorylated, proapoptotic form of BAD. Bcl-2 expression in the mitochondrial fraction of p185ΔBCR cells was also markedly diminished and mitochondrial RAF was undetectable. In p185ΔBCR cells transfected with a mitochondria-targeted, constitutively active RAF (M-Raf) BAD was expressed in the hyperphosphorylated form and released from the mitochondria into the cytosol. p185ΔBCR/M-Raf–transfected cells were completely resistant to apoptosis induced by growth factor deprivation in vitro. Moreover, constitutive expression of dominant-negative M-Raf (K375W) enhanced the susceptibility of 32Dcl3 cells expressing wild-type BCR/ABL to apoptosis. In severe combined immunodeficiency (SCID) mice, p185ΔBCR/M-Raf double transfectants were leukemogenic, whereas cells expressing only p185ΔBCR showed no leukemogenic potential. Together, these data support the existence of a BCR/ABL-dependent pathway that leads to expression of an active RAF in the mitochondria and promotes antiapoptotic and leukemia-inducing effects of BCR/ABL.
Keywords: oncogene, signal transduction, phosphorylation, apoptosis
B cr/abl is a chimeric oncogene generated from a chromosomal translocation between chromosomes 9 and 22 (Philadelphia chromosome), which juxtaposes sequences from the bcr gene with sequences upstream of the second exon of c-abl. bcr/abl genes encode two proteins: p185, which contains a smaller BCR portion (amino acids 1–426) and is associated with acute lymphocytic leukemia (1), and p210, which contains a larger BCR portion (amino acids 1–902 or –926) and is found in most cases of chronic myelogenous leukemia (CML1; reference 2). Both BCR/ABL chimeric proteins are constitutively active tyrosine kinases that can transform fibroblasts and hematopoietic cells in culture (3–5) and induce leukemia in mice (6, 7). The fused BCR sequences activate the tyrosine kinase, actin binding, and transforming functions of ABL. Activation of the ABL-transforming function has been shown to require two distinct domains of BCR: domain 1 (amino acids 1–63) and domain 2 (amino acids 176–242) (8). Sequences within the first exon of BCR (amino acids 192–242 and 298–413) have been implicated in the binding to the SH2 domain of ABL (9), and the deletion of domain 2 of BCR has been shown to decrease BCR/ABL-dependent transforming ability (9). The region spanning amino acids 243–923 does not seem to play a role in transformation, but is involved in the stabilization of actin fibers (10). BCR/ABL tyrosine kinase activity was recently shown to protect cells from apoptosis induced by various stimuli (11–13). The apoptotic process can be subdivided into three functional phases: initiation, wherein the death-inducing stimulus is applied; the effector phase, when the “decision to die” is made; and degradation, when overall cell entropy precludes further regulatory mechanisms and leads to the activation of metabolic enzymes (14). Although it is not yet clear which of these phases are affected by BCR/ABL, it has been shown that BCR/ABL can regulate the expression of the antiapoptotic gene bcl-2 (15, 16).
The bcl-2 gene, originally identified at the chromosomal breakpoint of t(14;18)-carrying B cell lymphomas, is the prototype of a growing family of apoptosis regulators consisting of both inhibitors and promotors. Many of the proteins belonging to this family are predominantly localized in the outer mitochondrial membrane. Bcl-2 itself shows a patchy distribution in contact areas between the outer and inner mitochondrial membrane (17, 18), and the mitochondrial localization of Bcl-2 family proteins is necessary for regulation of apoptosis (17, 19, 20). The ratio of antagonists (Bcl-2, Bcl-XL) and agonists (Bax, BAD, Bcl-XS) determines, in part, how cells will respond to an apoptotic stimulus (21, 22), but posttranslational modifications also play a major role in the regulation of apoptosis. Bcl-2 can be degraded by trypsin and chymotrypsin (23), and by caspases (24). Serine phosphorylation of Bcl-2 induced by treatment either with taxol, vinblastine, or vincristine has been correlated with loss of Bcl-2 activity (25); on the other hand, a recent study suggests that serine phosphorylation is an early event after IL-3 stimulation and is required for the antiapoptosis function of Bcl-2 (26). BAD has also been shown to be target of serine phosphorylation. In the presence of IL-3, BAD becomes phosphorylated on serine residues. Upon IL-3 withdrawal, BAD is unphosphorylated and binds Bcl-XL (20), thus inhibiting the survival-promoting effects of Bcl-XL (27). By contrast, the phosphorylated form is sequestered in the cytoplasm by 14-3-3, a protein which associates with many signaling molecules in a phosphoserine-dependent manner (28), but whose function in mammalian cells in not yet understood.
Recent reports have shown that the Ras-Raf-mitogen-activated protein (MAP) kinase pathway can play an important role in the regulation of the apoptotic process in hematopoietic cells. Expression of oncogenic Ras protein complemented the function of a mutant GM-CSF receptor that failed to activate the Ras-Raf-MAP kinase pathway and was unable to promote cell survival (29); expression of a dominant-negative Ras induced apoptotic cell death in BCR/ABL-expressing cells (30).
Active forms of a downstream effector of Ras, the serine-threonine kinase Raf-1, have also been shown to prevent apoptosis in hematopoietic cells (31). Moreover, inhibition of Raf-1 by cyclic AMP agonists in v-ABL– transformed cells or by antisense oligonucleotides in cells expressing BCR/ABL has been shown to induce apoptosis (32, 33). The antiapoptotic effects of Raf-1 might be, in part, mediated by the reported physical interaction between Raf-1 and Bcl-2 (34). Such interaction appears to target Raf-1 to mitochondria, where it may function to promote survival of growth factor–deprived cells, perhaps by phosphorylating and inactivating BAD (35). However, the physical interaction between Raf-1 and Bcl-2 was not confirmed in a more recent study (36).
In this report, we further assessed the role of BCR/ABL in the cellular responses to growth factor deprivation as the apoptotic stimulus. We demonstrate that Raf-1 kinase is constitutively localized to mitochondria in BCR/ABL-expressing cells, regardless of the presence of growth factors in the culture medium. Moreover, an intact BCR portion in p185 BCR/ABL is necessary for the mitochondrial localization of Raf-1 in IL-3–deprived BCR/ABL-expressing cells. This correlates with the inability of p185ΔBCR-expressing cells to survive upon IL-3 deprivation. Furthermore, the introduction of a mitochondria-targeted active form of Raf-1 into p185ΔBCR-expressing cells restores antiapoptotic function in vitro and leukemogenic potential in vivo.
Materials and Methods
Expression Vectors.
Plasmids LXSP/Mas70/Raf and LXSP/ Mas70/Raf K375W, containing the Mas70p TM domain (residues 1–30; reference 37) fused to the COOH-terminal region (amino acids 301–648) of Raf-1 (Δ1-300), were obtained as follows: (a) pcDNA3-Raf-1 and pcDNA3-Raf-1 (K375W) (gift of Dr. D. Morrison, National Cancer Institute, Frederick Cancer Research and Development Center, Frederick, MD) were used as templates for PCR amplification of wild-type and mutant Raf-1 using a forward primer (5′-CCAACAGGCTGGTCACAGCCGAAA-3′) and a reverse primer (5′-GAATTCGAATTC GCTTCTCTGAAAACATG-3′) containing two BamHI sites (underlined) in tandem; (b) an oligonucleotide encoding the Mas70p TM domain was synthesized immediately downstream of a XhoI restriction site; and (c) the PCR fragments and the Mas70p TM double-stranded oligonucleotide were ligated into the XhoI/BamHI-digested and dephosphorylated LXSP retroviral vector. Correct orientation and frame of both constructs was confirmed by DNA sequence analysis.
pSRα p185BCR/ABL, pSRα Y77F/R552L/Y793F p185BCR/ ABL, pSRα K671R p185BCR/ABL, and pSRαΔ176-426 p185BCR/ABL were obtained from Dr. A.M. Pendergast (Duke University Medical Center, Durham, NC).
Electroporation.
The murine IL-3–dependent 32Dcl3 myeloid precursor cell line (38) was maintained at 37°C in IMDM-CM (IMDM supplemented with 10% heat-inactivated fetal bovine serum [FBS], 2 mM l-glutamine, penicillin/streptomycin (100 μg/ml each), and 15% WEHI-conditioned medium [WEHI-CM] as a source of IL-3). Plasmids were introduced into 32Dcl3 cells by electroporation (200 mV, 960 μF; gene pulser, BioRad Labs., Hercules, CA). BCR/ABL-expressing clones were obtained after selection in G418-containing medium (1 mg/ml) and were maintained in IMDM-CM. Mitochondria-targeted (M)- Raf–expressing clones were selected in puromycin (2.5 μg/ml) containing IMDM-CM.
Western Blot Analyses and Immunoprecipitation.
Western blots were performed using anti–Raf-1, anti-Bax, anti-BAD, anti–Bcl-XL, anti-ERK2 (all from Santa Cruz Biotechnology, Santa Cruz, CA), antiphosphotyrosine (4G10; Upstate Biotechnology, Lake Placid, NY) anti–Bcl-2, anti-HSP90 (Transduction Laboratories, Lexington, KY), anti–cytochrome oxidase (COX) IV (Molecular Probes, Inc., Eugene, OR), and anti–BAG-1 (gift of Dr. J.C. Reed) antibodies. Immunoprecipitations of Raf-1, ERK2, Ras and BCR/ABL were carried out using anti-Raf-1, anti-ERK2, anti-Ras (all from Santa Cruz Biotechnology) and anti-ABL antibody (Oncogene Science, Cambridge, MA), respectively.
BAD Phosphorylation.
Parental and BCR/ABL-expressing cells were washed three times in phosphate-free RPMI medium (GIBCO BRL, Gaithersburg, MD) and incubated in the same medium for 3 h. Cells were then resuspended in phosphate-free RPMI medium containing 0.3 mCi/ml of [32P]orthophosphate (New England Nuclear, Boston, MA), 0.1% BSA, and 25 mM Hepes (pH 7.4), and incubated for 3 h. Cells were fractionated (see below, Preparation of Subcellular Fractions) and cytoplasmic fractions were precleared with preimmune serum and protein A–sepharose (Pharmacia, Piscataway, NJ). Endogenous BAD was immunoprecipitated with an anti-BAD polyclonal antibody (C20; Santa Cruz Biotechnology), previously coated with protein A–sepharose. Immunoprecipitates were resolved on a 12.5% polyacryamide gel, transferred to nitrocellulose filters, and exposed.
Enzyme Assays.
RAS activation was determined by measuring GTP-bound RAS as described (39). In brief, cells were washed in phosphate-free RPMI medium (ICN Pharmaceuticals Inc., Costa Mesa, CA) and incubated for 1 h in the same medium supplemented with 0.1% BSA (Sigma Chemical Co., St. Louis, MO), 25 mM Hepes (pH 7.4), and 2 mM l-glutamine. Cells were suspended in phosphate-free RPMI containing 0.25 mCi/ ml [32P]orthophosphate (New England Nuclear) and incubated at 37°C for 3 h. Cells were lysed in 1 ml of buffer containing 1% Triton X-100, 50 mM Hepes (pH 7.4), 1.0 mg/ml BSA, 5 mM MgCl2, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 mM benzamidine, 1 mM PMSF, and 0.2 mM sodium orthovanadate. Nuclei were pelleted by centrifugation (4 min, 12,000 g) and the remaining cell extract was solubilized in 1% Triton X-100, 50 mM Hepes, pH 7.4, 0.5% sodium deoxycholate, 1.0 mg/ml BSA, 0.5 M NaCl, 5 mM MgCl2, 10 μg/ml leupeptin, 10 mM benzamidine, 1 mM PMSF, and 0.2 mM sodium orthovanadate. Lysates were incubated with anti-RAS antibody-coated agarose beads (Santa Cruz Biotechnology) at 4°C for 1.5 h. Immunoprecipitates were washed with a buffer containing 50 mM Hepes (pH 7.4), 0.5 M NaCl, 5 mM MgCl2, 0.1% Triton X-100, 0.005% SDS, and 32P-containing GTP/GDP were eluted with 2 mM EDTA, 5 mM dithiothreitol (DTT), 0.2% SDS, 0.5 mM GDP, and 0.5 mM GTP at 68°C for 20 min. Samples were resolved by thin-layer chromatography, using 0.75 M KH2PO4 and visualized by autoradiography. The ratio of GTP-RAS to GTP + GDP-RAS was determined by liquid scintillation counting.
The in vitro Raf-1 kinase assay was performed as described (40). In brief, cells were lysed in 10 mM Hepes–150 mM NaCl, 1% NP-40, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 mM benzamidine, 1 mM PMSF, and 0.2 mM orthovanadate. Extracts were precleared using protein A–agarose beads (Oncogene Science) and then incubated with anti–Raf-1 antibody conjugated with protein A–agarose beads at 4°C for 2 h. Immunoprecipitates, washed four times with lysis buffer and once with kinase buffer (30 mM Hepes, pH 7.4, 1 mM DTT, 10 mM MgCl2, 10 mM MnCl2, and 15 μM ATP), were incubated at room temperature for 20 min in kinase buffer supplemented with 20 μCiγ-[32P]ATP and 5 μg histone H1 as substrate. The reactions were ended by adding equal volumes of gel loading buffer (4% SDS, 80 mM DTT, and 10% glycerol). Samples were resolved by SDS-PAGE and the phosphoproteins were visualized by autoradiography.
MAP kinase (MAPK) activity was evaluated in an in vitro kinase assay with anti–ERK-2 immunoprecipitates in kinase buffer (30 mM Hepes, pH 7.4, 1 mM DTT, 10 mM MgCl2, and 15 μM ATP), supplemented with 20 μCiγ-[32P]ATP and 2.5 μg myelin basic protein (MBP) as substrate (40a). Reactions were carried out at 37°C for 30 min and phosphoproteins were detected as described above.
BCR/ABL kinase activity was determined using 5 μg of enolase as substrate and anti-Abl immunoprecipitates from parental 32Dcl3 cells and wild-type or mutant BCR/ABL-expressing cells. Kinase reactions were carried out as described (13).
Cell Viability and Apoptosis Assays.
Relative numbers of viable cells were estimated by trypan blue exclusion assay. Apoptosis was evaluated by flow cytometry determination of DNA content in propidium iodide–stained nuclei (41).
Preparation of Subcellular Fractions.
Subcellular fractionation was performed as described (35). In brief, cells were lysed in hypotonic buffer (5 mM Tris, pH 7.4, 5 mM KCL, 1.5 mM MgCl2, 0.1 mM EGTA [pH 8.0], 1 mM DTT, 10 μg/ml aprotinin, 10 μg/ml of leupeptin, 10 mM benzamidine, 1 mM PMSF, and 0.2 mM sodium orthovanadate) at 4°C for 30 min. After homogenization (20–30 strokes with a Dounce homogenizer B pestle), samples were centrifuged (2,500 g for 5 min) to remove the nuclei, and centrifuged again (12,000 g for 30 min) to obtain the heavy membrane (HM) fraction (pellet). The supernatants were centrifuged at 150,000 g for 1.5 h to obtain light membrane (LM) and cytoplasmic (C) fractions. The HM and LM fractions were solubilized in Triton X-100, 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 mM benzamidine, 1 mM PMSF, and 0.2 mM sodium orthovanadate.
Leukemogenesis in SCID Mice.
5–7-wk-old male SCID mice (Taconic Farms, Inc., Germantown, NY) were injected intravenously with 5 × 106 cells/transfectant (10 mice/group), and 3 wk later organs were analyzed for the presence of leukemia. Tissue sections from bone marrow, spleen, lung, liver, kidney, and brain were fixed in phosphate-buffered formalin and embedded in paraffin blocks. Two levels of each block were cut and slides were stained with hematoxylin and eosin.
Results
An Intact BCR Segment Is Required for the Antiapoptosis Function of BCR/ABL.
Although a functional BCR/ABL tyrosine kinase is absolutely required for protection from apoptosis (13), the consequences of disrupting other domains or mutating residues important for BCR/ABL-dependent signaling are much less severe, unless multiple residues are involved (13). One exception is the p185ΔBCR mutant, which lacks 251 amino acids (176 to 426) of the BCR domain and is reportedly defective in protecting growth factor–dependent cells from apoptosis induced by IL-3 withdrawal (13). To investigate mechanisms associated with apoptosis susceptibility upon disruption of the BCR portion of the BCR/ABL oncoprotein, growth factor–dependent 32Dcl3 myeloid precursor cell derivatives were established by electroporation of plasmids carrying either wild-type BCR/ABL (p185WT) or a mutant lacking amino acids 176–426 of the BCR portion (p185ΔBCR). Selected clones of the transfected cells expressed comparable levels of wild-type and mutant BCR/ABL protein (Fig. 1 A); moreover, the ectopically expressed proteins were similarly potent in their tyrosine kinase activity, as indicated by phosphorylation of the enolase substrate by anti-ABL immunoprecipitates (Fig. 1 B). Upon IL-3 deprivation, parental cells lost viability very rapidly and were all dead after 48 h; p185WT-expressing cells were all viable, whereas a marked decrease in the frequency of viable cells was noted in the p185ΔBCR cell cultures after a 48-h IL-3 deprivation (data not shown). Loss of viability in cultures of parental and p185ΔBCR-expressing cells was due to apoptosis, as indicated by flow cytometry detection of cells with hypodiploid DNA content (Fig. 1 C).
Figure 1.



(A) Expression of BCR/ABL proteins in 32Dcl3 cell clones. 32Dcl3 cells were electroporated with the pSRα constructs encoding the indicated BCR/ABL proteins. Western blots show expression of wild-type or mutant BCR/ABL protein in representative clones. Levels of HSP90 were detected as control for protein loading. (B) In vitro kinase activity of wild-type (p185WT) and mutant (p185ΔBCR) BCR/ABL. Phosphorylation of enolase (5 μg) used as a substrate was examined in anti-ABL immunoprecipitates from parental and BCR/ABL-expressing 32Dcl3 cells. Wild-type and mutant BCR/ABL were detected in the immunoprecipitates by Western blot with the Ab-3 anti-ABL antibody. Low levels of c-abl expression in 32Dcl3 immunoprecipitates were detected only after long exposure. (C) Apoptosis in cultures of IL-3–deprived parental and BCR/ABL-expressing 32Dcl3 cells. Apoptotic cells were evaluated cytometrically (propidium iodide staining) in cells cultured in the absence of IL-3–containing medium for 24 and 48 h. Representative of three independent experiments with similar results.
Activation of the Ras-Raf-MAPK Pathway in p185WT- and p185ΔBCR-expressing Cells.
Expression of BCR/ABL leads to constitutive activation of RAS (42, 39, 13), and apoptosis of BCR/ABL-expressing cells is induced upon conditional expression of dominant-negative RAS (30). To determine whether p185ΔBCR-expressing cells can activate the Ras-Raf-MAPK pathway, we analyzed RAS activation by direct examination of RAS-GTP levels. In parental 32Dcl3 cells, RAS was detected in the GTP-bound form only upon culture with IL-3–supplemented medium, whereas both p185WT- and p185ΔBCR-expressing cells displayed constitutive levels of RAS activation, although the relative amount of RAS-GTP in p185ΔBCR-expressing cells was somewhat lower than that in p185WT-expressing cells (Fig. 2 A). To determine whether the activation of RAS by p185ΔBCR correlated with that of RAS downstream effectors in the MAPK pathway, the levels of Raf-1 activity were analyzed by in vitro kinase assay, using histone H1 as substrate. After starvation, low levels of Raf-1 activity were detected in parental cells, higher levels were found in p185ΔBCR cells, and p185WT cells showed the highest Raf-1 activity (Fig. 2 B). A decrease in the level of Raf-1 expression was noted in IL-3–starved 32Dcl3 cells, consistent with a previous report (31). Since tyrosine phosphorylation of Raf-1 is postulated as an important mechanism for RAF activation (43), we assessed RAF phosphorylation using antiphosphotyrosine immunoblotting of anti-RAF immunoprecipitates (Fig. 2 C), and by phosphoaminoacid analysis (data not shown). Both assays revealed equal levels of RAF tyrosine phosphorylation in p185WT- and in p185ΔBCR-expressing cells.
Figure 2.


RAS, RAF, and MAPK activation in BCR/ABL-expressing 32Dcl3 cells. (A) GTP-bound RAS was measured as described (39) using serum– and IL-3–starved (5 h) BCR/ABL- expressing cells. (B) c-Raf-1 activity was measured as described (40) using histone H1 as substrate. (C) c-Raf-1 tyrosine phosphorylation in BCR/ABL-expressing cells. Tyrosine phosphorylated c-Raf-1 was detected by antiphosphotyrosine blotting of anti–c-Raf-1 immunoprecipitates from BCR/ABL-expressing cells. (D) MAPK activity was measured as described (51), using MBP as substrate. Representative of three independent experiments.
One effect of Raf-1 activation is the induction of MAPK kinase kinase activity, which in turn activates the MAPK. MAPK activity is dependent on growth factor stimulation and provides an index of the proliferative capacity of the cell. Thus, we analyzed the levels of activation of MAPK by in vitro kinase assay, using MBP as substrate. In parental 32Dcl3 cells, withdrawal of IL-3 was associated with downregulation of MAPK activity, whereas in p185WT-expressing cells, levels of activation were similar to those in parental 32Dcl3 cells growing in the presence of IL-3. In p185ΔBCR-expressing cells, activation of MAPK was approximately twofold lower than in p185WT-expressing cells, but clearly higher than in starved parental cells. Thus, IL-3–deprived p185ΔBCR-expressing cells retain the ability to activate the Ras-Raf-MAPK pathway, but the activation of each of these signal transducing molecules is less robust than in cells carrying wild-type BCR/ABL.
Expression of Anti- and Proapoptosis Proteins in p185ΔBCR-expressing Cells.
Activation, albeit at a reduced level, of the Ras-Raf-MAPK pathway in p185ΔBCR-expressing cells suggests that the enhanced susceptibility of these cells to apoptosis involves the disruption of other signaling pathways required for cell survival. Since enhanced expression of Bcl-2 is induced by BCR/ABL expression in hematopoietic cells (15), expression levels of several proapoptotic (Bax and BAD) and antiapoptotic proteins (BAG-1, Bcl-XL, and Bcl-2) were compared in parental and BCR/ABL-expressing cells. In parental 32Dcl3 cells, the expression of Bax, BAG-1, and Bcl-XL was not affected by IL-3 deprivation (data not shown), whereas Bcl-2 levels were clearly diminished and BAD was detected at lower levels and only in its fast-migrating (presumably due to hypophosphorylation), proapoptotic form (Fig. 3 A). In p185WT-expressing cells, Bcl-2 and BAD levels were enhanced, but the latter was detected predominantly in its slow-migrating, nonfunctional form (Fig. 3 A). Compared to p185WT-expressing cells, p185ΔBCR-expressing cells showed markedly decreased Bcl-2 levels; moreover, BAD was expressed in its fast-migrating, proapoptotic form (Fig. 3 A). BAD phosphorylation was directly assessed in anti-BAD immunoprecipitates from cells metabolically labeled with [32P]orthophosphate. As shown in Fig. 3 B, BAD phosphorylation was induced upon IL-3 treatment of parental 32Dcl3 cells and was constitutive in cells expressing wild-type BCR/ ABL, but was not detectable in p185ΔBCR-expressing cells. Together, these results suggest that an intact BCR domain is necessary for a BCR/ABL-dependent pathway that maintains elevated levels of Bcl-2 and promotes BAD phosphorylation.
Figure 3.
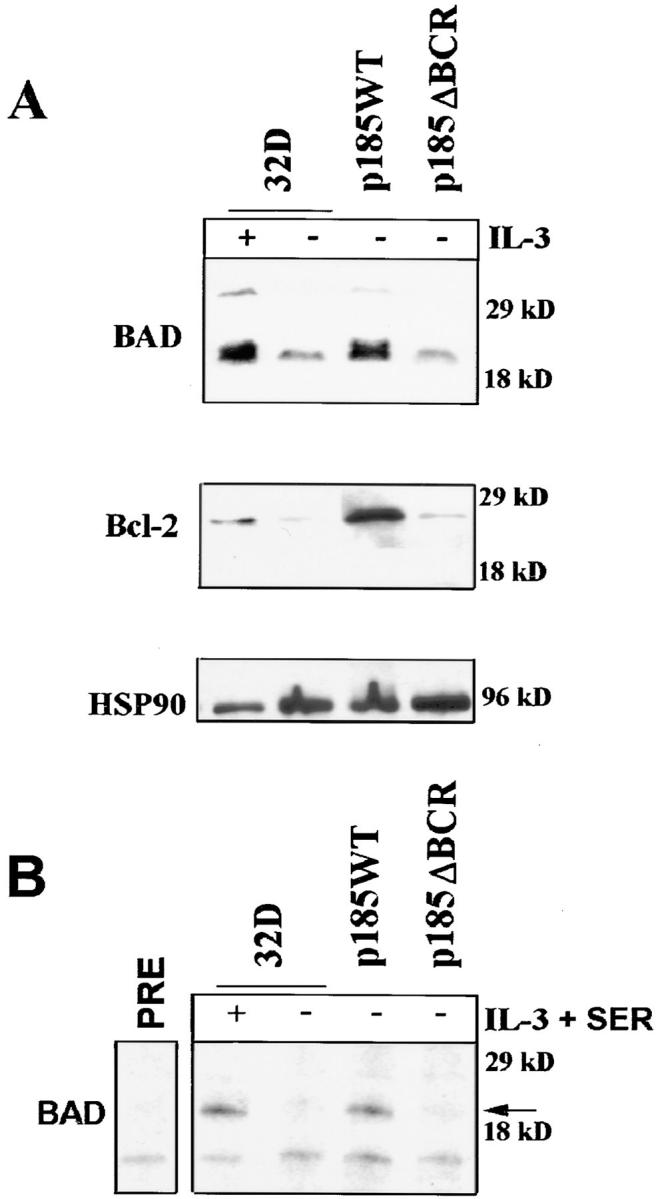
(A) Expression of anti- and proapoptosis proteins in BCR/ ABL-expressing cells. Western blots show expression of BAD and Bcl-2 proteins in parental and BCR/ABL-expressing 32Dcl3 cells. Cells were starved of IL-3–containing medium for 16 h. (B) Phosphorylation of BAD in BCR/ABL-expressing cells. Parental and BCR/ABL-expressing cells were starved from both IL-3 and serum (3 h) in [32P]orthophosphate-containing medium for 3 h. After starvation, parental cells were either stimulated for 30 min with IL-3 (150 mg/ml) and 0.1% FBS or left untreated. The arrow indicates phosphorylated BAD (∼24 kD). As control, labeled extracts were incubated with preimmune serum and protein A–sepharose beads (PRE). Representative of two different experiments.
BCR/ABL Induces the Constitutive Localization of Raf-1 to Mitochondria.
Previous studies have suggested that Raf-1 is a potential regulator of the apoptotic pathway because of its coimmunoprecipitation with Bcl-2 and its localization to mitochondria (34, 35, 44). Moreover, Raf-1 appears to protect 32Dcl3 cells from apoptosis induced by IL-3 deprivation if constitutively expressed in the mitochondria (35). Thus, we investigated whether Bcl-2 expression correlates with levels of mitochondrial Raf-1 in parental and BCR/ ABL-expressing cells. By subcellular fractionation and Western blot analysis, low levels of Raf-1 were detected in mitochondrial and membrane fractions (HM and LM, respectively) of parental 32Dcl3 cells cultured in the presence of IL-3 (Fig. 4 A). After IL-3 removal, Raf-1 protein was no longer detectable in the mitochondrial fraction of parental cells or of p185ΔBCR-expressing cells, but was readily detectable in the mitochondrial fraction of IL-3–starved p185WT-expressing cells (Fig. 4 A). The levels of mitochondrial Raf-1 in parental and BCR/ABL-expressing 32Dcl3 cells were correlated well with those of Bcl-2. In IL-3–deprived parental cells, the decline of mitochondrial Bcl-2 expression correlated with the decrease in Bcl-2 levels observed in total lysates (Fig. 3). In p185WT-expressing cells, levels of mitochondrial Bcl-2 were similar to those detected in parental cells growing in the presence of IL-3, whereas in p185ΔBCR-expressing cells, Bcl-2 levels were comparable with those observed in growth factor–starved 32Dcl3 cells (Fig. 4 B).
Figure 4.


c-Raf-1 and Bcl-2 expression in subcellular fractions of parental and BCR/ABL-expressing 32Dcl3 cells. (A) Western blots show c-Raf-1 levels in fractions enriched for mitochondria (HM), cytoplasmic membrane (LM), and cytoplasm (C) from parental and BCR/ABL-expressing cells. Cells were starved of IL-3–containing medium for 16 h. (B) Western blots show Bcl-2 levels in the mitochondrial fraction of parental and BCR/ABL-expressing cells cultured as described above. Levels of the subunit IV of mitochondrial COX were measured as control of equal loading.
Mitochondrial Targeting of Constitutively Active Raf-1 Rescues p185ΔBCR-expressing Cells from Apoptosis Induced by IL-3 Deprivation.
In p185ΔBCR-expressing cells, Raf-1 was not detectable in the mitochondria, likely as a consequence of the reduced expression of Bcl-2; however, it is unclear whether lack of mitochondrial Raf-1 expression and susceptibility of p185ΔBCR-expressing cells to apoptosis are causally linked. To address this issue, we generated an expression plasmid in which human Raf-1 was fused with the transmembrane domain of the yeast outer mitochondrial membrane protein MAS70p (37). Moreover, to prevent Raf-1 targeting to the plasma membrane and to obtain a constitutively active mutant, we deleted the NH2-terminal domain of Raf-1, which mediates binding to Ras and is known to exert a negative regulatory function (45). After transfection of p185ΔBCR cells, ectopic expression of M-Raf was detected by Western blot either in total lysates (Fig. 5 A), or in the mitochondrial fraction of selected clones (Fig. 5 B). Indeed, M-Raf was highly expressed and restricted to the mitochondrial fraction. As expected, the decrease in Bcl-2 levels detected in the mitochondrial fraction of IL-3–starved p185ΔBCR-expressing cells (compared to p185WT cells, Fig. 4 B) was not reversed by ectopic expression of M-Raf (Fig. 5 C). By contrast, ectopic expression of M-Raf induced hyperphosphorylation of BAD (Fig. 5 C) and its release from the mitochondria into the cytosol (Fig. 5 D). Analysis of the kinetics of cell death, upon IL-3 removal, in p185ΔBCR-expressing cells and in selected clones expressing p185ΔBCR and constitutively active M-Raf indicated that p185ΔBCR/M-Raf–expressing cells were protected from apoptosis induced by IL-3 deprivation (Fig. 6). Moreover, these cells were able to proliferate in the absence of IL-3 (data not shown). These experiments support the hypothesis that the apoptosis susceptibility of p185ΔBCR-expressing cells is causally linked to the inability of the p185ΔBCR mutant to induce mitochondrial targeting of Raf-1. To determine whether the expression of constitutively active M-Raf specifically rescues p185ΔBCR-expressing cells from apoptosis or whether 32Dcl3 cells expressing other BCR/ABL mutants defective in apoptosis protection can be rescued as well, the effect of M-Raf was studied on cells expressing either a kinase-deficient mutant (K671R) or a triple mutant (Y177F/R552L/ Y793F) lacking a functional phosphotyrosine binding motif in the SH2 domain, and GRB2 binding and autophosphorylation sites. Ectopic expression of M-Raf in both cell transfectants (Fig. 7 A) induced only a modest protection from cell death after 24 h, similar to that observed in parental cells expressing M-Raf (compare Fig. 7, B and C). Moreover, after 48 h, <10% of the transfected cells were viable (Fig. 7 C).
Figure 5.

(A) RAF expression in total extracts of p185ΔBCR/ M-Raf transfectants. Western blot shows expression of endogenous and mitochondria-targeted Raf-1 in total extracts of selected clones. (B) RAF expression in subcellular extracts of p185ΔBCR/ M-Raf transfectants. Western blot shows expression of endogenous (Raf-1) and mitochondria-targeted (M-Raf) Raf-1 in subcellular extracts from p185ΔBCR/M-Raf selected clones. Levels of the subunit IV of mitochondrial COX and of HSP90 were measured as control of equal loading and of appropriate subcellular fractionation. (C) Bcl-2 and BAD expression in total cell lysate of p185ΔBCR– and p185ΔBCR/ M-Raf–expressing cells. (D) BAD expression in subcellular fractions of p185WT–, p185ΔBCR–, and p185ΔBCR/M-Raf – expressing cells. Western blots show BAD expression in mitochondria- (HM), cytoplasmic membrane– (LM), and cytoplasm- (C) enriched fractions. Arrows, hypo- and hyperphosphorylated BAD. Cells were starved of IL-3–containing medium for 16 h. Levels of the subunit IV of mitochondrial COX and of HSP90 were measured as control of equal loading and of appropriate subcellular fractionation.
Figure 6.

Viability of p185ΔBCR/M-Raf transfectants in IL-3–deprived cultures. Numbers of viable cells from triplicate wells were estimated for each transfectant at the indicated times by trypan blue exclusion assay. Representative of three independent experiments.
Figure 7.


(A) BCR/ABL and RAF expression in 32Dcl3 cells expressing a kinase-deficient or triple mutant p185BCR/ABL. Western blots show expression of mutant BCR/ABL (KD, K671R kinase deficient; TM, Y177F/R552L/Y793F triple mutant) and RAF (endogenous and mitochondria targeted) in a representative clone per each transfectant. Levels of HSP90 were determined as control for protein loading. (B and C) Viability of mutant (KD and TM) p185BCR/ABL- and mutant (KD and TM) p185BCR/ABL/M-Raf transfectants in IL-3–deprived cultures. Numbers of viable cells from triplicate wells were estimated for each transfectant at the indicated times by trypan blue exclusion assay. Representative of three independent experiments using four different clones per each transfectant.
Mitochondrial Targeting of Constitutively Active Raf-1 Rescues the Leukemogenic Potential of p185ΔBCR-expressing Cells.
To determine whether expression of constitutively active Raf-1 in the mitochondria can activate a survival pathway required for the leukemia-inducing effect of BCR/ABL, immunocompromised SCID mice (10–15/group) were injected with an equal number (5 × 106/mouse) of 32Dcl3 cells expressing wild-type BCR/ABL, the p185ΔBCR mutant, or coexpressing p185ΔBCR and constitutively active M-Raf. As an additional control, mice were also injected with 32Dcl3 cells expressing the constitutively active M-Raf. 3 wk after cell injection, various organs from the SCID mice (three per group) were evaluated by visual inspection and light microscopy (Fig. 8). As expected, injection of wild-type BCR/ABL-expressing cells resulted in splenomegaly and development of myelogenous leukemia that involved several organs. The spleen showed acute myelogenous leukemia (AML) without overt maturation. AML focally involved the liver and other nonhematopoietic organs such as kidneys and meninges. The bone marrow showed focal areas of blast accumulation, but most marrow cells exhibited features of initial granulocytic differentiation. Essentially normal organ histology was seen in the mice injected with p185ΔBCR-expressing cells, except for splenic extramedullary hematopoiesis, congestion, and iron deposition; no overt leukemia was seen in any of the organs. In contrast, injection of 32Dcl3 cells coexpressing the nononcogenic p185ΔBCR mutant and the constitutively active mitochondrial Raf-1 resulted in mild to moderate splenomegaly in all three mice tested. Hematoxylin and eosin–stained sections demonstrated patchy to diffuse involvement of the spleens by poorly differentiated AML. AML also involved livers (two of three animals), kidneys, and meninges. Bone marrows showed either CML-like myeloproliferation with various degrees of fibrosis or an overt AML. Although abnormal, the bone marrows usually demonstrated a higher degree of myeloid maturation than other organs. Leukemic infiltrates in all the organs appeared similar to those in mice injected with cells expressing wild-type BCR/ABL, although the extent of the disease was less. Injection of parental 32Dcl3 cells (data not shown) or 32Dcl3 cells expressing M-Raf only (Fig. 8) did not lead to development of leukemia. These differences in histopathologic findings were reflected in the survival of the various groups; mice (10/group) injected with parental 32Dcl3 cells, p185ΔBCR–, and M-Raf–expressing cells never developed leukemia, whereas mice (10 and 15 [five/each clone]) injected with p185WT– and p185ΔBCR/M-Raf– expressing cells died of leukemia 4–6 and 6–9 wk after injection, respectively (Fig. 9).
Figure 8.
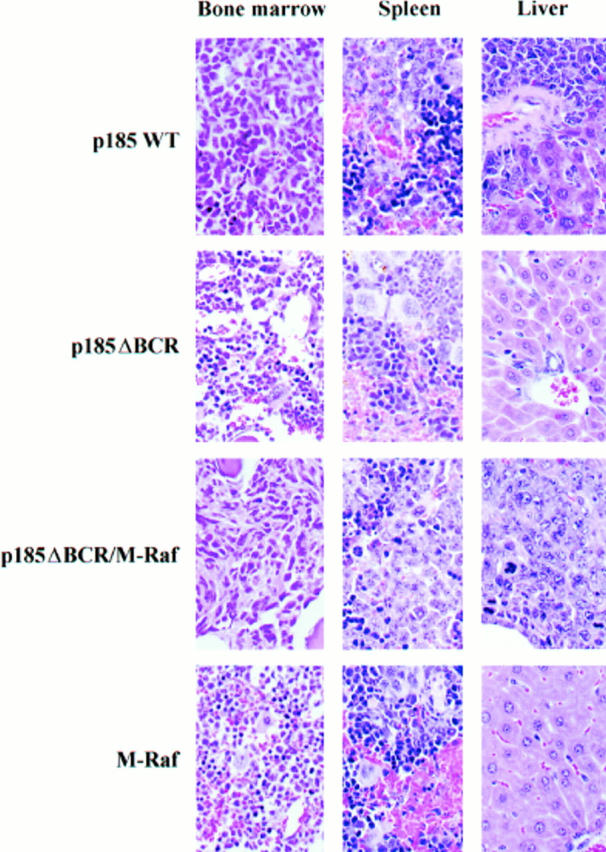
Histologic findings in mice injected with BCR/ABL– M-Raf–, or BCR/ABL/M-Raf– expressing 32Dcl3 cells. Tissue sections from various organs were fixed in phosphate-buffered formalin, embedded in paraffin, and stained with hematoxylin/ eosin. Representative microphotographs from bone marrow, spleen, and liver (original magnification: 600) from groups of three mice of each phenotype.
Figure 9.

Survival curves of mice injected with BCR/ABL–, M-Raf–, or BCR/ABL/M-Raf–expressing 32Dcl3. SCID mice were injected intravenously with 5 × 106 cells from parental or transfected 32Dcl3 cells. 10 mice were included in the parental, p185WT, p185ΔBCR (five mice per two clones), and M-Raf (five mice per two clones) group, whereas 15 mice were included in the p185ΔBCR/M-Raf (five mice per three clones) group.
A Dominant Negative Raf-1 Targeted to Mitochondria Induces Apoptosis in p185WT Cells.
To further demonstrate the importance of mitochondrial expression of a functional Raf-1 in BCR/ABL-mediated leukemogenesis, a dominant-negative Raf-1 mutant (K375W) was targeted to the mitochondria and assessed for its ability to induce apoptosis of IL-3–deprived p185WT-expressing 32Dcl3 cells. Upon IL-3 deprivation, expression of the mitochondria-targeted M-Raf (K375W) mutant (Fig. 10 A) slightly reduced the viability of p185WT-expressing cells (Fig. 10 B). However, the effect became more pronounced when cells were deprived of both IL-3 and serum (Fig. 10 C). After 24 h, ∼65% of p185WT/M-Raf (K375W) cells were dead, whereas <20% of p185WT-expressing cells were dead. A similar difference in the viability of the transfected cells was observed after 48 h in IL-3– and serum–starved cultures (Fig. 10 C).
Figure 10.


(A) RAF expression in subcellular extracts of p185WT– and p185WT/K375W M-Raf–transfected cells. Western blot shows expression of endogenous (Raf-1) and mitochondria-targeted (M-Raf) in subcellular extracts of selected clones. Levels of the subunit IV of mitochondrial COX and of HSP90 were measured as control of equal loading and of appropriate subcellular fractionation. (B and C) Viability of p185WT BCR/ABL– and p185WT BCR/ABL/K375W M-Raf–expressing 32Dcl3 cells. Numbers of viable cells (triplicate wells per transfectant) in IL-3– (B) or IL-3– and serum– (C) deprived cultures were estimated at the indicated times by trypan blue exclusion assay. Representative of three independent experiments.
Discussion
Reduced susceptibility to apoptosis is one of the characteristics of BCR/ABL-expressing cells. Although this property is especially manifested by growth factor–dependent hematopoietic cell lines ectopically expressing BCR/ABL and by CML cells in blast transformation (11, 13, 46), progenitor cells from CML in chronic phase also exhibit a prolonged survival in serum-free cultures (12). By contrast, growth factor–independent proliferation and differentiation arrest are typical features of BCR/ABL-expressing hematopoietic cell lines and CML-blast crisis cells, but not of CML-chronic phase progenitors (47). Thus, enhanced cell survival, rather than proliferative advantage, might be the primary and most direct consequence of BCR/ABL expression in hematopoietic progenitor cells.
The mechanisms underlying BCR/ABL-dependent protection from apoptosis induced by growth factor deprivation and by other death-inducing stimuli are not well understood. A functional tyrosine kinase is an absolute requirement for apoptosis protection (13), and recent data also suggest that disruption of RAS activity promotes apoptotic cell death of BCR/ABL-expressing myeloid precursor cells (30). Moreover, BCR/ABL activates the Akt serine/ threonine kinase (48), which promotes survival in many cell types by phosphorylation and consequent inactivation of the proapoptotic BAD (49, 50) and, at least in hematopoietic cells, by enhancing Bcl-2 expression (48, 51). Increased Bcl-2 levels in BCR/ABL-expressing cells have also been reported (15, 16); however, no information is available about the mechanisms and pathways leading to such an increase.
Mechanisms of Apoptosis Susceptibility in p185ΔBCR-expressing 32Dcl3 Cells.
In addition to kinase-deficient BCR/ABL, the ΔBCR (176–426) BCR/ABL (p185ΔBCR) mutant was also unable to protect transfected 32Dcl3 cells from apoptosis (reference 13, and this study). Apoptosis susceptibility in 32Dcl3 cells expressing the p185ΔBCR mutant was associated with reduced Bcl-2 levels and expression of nonphosphorylated, proapoptotic BAD. The reduced levels of Bcl-2 expression in p185ΔBCR-expressing cells were not due to a transcriptional mechanism since Bcl-2 mRNA levels were not diminished (data not shown); on the other hand, the rate of Bcl-2 synthesis was decreased and Bcl-2 half-life was shorter (data not shown), possibly explaining the reduced Bcl-2 levels in p185ΔBCR-expressing cells. The expression of nonphosphorylated, proapoptotic BAD might reflect the reduced ability of the p185ΔBCR mutant to activate the Akt serine/threonine kinase (48), which, in turn, phosphorylates BAD (49, 50). However, it seems unlikely that this is the only mechanism, since SH2 domain BCR/ABL mutants are even less capable of Akt activation than the p185ΔBCR mutant (48), and yet can protect 32Dcl3 cells from apoptosis much more efficiently (reference 13; and our unpublished results).
In addition to the changes in Bcl-2 and BAD, p185ΔBCR-expressing cells also failed to express detectable levels of c-Raf-1 in the mitochondria. This might be the consequence of the decrease in the levels of Bcl-2, which has been shown to target c-Raf-1 to the mitochondria (35); however, the undetectable expression of c-Raf-1 in the mitochondrial fraction of p185ΔBCR-expressing cells contrasts with the reduced but readily detectable Bcl-2 levels, raising the possibility that BCR/ABL might regulate the targeting of c-Raf-1 to mitrochondria in a Bcl-2-independent manner.
Complementation of the p185ΔBCR-dependent Phenotype by Constitutively Active Mitochondria-targeted c-Raf-1.
In a recent study (35), susceptibility of IL-3–deprived 32Dcl3 cells to apoptosis was alleviated by ectopic expression of mitochondria-targeted, constitutively active c-Raf-1. In our study, apoptosis susceptibility and leukemogenic potential of p185ΔBCR-expressing cells were rescued by the constitutively active M-Raf. Interestingly, apoptosis susceptibility of cells expressing a kinase-deficient (K671R) BCR/ABL mutant or a triple mutant (Y177F/R552L/ Y793F) BCR/ABL defective in the SH2 domain function, and in autophosphorylation and GRB-2 binding activity was not rescued by M-Raf. These findings are most likely explained by the severity of the apoptosis-susceptible phenotype of kinase deficient– and triple mutant–transfected cells. In this regard, the Y177F/R552L/Y793F BCR/ABL mutant retained its tyrosine kinase activity, but did not activate RAS in transfected 32Dcl3 cells (13). Constitutively active M-Raf did not exert strong antiapoptosis effects in these cells, but was able to correct the apoptosis susceptibility of cells expressing the p185ΔBCR mutant which, instead, is competent for Ras-Raf-1-MAP kinase activation. Thus, RAS-dependent Raf-1/MAP kinase activation and mitochondria-targeting of Raf-1 are independently regulated by BCR/ABL, but are both necessary for protection from apoptosis induced by growth factor deprivation. The biological relevance of a BCR/ABL-dependent pathway of mitochondrial Raf-1 targeting is also supported by the observation that expression of a mitochondria-targeted dominant-negative Raf-1 in p185WT 32Dcl3 cells was associated with increased susceptibility to apoptosis, especially evident in the absence of both IL-3 and serum. Susceptibility to apoptosis associated with expression of a dominant-negative RAS was more pronounced (30), probably due to the functional inhibition of both plasma membrane–associated and mitochondrial Raf-1.
Constitutively active M-Raf restored the leukemogenic potential of p185ΔBCR-expressing cells, underscoring the importance of the activation of antiapoptosis mechanisms in BCR/ABL leukemogenesis. p185ΔBCR/M-Raf–expressing cells were not only resistant to apoptosis induced by IL-3 deprivation, but were also able to proliferate, which may explain their ability to induce leukemia in mice. Compared to p185WT-expressing cells, p185ΔBCR/M-Raf–expressing cells caused a less bulky leukemia, but with similar widespread organ distribution. This probably reflects the reduced proliferation rate of p185ΔBCR/M-Raf–expressing cells, compared to p185WT-expressing cells. In vivo, p185ΔBCR/M-Raf 32Dcl3 cells in liver and spleen did not undergo differentiation, whereas these cells showed a more differentiated morphology in the bone marrow, most likely due to the exposure to more potent differentiation stimuli in that microenvironment. However, there were no detectable morphological differences between p185WT and p185ΔBCR/M-Raf cells, suggesting that both wild-type and mutant p185ΔBCR are equally potent in blocking hematopoietic differentiation, provided that an antiapoptosis signal (M-Raf expression) permits the survival of p185ΔBCR-expressing cells.
Potential Mechanisms of M-Raf Requirement in BCR/ABL Leukemogenesis.
The importance of antiapoptosis pathways in BCR/ABL tumorigenesis was first demonstrated in a study showing that suppression of Bcl-2 expression inhibits the tumorigenic potential of wild-type BCR/ABL-expressing hematopoietic cells (15).
Our data further support the role of survival factors in BCR/ABL leukemogenesis, but the mechanisms whereby M-Raf expression promotes survival and favors the leukemia-inducing effects of p185ΔBCR-expressing cells remain unclear. Upon M-Raf expression in p185ΔBCR-expressing cells, Bcl-2 levels were unaffected, remaining identical to those in cells expressing p185ΔBCR only. However, M-Raf expression induced phosphorylation of BAD, consistent with the results of a previous study (35) in which phosphorylated BAD was detected in cells transiently transfected with M-Raf. Moreover, phosphorylated BAD was detected in the cytosolic fraction; this suggests that Raf-1– dependent phosphorylation of BAD in the mitochondria might serve as a “rescue” mechanism to inactivate proapoptotic BAD that was not sequestered in the cytosol upon phosphorylation and interaction with 14-3-3 (20). The role of Raf-1 in BAD phosphorylation remains unclear; the major sites of BAD serine phosphorylation in vivo are within a motif recognized by Akt, and their phosphorylation is Akt dependent as indicated by its inhibition in cells treated with the PI-3k/Akt pathway inhibitor wortmannin or upon expression of dominant-negative Akt (49, 50). Moreover, Akt phosphorylates BAD in vitro at the same sites phosphorylated in vivo. By contrast, Raf-1 appears to phosphorylate BAD in vitro at sites distinct from those phosphorylated by Akt and required for the interaction with 14-3-3 (20).
Thus, M-Raf–dependent phosphorylation of BAD might involve residues distinct from those phosphorylated by Akt, yet resulting in the inhibition of the proapoptotic function of BAD; alternatively, the serine residues of BAD that are phosphorylated by Akt may also undergo Raf-1–dependent phosphorylation in the mitochondria.
In conclusion, we have identified mitochondrial Raf-1 as an effector of BCR/ABL in apoptosis resistance and leukemogenesis. An understanding of the precise mechanisms whereby M-Raf is activated by BCR-ABL and promotes cell survival awaits further experimentation.
Acknowledgments
We thank Margaret Nieborowska-Skorska for establishing the cell lines expressing p185WT and p185ΔBCR BCR/ABL, and Danilo Perrotti and Giorgia Gri for helpful discussion and advice.
Footnotes
P. Salomoni was supported in part by a postdoctoral fellowship from the University of Modena Medical School, Modena, Italy. T. Skorski was supported in part by R29 CA70815. This work was supported in part by National Institutes of Health and American Cancer Society grants to B. Calabretta.
Abbreviations used in this paper: AML, acute myelogenous leukemia; C, cytoplasmic; COX, cytochrome oxidase; CML, chronic myelogenous leukemia; DTT, dithiothreitol; HM, heavy membrane; LM, light membrane; M, mitochondria-targeted; MAP, mitogen-activated protein; MAPK, MAP kinase; MBP, myelin basic protein.
References
- 1.Clark SS, McLaughlin J, Timmonis M, Pendergast AM, Ben-Neriah Y, Dow L, Rovera G, Smith SD, Witte ON. Expression of a distinctive bcr-abl oncogene in Ph1-positive acute lymphoblastic leukemia (ALL) Science. 1988;239:775–779. doi: 10.1126/science.3422516. [DOI] [PubMed] [Google Scholar]
- 2.Shtivelman E, Lifshitz B, Gale RP, Roe BA, Canaani E. Alternative splicing of RNAs transcribed from the human abl gene and from bcr-abl fused gene. Cell. 1986;47:277–286. doi: 10.1016/0092-8674(86)90450-2. [DOI] [PubMed] [Google Scholar]
- 3.Daley GR, Baltimore D. Transformation of an interleukin 3–dependent hematopoietic cell line by the chronic myelogenous leukemia–specific p210bcr/ablprotein. Proc Natl Acad Sci USA. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr/abl oncogenic products. Science. 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 5.Gishizki ML, Witte ON. Initiation of deregulated growth of multipotent progenitor cells by bcr-abl in vitro. Science. 1992;256:836–839. doi: 10.1126/science.1375394. [DOI] [PubMed] [Google Scholar]
- 6.Daley GR, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the p210bcr/ablgene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 7.Heisterkamp N, Jenster G, Hoeve JT, Zovich D, Pattengale PK, Graffen J. Acute leukemia in bcr/abl transgenic mice. Nature. 1990;344:251–253. doi: 10.1038/344251a0. [DOI] [PubMed] [Google Scholar]
- 8.McWhirter JR, Glasso DL, Wang JY. A colied-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol. 1993;13:7587–7595. doi: 10.1128/mcb.13.12.7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pendergast AM, Muller AJ, Havlik MH, Maru Y, Witte ON. BCR sequences essential for transformation by the BCR/ABL oncogene bind to the ABL SH2 regulatory domain in a non-phosphotyrosine–dependent manner. Cell. 1991;66:161–171. doi: 10.1016/0092-8674(91)90148-r. [DOI] [PubMed] [Google Scholar]
- 10.McWhirter JR, Wang JY. Effect of Bcr sequences on the cellular function of the Bcr-Abl oncoprotein. Oncogene. 1997;15:1625–1634. doi: 10.1038/sj.onc.1201342. [DOI] [PubMed] [Google Scholar]
- 11.McGahon A, Bissonnette R, Schmitt M, Cotter KM, Green DR, Cotter TG. BCR/ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83:1179–1187. [PubMed] [Google Scholar]
- 12.Bedi A, Zehnbauer BA, Barber JP, Sharkis SJ, Jones RJ. Inhibition of apoptosis by BCR/ABL in chronic myeloid leukemia. Blood. 1994;83:2038–2044. [PubMed] [Google Scholar]
- 13.Cortez D, Kadlec L, Pendergast AM. Structural and signalling requirements for BCR-ABL–mediated transformation and inhibition of apoptosis. Mol Cell Biol. 1995;15:5531–5541. doi: 10.1128/mcb.15.10.5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 15.Sanchez-Garcia I, Grütz G. Tumorigenic activity of the BCR/ABL oncogenes is mediated by Bcl-2. Proc Natl Acad Sci USA. 1995;92:5287–5291. doi: 10.1073/pnas.92.12.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu J, Nabissa PM, Hoffman B, Lieberman DA, Shore SK. Activated abloncogenes and apoptosis: differing responses of transformed myeloid progenitor cell lines. Blood. 1996;87:4368–4375. [PubMed] [Google Scholar]
- 17.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang E, Korsmeyer SJ. Molecular thanatopsis: a discourse on the BCL-2 family and cell death. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- 19.Tanaka S, Saito K, Reed JC. Structure–function analysis of the Bcl-2 oncoprotein. Addition of a heterologous transmembrane domain to portions of the Bcl-2 beta protein restores function as a regulator of cell survival. J Biol Chem. 1993;268:10920–10926. [PubMed] [Google Scholar]
- 20.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL . Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 21.Oltvai Z, Korsmeyer S. Checkpoints of dueling dimers foil death wishes. Cell. 1994;79:189–192. doi: 10.1016/0092-8674(94)90188-0. [DOI] [PubMed] [Google Scholar]
- 22.Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci USA. 1995;92:7834–7838. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strack PR, Frey MW, Rizzo CJ, Cordova B, George HJ, Meade R, Ho SP, Corman J, Tricht R, Korant BD. Apoptosis mediated by HIV proteases is preceded by cleavage of Bcl-2. Proc Natl Acad Sci USA. 1996;93:9571–9576. doi: 10.1073/pnas.93.18.9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng EHY, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- 25.Haldar S, Jena N, Croce CM. Inactivation of Bcl-2 by phosphoryation. Proc Natl Acad Sci USA. 1995;92:4507–4511. doi: 10.1073/pnas.92.10.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito T, Deng X, Can B, May SW. Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem. 1997;272:11671–11673. doi: 10.1074/jbc.272.18.11671. [DOI] [PubMed] [Google Scholar]
- 27.Kelekar M, Chang BS, Harlan JE, Fesik SW, Thompson CB. Bad is a BH3 domain containing protein that forms an inactivating dimer with Bcl-XL . Mol Cell Biol. 1997;17:7040–7046. doi: 10.1128/mcb.17.12.7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling molecules is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 29.Kinoshita T, Yokata T, Arai K, Miyajima A. Suppression of apoptotic death in hematopoietic cells by signalling through the IL-3/GM-CSF receptors. EMBO (Eur Mol Biol Organ) J. 1995;14:266–275. doi: 10.1002/j.1460-2075.1995.tb07000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cortez D, Stoica G, Pierce JH, Pendergast AM. The BCR-ABL tyrosine kinase inhibits apoptosis by activating a Ras-dependent signaling pathway. Oncogene. 1996;13:2589–2594. [PubMed] [Google Scholar]
- 31.Cleveland JL, Troppmair J, Packham G, Askew DS, Lloyd P, Gonzales-Garcia M, Nunez G, Ihle JN, Rapp UR. v-Raf suppresses apoptosis and promotes growth of interleukin-3–dependent myeloid cells. Oncogene. 1994;9:2217–2226. [PubMed] [Google Scholar]
- 32.Weissinger EM, Eissner U, Grammer C, Fackler S, Haefner B, Yoon LS, Lu KS, Bazarov A, Sedivy JM, Mischak H, Kolch W. Inhibition of the Raf-1 kinase by cyclic AMP agonists causes apoptosis of v-abl–transformed cells. Mol Cell Biol. 1997;17:3229–3241. doi: 10.1128/mcb.17.6.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skorski T, Nieborowska-Skorska M, Szczylik C, Kanakaraj P, Perrotti D, Zon G, Gewirtz A, Perussia B, Calabretta B. c-Raf-1 serine/threonine kinase is required in BCR/ABL-dependent and normal hematopoiesis. Cancer Res. 1995;55:2275–2278. [PubMed] [Google Scholar]
- 34.Wang H-G, Miyashita T, Takayama S, Sato T, Torigol T, Krajewski S, Tanaka S, Hovey L, III, Troppmair J, Rapp U, Reed J. Apoptosis regulation by interaction of bcl-2 protein and Raf-1 kinase. Oncogene. 1994;9:2751–2756. [PubMed] [Google Scholar]
- 35.Wang H-G, Rapp UR, Reed J. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87:629–638. doi: 10.1016/s0092-8674(00)81383-5. [DOI] [PubMed] [Google Scholar]
- 36.Olivier R, Otter I, Monney L, Wartmann M, Borner C. Bcl-2 does not require Raf kinase activity for its death-protective function. Biochem J. 1997;324:75–83. doi: 10.1042/bj3240075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hase T, Muller V, Riezman H, Schatz G. The 70-kd protein of the yeast mitochondrial outer membrane is targeted and anchored via its extreme amino terminus. EMBO (Eur Mol Biol Organ) J. 1984;3:3157–3164. doi: 10.1002/j.1460-2075.1984.tb02274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valtieri M, Tweardy DJ, Caracciolo D, Johnson K, Mavilio F, Altmann S, Santoli D, Rovera G. Cytokine-dependent granulocytic differentiation: regulation of proliferative and differentiative responses in a murine progenitor cell line. J Immunol. 1987;138:3829–3835. [PubMed] [Google Scholar]
- 39.Skorski T, Kanakaraj P, Ku DH, Nieborowska-Skorska M, Canaani E, Zon G, Perussia B, Calabretta B. Negative regulation of p120GAP GTPase promoting activity by p210bcr/abl: implication for RAS-dependent Philadelphia chromosome positive cell growth. J Exp Med. 1994;179:1855–1865. doi: 10.1084/jem.179.6.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fabian JR, Daar IO, Morrison DK. Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol Cell Biol. 1993;13:7170–7179. doi: 10.1128/mcb.13.11.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40a.Fabian JR, Morrison DK, Daar IO. Requirement for Raf and MAP kinase function during the meiotic maturation of Xenopusoocytes. J Cell Biol. 1993;122:645–652. doi: 10.1083/jcb.122.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salomoni P, Perrotti D, Martinez R, Franceschi C, Calabretta B. Resistance to apoptosis in CTLL-2 cells constitutively expressing C-Myb is associated with induction of BCL-2 expression and Myb-dependent regulation of bcl-2 promoter activity. Proc Natl Acad Sci USA. 1997;96:3296–3301. doi: 10.1073/pnas.94.7.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandanas RA, Leibowitz DS, Gharehbaghi K, Tauchi T, Burgess GS, Miyazawa K, Jayaram HN, Boswell HS. Role of p21 RAS in p210bcr/abltransformation of murine myeloid cells. Blood. 1993;82:1838–1847. [PubMed] [Google Scholar]
- 43.Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO (Eur Mol Biol Organ) J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blagosklomy M, Schulte T, Nguyen P, Trepel J, Neckers L. Taxol-induced apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a novel c-Raf-1 signal transduction pathway. Cancer Res. 1996;56:1851–1854. [PubMed] [Google Scholar]
- 45.Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol. 1997;9:174–179. doi: 10.1016/s0955-0674(97)80060-9. [DOI] [PubMed] [Google Scholar]
- 46.Skorski T, Nieborowska-Skorska M, Wlodarski P, Zon G, Iozzo R, Calabretta B. Antisense oligodeoxynucleotide combination therapy of primary chronic myelogenous leukemia blast crisis in SCID mice. Blood. 1996;88:1005–1012. [PubMed] [Google Scholar]
- 47.Kantarjan HM, Deisseroth A, Kurzrok R, Esrov Z, Talpaz M. Chronic myelogenous leukemia: a concise update. Blood. 1993;82:691–703. [PubMed] [Google Scholar]
- 48.Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, Trotta R, Wlodarski P, Perrotti D, Chan TO, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/ Akt-dependent pathway. EMBO (Eur Mol Biol Organ) J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg M. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 50.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3–induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 51.Ahmed NN, Grimes HL, Bellacosa A, Chan TO, Tsichlis PN. Transduction of IL-2 anti-apoptotic and proliferative signals via Akt. Proc Natl Acad Sci USA. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]