

Abstract
Bacterial lipopolysaccharide (LPS) induces activation of the transcription factor nuclear factor κB (NF-κB) in host cells upon infection. LPS binds to the glycosylphosphatidylinositol (GPI)- anchored membrane protein CD14, which lacks an intracellular signaling domain. Here we investigated the role of mammalian Toll-like receptors (TLRs) as signal transducers for LPS. Overexpression of TLR2, but not TLR1, TLR4, or CD14 conferred LPS inducibility of NF-κB activation in mammalian 293 cells. Mutational analysis demonstrated that this LPS response requires the intracellular domain of TLR2. LPS signaling through TLR2 was dependent on serum which contains soluble CD14 (sCD14). Coexpression of CD14 synergistically enhanced LPS signal transmission through TLR2. In addition, purified recombinant sCD14 could substitute for serum to support LPS-induced TLR2 activation. LPS stimulation of TLR2 initiated an interleukin 1 receptor–like NF-κB signaling cascade. These findings suggest that TLR2 may be a signaling component of a cellular receptor for LPS.
Keywords: lipopolysaccharide, CD14, interleukin 1 receptor, nuclear factor κB, signal transduction
Bacterial LPS (also known as endotoxin), a constituent of the outer membrane of the cell wall of Gram-negative bacteria, is one of the main causative agents of septic shock in humans (1, 2). Recognition of LPS is a key event in host antimicrobial defense reactions (1, 2). LPS is a complex glycolipid composed of a hydrophilic polysaccharide portion and a hydrophobic domain known as lipid A (1). The conserved lipid A structure has been identified as the LPS component responsible for LPS-induced biological effects (1). LPS elicits its responses through stimulation of host cells such as monocytes and macrophages to produce and release endogenous mediators including the proinflammatory cytokines IL-1, IL-6, and TNF (1).
LPS-induced signal transmission is thought to entail its binding to specific cellular receptors, which trigger intracellular signaling cascades leading to activation of the transcription factor nuclear factor κB (NF-κB)1 and p38 mitogen-activated protein kinases (MAP kinases), among others (2). The 55-kd glycoprotein CD14 binds LPS with high affinity and is involved in mediating LPS responses (1, 2). Binding of LPS to CD14 requires the serum factor, LPS-binding protein (LBP), which delivers LPS to CD14- expressing monocytes/macrophages (1, 2). In CD14− cells such as endothelial cells, granulocytes, and lymphocytes, soluble CD14 (sCD14) found in serum is thought to functionally replace membrane-bound CD14 (1, 2). Since CD14 is a glycosylphosphatidylinositol (GPI)-anchored membrane protein devoid of a cytoplasmic domain, it presumably does not elicit intracellular signaling events directly (1, 2). To date, the identification of a bona fide signal-transducing component of the putative LPS receptor complex has remained elusive.
The toll gene controls dorsoventral pattern formation during the early embryonic development of Drosophila melanogaster (3). Interestingly, toll participates in antimicrobial immune responses upon infection in adult Drosophila (4). The Toll protein is a transmembrane receptor that displays resemblance in its intracellular portion to the signaling domains of members of the IL-1R family which mediate activation of NF-κB (3). Indeed, Toll initiates a signaling cascade homologous to mammalian NF-κB activation pathways (3).
Recently, several members of a mammalian Toll-like receptor (TLR) family have been identified (5–8). In addition to homology within their cytoplasmic domains, both mammalian and Drosophila Toll receptors share a characteristic repeating leucine-rich motif in their extracellular regions. Similar to IL-1Rs and Drosophila Toll, several TLRs have been shown to signal through the NF-κB pathway (6, 8). In particular, functional analysis of TLR4 (also designated hToll) demonstrated that it is capable of inducing the expression of NF-κB–controlled genes for the inflammatory cytokines IL-1, IL-6, and IL-8, as well as expression of the costimulatory molecule B7.1, which is required for the activation of naive T cells (6). These findings provided evidence that TLR4 functions in vertebrates as a nonclonal receptor of the innate immune system that signals activation of adaptive immunity (6). To further investigate a conserved function for TLRs in innate immune responses, we examined if mammalian members of this receptor family might be involved in LPS signal transduction.
Materials and Methods
Cell Culture and Biological Reagents.
A subclone of the human embryonic 293 cell line has been maintained at Tularik, Inc., since 1993 as described (9). Mono-Mac-6 cells were licensed from Dr. H.W.L. Ziegler-Heitbrock (University of Munich, Munich, Germany). All other cell lines were obtained directly from the American Type Culture Collection (Rockville, MD). Tissue culture experiments were performed in the presence of 10% certified fetal bovine serum (GIBCO BRL, Gaithersburg, MD) unless otherwise stated. The serum was not heat inactivated and contained endotoxin at <10 EU/ml as specified by the manufacturer. For stimulation of tissue culture cells, LPS from Escherichia coli serotype 0111:B4 prepared by phenol extraction was purchased from Sigma Chemical Co. (St. Louis, MO). Further purification of LPS by ion exchange chromatography or gel filtration (Sigma Chemical Co.) had no effect on the biological activity. Similar results were obtained with LPS preparations from various other E. coli strains as well as from Salmonella minnesota, Salmonella typhimurium, and Shigella flexneri obtained from Sigma Chemical Co., Difco Laboratories, Inc. (Detroit, MI), or List Biological Laboratories, Inc. (Campbell, CA). Lipid A from E. coli K12 D31m4 LPS was from List Biological Laboratories, Inc. Recombinant human TNF-α and IL-1β were provided by Genentech Inc. (South San Francisco, CA). Anti-Flag epitope mAb M2 was from Eastman Kodak Co. (New Haven, CT) and anti-CD14 mAb MEM18 from Caltag Laboratories (Burlingame, CA). Rabbit anti-Flag polyclonal antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit polyclonal antibodies against human CD14 were raised against a 33-mer peptide, VEVEIHAGGLNLEPFLKRVDADADPRQYADTVK, by Berkeley Antibody Company (Richmond, CA).
Expression Plasmids.
The coding region of human CD14 was amplified by reverse transcription (RT)-PCR from mRNA isolated from THP-1 cells and inserted into the mammalian expression plasmid pRK (10), downstream of the CMV immediate-early promoter and enhancer. The coding regions of TLR1, TLR2, and TLR4 minus the respective NH2-terminal signal sequences were amplified by PCR from a human leukocyte cDNA library (Clontech, Palo Alto, CA) and cloned into the expression vector pFlag-CMV-1 (Eastman Kodak Co.), in which the preprotrypsin leader precedes an NH2-terminal Flag epitope. Epitope tagging had no influence on the activity of TLRs in tissue culture. Expression plasmids for TLR2 truncation mutants were generated by subcloning PCR-amplified TLR2 cDNA fragments.
NF-κB Reporter Assay.
Human embryonic kidney 293 cells (3 × 105) were plated in 3.5-cm dishes and transfected on the following day by the calcium phosphate precipitation method (11, 12) with the indicated amounts of expression vectors and 0.1 μg ELAM-1 luciferase reporter plasmid (13). The cells were lysed for measurement of luciferase activity using reagents from Promega Corp. (Madison, WI). An RSV-β-galactosidase control plasmid (1 μg) was used for normalizing transfection efficiencies. All bar diagrams are shown as mean ± SEM for one representative experiment in which each transfection was performed in duplicate. Each experiment was repeated at least twice in all cases.
Electrophoretic Mobility Shift Assay.
293 cell clones stably expressing Flag epitope–tagged TLRs or CD14 were generated by selection with G418 (GIBCO BRL) and subsequent FACS® analysis as described (14). Nuclear extracts from 2 × 106 cells were prepared (15) and analyzed for NF-κB DNA-binding activity with a radiolabeled double-stranded oligonucleotide containing two NF-κB binding sites (15).
Immunoprecipitation and Immunoblotting.
For the detection of transiently overexpressed CD14 and TLRs, 4 μg of expression plasmids was transfected into 293 cells (3 × 106) seeded in 100-mm dishes. Total cell lysates were prepared and incubated with anti-CD14 mAb MEM18 or anti-Flag mAb M2 and protein G–agarose beads (Oncogene Science, Inc., Manhasset, NY) as described (12). The immune complexes were washed, fractionated by SDS-PAGE, and transferred to nitrocellulose membrane. Immunoblot analysis was performed with rabbit polyclonal antibodies against CD14 or the Flag epitope using enhanced chemiluminescence detection (Amersham Pharmacia Biotech, Inc., Piscataway, NJ).
RT-PCR Analysis.
Poly(A)+ RNA was isolated from tissue culture cells using the Oligotex™ Direct mRNA kit (QIAGEN, Inc., Chatsworth, CA) and treated with RNase-free DNase I (Roche Molecular Biochemicals, Indianapolis, IN). RT was performed using the SuperScript™ Preamplification System (GIBCO BRL) followed by PCR amplification with Taq polymerase (QIAGEN, Inc.) for 24 cycles at 95°C for 40 s, 54°C for 40 s, and 72°C for 1 min. PCR primers for TLR2 were 5′-GCCAAAGTCTTGATTGATTGG and 5′-TTGAAGTTCTCCAGCTCCTG. GAPDH primers were obtained from Clontech.
Purification of Recombinant sCD14.
The coding region of human CD14 lacking the COOH-terminal four amino acids was fused in frame to a COOH-terminal Flag epitope in pRK (10). For transient expression of sCD14, 293 cells were cultured in serum-free CHO-S-SFM II medium (GIBCO BRL) supplemented with 10 ng/ml EGF (GIBCO BRL). sCD14 was purified to homogeneity from tissue culture supernatants by chromatography with anti-Flag M2 affinity resin (Eastman Kodak Co.) and Flag peptide elution as described (12).
Results
Effect of TLRs on LPS-induced NF-κB Activation.
We initially established a tissue culture system suitable for analyzing LPS signaling components. Human embryonic kidney 293 cells were transiently transfected with an NF-κB– dependent E-selectin (ELAM-1) promoter luciferase reporter gene (13). No significant induction of reporter gene activity was observed upon stimulation of transfected cells with LPS (Fig. 1 A). Similarly, transient as well as stable transfection of an expression vector for CD14 did not mediate LPS induction of the reporter gene (Fig. 1 A, and data not shown). This indicates that the examined 293 cells are either defective in or express functionally insufficient levels of one or more components of the LPS signal recognition and/or signal transduction pathway other than CD14.
Figure 1.


Effect of CD14 and TLRs on LPS-induced NF-κB activation. (A) TLR2-mediated activation of NF-κB by LPS. Luciferase activities were determined in 293 cells transfected with the indicated expression plasmids (1 μg) together with ELAM-1 luciferase reporter plasmid for 24 h, and either left untreated (white bars) or stimulated with LPS (10 μg/ml) for 6 h (black bars) before harvest. (B) Expression of CD14 and TLRs. 293 cells were transiently transfected with aliquots from the same transfection mixtures as in A containing 4 μg of expression plasmid as described in Materials and Methods. Flag epitope–tagged TLRs and CD14 were immunoprecipitated and detected by immunoblotting with anti-Flag antibodies ( lanes 1, 3–5; upper panel ) and anti-CD14 antibodies (lanes 1 and 2; lower panel), respectively. (C) LPS induction of NF-κB DNA-binding activity by TLR2. EMSA was performed with nuclear extracts from 293 cells and cells stably expressing TLR2 (293-TLR2) that were either left untreated (lanes 1 and 4) or stimulated with LPS (10 μg/ml; lanes 2 and 4) or IL-1 (20 ng/ml; lanes 3 and 6) for 20 min before harvest.
We next tested the effect of transiently transfected TLR expression vectors on NF-κB–dependent reporter gene activity. Overexpression of TLR4 constitutively activated the reporter gene (Fig. 1 A). Similar results had previously been observed for a mutant TLR4 protein, in which the native extracellular domain was replaced by the extracellular domain of CD4 (6). LPS stimulation did not elicit additional reporter gene activation by TLR4 (Fig. 1 A). In contrast to TLR4, overexpression of TLR1 and TLR2 caused only marginal activation of the reporter gene in unstimulated cells despite comparable expression levels of all three receptors (Fig. 1, A and B). Although TLR1-expressing cells did not respond to LPS stimulation, cells expressing TLR2 strongly activated the NF-κB–dependent reporter gene upon stimulation with LPS (Fig. 1 A). This stimulation occurred in a dose-dependent manner with respect to LPS concentration (see Fig. 4) and the amount of transiently transfected TLR2 expression plasmid (data not shown). LPS-induced reporter gene activity through TLR2 consistently exceeded by three- to fourfold that produced by TLR4 overexpression (Fig. 1 A).
Figure 4.

Serum dependence of TLR2-mediated NF-κB activation by LPS. 293 cells were transfected with TLR2 expression plasmid (1 μg) and ELAM-1 luciferase reporter plasmid. After 24 h, cells were stimulated with the indicated concentrations of LPS for 6 h either in the presence (white bars) or absence of 10% fetal bovine serum (black bars) before harvest. Luciferase activities were determined relative to empty vector control.
NF-κB–dependent reporter gene activation by LPS was also observed in a similar manner in 293 cell clones stably expressing TLR2 (293-TLR2) but not TLR1 or TLR4 (data not shown). Furthermore, electrophoretic mobility shift assays (EMSAs) demonstrated that nuclear extracts from LPS-treated 293-TLR2 cells contained a significant amount of NF-κB DNA-binding activity (Fig. 1 C). Thus, overexpression of TLR2 is sufficient to confer LPS inducibility of NF-κB activation in mammalian 293 cells.
TLR2-mediated NF-κB Activation by Lipid A.
Since the lipid A portion of LPS is sufficient to induce LPS responses, we tested the effect of lipid A on TLR2-mediated NF-κB activation in transient reporter gene assays. Although 293 cells transfected with empty control vector could not be stimulated with lipid A, TLR2-expressing cells responded to lipid A in a dose-dependent manner similar to stimulation with LPS (Fig. 2). Thus, TLR2 overexpression mediates responsiveness to the biologically active moiety of LPS, lipid A.
Figure 2.

TLR2-mediated activation of NF-κB by lipid A. 293 cells were transfected with 200 ng empty control vector (white bars) or TLR2 expression plasmid (black bars) and ELAM-1 luciferase reporter plasmid. After 24 h, cells were stimulated with the indicated concentrations of LPS or lipid A for 6 h before determination of luciferase activities.
Mutational Analysis of TLR2.
We next investigated the contribution of the intracellular domain of TLR2 to LPS-induced NF-κB activation by mutational analysis. In contrast to wild-type TLR2, deletion of the majority of the cytoplasmic portion of TLR2 created a mutant TLR2 protein that was defective in LPS-stimulated NF-κB activation (Fig. 3). This indicates that TLR2-mediated NF-κB activation in response to LPS occurs via an intracellular signaling cascade initiated at the cytoplasmic domain of TLR2. Subsequently, mutant versions of TLR2 were constructed by successive COOH-terminal truncation of the receptor cytoplasmic domain. Analysis in transient transfection assays revealed that deletion of the COOH-terminal 15 amino acids of the cytoplasmic domain of TLR2 was sufficient to abrogate LPS-induced NF-κB reporter gene activation (Fig. 3). This mutation precisely eliminates the most COOH-terminal block of amino acid homology within the cytoplasmic domains of members of the Toll and IL-1R families (7).
Figure 3.

Analysis of TLR2 deletion mutants. The horizontal bars represent the sequence of TLR2 with transmembrane domain (TM) and intracellular domain (IC) indicated. The amino acids contained in each deletion mutant are stated. 293 cells were transiently cotransfected with ELAM-1 luciferase reporter plasmid and TLR2 expression vectors (1 μg) as indicated. After 24 h, cells were either stimulated with LPS (1 μg/ml) for 6 h or left untreated before harvest. Values shown represent luciferase activities determined relative to empty vector control.
Serum Dependence of LPS Signaling through TLR2.
To investigate the serum dependence of LPS signaling through TLR2, 293 cells transiently expressing TLR2 were stimulated with LPS over a wide concentration range either in the presence or absence of serum. When serum was present reporter gene activation was detected at low LPS doses (Fig. 4). In contrast, in the absence of serum low concentrations of LPS were ineffective in stimulating reporter gene activity (Fig. 4). Only at much higher LPS concentrations did reporter gene activation become apparent under serum-free conditions (Fig. 4). Overall, the dose response to LPS was shifted by approximately three orders of magnitude depending on the serum conditions (Fig. 4). Thus, efficient LPS signal transmission by TLR2 requires serum components which presumably include sCD14 and/or LBP. At very high concentrations of LPS, reporter gene induction was independent of serum conditions (Fig. 4). This is in agreement with observations that responsiveness to LPS at high concentrations bypasses the requirement for LBP and CD14 (16).
Role of CD14 in LPS Signaling by TLR2.
The effect of CD14 on LPS signaling through TLR2 was further examined in transient transfection assays in 293 cells. When CD14 was coexpressed with TLR2, reporter gene activation at a low LPS dose was increased by approximately five- to sevenfold compared with its induction in cells expressing TLR2 alone (Fig. 5 A). This stimulatory activity of CD14 decreased at higher LPS concentrations and was undetectable at a very high LPS dose (Fig. 5 A). Thus, at low levels of LPS CD14 exerts a synergistic effect on LPS- induced signal transduction through TLR2.
Figure 5.
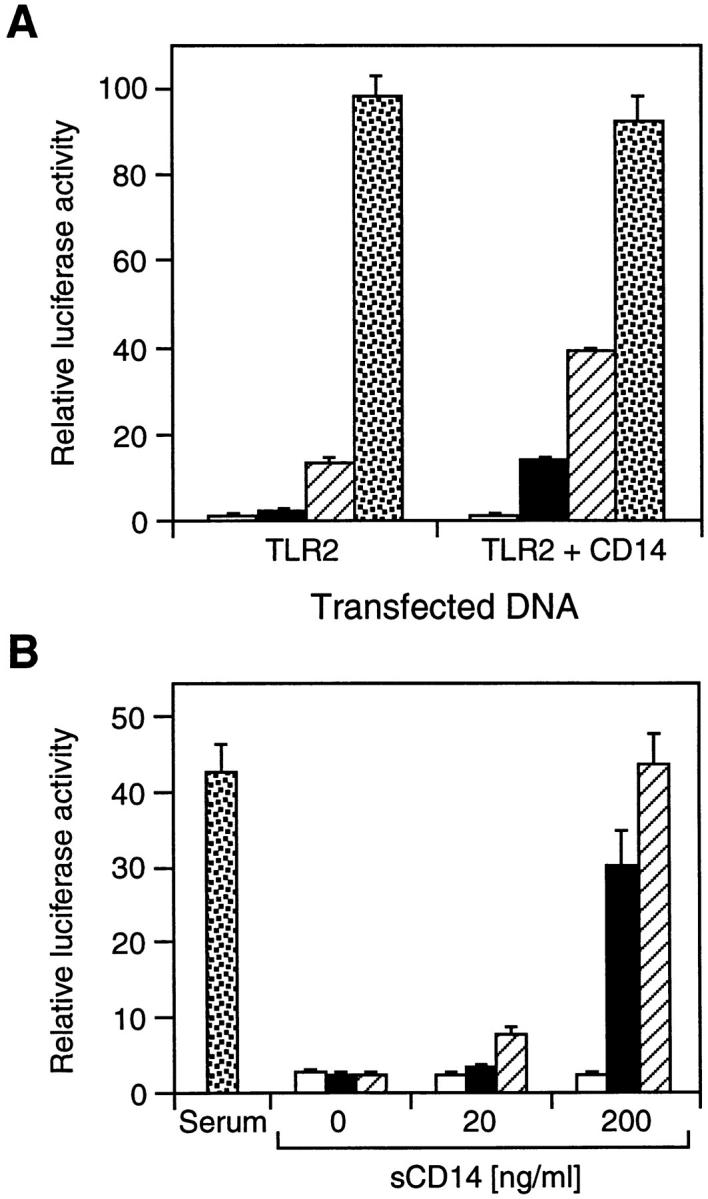
Effect of CD14 on TLR2-mediated NF-κB activation by LPS. (A) Synergistic activation of NF-κB by coexpression of CD14 and TLR2. 293 cells were transfected with ELAM-1 luciferase reporter plasmid (reference 13) and expression vectors for TLR2 (50 ng) and CD14 (10 ng) as indicated. After 24 h, cells were either left untreated (white bars) or stimulated for 6 h with LPS at 1 ng/ml (black bars), 10 ng/ml (hatched bars), and 10 μg/ml (stippled bars) before harvest. Luciferase activities were determined relative to empty vector control. (B) Effect of purified recombinant sCD14 on LPS-induced NF-κB activation by TLR2. 293 cells were transfected with 200 ng TLR2 expression plasmid and ELAM-1 luciferase reporter plasmid. After 24 h, cells were left untreated (white bars) or stimulated for 6 h with LPS at 10 ng/ml (black bars), 100 ng/ml (hatched bars), or 1 μg/ml (stippled bars) in medium containing either serum or purified recombinant sCD14 as indicated. Luciferase activities were determined relative to empty vector control.
We further addressed the contribution of CD14 to TLR2-mediated LPS signaling by using purified recombinant sCD14. In transient reporter gene assays, sCD14 was able to substitute dose-dependently for serum in supporting LPS signaling by TLR2 (Fig. 5 B). This indicates that CD14 is required for activation of TLR2 at low concentrations of LPS.
Involvement of TNF and IL-1 Signal Transducers in LPS Signaling by TLR2.
To examine the signaling pathway initiated by TLR2 in response to LPS, we used dominant-negative versions of components of the NF-κB signaling cascades for IL-1 and TNF in transient cotransfection assays in 293 cells. Catalytically inactive mutants of the two IκB kinases, IKK-α and IKK-β (12, 17–20), as well as the NF-κB–inducing kinase, NIK (21), inhibited TLR2-mediated reporter gene activation by LPS similar to IL-1– and TNF-induced activation (Fig. 6), suggesting that these kinases represent common components in the respective NF-κB pathways. TLR2-induced NF-κB activation was not inhibited by a dominant-negative mutant of TNF receptor– associated factor (TRAF)2, which suppresses NF-κB activation by TNF but not IL-1 in mammalian cells (22; Fig. 6). Conversely, a dominant-negative version of TRAF6, which inhibits NF-κB activation by IL-1 but not TNF (22), also suppressed NF-κB activation by TLR2 (Fig. 6). In addition, a truncated version of MyD88, a constituent of the IL-1R complex (23, 24), inhibited both TLR2- and IL-1R–mediated NF-κB activation but not induction by TNF (Fig. 6). Thus, TLR2 stimulation by LPS triggers an IL-1R–like signaling cascade leading to activation of NF-κB.
Figure 6.

Involvement of IL-1 and TNF signaling molecules in TLR2-mediated NF-κB activation by LPS. 293–IL-1RI cells (reference 14) were transfected with ELAM-1 luciferase reporter plasmid and expression vectors (0.3 μg) for TLR2 and dominant-negative mutants of components of the IL-1– and TNF-induced NF-κB pathways as indicated. After 24 h, cells were stimulated for 6 h with LPS (10 μg/ml; black bars), IL-1β (20 ng/ml; hatched bars), or TNF-α (100 ng/ml; white bars) before harvest. Luciferase values were determined and are expressed as percent activation for each stimulus relative to cells transfected with TLR2 alone. LPS, IL-1, and TNF induced 117-, 436-, and 28-fold activation of the reporter gene, respectively, compared with untreated cells.
Distribution of TLR2 Transcripts.
Expression of TLR2 has been detected predominantly in peripheral blood leukocytes, spleen, and lung (7, 8). We used RT-PCR to further analyze TLR2 expression in LPS-responsive cell lines of monocytic origin. Human THP-1, U937, and Mono-Mac-6 cells were found to express significant levels of TLR2 mRNA (Fig. 7). In contrast, TLR2 message could not readily be detected in our LPS-resistant 293 cells (Fig. 7). These results substantiate the requirement for TLR2 in LPS signaling and are consistent with the profound effects of LPS on cells of myeloid lineage.
Figure 7.

TLR2 expression in tissue culture cells. Expression of TLR2 mRNA in THP-1 (lane 1), U937 (lane 2), Mono-Mac-6 (lane 3), and 293 cells was analyzed by PCR after RT (top) or without RT (middle). RT-PCR analysis of GAPDH expression was used as control (bottom).
Discussion
The recent identification of several members of a mammalian TLR family raised the issue of their functional role in the vertebrate immune system. In Drosophila, Toll is involved in the rapid and transient transcriptional induction of several genes encoding potent antimicrobial peptides upon septic injury (4). Conserved throughout evolution as innate immunity, this type of activation of nonspecific defense mechanisms by infectious microorganisms uses germline-encoded receptors for the recognition of microbial pathogens (25). A prominent example of such “pattern recognition receptors” is CD14, which is activated during the initial phase of bacterial infection in vertebrates (1, 2). CD14 binds LPS, which possesses a general structure shared by all Gram-negative bacteria. However, CD14 presumably does not trigger intracellular signaling events directly, since it lacks a receptor cytoplasmic domain. The observation that human TLR4 (hToll) may function in the innate immune system in vertebrates similar to Toll in Drosophila (6) prompted us to investigate if members of the mammalian TLR family might participate in the recognition of bacterial LPS. Our results demonstrate specifically that overexpression of TLR2 confers responsiveness to LPS stimulation in mammalian 293 cells. The identification of TLR2 as a signal transducer for LPS suggests that TLR2 may be a component of a cellular LPS receptor.
The proinflammatory cytokines IL-1 and TNF as well as bacterial LPS are potent external stimuli regulating the activity of NF-κB transcription factors, which control the expression of numerous immune and inflammatory response genes (26, 27). Members of the Toll receptor family share homology in their intracellular portions with the signaling domains of the IL-1R family, and Drosophila Toll and several mammalian TLRs have been shown to activate NF-κB (3, 6–8). Our mutational analysis of TLR2 illustrates the importance of the conserved motif of Toll- and IL-1–like receptors in signaling NF-κB activation. Elucidation of the NF-κB signaling cascades triggered by TNF and IL-1 has identified a number of distinct and shared components in both pathways (28). Our findings indicate that LPS stimulation of TLR2 initiates an IL-1– rather than a TNF-like NF-κB activation pathway. This is in agreement with Toll signaling in Drosophila (3). In addition, overexpression of TLR4 (hToll) in 293 cells, which constitutively activates NF-κB, was recently found to involve IL-1 signaling components (29, 30).
In contrast to CD14, binding studies indicate that TLR2 does not bind LPS with high affinity (our unpublished observation). Concurrently, we demonstrate that TLR2 signaling requires CD14 to increase LPS sensitivity. Conceptually, LPS signal transmission through TLR2 may occur in a manner analogous to the receptor signaling system used by glial cell line–derived neurotrophic factor (GDNF), a member of the TGF-β ligand family. GDNF binds to the GPI-anchored GDNF receptor α and signals via the transmembrane receptor tyrosine kinase, RET, which itself does not appreciably bind GDNF (31). Alternatively, the putative cellular LPS receptor may comprise additional components, or LPS receptors of distinct composition may exist on different cell types. Recently, a biophysical model of LPS action has also been proposed which is dependent on LPS interactions with membrane lipids and endocytic movement (32). It remains to be investigated if and how TLR2 might participate in such a signaling mechanism. Regardless of these considerations, the implication of TLR2 in LPS signaling supports an important role for the evolutionary conserved Toll receptor family in innate immune responses and provides further insights into the molecular mechanisms underlying Gram-negative sepsis.
Acknowledgments
We thank Keith Williamson and Joanne Waszuck for DNA sequencing and Ping Jiang for providing plasmids. We also thank Zhaodan Cao and Dave Goeddel for helpful suggestions and continuing support during this project.
Abbreviations used in this paper
- EMSA
electrophoretic mobility shift assay
- GNDF
glial cell line–derived neurotrophic factor
- GPI
glycosylphosphatidylinositol
- IKK
IκB kinase
- LBP
LPS binding protein
- MAP kinase
mitogen-activated protein kinase
- NF-κB
nuclear factor κB
- NIK
NF-κB–inducing kinase
- RT
reverse transcription
- sCD14
soluble CD14
- TLR
Toll-like receptor
- TRAF
TNF receptor–associated factor
References
- 1.Schletter J, Heine H, Ulmer AJ, Rietschel ET. Molecular mechanisms of endotoxin activity. Arch Microbiol. 1995;164:383–389. doi: 10.1007/BF02529735. [DOI] [PubMed] [Google Scholar]
- 2.Ulevitch RJ, Tobias PS. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 3.Belvin MP, Anderson KV. A conserved signaling pathway: the Drosophilatoll-dorsal pathway. Annu Rev Cell Dev Biol. 1996;12:393–416. doi: 10.1146/annurev.cellbio.12.1.393. [DOI] [PubMed] [Google Scholar]
- 4.Hoffmann JA, Reichhart J-M. Drosophilaimmunity. Trends Cell Biol. 1997;7:309–316. doi: 10.1016/S0962-8924(97)01087-8. [DOI] [PubMed] [Google Scholar]
- 5.Mitcham JL, Partnet P, Bonnert TP, Garka KE, Gerhart MJ, Slack JL, Gayle MA, Dower SK, Sims JE. T1/ST2 signaling establishes it as a member of an expanding interleukin-1 receptor family. J Biol Chem. 1996;271:5777–5783. doi: 10.1074/jbc.271.10.5777. [DOI] [PubMed] [Google Scholar]
- 6.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the DrosophilaToll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 7.Rock FL, Hardiman G, Timans JC, Kastelein R, Bazan JF. A family of human receptors structurally related to DrosophilaToll. Proc Natl Acad Sci USA. 1998;95:588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaudhary PM, Ferguson C, Nguyen V, Nguyen O, Massa HF, Eby M, Jasmin A, Trask BJ, Hood L, Nelson PS. Cloning and characterization of two Toll/Interleukin-1 receptor-like genes TIL3 and TIL4: evidence for a multi-gene receptor family in humans. Blood. 1998;91:4020–4027. [PubMed] [Google Scholar]
- 9.Hsu H, Xiong J, Goeddel DV. The TNF receptor-1 associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 10.Schall TJ, Lewis M, Koller KJ, Lee A, Rice GC, Wong GHW, Gatanaga T, Granger GA, Lentz R, Raab H, et al. Molecular cloning and expression of a receptor for human tumor necrosis factor. Cell. 1990;61:361–370. doi: 10.1016/0092-8674(90)90816-w. [DOI] [PubMed] [Google Scholar]
- 11.Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1987. Current Protocols in Molecular Biology. John Wiley & Sons, Inc., New York.
- 12.Régnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IκB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- 13.Schindler U, Baichwal VR. Three NF-κB binding sites in the human E-selectin gene required for maximal tumor necrosis factor alpha-induced expression. Mol Cell Biol. 1994;14:5820–5831. doi: 10.1128/mcb.14.9.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996;271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 15.Schütze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Krönke M. TNF activates NF-κB by phosphatidylcholine-specific phospholipase c-induced “acidic” sphingomyelin breakdown. Cell. 1992;71:765–776. doi: 10.1016/0092-8674(92)90553-o. [DOI] [PubMed] [Google Scholar]
- 16.Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukocyte Biol. 1996;60:8–26. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 17.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 18.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 19.Woronicz JD, Gao X, Cao Z, Rothe M, Goeddel DV. IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science. 1997;278:866–869. doi: 10.1126/science.278.5339.866. [DOI] [PubMed] [Google Scholar]
- 20.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 21.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 22.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 23.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- 24.Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science. 1997;278:1612–1615. doi: 10.1126/science.278.5343.1612. [DOI] [PubMed] [Google Scholar]
- 25.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 26.Lenardo M, Baltimore D. NF-κB: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 27.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 28.May MJ, Gosh S. Signal transduction through NF-κB. Immunol Today. 1998;19:80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 29.Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. The human Toll signaling pathway: divergence of nuclear factor κB and JNK/SAPK activation upstream of tumor necrosis factor receptor–associated factor 6 (TRAF6) J Exp Med. 1998;187:2097–2101. doi: 10.1084/jem.187.12.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Gosh S, Janeway CA., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 31.Robertson K, Mason I. The GDNF-RET signalling partnership. Trends Genet. 1997;13:1–3. doi: 10.1016/s0168-9525(96)30113-3. [DOI] [PubMed] [Google Scholar]
- 32.Thieblemont N, Thieringer R, Wright SD. Innate immune recognition of bacterial lipopolysaccharide: dependence on interactions with membrane lipids and endocytic movement. Immunity. 1998;8:771–777. doi: 10.1016/s1074-7613(00)80582-8. [DOI] [PubMed] [Google Scholar]