

Abstract
Chemotherapeutic drugs cause DNA damage and kill cancer cells mainly by apoptosis. p53 mediates apoptosis after DNA damage. To explore the pathway of p53-dependent cell death, we investigated if p53-dependent apoptosis after DNA damage is mediated by the CD95 (APO-1/Fas) receptor/ligand system. We investigated hepatoma, gastric cancer, colon cancer, and breast cancer cell lines upon treatment with different anticancer agents known to act via p53 accumulation. Cisplatin, mitomycin, methotrexate, mitoxantrone, doxorubicin, and bleomycin at concentrations present in the sera of patients during therapy led to an upregulation of both CD95 receptor and CD95 ligand. Induction of the CD95 ligand occurred in p53 wild-type (wt), p53 mutant (mt), and p53 deficient (p53−/−) cell lines and at wt and mt conformation of temperature-sensitive p53 mutants. In contrast, upregulation of the CD95 receptor was observed only in cells with wt p53, not in cells with mt or without any p53. Restitution of inducible wt p53 function restored the ability of p53−/− Hep3B cells to upregulate the CD95 receptor in response to anticancer drugs. This rendered the cells sensitive to CD95-mediated apoptosis. In an attempt to understand how CD95 expression is regulated by p53, we identified a p53-responsive element within the first intron of the CD95 gene, as well as three putative elements within the promoter. The intronic element conferred transcriptional activation by p53 and cooperated with p53-responsive elements in the promoter of the CD95 gene. wt p53 bound to and transactivated the CD95 gene, whereas mt p53 failed to induce apoptosis via activation of the CD95 gene. These observations provide a mechanistic explanation for the ability of p53 to contribute to tumor progression and to resistance of cancer cells to chemotherapy.
Keywords: apoptosis, CD95 (APO-1/Fas), p53, cancer therapy, drug resistance
Resistance of tumor cells to chemotherapy remains an obstacle to the successful treatment of human cancer. Combination chemotherapy has had a significant impact on survival from malignancies such as Hodgkin's disease, testicular cancer, and childhood acute leukemias. However, the majority of cancers are either initially resistant to chemotherapy, e.g., colon, renal, and hepatocellular cancer, or are initially chemosensitive but acquire resistance to a broad spectrum of drugs during treatment, such as lymphoma or breast cancer. One explanation for multidrug resistance in some tumors is the increased activity of the p-glycoprotein, a product of the mdr1 gene and an energy-dependent multidrug transporter that acts to decrease the cellular efflux of many drugs. However, amplification of mdr1 accounts for only a small percentage of cancers with a multidrug resistance phenotype. Drugs found to be efficacious in the treatment of cancer include diverse chemical compounds such as antimetabolites (e.g., methotrexate and fluorouracil), DNA-damaging agents (e.g., cyclophosphamide, cisplatin, and doxorubicin), mitotic inhibitors (e.g., vincristine), nucleotide analogues (e.g., 6-mercaptopurine), inhibitors of topoisomerases involved in DNA repair (e.g., etoposide), inhibitors of DNA polymerase (e.g., bleomycin), or intercalating agents like mitoxantrone. For years it has been known that these diverse drugs can induce apoptosis (1, 2). However, the mechanism of apoptosis induced by the drugs was not known. Recent evidence (3–6) suggests that genes that regulate apoptotic cell death may play an important role in determining the sensitivity of tumor cells to chemotherapy.
Friesen et al. (3) and Müller et al. (6) have shown that death induced in tumor cells by anticancer treatment is an active program of the cell which involves the CD95 system, one of the key regulatory systems of apoptosis. Treatment of leukemic and hepatocellular carcinoma cell lines, respectively, in vitro with chemotherapeutic agents causes upregulation of the CD95 ligand (CD95L), activating both an autocrine suicide and a fratricide death system. CD95L is expressed in a membrane form or is released by the tumor cells exposed to the drug. Binding of CD95L to the CD95 receptor then initiates the apoptotic signal in chemosensitive cells. Furthermore, we have shown for hepatoma cells that in addition to the expression of CD95L, cells treated with anticancer drugs upregulate CD95 receptor expression (6).
The presence of functional wild-type (wt)1 p53 is closely coupled with efficient induction of CD95-mediated apoptosis in many (6–8) but not all (9, 10) cell types. Upregulation of the CD95 receptor—after anticancer therapy—in hepatocellular carcinomas was preceded and directed by upregulated p53 and, thus, only occurred in hepatoma cell lines that express wt p53 (6). p53 has multiple functions (11–13), including cell cycle control in response to DNA damage (14, 15), induction of apoptosis (16–19), and DNA repair (20–22). Anticancer therapy causes damage of the DNA in the treated cells. p53 modulates cellular responses to DNA damage in mammals, affecting cell cycle progression and/or programmed cell death. Although the cyclin-dependent kinase inhibitor CDKN1A, also known as p21, appears to be the major effector of p53-mediated G1 cell cycle arrest after DNA damage (23–26), the mechanism by which p53 signals influence the apoptotic machinery after DNA damage remains unclear. p53 also acts as a sequence-specific DNA binding protein which activates the transcription of target genes. The proapoptotic protein BAX appears to be transcriptionally induced by p53 after DNA damage in certain cell types (27). However, BAX appears to contribute only in part to p53-mediated cell death (28).
Based on our observation that the CD95 receptor was only upregulated in cancer cells carrying wt p53 after DNA damage and on the fact that forced overexpression of wt p53 can stimulate CD95 gene transcription (29), we investigated if the CD95 system is activated and/or regulated by p53 in a variety of human solid cancer cell lines. These cell lines are representative of the “major killers” among human tumors, in which p53 mutations are known to play an important role. Thus, colon, stomach, hepatoma, and breast cancer cell lines with different p53 status were evaluated. We applied anticancer drugs with different mechanisms of action but concentrated on drugs that use the p53 pathway. Our data demonstrate that drug-induced p53 upregulation is involved in CD95 gene induction and apoptosis. Induction of CD95 gene transcription by p53 is mediated through a strong p53-responsive element located within the first intron of the gene. This element cooperates with sequences in the CD95 promoter to achieve maximal transactivation by wt p53.
Materials and Methods
Cell Lines.
The following cell lines were used: (a) HepG2 cells derived from a human hepatoblastoma (30) only expressing small amounts of wt p53 (31; our unpublished sequence data); (b) Huh7 cells derived from a hepatocellular carcinoma (32) and shown to express p53 with increased half-life as a result of a point mutation at codon 220 (33); (c) Hep3B cells (30) deficient of p53 (31); (d) Hs746T gastric cancer cells expressing wt p53; (e) HT29 colon cancer cells with mt p53 (34, 35); ( f ) AGS colon cancer cells with wt p53; (g) MCF7 breast cancer cells expressing wt p53 (36, 37); and (h) H1299 human lung adenocarcinoma cells deficient of p53 (38). HepG2, Huh7, and Hs746T cells were maintained in DME (GIBCO BRL, Eggenstein, Germany) containing 10% FCS, 5 mM l-glutamine, and 100 μg/ml penicillin (GIBCO BRL). Hep3B cells were grown in MEM (Laboratoires Eurobio, Raunheim, Germany). HT29, MCF7, and H1299 cells were cultured in RPMI 1640 medium (GIBCO BRL) supplemented with 10% heat-inactivated FCS (GIBCO BRL), 10 mM Hepes, pH 7.3 (GIBCO BRL), 100 U/ml streptomycin (GIBCO BRL), and 2 mM l-glutamine (GIBCO BRL). AGS cells were propagated in Ham's F-12 medium (GIBCO BRL) supplemented with 10% heat-inactivated FCS, 10 mM Hepes, pH 7.3, 100 U/ml streptomycin, and 2 mM l-glutamine.
Transfections.
Before transfection, H1299 and Hep3B cells were seeded at 0.6 × 106 cells/6-cm dish. The medium was changed to DME supplemented with 10% FCS, and cells were transfected by the calcium phosphate method with the precipitate left on cells for 16 h. Next, cells were glycerol shocked for 1 min and plated in RPMI medium supplemented with 10% FCS. Cells were harvested 24 h later and assayed for luciferase activity as described previously (39).
For preparation of cell extracts for immunoselection assays, H1299 cells were plated at a density of 1.2 × 106/10-cm dish and transfected as above with 5 μg pCMVp53wt (40) DNA.
Generation of Stable Hep3B-derived Clones.
Stable Hep3B- derived clones were generated in which wt p53 activity can be deliberately induced. Hep3B cells are particularly appropriate for our studies because they lack p53 (30, 31, 41). Stable Hep3B clones were generated expressing either puromycin resistance alone (control cell line: BT-2E) or in conjunction with either the temperature-sensitive mutant p53val135 (42) or a p53 (modified)–estrogen receptor chimera in which p53 activity is induced posttranslationally by addition of the specific ligand 4-OH tamoxifen (cell line: BT-4P) (43). These cell lines have been described (41).
A third type of stably transfected Hep3B clones was established expressing the temperature-sensitive mutant p53ala143 (44). Transfection was performed by calcium phosphate precipitation with p53ala143. 2 d later G418 (Sigma Chemical Co., Munich, Germany) was added, and cells were maintained under G418 selection. After ∼14–21 d, G418-resistant colonies were recovered and expanded.
4-OH tamoxifen was purchased from Sigma Chemical Co. and stored at –20°C as a 1 mM stock solution in DMSO. A 100 μM working stock in DMSO was used for direct addition to cultured cells. Puromycin was purchased from Sigma Chemical Co. and stored at –20°C as a sterile solution of 1 mg/ml in water.
Treatment with Anticancer Agents.
The different cell lines were treated with bleomycin (Heinrich Mack Nachf., Illertissen, Germany) at a dose range of 60 ng/ml to 6 mg/ml, or with doxorubicin (Rhône-Poulenc Rorer GmbH, Köln, Germany) at a dose range of 0.4 ng/ml to 4 μg/ml for 3–72 h. Additionally, cisplatin at a dose range of 0.1 ng/ml to 0.1 mg/ml or methotrexate at a dose range of 0.1 ng/ml to 1 mg/ml was applied. The concentrations relevant for therapy are 1.5–3 μg/ml for bleomycin (45), 0.4– 1.6 μg/ml for cisplatin (46, 47), 7–12 μg/ml for methotrexate (48), and 0.001–0.02 μg/ml for doxorubicin (49) in patients' sera.
Treatment with IgG3 Anti–APO-1.
The CD95 receptor was stimulated with the mAb IgG3 anti–APO-1 at concentrations of 100 ng/ml as described (50–52).
Detection of Apoptosis.
Apoptosis was assessed by FACS® analysis carried out in a FACScan® flow cytometer (Becton Dickinson GmbH, Heidelberg, Germany) using CellQuest software.
Quantification of DNA fragmentation was performed by FACS® analysis of propidium iodide–stained nuclei as described previously (53). Hepatocytes floating in the culture medium were collected by centrifugation at 200 g. Adherent hepatocytes were harvested by incubation with 1% trypsin for 1 min. The cells were washed with PBS, suspended in hypotonic lysis buffer (0.1% sodium citrate; Merck, Darmstadt, Germany), 0.1% Triton X (Serva Feinbiochemica, Heidelberg, Germany), and 50 ng/ml propidium iodide (Sigma Chemical Co.) and incubated at 4°C for 6 h. Cells were then analyzed for DNA content by flow cytometry.
Early apoptotic changes were identified by using Annexin-V-Fluos (Boehringer Mannheim GmbH, Mannheim, Germany), which binds to phosphatidylserine exposed on the outer leaflet of apoptotic cell membranes. Propidium iodide was used for the discrimination of necrotic cells from the annexin V positively stained cell cluster. Cells were trypsinized, washed with PBS, centrifuged at 200 g for 5 min, and resuspended in 100 μl Annexin-V-Fluos labeling solution containing 20 μl Annexin-V-Fluos labeling reagent in 1,000 μl Hepes buffer (10 mM Hepes/ NaOH, pH 7.4, 140 mM NaCl, 5 mM CaCl2) and 20 μl propidium iodide. Cells were incubated for 10–15 min and analyzed on a flow cytometer using CellQuest software. A 488-nm excitation and a filter >560 nm for propidium iodide detection were used.
Detection of the CD95 (APO-1/Fas) Receptor.
Cell surface expression of the CD95 receptor was assessed by FACScan®. Anti– APO-1 (IgG3, κ) was used as purified biotinylated antibody. Quantum Red streptavidin (Sigma Chemical Co.) was used as secondary reagent for indirect immunofluorescence. Hepatoma cells were incubated in 50 μl culture medium with biotinylated anti–APO-1. After 30 min incubation, cells were washed twice, incubated for 30 min with Quantum Red streptavidin, washed twice again, and assayed. Upon data acquisition a gate was set on intact cells by forward/side scatter analysis, and 104 viable cells were analyzed. Percent enhanced CD95 expression was calculated according to the formula (% CD95+ treated cells − % Quantum Red+ treated cells) − (% CD95+ control cells − % Quantum Red+ control cells).
Detection of CD95L mRNA Expression by PCR.
Total cellular RNA was prepared from 3 × 106 cells treated with different anticancer drugs, using the RNeasy Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer's instructions. Expression of β-actin (MWG Biotech GmbH, Ebersberg, Germany) was used as an internal standard for RNA integrity and equal gel loading. 1 μg of total cellular RNA was retrotranscribed after heat denaturation (3 min, 60°C) and annealing with oligo (dT) primers (16-mer; Perkin Elmer, Weiterstadt, Germany) in the presence of 75 U MnLV RT (Perkin Elmer), 67 μM MgCl2, and 63 μM of each dNTP in 20 μl for 45 min at 42°C. Reactions were stopped by heat inactivation for 5 min at 90°C. Aliquots of 10 μl of the cDNA were then amplified in a DNA thermocycler (Stratagene Inc., Heidelberg, Germany) with 2.5 U of Ampli Taq DNA polymerase AS (Perkin Elmer), 10 pM of both upstream and downstream APO-1L primers, and 2.5 μM of both upstream and downstream human β-actin primers in a 50 μl vol. Each PCR cycle consisted of a denaturation step (94°C, 1 min), an annealing step (56°C, 1 min), and an elongation step (72°C, 1 min). The primers used for amplification of the CD95L have been described (54). The PCR products were analyzed on a 1.4% TBE (Tris-borate-EDTA) agarose gel.
Cytotoxicity Assay: MTT Assay.
The MTT assay is a colorimetric assay based on the ability of the viable cells to reduce a soluble yellow tetrazolium salt (MTT) to blue formazan crystals. A 100-μl suspension of 7 × 103 cells was added to each well of 96-well plates 24 h before the assay. Various concentrations of the different anticancer drugs, IgG3 anti–APO-1, and F(ab′)2 anti– APO-1 fragments were added to the cells. After 3–72 h, 5 mg/ml of MTT dye was added and the plates were incubated for 12 h. OD was determined by eluting the dye with isopropanol/formic acid, and absorbance was measured at 540 nm.
Results are shown as the mean of data from six independent wells ± SD. To rule out that drug-treated cells may simply arrest growth, making it difficult to determine what percentage of the reduced MTT is due to apoptosis, all cytotoxicity assays were verified by FACS® analysis using the method of Nicoletti et al. (53) to assess the subdiploid DNA content and specific apoptosis.
Immunoselection of p53-binding DNA Fragments.
DNA of cosmid cAPO-1 (55) was digested to completion with Sau3A1. The mixture of small DNA fragments was ligated into the BamHI site of pBlueScript II KS+, and the ligation products were transformed into Escherichia coli. Transformants were selected in liquid culture containing ampicillin, and DNA was prepared for the drug-resistant culture. The DNA was taken through three consecutive rounds of p53 immunoselection. The procedure was essentially as described in Zauberman et al. (56), except that the extracts used for selection were prepared from H1299 cells transiently transfected with a p53 expression plasmid (pCMVp53wt) and harvested 40 h after transfection. At the end of the third selection cycle, digestion of the enriched plasmid DNA revealed only a single insert band of 0.7 kb. Single colonies were obtained from the enriched plasmid population, the corresponding plasmids were extracted, and each was confirmed for the presence of the expected 0.7-kb insert. The insert was then subjected to DNA sequencing, and used for construction of luciferase-based reporter plasmids.
Plasmids.
Plasmid pCMVp53wt, encoding mouse wt p53, has been described previously (40). Plasmid CD95(Ps)-luc was constructed by ligating a 1.71-kb HindIII-SacII fragment of cosmid cAPO-1 (55), containing the 1.43-kb 3′ end of the human CD95 gene promoter and the 5′ end of exon 1 (0.28 kb) (57), into pGL3-Basic (Promega Corp., Madison, WI) or into pTATA-Luc (a gift of T. Wirth, Institut für Medizinische Strahlen-und Zellforschung, Würzburg, Germany). Plasmid CD95(P)-luc, containing an extended 5′ version of the CD95 promoter (1.9 kb) and the 5′ end of exon 1 (0.28 kb) (57), was constructed by cloning a 450-bp PCR fragment of the 5′ part of the CD95 promoter (58) in the HindIII site of CD95(Ps)-luc. Orientation of the PCR fragment and sequence was checked by sequencing. Plasmid CD95(I+SV)-luc was constructed by ligating a 0.7-kb CD95 gene fragment of intron 1 (excised from the pBlueScript vector with SacI+SalI) between the SacI and XhoI sites of pGL3-Promoter (Promega Corp.); this places the intronic CD95 fragment upstream of a minimal SV40 promoter, followed by the luciferase reporter. CD95(Ps+I)-luc and CD95(Ps+Is)-luc were generated by inserting the above 0.7-kb fragment or a HindIII-cleaved 0.6-kb subfragment thereof, respectively, downstream of the CD95 promoter in plasmid CD95(Ps)-luc. Cyclin G luciferase contains the cyclin G gene promoter in front of a luciferase reporter (59).
Statistical Analysis.
To examine whether synergy (60, 61) between APO-1 stimulation and concurrent chemotherapeutic treatment is observed, a balanced two-way ANOVA (model with fixed effects) was performed. In addition, we applied MANOVA (multivariate analysis of variance) to test for statistical significance. Statistical analysis was carried out with SAS software (SAS Institute, Inc., Cary, NC).
Results
Induction of Apoptosis by Anticancer Drugs
The anticancer drugs 5-fluorouracil, methotrexate, mitomycin, cisplatin, mitoxantrone, doxorubicin, etoposide, cyclophosphamide, and bleomycin have been shown to trigger nuclear accumulation of the tumor suppressor protein p53 (6, 62). To analyze whether these drugs use the CD95 system to exert their cytotoxic effects, we first investigated if they induce apoptosis. We found that all of the above drugs irrespective of their intracellular target cause death of sensitive target cells by inducing apoptosis (Table 1). Having established that the accumulation of p53 by DNA damage upon treatment with anticancer drugs resulted in induction of apoptosis, we investigated if the CD95 receptor/ligand system is involved in p53-mediated apoptosis.
Table 1.
Induction of CD95 and Apoptosis by Anticancer Drugs in Cell Lines with wt p53
| Drug* | Mechanism of action | Concentration tested for CD95 receptor induction‡ | CD95 receptor induction, increase above controls§ | Increased responsiveness towards induction of apoptosis by CD95 receptor stimulation‖ | ||||
|---|---|---|---|---|---|---|---|---|
| μg/ml | % | % | ||||||
| Fluorouracil | Pyrimidine antagonist | 2.5–2,500 | 5–62¶ | 46–55 | ||||
| Methotrexate | Folic acid antagonist | 0.25–2,500 | 8–65¶ | 28–70 | ||||
| Mitomycin | Alkylation | 0.01–100 | 3–56¶ | 0–53 | ||||
| Cisplatin | Alkylation | 0.1–100 | 0–60** | 0–64 | ||||
| Cyclophosphamide | Alkylation | 0.001–100 | 28–40†† | 39–45 | ||||
| Mitoxantrone | Intercalation | 0.0001–1 | 8–52†† | 43–51 | ||||
| Doxorubicin | Intercalation | 0.002–2 | +§§ | 18–43 | ||||
| Etoposide | Inhibits topoisomerase II | 0.001–1 | 22–55†† | 45–62 | ||||
| Bleomycin¶¶ | Inhibits DNA polymerase | 0.3–3,000 | 15–78‖‖ | 27–73 |
Anticancer drugs with different mechanisms of action were analyzed for their ability to induce the CD95 receptor. Different solid human cancer cell lines (HepG2, Hs746T, AGS, MCF7, Huh7, and HT29) were treated with the specific anticancer drug for 48 h.
A wide range of drug concentrations were tested. Clinically relevant concentrations of the chemotherapeutic drugs are marked with an asterisk in Fig. 2.
Treatment with 5-fluorouracil, methotrexate, mitomycin, cisplatin, mitoxantrone, doxorubicin, etoposide, cyclophosphamide, and bleomycin led to upregulation of the CD95 receptor in HepG2 cells (wt p53), in AGS cells (wt p53), in Hs746T cells (wt p53), and in MCF7 cells (wt p53). No induction or only weak induction of the CD95 receptor was observed in cell lines with mutant p53 (Huh7 and HT29) and in cell lines lacking p53 (Hep3B). Cell surface expression of the CD95 receptor was assessed by FACScan®. Percent CD95 induction was calculated as (% CD95+ treated cells − % Quantum Red+ treated cells) − (% CD95+ control cells − % Quantum Red+ control cells).
The upregulation of the CD95 receptor after cytostatic treatment is functional. It provides the cell with an increased responsiveness towards CD95-mediated apoptosis. Hepatoma cells with different p53 mutational status were treated with diverse anticancer drugs alone (for 48 h) and in combination with or without the agonistic apoptosis-inducing antibody IgG3 anti–APO-1, 100 ng/ml, for an additional 24 h. The rate of cell death was assessed by cytotoxicity assay (MTT). Data are expressed as 100 − fraction of the living cells treated with the specific anticancer drug only. A balanced two-way ANOVA revealed a synergistic interaction between anti–APO-1– and chemotherapy-induced cell death for all the drugs tested (P < 0.0001, tested in HepG2).
Range of data from tests in HepG2, Hs746T, AGS, and MCF7.
Range of data from tests in HepG2, Hs746T, and MCF7.
Range of data from tests in HepG2.
Could not exactly be quantified by FACScan® due to autofluorescence of doxorubicin.
Range of data from tests in HepG2, Hs746T, and AGS.
These data have been published in part (for HepG2) in reference 6.
Induction of the CD95 Receptor by Anticancer Drugs
Treatment with 5-fluorouracil, methotrexate, mitomycin, cisplatin, mitoxantrone, doxorubicin, etoposide, cyclophosphamide, and bleomycin led to upregulation of CD95 mRNA (6) and of the CD95 receptor protein in HepG2 cells (wt p53), in AGS cells (wt p53), in HS746T cells (wt p53), and in MCF-7 cells (wt p53) (Table 1, and Figs. 1 and 2). In contrast, no induction or only weak induction of the CD95 receptor was observed in Huh7 and HT29 cells with mt p53, and in Hep3B cells lacking p53 altogether. All drugs tested induced the CD95 receptor in cells with wt p53 independent of their mechanism of action (Table 1). A wide range of drug concentrations were tested, and it is of note that clinically relevant concentrations were effective in upregulation of the CD95 receptor with all drugs tested.
Figure 1.

FACS® analysis of CD95 receptor expression in AGS (wt p53) and HT29 cells (mt p53). Clinically relevant concentrations of diverse anticancer drugs with different mechanisms of action induce CD95 receptor expression (dependent on the p53 status). As an example for the investigated cell lines expressing either wt or mt p53, AGS colon cancer cells and HT29 colon cancer cells are shown here. Enhanced CD95 receptor expression was observed in hepatoma, gastric, colon, and breast cancer cell lines with wt p53 only. Percent enhanced CD95 expression was calculated as (% CD95+ treated cells – % Quantum Red+ treated cells) – (% CD95+ control cells – % Quantum Red+ control cells). AGS cells, for example, displayed enhanced CD95 receptor expression: up to 60% positive cells in response to mitomycin, up to 20% in response to 5-fluorouracil, and up to 50% positive cells in response to bleomycin.
Figure 2.

Quantitative flow cytometry analysis of CD95 receptor expression in different solid human tumor cell lines after treatment with diverse anticancer drugs. *Clinically relevant concentrations of the cytostatic drugs. Percent CD95 expression was calculated as % CD95+ cells − % Quantum Red+ cells. Only cell lines with wt p53 expression, HepG2, AGS, HS746T, and MCF-7 ( filled symbols), displayed upregulated CD95 receptor expression up to >80% positive cells in response to drug treatment. In contrast, neither Hep3B cells lacking p53 nor Huh7 and HT29 cells (open symbols) with mt p53 responded to cytotoxic treatment with upregulation of the CD95 receptor.
Increased Responsiveness towards Induction of Apoptosis by Anti–APO-1 Antibodies
Upregulation of CD95 receptor expression after cytostatic treatment resulted in increased responsiveness towards CD95-mediated apoptosis. Hepatoma cells with different p53 mutational status were treated with diverse anticancer drugs alone or in combination with the agonistic apoptosis-inducing antibody IgG3 anti–APO-1. In HepG2 cells, treatment with anticancer drugs for 48 h followed by stimulation of the CD95 receptor with anti–APO-1 resulted in induction of apoptosis in up to 70% of the cells, dependent on the concentration and the particular drug applied (Fig. 3). Thus, a statistically significant (P < 0.0001) synergy in induction of apoptosis by anticancer drugs and by anti–APO-1 was seen for all the drugs tested (Table 1).
Figure 3.
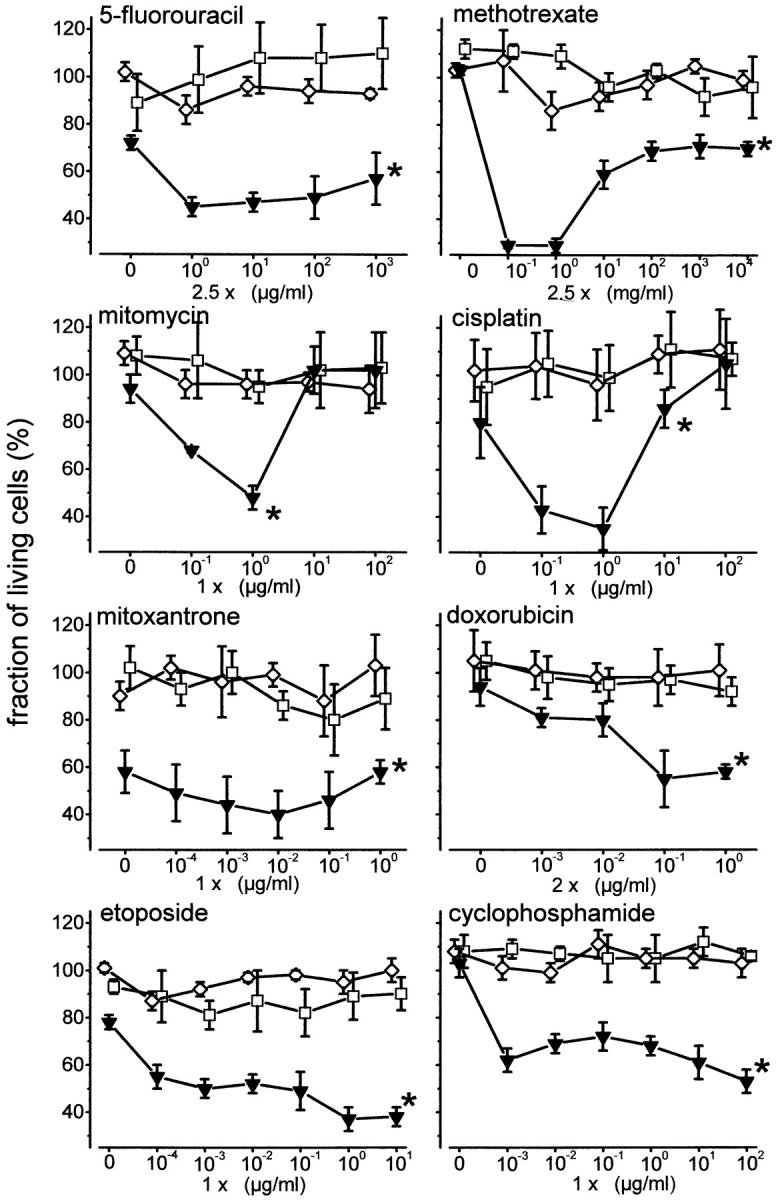
Increased responsiveness towards induction of apoptosis by CD95 receptor stimulation after treatment with anticancer drugs. Cytotoxicity assay with MTT staining of viable cells. HepG2 cells (wt p53, inverted triangles), Huh7 cells (mt p53, squares), and Hep3B cells (p53−/−, diamonds) were treated with different doses of 5-fluorouracil, methotrexate, mitomycin, cisplatin, mitoxantrone, doxorubicin, etoposide, and cyclophosphamide alone for 48 h and in combination with or without IgG3 anti–APO-1, 100 ng/ml, for an additional 24 h. Data are expressed as the fraction of living cells treated with specific anticancer drug only (mean ± SD, n = 6 wells). Only HepG2 cells with wt p53 exhibited an increased responsiveness towards induction of apoptosis by agonistic anti–APO-1 antibodies after cytostatic treatment. Anti–APO-1 treatment did not induce further toxicity in Huh7 cells (mt p53) or Hep3B cells (p53−/−). * By MANOVA, between-subject effect P < 0.0001 compared with Hep3B, P < 0.0001 compared with Huh7.
Induction of CD95L by Anticancer Drugs
CD95L mRNA was induced upon cytostatic treatment in HepG2 cells (wt p53) as well as in Hep3B (p53−/−) and Huh7 cells (mt p53). Thus, cytostatic treatment stimulated CD95L mRNA expression in different hepatoma cell lines independent of the p53 status of the cells. The ability to induce CD95L mRNA expression occurred regardless of the mechanism of action of the anticancer drug (Fig. 4). However, it is of note that there was a high variability in the extent of CD95L induction dependent on the chemotherapeutic agent used and the cell line tested. Furthermore, anticancer drugs with clinically known therapeutic effectivity for the specific tumor were observed to be most effective in CD95L induction, e.g., mitoxantrone in hepatomas and hepatoblastomas (Fig. 4) or 5-fluorouracil in cell lines from colorectal cancer (data not shown). Anticancer drugs with different mechanisms of action also upregulated CD95L mRNA expression in the gastric cancer cell line Hs746T (wt p53) and in the colon cancer cell line HT29 (mt p53), independent of the p53 status of the tumor cells (data not shown).
Figure 4.

Induction of the CD95L by cytostatic drugs with different mechanisms of action. Semiquantitative PCR analysis of CD95L mRNA expression in HepG2, Hep3B, and Huh7 cells upon treatment with cyclophosphamide (cyclo), 5-fluorouracil (5-FU), doxorubicin (doxo), mitomycin (mitom), mitoxantrone (mitox), and actinomycin (actino). CD95L mRNA expression was induced in HepG2 (wt p53), Hep3B (p53−/−), and Huh7 cells (mt p53). Densitometry was performed to analyze CD95L expression in relation to β-actin expression. CD95L mRNA induction was independent of the p53 status of the cells. The extent of CD95L expression upon chemotherapeutic treatment showed variability dependent on the agent and the cell line tested.
These data are supported by the fact that CD95L mRNA was highly elevated upon bleomycin treatment of Hep3B cells stably transfected with the temperature-sensitive mutants p53ala143 or p53val135. CD95L mRNA upregulation occurred both at 32°C and at 37°C, i.e., at both wt and mt conformations (Fig. 5).
Figure 5.

Induction of the CD95L by bleomycin (bleo) in the temperature-sensitive (ts) mutants p53ala143 and p53val135. Semiquantitative PCR analysis of CD95L mRNA expression in Hep3B cells stably expressing the temperature-sensitive mutant p53ala143 or p53val135. Densitometry was performed to quantify CD95L expression in relation to β-actin expression. Bleomycin treatment induced CD95L mRNA expression in the temperature-sensitive mutants p53ala143 and p53val135 at both temperatures, independent of their p53 mutational status.
Our data in the above experiments suggest that cytostatic drugs upregulate both CD95 and CD95L, the former dependent and the latter independent of the p53 status.
Restitution of wt p53 Induces the CD95 Receptor
Our data show a strong correlation between wt p53 status and induction of CD95 receptor expression in cancer cells exposed to chemotherapeutic drugs. This relationship was further characterized using Hep3B cells transfected (a) transiently with wt p53 cDNA, (b) stably with a tamoxifen-regulated p53–estrogen receptor chimera, (c) stably with the temperature-sensitive mutant p53val135, and (d) stably with the temperature-sensitive mutant p53ala143.
Transient Transfection with wt p53 cDNA.
Cells transfected with wt p53 cDNA responded with upregulation of the CD95 receptor upon bleomycin treatment. In contrast, bleomycin treatment did not lead to an upregulation of the CD95 receptor in mock-transfected Hep3B cells (Fig. 6).
Figure 6.

Restitution of a wt p53 status restores the ability of the p53−/− cell line Hep3B to increase the CD95 receptor upon anticancer treatment (FACS® analysis). Transient transfection of p53−/− Hep3B cells with wt p53. Either a wt p53 expression plasmid in which expression of the p53 gene is under the control of the CMV promoter, or the corresponding control plasmid without the p53 insert (mock) was transiently transfected into Hep3B cells. Upon transient transfection with wt p53 cDNA and bleomycin treatment, p53−/− Hep3B cells increased CD95 receptor expression.
Stable Transfection with a Tamoxifen-regulated p53–estrogen Receptor Chimera.
Hep3B cells stably transfected with a p53–estrogen receptor chimera (BT-4P) were treated with 100 nM 4-OH tamoxifen for 24 h to activate p53. BT-2E control cells transfected with the puromycin resistance gene only were treated with bleomycin for 48 h. Induction of p53 upon tamoxifen treatment led to upregulation of the CD95 receptor from 0–8% of the cells. When bleomycin treatment was also included, the CD95 receptor expressing cells increased to 30% (Fig. 7 A). In contrast, BT-2E cells did not exhibit CD95 receptor expression either before or after tamoxifen treatment (Fig. 7 B). Significantly, upregulation of CD95 receptor expression after reconstitution of wt p53 activity was of functional consequence. Thus, stimulation of the upregulated CD95 receptor in BT-4P cells with the agonistic antibody IgG3 anti–APO-1 resulted in greatly enhanced apoptosis (Fig. 7 C).
Figure 7.
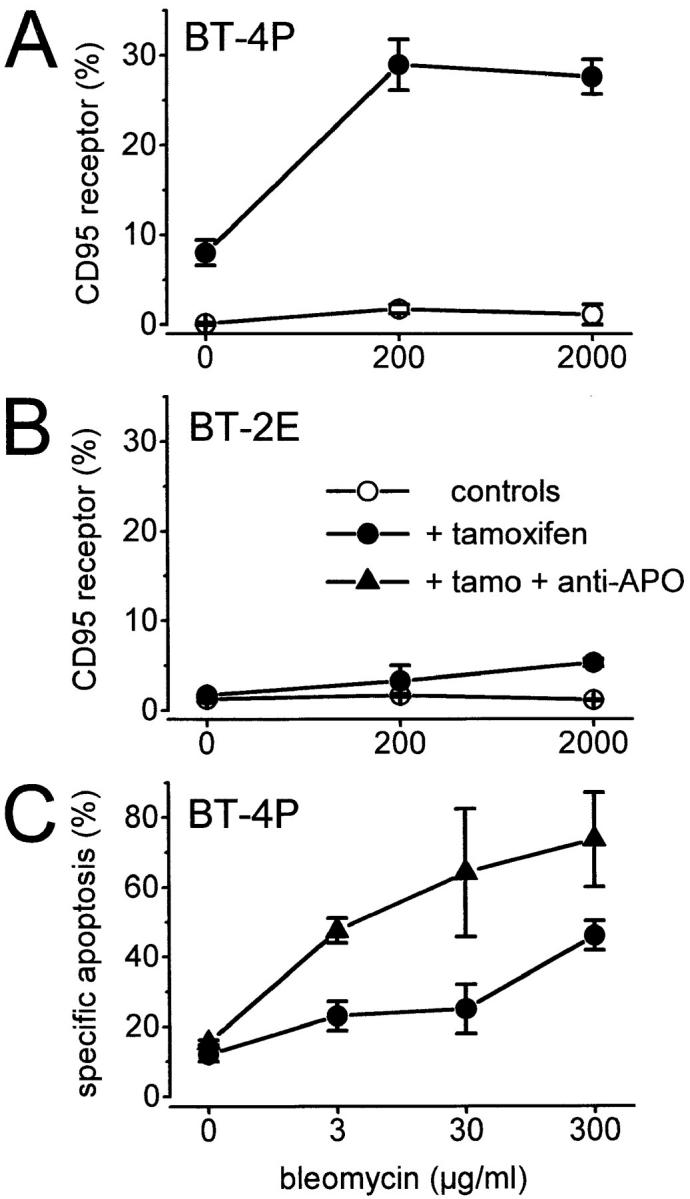
Induction of the CD95 receptor by tamoxifen activation of a p53–estrogen receptor fusion protein in stably transfected p53−/− Hep3B cells (FACS® analysis). The p53−/− cell line Hep3B was stably transfected with either puromycin resistance alone (control cell line: BT-2E) or with a p53 (modified)–estrogen receptor chimera (cell line: BT-4P). In BT-4P cells, p53 activity is induced by addition of the specific ligand 4-OH tamoxifen. The tamoxifen-activated p53 induces CD95 receptor expression in BT-4P cells. A further increase in CD95 receptor expression up to 30% of the cells is induced by bleomycin treatment (A). BT-2E did not exhibit CD95 receptor expression either before or after tamoxifen treatment (B). The functionality of the upregulated CD95 receptor in BT-4P cells is evident upon treatment of BT-4P with IgG3 anti–APO-1. Stimulation of the CD95 receptor by IgG3 anti–APO-1 and concurrent bleomycin treatment lead to an increased rate of apoptosis, after tamoxifen activation of p53 in BT-4P cells (C, + tamo + anti-APO).
Stable Transfection with the Temperature-sensitive p53 Mutants.
At 37°C p53val135 and p53ala143 are in an mt conformation and can transform cells through a negative dominant mechanism, whereas at the permissive temperature of 32°C they become transcriptionally active and regain wt activity. 40% of Hep3B cells, stably transfected with the temperature-sensitive mutant p53val135, exhibited CD95 receptor expression at 32°C (wt p53 conformation). In contrast, at 37°C (mt p53 conformation) no CD95 receptor expression could be detected. Addition of bleomycin led to a further increase in CD95 receptor–expressing cells up to 70% (Fig. 8 A). Likewise, Hep3B cells stably transfected with the temperature-sensitive mutant p53ala143 only showed upregulation of the CD95 receptor at the permissive temperature. Upon addition of bleomycin, CD95 receptor–expressing cells increased to 30% (Fig. 8 B). Taken together, these data support the conclusion that wt p53 activity is essential for drug-induced CD95-mediated apoptosis. Furthermore, these data prompted us to investigate the molecular basis for the regulation of CD95 gene expression by p53.
Figure 8.

Restitution of inducible wt p53 function restores the ability of p53−/− Hep3B cells to upregulate the CD95 receptor in response to anticancer drugs. FACS® analysis of CD95 receptor expression in Hep3B clones stably expressing the temperature-sensitive (ts) mutant p53val135 (A) or p53ala143 (B). These temperature-sensitive mutants show mt p53 conformation at 37°C; at 32°C, the permissive temperature, they regain wt p53 characteristics. The CD95 receptor is inducible only at the permissive temperature, 32°C. In the temperature-sensitive mutant p53val135, temperature down-shift induces the CD95 receptor in up to 40% of the cells; additional treatment with bleomycin leads to CD95 expression in up to 70% of the cells (A). Likewise, in the temperature-sensitive mutant p53ala143, CD95 receptor expression increased to 30% of the cells upon temperature down-shift and additional bleomycin treatment (B).
Analysis of Putative p53-response Elements in the CD95 Gene
To further investigate whether p53 interacts directly with the CD95 gene, we first performed a computer search for potential p53-responsive elements within the promoter of the CD95 receptor gene. This search indicated the presence of three putative elements within the CD95 promoter which showed limited homology with the p53 consensus binding site (63). However, it is of note that each of these putative elements diverges from the consensus in at least one of the positions corresponding to critical DNA–protein contact residues, questioning their ability to function as effective p53 response elements (Fig. 9, A and C). Therefore, a more comprehensive, experimental approach was undertaken to search for possible p53-binding elements within the CD95 gene. A recombinant cosmid comprising the entire human CD95 locus (cAPO-1 [55]) was subjected to an immunoselection protocol, which specifically enriches DNA fragments containing high-affinity p53-binding sites (56). This selection procedure resulted in efficient enrichment of a single p53-binding plasmid clone, carrying a human genomic DNA insert of 0.7 kb (data not shown); no other clones were enriched, suggesting that the isolated clone may represent the only high-affinity p53-binding element within the CD95 genomic cosmid.
Figure 9.

(A) Map of the human CD95 gene. Exons 1–9 are numbered and represented by black boxes. (B) Position of the putative p53 binding elements (p53-BE) within the CD95 gene promoter, suggested by computer analysis. Numbering is according to Wada et al. (reference 57). Shown also is a comparison between each element and the consensus p53-binding site (bottom [reference 63]). R, purine; Y, pyrimidine; W, A or T. *Putative. Lower case letters indicate deviations from the consensus.
The sequence of the first 266 nucleotides of the cloned 0.7-kb insert is shown in Fig. 10 A. It is derived from within the first intron of the human CD95 gene; its 5′ end is located 142 nucleotides downstream of the 3′ end of exon 1 (Fig. 10 B). The cloned DNA fragment contains a stretch of 20 contiguous nucleotides (underlined) exhibiting a high degree of homology (18/20, including full conservation of all critical core nucleotides) with the p53 consensus binding site (63; Fig. 10 C).
Figure 10.

The intronic p53-binding site within the CD95 gene. (A) Sequence of the first 266 nucleotides of the Sau3A1 fragment, which contains the p53-binding site in the first intron of the CD95 gene (sequence data available from EMBL/GenBank/DDBJ under accession no. AJ011034). Numbering is according to Wada et al. (reference 57). p53BE, the putative p53-binding element. (B) Position of the putative p53-binding element within the CD95 gene. Numbering is as in Wada et al. (reference 57); the first ATG of the CD95 protein coding region is indicated. (C) Comparison between the CD95 intronic putative p53-binding element (top line) and the consensus p53-binding site (bottom line; reference 63). R, purine; Y, pyrimidine; W, A or T. Lower case letters indicate deviations from the consensus.
The CD95 p53-binding Element Confers Transcriptional Activation by p53, and Cooperates with the CD95 Promoter
To test whether the intronic p53-binding fragment of the CD95 gene can mediate p53-dependent transcriptional activation, several luciferase-based reporter plasmids were constructed and assayed by transient transfection. A schematic drawing of the relevant plasmids used for this purpose is presented in Fig. 11. In plasmid CD95(Ps)-luc the luciferase gene is preceded by the 1.43-kb CD95 promoter and the 5′ end of exon 1 (57; HindIII-SacII fragment, see Fig. 10 B). In plasmid CD95(P)-luc the luciferase gene is preceded by the 1.9-kb CD95 promoter and the 5′ end of exon 1 (58) comprising the three computer-suggested putative p53-binding elements (Fig. 9 B). In CD95(I+SV)-luc, the 0.7-kb CD95 intronic DNA fragment is positioned in front of a minimal SV40 promoter. CD95(Ps+I)-luc contains the CD95 promoter upstream of the CD95 intronic region.
Figure 11.

The p53-binding intronic CD95 region confers p53-dependent transcriptional activation. (Left) Schematic diagram of relevant luciferase reporter constructs used for transcriptional analysis. Ps, 1.43-kb CD95 promoter region (reference 57); P, 1.9-kb CD95 promoter region with the three putative (computer-identified) p53-binding sites (reference 58); I, CD95 0.7-kb intronic region selected for p53 binding; SV, SV40 minimal promoter; Luc, luciferase gene; p53-BE, p53-binding element. (Right) Analysis of p53-dependent luciferase activity. Hep3B cells were transfected with 1 μg of each of the indicated reporter plasmids together with 100 ng of either a wt p53 expression plasmid, pCMVp53wt, or an equivalent amount of empty vector. Shown is the fold p53-dependent activation of each reporter plasmid, calculated relative to the value obtained with the same reporter in the absence of p53. These results were supported in H1299 human lung cancer carcinoma cells: the CD95 promoter alone was only minimally stimulated by wt p53, highly activated (34-fold) in conjunction with the p53-binding intronic region in the long (Ps+I) or short (Ps+Is) version downstream of the CD95 promoter. The effect was CD95 promoter specific, not seen with the RSV promoter (devoid of a p53- responsive element, 0.95-fold activation), and comparable to activation (36-fold) with a natural p53-responsive promoter of the cyclin G gene.
Reporter plasmids were transfected transiently into Hep3B human hepatoma cells (Fig. 11). As seen in Fig. 11, the CD95 promoter alone (Ps and P) was only minimally (up to twofold) stimulated by wt p53, irrespective of whether the 1.43- or 1.9-kb version of the CD95 promoter was used. These data suggest that this promoter contains only weak p53-responsive elements. On the other hand, when the CD95 promoter was placed in conjunction with the p53-binding intronic CD95 DNA region (Ps+I), transcriptional activity became strongly stimulated by wt p53 (Fig. 11) but not by a tumor-derived p53 mutant (data not shown). It is of note that the basal activity of this plasmid, in the absence of p53, was very low and almost similar to that of the CD95 promoter alone; this combination of control elements resulted in substantial transcriptional activity only in the presence of p53. Of note, when the intronic p53-binding fragment was placed in front of an irrelevant SV40 promoter, activation by p53 was significantly less dramatic than in the context of the natural CD95 promoter (I+SV, Fig. 11). These results were supported by data in a different cellular context, in H1299 human lung adenocarcinoma cells (see legend to Fig. 11). A similar ability to confer p53-dependent transcriptional activation was also provided when a shorter version of the intronic region was placed downstream of the CD95 promoter (Ps+Is; 34-fold activation). In addition, the effect of p53 was specific for CD95, and was not exerted on the RSV promoter, which does not contain any p53-responsive elements (0.95-fold activation). Furthermore, the extent of stimulation by p53 (fold activation) of the CD95 promoter plus intronic region was comparable to that seen with a natural p53-responsive promoter derived from the cyclin G gene (36-fold activation).
Taken together, these data imply that the first intron of the CD95 gene harbors a p53-responsive enhancer element. This element cooperates preferentially with its authentic promoter, and less so with an irrelevant viral promoter. Hence, cooperativity exists between this intronic p53 response element and one or more elements residing within the promoter region of the CD95 gene.
Discussion
The data in this paper show that the CD95 system plays a general role in induction of cytotoxicity by anticancer drugs in a variety of cells of different histotype. Clinically relevant concentrations of diverse anticancer drugs such as cisplatin, bleomycin, methotrexate, doxorubicin, cyclophosphamide, etoposide, and mitoxantrone induce CD95 receptor expression in hepatoma, gastric cancer, colon cancer, and breast cancer cell lines, thereby strongly increasing the sensitivity towards CD95-induced apoptosis. Thus, chemotherapy may sensitize tumor cells by upregulating expression of death regulators such as the CD95 receptor. Most notably, drug-induced upregulation of the CD95 receptor is dependent on the p53 status of the tumor cell. Upregulation of the CD95 receptor occurred only in cell lines with intact p53. This upregulation is dependent on accumulation of endogenous p53 after DNA damage and can be reconstituted in p53 null cell lines by exogenous wt p53 transfection. Caspase-8 cleavage was observed in cell lines of solid human tumors upon treatment with anticancer drugs, irrespective of whether or not apoptosis was dependent on the CD95 system (data not shown). Hence, additional effector pathways besides CD95/CD95L signaling are likely to contribute to drug-induced apoptosis. We further show for different solid tumors that anticancer drugs at concentrations measured during chemotherapy in patients' sera lead to upregulation of both the CD95 receptor and CD95L. Upregulation of CD95L upon treatment with anticancer drugs was demonstrated in cell lines containing either wt p53, mt p53, or no p53 at all. Thus, the regulation of CD95L clearly involves p53-independent mechanisms.
Upregulation of the CD95 receptor might render a tumor cell chemotherapy sensitive (and sensitive towards CD95L-expressing antitumor T cells). Upregulated CD95L might bind to an increased number of CD95 receptors. On the contrary, drug resistance could result from downregulation of the CD95 receptor. In fact, we have previously demonstrated loss of CD95 receptor and gain of CD95L expression in hepatocellular carcinomas in patients (64).
Constitutive CD95L expression has also been shown for melanoma (65), lung cancer (66), and colon cancer (67). In addition, cytostatic drug treatment can induce production of CD95L. However, drug-induced expression of CD95L in CD95 receptor–negative tumors cannot be effective and, furthermore, could result in selective elimination of antitumor lymphocytes. Thus, chemotherapy might render the tumor an immune-privileged site. Invading T cells, normally engaged in destruction of the tumor tissue, might be eliminated through CD95L produced by the target cells. There are several reports demonstrating that CD95L might be useful for creating immune-privileged tissues in the context of tumor immune privilege and organ and tissue transplantation (68–71). On the other hand, CD95L-expressing fibroblasts transplanted into nude mice were readily rejected as a result of massive neutrophilic infiltration of the graft (72). This demonstrates that CD95L expression might not always support survival of the respective tissue.
On the other hand, tumor cells that express the CD95 receptor upon induction by anticancer therapy may be turned into susceptible targets for killer cells. Clinical outcome, i.e., death or survival, might be determined by a balance between CD95 receptor and CD95L expression on tumor and immune cells. Upregulation of the CD95 receptor may prepare the tumor cells to be eliminated by the immune system using a CD95-dependent pathway. Thus, in addition to their direct cytotoxic effects, chemotherapeutic drugs sensitize tumor cells to CD95-mediated cytotoxicity and CD95-dependent immune clearance.
Most importantly, our data suggest a potential mechanism by which p53-dependent cell death may be mediated. We show a strong correlation between p53 status and induction of CD95 receptor in response to DNA damage. Both DNA damage and transfection/restitution of wt p53 caused induction of the CD95 receptor, and DNA damage failed to induce the CD95 receptor in tumor cells with mt or deficient p53. The hypothesis that the CD95 gene is p53 regulated was further confirmed in several systems with stably transfected inducible p53.
To investigate if p53 directly transactivates the CD95 gene in a manner similar to its effects on CDKN1A (23– 26), gadd45 (20, 73), mdm2 (56, 74), cyclin G (59, 75), and BAX (27), we looked for p53 response elements in the CD95 gene. Computer analysis suggested three potential p53 response elements within the CD95 promoter. However, the CD95 promoter alone was only minimally stimulated by wt p53, and activation of no more than twofold was obtained. On the other hand, direct cloning of p53-binding elements from the entire human CD95 locus yielded a DNA fragment containing a strong p53-binding site. This site, derived from the first intron of the human CD95 gene, displays a high degree of homology with the p53 consensus binding site (63). When the CD95 promoter was then placed in conjunction with this p53-binding DNA region, transcriptional activity became strongly stimulated (up to 50-fold) in transfected hepatoma and lung carcinoma cells. This suggests a cooperativity between the intronic element and putative p53 response elements within the CD95 promoter. Further evidence for such cooperativity is provided by the observation that the intronic CD95 element is far less potent in conferring p53-responsiveness in conjunction with an irrelevant SV40 promoter.
Interestingly, there is a remarkably high similarity between the two adjacent 10-mer half-sites of this putative p53-binding element; only the first and tenth nucleotides are not identical between the two half sites. This argues for the possibility that the CD95 intronic p53-binding site may be recognized by a specific form of p53 with distinct binding site preferences.
The observation that the CD95 gene can only be activated by wild-type but not by the p53 mutants tested would also argue for a differential regulation of the CD95 gene by wt and mt p53. We hypothesize that wt p53 can stimulate CD95 gene transcription, whereas the mt p53 protein or specific mutants of p53 protein fail to function properly in transcription of the CD95 gene. A differential regulation by wt and mt p53 is also known for the IGF-IR (insulin-like growth factor I receptor) promoter. wt p53 has the potential to suppress the IGF-IR promoter, whereas mutant versions of the p53 protein can derepress the IGF-IR promoter (76). Further studies are needed in view of the more than 150 different p53 mutations described in human cancers so far to characterize the kind of mutations that lead to loss of the ability of p53 to act as a transcription factor of the CD95 gene.
The fact that wt p53 has a stimulatory effect on CD95 gene activity, whereas the investigated mt p53 proteins are not capable of activating the CD95 gene, may explain why mutations in p53 contribute to tumor progression and resistance of cancer cells to chemotherapy. Clinical studies will have to show if p53 and CD95 status are correlated in primary human tumors and if treatment response to cancer chemotherapeutic drugs and prognosis are dependent on p53 and the CD95 apoptotic pathway. Preliminary data from ongoing clinical studies in patients with hepatocellular carcinoma show that the majority of the tumors with wt p53 express the CD95 receptor, whereas only a minority of the hepatocellular carcinomas with mutant p53 display CD95 expression (our unpublished observations).
Furthermore, the ability of wt p53 to activate CD95 gene expression and confer sensitivity to CD95-mediated apoptosis in Hep3B cells may have implications for wt p53 gene therapy. Our data suggest that upregulation of the CD95 receptor contributes to the apoptosis-inducing effect of wt p53. These findings may provide an explanation for the tumor regression observed in non-small cell lung cancer patients after wt p53 gene transfer (77). Upregulation of the CD95 receptor in CD95L-positive tumors may account for the increased rate of apoptosis reported to occur in these tumors after wt p53 gene transfer. CD95L expression may have been acquired during the course of tumor development as an immune-privileged site or may have been induced in the tumor cells upon cytostatic treatment. Thus, wt p53 gene transfer and chemotherapy could synergize in their action on CD95 receptor and CD95L expression and induce autocrine and paracrine apoptosis. A synergistic effect of wt p53 gene transfer and chemotherapeutic treatment on induction of apoptosis has already been reported in vitro for lung cancer cell lines (77–79). Our data provide evidence that activation of the CD95 system may be the molecular basis for this enhancement of apoptosis. Thus, reconstitution of p53 function with or without combined chemotherapy is an attractive goal for somatic gene therapy in cancer.
We have shown that p53 directly transactivates the CD95 gene. A critical question is whether p53-induced apoptosis is functionally dependent on the induction of the CD95 gene. With the identification of killer/DR5 (80) as another pathway of p53-dependent apoptosis after DNA damage, it is evident that p53-dependent apoptosis is not mediated solely by the CD95 system. The observation that p53-dependent cell death after DNA damage is mediated by TNF-R family members could have significant implications for manipulating apoptosis and therapy.
Interference of anticancer drugs with apoptosis pathways can take place at several levels, including accumulation of wt p53 protein, triggering of CD95L–CD95 receptor interaction, triggering of other death receptors, stimulation of signaling cascades, and activation of death effector molecules including caspases, thus influencing the balance of proapoptotic and antiapoptotic programs. Therefore, drug sensitivity and resistance will likely be affected by alterations of any of these components of the apoptosis pathways.
Acknowledgments
We thank Sybille Klevenz, Susanne Allgäuer, and Concepcion Sainz-Rueda for expert technical assistance. We thank T. Wirth for providing pTATA-Luc and A. Hekele for helpful discussions.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Mu 1280/1-1), the Medizinische Forschungsförderung Heidelberg (121/1997), the Sonderforschungsbereich 601 (Molekulare Pathogenese Hepato-gastroenterologischer Erkrankungen), the Tumorzentrum Heidelberg/Mannheim, and the National Institutes of Health (DK37340;2 to S.L. Friedman), and by grants from the National Cancer Institute (ROI CA 40099) and the Israel-USA Binational Science Foundation (to M. Oren). S.L. Friedman acknowledges a Fulbright Senior Scholar Award.
Abbreviations used in this paper
- IGF-IR
insulin-like growth factor I receptor
- MANOVA
multivariate analysis of variance
- mt
mutated
- wt
wild-type
References
- 1.Fisher DE. Apoptosis in cancer therapy: crossing the threshold. Cell. 1994;78:539–542. doi: 10.1016/0092-8674(94)90518-5. [DOI] [PubMed] [Google Scholar]
- 2.Fung CY, Fisher DE. Shifting the cancer paradigm: must we kill to cure? . J Clin Oncol. 1995;13:801–807. doi: 10.1200/JCO.1995.13.4.801. [DOI] [PubMed] [Google Scholar]
- 3.Friesen C, Herr I, Krammer PH, Debatin KM. Involvement of the CD95 (APO-1/Fas) receptor/ ligand system in drug-induced apoptosis in leukemia cells. Nat Med. 1996;2:574–580. doi: 10.1038/nm0596-574. [DOI] [PubMed] [Google Scholar]
- 4.Micheau O, Solary E, Hammann A, Martin F, Dimanche BM. Sensitization of cancer cells treated with cytotoxic drugs to fas-mediated cytotoxicity. J Natl Cancer Inst. 1997;89:783–789. doi: 10.1093/jnci/89.11.783. [DOI] [PubMed] [Google Scholar]
- 5.Fulda S, Sieverts H, Friesen C, Herr I, Debatin KM. The CD95 (APO-1/Fas) system mediates drug- induced apoptosis in neuroblastoma cells. Cancer Res. 1997;57:3823–3829. [PubMed] [Google Scholar]
- 6.Müller M, Strand S, Hug H, Heinemann E-M, Walczak H, Hofmann WJ, Stremmel W, Krammer PH, Galle PR. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clin Invest. 1997;99:403–413. doi: 10.1172/JCI119174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamura T, Aoyama N, Saya H, Haga H, Futami S, Miyamoto M, Koh T, Ariyasu T, Tachi M, Kasuga M, Takahashi R. Induction of Fas-mediated apoptosis in p53-transfected human colon carcinoma cells. Oncogene. 1995;11:1939–1946. [PubMed] [Google Scholar]
- 8.Miyake H, Hara I, Gohji K, Arakawa S, Kamidono S. p53 modulation of Fas/Apo-1 mediated apoptosis in a human renal cell carcinoma cell line. Int J Oncol. 1998;12:469–473. doi: 10.3892/ijo.12.2.469. [DOI] [PubMed] [Google Scholar]
- 9.Egle A, Villunger A, Marschitz I, Kos M, Hittmair A, Lukas P, Grunewald K, Greil R. Expression of Apo-1/Fas (CD95), Bcl-2, Bax and Bcl-x in myeloma cell lines: relationship between responsiveness to anti-Fas mab and p53 functional status. Br J Haematol. 1997;97:418–428. doi: 10.1046/j.1365-2141.1997.382680.x. [DOI] [PubMed] [Google Scholar]
- 10.Fuchs EJ, McKenna KA, Bedi A. p53-dependent DNA damage-induced apoptosis requires Fas/APO-1-independent activation of CPP32β. Cancer Res. 1997;57:2550–2554. [PubMed] [Google Scholar]
- 11.Harris CC. p53 tumor suppressor gene: from the basic research laboratory to the clinic—an abridged historical perspective. Carcinogenesis. 1996;17:1187–1198. doi: 10.1093/carcin/17.6.1187. [DOI] [PubMed] [Google Scholar]
- 12.Harris CC. Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies. J Natl Cancer Inst. 1996;88:1442–1455. doi: 10.1093/jnci/88.20.1442. [DOI] [PubMed] [Google Scholar]
- 13.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 14.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 15.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 17.Lotem J, Sachs L. Hematopoietic cells from mice deficient in wild-type p53 are more resistant to induction of apoptosis by some agents. Blood. 1993;82:1092–1096. [PubMed] [Google Scholar]
- 18.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 19.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 20.Smith ML, Chen IT, Zhan Q, Bae I, Chen CY, Gilmer TM, Kastan MB, O'Connor PM, Fornace AJ., Jr Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science. 1994;266:1376–1380. doi: 10.1126/science.7973727. [DOI] [PubMed] [Google Scholar]
- 21.Smith ML, Chen IT, Zhan Q, O'Connor PM, Fornace AJ., Jr Involvement of the p53 tumor suppressor in repair of u.v.-type DNA damage. Oncogene. 1995;10:1053–1059. [PubMed] [Google Scholar]
- 22.Wang XW, Yeh H, Schaeffer L, Roy R, Moncollin V, Egly JM, Wang Z, Freidberg EC, Evans MK, Taffe BG, et al. p53 modulation of TFIIH-associated nucleotide excision repair activity. Nat Genet. 1995;10:188–195. doi: 10.1038/ng0695-188. [DOI] [PubMed] [Google Scholar]
- 23.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 24.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- 25.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 26.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 27.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 28.Yin C, Knudson CM, Korsmeyer SJ, Van DT. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature. 1997;385:637–640. doi: 10.1038/385637a0. [DOI] [PubMed] [Google Scholar]
- 29.Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E, Radinsky R. Wild-type human p53 and a temperature-sensitive mutant induce FAS/APO-1 expression. Mol Cell Biol. 1995;15:3032–3040. doi: 10.1128/mcb.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature. 1979;282:615–616. doi: 10.1038/282615a0. [DOI] [PubMed] [Google Scholar]
- 31.Ponchel F, Puisieux A, Tabone E, Michot JP, Froschl G, Morel AP, Frebourg T, Fontaniere B, Oberhammer F, Ozturk M. Hepatocarcinoma-specific mutant p53-249ser induces mitotic activity but has no effect on transforming growth factor beta 1-mediated apoptosis. Cancer Res. 1994;54:2064–2068. [PubMed] [Google Scholar]
- 32.Nakabayashi H, Taketa K, Yamane T, Miyazaki M, Miyano K, Sato J. Phenotypical stability of a human hepatoma cell line, HuH-7, in long-term culture with chemically defined medium. Jpn J Cancer Res. 1984;75:151–158. [PubMed] [Google Scholar]
- 33.Hsu IC, Tokiwa T, Bennett W, Metcalf RA, Welsh JA, Sun T, Harris CC. p53 gene mutation and integrated hepatitis B viral DNA sequences in human liver cancer cell lines. Carcinogenesis. 1993;14:987–992. doi: 10.1093/carcin/14.5.987. [DOI] [PubMed] [Google Scholar]
- 34.Huang F, Hsu S, Yan Z, Winawer S, Friedman E. The capacity for growth stimulation by TGF beta 1 seen only in advanced colon cancers cannot be ascribed to mutations in APC, DCC, p53 or ras. Oncogene. 1994;9:3701–3706. [PubMed] [Google Scholar]
- 35.Little JB, Nagasawa H, Keng PC, Yu Y, Li CY. Absence of radiation-induced G1 arrest in two closely related human lymphoblast cell lines that differ in p53 status. J Biol Chem. 1995;270:11033–11036. doi: 10.1074/jbc.270.19.11033. [DOI] [PubMed] [Google Scholar]
- 36.Plummer SJ, Adams L, Simmons JA, Casey G. Localization of a growth suppressor activity in MCF7 breast cancer cells to chromosome 17q24-q25. Oncogene. 1997;14:2339–2345. doi: 10.1038/sj.onc.1201073. [DOI] [PubMed] [Google Scholar]
- 37.Blagosklonny MV, Schulte TW, Nguyen P, Mimnaugh EG, Trepel J, Neckers L. Taxol induction of p21WAF1 and p53 requires c-raf-1. Cancer Res. 1995;55:4623–4626. [PubMed] [Google Scholar]
- 38.Mitsudomi T, Steinberg SM, Nau MM, Carbone D, D'Amico D, Bodner S, Oie HK, Linnoila RI, Mulshine JL, Minna JD. p53 gene mutations in non-small-cell lung cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene. 1992;7:171–180. [PubMed] [Google Scholar]
- 39.Haupt Y, Rowan S, Oren M. p53-mediated apoptosis in HeLa cells can be overcome by excess pRB. Oncogene. 1995;10:1563–1571. [PubMed] [Google Scholar]
- 40.Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci USA. 1989;86:8763–8767. doi: 10.1073/pnas.86.22.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedman SL, Shaulian E, Littlewood T, Resnitzky D, Oren M. Resistance to p53-mediated growth arrest and apoptosis in Hep 3B hepatoma cells. Oncogene. 1997;15:63–70. doi: 10.1038/sj.onc.1201149. [DOI] [PubMed] [Google Scholar]
- 42.Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62:671–680. doi: 10.1016/0092-8674(90)90113-s. [DOI] [PubMed] [Google Scholar]
- 43.Vater CA, Bartle LM, Dionne CA, Littlewood TD, Goldmacher VS. Induction of apoptosis by tamoxifen-activation of a p53-estrogen receptor fusion protein expressed in E1A and T24 H-ras transformed p53−/−mouse embryo fibroblasts. Oncogene. 1996;13:739–748. [PubMed] [Google Scholar]
- 44.Zhang W, Guo XY, Hu GY, Liu WB, Shay JW, Deisseroth AB. A temperature-sensitive mutant of human p53. EMBO (Eur Mol Biol Organ) J. 1994;13:2535–2544. doi: 10.1002/j.1460-2075.1994.tb06543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hall SW, Strong JE, Broughton A, Frazier ML, Benjamin RS. Bleomycin clinical pharmacology by radioimmunoassay. Cancer Chemother Pharmacol. 1982;9:22–25. doi: 10.1007/BF00296756. [DOI] [PubMed] [Google Scholar]
- 46.Shea TC, Flaherty M, Elias A, Eder JP, Antman K, Begg C, Schnipper L, Frei E, III, Henner WD. A phase I clinical and pharmacokinetic study of carboplatin and autologous bone marrow support. J Clin Oncol. 1989;7:651–661. doi: 10.1200/JCO.1989.7.5.651. [DOI] [PubMed] [Google Scholar]
- 47.Kurihara N, Kubota T, Hoshiya Y, Otani Y, Ando N, Kumai K, Kitajima M. Pharmacokinetics of cis- diamminedichloroplatinum (II) given as low-dose and high-dose infusions. J Surg Oncol. 1996;62:135–138. doi: 10.1002/(SICI)1096-9098(199606)62:2<135::AID-JSO10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 48.Evans WE, Crom WR, Abromowitch M, Dodge R, Look AT, Bowman WP, George SL, Pui C-H. Clinical pharmacodynamics of high-dose methotrexate in acute lymphocytic leukemia. N Engl J Med. 1986;314:471–477. doi: 10.1056/NEJM198602203140803. [DOI] [PubMed] [Google Scholar]
- 49.Muller C, Chatelut E, Gualano V, De Forni M, Huguet F, Attal M, Canal P, Laurent G. Cellular pharmacokinetics of doxorubicin in patients with chronic lymphocytic leukemia: comparison of bolus administration and continuous infusion. Cancer Chemother Pharmacol. 1993;32:379–384. doi: 10.1007/BF00735923. [DOI] [PubMed] [Google Scholar]
- 50.Trauth BC, Klas C, Peters AJM, Matzku S, Möller P, Falk W, Debatin KM, Krammer PH. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245:301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- 51.Dhein J, Daniel PT, Trauth BC, Oehm A, Möller P, Krammer PH. Induction of apoptosis by monoclonal antibody anti-APO-1 class switch variants is dependent on cross-linking of APO-1 cell surface antigens. J Immunol. 1992;149:3166–3173. [PubMed] [Google Scholar]
- 52.Dhein J, Walczak H, Bäumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 53.Nicoletti I, Migliorati G, Paggliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 54.Peter ME, Dhein J, Ehret A, Hellbardt S, Walczak H, Moldenhauer G, Krammer PH. APO-1(CD95)- dependent and -independent antigen receptor-induced apoptosis in human T and B cell lines. Int Immunol. 1995;7:1873–1877. doi: 10.1093/intimm/7.11.1873. [DOI] [PubMed] [Google Scholar]
- 55.Behrmann I, Walczak H, Krammer PH. Structure of the human APO-1 gene. Eur J Immunol. 1994;24:3057–3062. doi: 10.1002/eji.1830241221. [DOI] [PubMed] [Google Scholar]
- 56.Zauberman A, Barak Y, Ragimov N, Levy N, Oren M. Sequence-specific DNA binding by p53: identification of target sites and lack of binding to p53-MDM2 complexes. EMBO (Eur Mol Biol Organ) J. 1993;12:2799–2808. doi: 10.1002/j.1460-2075.1993.tb05941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wada N, Matsumura M, Ohba Y, Kobayashi N, Takizawa T, Nakanishi Y. Transcription stimulation of the Fas-encoding gene by nuclear factor for interleukin-6 expression upon influenza virus infection. J Biol Chem. 1995;270:18007–18012. doi: 10.1074/jbc.270.30.18007. [DOI] [PubMed] [Google Scholar]
- 58.Rudert F, Visser E, Forbes L, Lindridge E, Wang Y, Watson J. Identification of a silencer, enhancer, and basal promoter region in the human CD95 (Fas/APO-1) gene. DNA Cell Biol. 1995;14:931–937. doi: 10.1089/dna.1995.14.931. [DOI] [PubMed] [Google Scholar]
- 59.Zauberman A, Lupo A, Oren M. Identification of p53 target genes through immune selection of genomic DNA: the cyclin G gene contains two distinct p53 binding sites. Oncogene. 1995;10:2361–2366. [PubMed] [Google Scholar]
- 60.Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int J Radiat Oncol Biol Phys. 1979;5:85–91. doi: 10.1016/0360-3016(79)90044-0. [DOI] [PubMed] [Google Scholar]
- 61.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 62.Fritsche M, Haessler C, Brandner G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents [erratum in 8:2605] Oncogene. 1993;8:307–318. [PubMed] [Google Scholar]
- 63.El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 64.Strand S, Hofmann WJ, Hug H, Müller M, Otto G, Strand D, Mariani SM, Stremmel W, Krammer PH, Galle PR. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells—a mechanism of immune evasion? . Nat Med. 1996;2:1361–1366. doi: 10.1038/nm1296-1361. [DOI] [PubMed] [Google Scholar]
- 65.French LE, Hahne M, Viard I, Radlgruber G, Zanone R, Becker K, Muller C, Tschopp J. Fas and Fas ligand in embryos and adult mice: ligand expression in several immune-privileged tissues and coexpression in adult tissues characterized by apoptotic cell turnover. J Cell Biol. 1996;133:335–343. doi: 10.1083/jcb.133.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Niehans GA, Brunner T, Frizelle SP, Liston JC, Salerno CT, Knapp DJ, Green DR, Kratzke RA. Human lung carcinomas express Fas ligand. Cancer Res. 1997;57:1007–1012. [PubMed] [Google Scholar]
- 67.O'Connell J, O'Sullivan GC, Collins JK, Shanahan F. The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med. 1996;184:1075–1082. doi: 10.1084/jem.184.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stuart PM, Griffith TS, Usui N, Pepose J, Yu X, Ferguson TA. CD95 ligand (FasL)-induced apoptosis is necessary for corneal allograft survival. J Clin Invest. 1997;99:396–402. doi: 10.1172/JCI119173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 70.Bellgrau D, Gold D, Selawry H, Moore J, Franzusoff A, Duke RC. A role for CD95 ligand in preventing graft rejection. Nature. 1995;377:630–632. doi: 10.1038/377630a0. [DOI] [PubMed] [Google Scholar]
- 71.Griffith TS, Yu X, Herndon JM, Green DR, Ferguson TA. CD95-induced apoptosis of lymphocytes in an immune privileged site induces immunological tolerance. Immunity. 1996;5:7–16. doi: 10.1016/s1074-7613(00)80305-2. [DOI] [PubMed] [Google Scholar]
- 72.Seino K, Kayagaki N, Okumura K, Yagita H. Antitumor effect of locally produced CD95 ligand. Nat Med. 1997;3:165–170. doi: 10.1038/nm0297-165. [DOI] [PubMed] [Google Scholar]
- 73.Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 74.Zauberman A, Flusberg D, Haupt Y, Barak Y, Oren M. A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res. 1995;23:2584–2592. doi: 10.1093/nar/23.14.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okamoto K, Beach D. Cyclin G is a transcriptional target of the p53 tumor suppressor protein. EMBO (Eur Mol Biol Organ) J. 1994;13:4816–4822. doi: 10.1002/j.1460-2075.1994.tb06807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Werner H, Karnieli E, Rauscher FJ, III, Leroith D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc Natl Acad Sci USA. 1996;93:8318–8323. doi: 10.1073/pnas.93.16.8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roth JA, Nguyen D, Lawrence DD, Kemp BL, Carrasco CH, Ferson DZ, Hong WK, Komaki R, Lee JJ, Nesbitt JC, et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat Med. 1996;2:985–991. doi: 10.1038/nm0996-985. [DOI] [PubMed] [Google Scholar]
- 78.Fujiwara T, Grimm EA, Mukhopadhyay T, Zhang WW, Owen SL, Roth JA. Induction of chemosensitivity in human lung cancer cells in vivo by adenovirus-mediated transfer of the wild-type p53 gene. Cancer Res. 1994;54:2287–2291. [PubMed] [Google Scholar]
- 79.Nguyen DM, Spitz FR, Yen N, Cristiano RJ, Roth JA. Gene therapy for lung cancer: enhancement of tumor suppression by a combination of sequential systemic cisplatin and adenovirus-mediated p53 gene transfer. J Thorac Cardiovasc Surg. 1996;112:1372–1376. doi: 10.1016/S0022-5223(96)70154-X. [DOI] [PubMed] [Google Scholar]
- 80.Wu GS, Burns TF, McDonald ER, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–143. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]