

Abstract
Fas and Fas-associated death domain (FADD) play a critical role in the homeostasis of different cell types. The regulation of Fas and FADD-mediated cell death is pivotal to many physiological functions. The activation of T lymphocytes by concanavalin A (Con A) inhibited Fas-mediated cell death. We identified that among the several activation signals downstream of Con A stimulation, mitogen-activated protein (MAP) kinase kinase (MKK) was the major kinase pathway that antagonized Fas-triggered cell death. MKK1 suppressed FADD- but not caspase-3– induced apoptosis, indicating that antagonism occurred early along the Fas-initiated apoptotic cascade. We further demonstrated that activation of MKK1 led to expression of FLIP, a specific inhibitor of FADD. MKK1 inhibition of FADD-induced cell death was abrogated if induction of FLIP was prevented, indicating that FLIP mediates MKK1 suppression of FADD-mediated apoptosis. Our results illustrate a general mechanism by which activation of MAP kinase attenuates apoptotic signals initiated by death receptors in normal and transformed cells.
Keywords: mitogen-activated protein kinase kinase, Fas-associated death domain protein, Fas, FLIP, apoptosis
Fas (APO-1, CD95) is a 45-kD transmembrane protein (1) which transmits apoptotic signals upon engagement by Fas ligand or by specific anti-Fas antibodies. The death pathway initiated from Fas involves a series of death- induced molecules (1). Fas-associated death domain protein (FADD; MORT1)1 is directly recruited to Fas upon Fas engagement (2, 3). FADD then binds caspase-8 (FADD-like IL-1β–converting enzyme [FLICE] or MACH; references 4–6), followed by eventual activation of caspase-3 (CPP32 [7, 8]). FADD is also recruited to TNFR1 by TNFR1-associated death domain protein (TRADD) adapter, and transmits the death signal downstream of TNF binding (9). More recently, FADD was identified as the common mediator for apoptosis triggered by death receptor (DR)3 and DR5 (10–14). In addition, FADD is essential for embryo development (14, 15).
Fas-initiated apoptosis is regulated by a few molecules, including Bcl-2, Bcl-XL, crmA, and inhibitor of apoptosis protein (IAP) (1). Of these regulatory molecules, one that interferes most upstream in the Fas-mediated death cascade is FLIP (FLAME-1, I-FLICE, CASH, casper), a homologue of FLICE (16–20). Both the short form and long form of FLIP (FLIPS and FLIPL) interact with FADD and FLICE. Overexpression of FLIP suppresses Fas- and TNF-induced apoptosis (16–18, 20). Increased expression of FLIP is found in Fas ligand–resistant melanoma cell lines and in melanoma tumors (16). A viral homologue of FLIP also prevents DR-induced apoptosis (21).
Fas-induced apoptosis in T lymphocytes is known to be antagonized by activation through TCRs (22–24). In search of an activation signal that interferes with Fas-triggered apoptosis, we identified mitogen-activated protein kinase (MAPK) as the major kinase pathway which inhibits FADD-mediated cell death. We also demonstrated that MAPK kinase (MKK) abrogated Fas-initiated apoptosis through the induction of FLIP expression. Our results reveal the molecular mechanism by which activation of MAPK, mostly through tyrosine kinase receptors and G protein–coupled receptors, attenuates apoptotic signals initiated by DRs in normal and transformed cells.
Materials and Methods
Reagents and Cell Lines.
TPA, A23187, and Con A were purchased from Sigma Chemical Co. (St. Louis, MO). PD 098059, SB 203580, wortmannin, rapamycin, and calphostin c were purchased from Calbiochem Corp. (San Diego, CA). Cyclosporin A was a gift of Sandoz Pharmaceutical Co. (Taipei, Taiwan). Anti– human Fas antibody CH-11 (25) was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY). Anti–mouse Fas antibody Jo2 was obtained from PharMingen (San Diego, CA). Human T cell leukemia Jurkat cells (TIB 152) were obtained from the American Type Culture Collection (Rockville, MD). Thymocytes and splenocytes were isolated from 8-wk-old MRL +/+ and MRL lpr/lpr mice (The Jackson Laboratory, Bar Harbor, ME). Splenic T cells were purified as described previously (26) and activated with TPA/A23187 for 24 h. Activated splenic T cells were then washed, and incubated in the presence of IL-2 (10 U/ml) for another 3 d before anti-Fas treatment. For stimulation of splenic T cells with Con A, glutaraldehyde-fixed B cells TA3 (27) were added as presenting cells.
Plasmids.
pcDNA3-AU1-FADD (2) and caspase-3 (CPP32/ Yama [28]) were gifts of Dr. Vishva Dixit (University of Michigan, Ann Arbor, MI). The constitutively active form of MKK1, pMCL-MKK1-N3/S218E/S222D (29), was a gift of Dr. Natalie G. Ahn (University of Colorado, Boulder, CO). The active mutant of MKK3b [MKK3b(Glu189, Glu193); 30] was a gift of Dr. Jiahuai Han (Scripps Research Institute, La Jolla, CA). The full length of FLIP long form (−74 to +1515 nucleotides [nt]) was isolated by reverse-transcription (RT)-PCR, blunt ended, partially sequenced, and subcloned into the EcoRV site of pcDNA3 (Invitrogen Corp., Leek, The Netherlands) with an HA tag. FLIP antisense construct contained the antisense sequence of FLIP −74 to +615 nt in pcDNA3. Green fluorescence protein expression vector pGreen Lantern-1 was purchased from GIBCO BRL (Gaithersburg, MD).
Quantitation of FLIP mRNA.
2 μg of total RNA was used for cDNA synthesis by using oligo-dT as primer (31). 1/10 of the cDNA synthesized was then amplified by using the following primers: human (h)FLIPL/S 5′, TGT TGC TAT AGA TGT GG; hFLIPL/S 3′, CAG GTC TAT TCT GTG GA; hFLIPL 5′, ACT ATG TGG TGT CAG AGG GCC AG; hFLIPL 3′ is the same as hFLIPL/S 3′; murine (m)FLIP 5′, GTC ACA TGA CAT AAC CCA GAT TGT; and mFLIP 3′, GTA CAG ACT GCT CTC CCA AGC.
Transfection, Immunoblot, and MAPK Assay.
Jurkat T cells and activated splenic T cells were transfected with the DEAE-dextran method (32). For 293T cells, transfection was performed using the calcium phosphate method. Immunoblot was performed according to the method described previously (32). MAPK activity was analyzed by immunoprecipitation of cell lysate with anti-ERK2 C-14 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and the kinase activity of the immune complex was determined as described previously (26).
Cell Death Measurement.
All cultures were performed in RPMI with 10% FCS (GIBCO BRL), 10 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 × 10−5 M 2-ME. The extent of apoptosis was determined by propidium iodide (PI) staining. At the indicated times after treatment with different agents, cells were harvested, washed twice in PBS, and resuspended in hypotonic fluorochrome solution (50 μg/ml PI, 0.1% sodium citrate, 0.1% Triton X-100 [33]). Cells were placed at 4°C in the dark overnight, and DNA content was analyzed by FACScan® (Becton Dickinson, Mountain View, CA). The fraction of cells with sub-G1 DNA content was assessed using the CellFit program (Becton Dickinson). For cells transiently transfected with FADD/MKK1, survival was monitored with cotransfection of green fluorescence protein expression vector pGreen Lantern-1 (GIBCO BRL [34]). Treated cells were examined using a fluorescence microscope (Nikon Inc., Melville, NY).
Results
Inhibition of Fas-mediated Apoptosis in Jurkat T Cells by Con A Activation Was Mediated by MKK.
Anti-Fas antibody CH-11 (25) induced extensive cell death in Jurkat cells (Fig. 1 A). Activation of Jurkat cells by Con A reduced Fas-activated cell death from 75 to 20% (Fig. 1 A). The suppression of cell death was not due to activation-induced secretion of IL-2, as IL-2 (50 U/ml) by itself did not prevent Fas-activated apoptosis (not shown). The inhibition of Fas-triggered apoptosis must be due to the activation signal downstream of TCR engagement. We tested several signaling pathways that may be involved in the antagonism of Fas-induced cell death by using specific inhibitors for each pathway. The addition of PD 098059, a selective inhibitor of MKK (35), effectively abrogated the preventive effect of Con A (Fig. 1 A). Only a weak or minimal reversal effect was observed from inhibition of other signaling molecules on Con A–activated Jurkat T cells. The inhibitors tested included calphostin c, cyclosporin A, SB 203580, wortmannin, and rapamycin. Protein kinase C, phosphatidylinositide 3-kinase, and protein kinase B have been implicated to protect against Fas-mediated apoptosis (36, 37), yet MKK was the major kinase activated by Con A stimulation that antagonized Fas-initiated cell death. A role of MAPK in the suppression of Fas-induced T cell death by lectin has also been recently reported (23). The antiapoptotic activity of MKK was further confirmed by transient expression of active MKK1 (29) in Jurkat cells. Transfection of the vector alone (pcDNA3) did not interfere with Fas-induced apoptosis. The induction of MAPK (see below) by MKK1 prevented apoptosis of Jurkat cells triggered by anti-Fas antibody (Fig. 1 B). In contrast, the expression of active MKK3b (30), which led to activation of p38 MAPK (Fig. 1 C), did not increase resistance to Fas-induced apoptosis in Jurkat cells. Neither MKK1 nor MKK3b alone induces cell death of Jurkat cells (Fig. 1 B).
Figure 1.

Inhibition of anti-Fas–induced apoptosis in Jurkat T cells by Con A activation was mediated by MKK. (A) Jurkat cells were stimulated with anti-Fas antibody CH11 (125 ng/ml) and/or Con A (4 μg/ml) in the absence or presence of the inhibitors for 16 h. DNA content was determined by staining with 20 μg/ml PI and analyzed by FACScan® (Becton Dickinson). The fraction of cells with sub-G1 DNA content was assessed by the CellFit program (Becton Dickinson) and used as percent cell death. The inhibitors used were calphostin C (CC, 0.5 μM), cyclosporin A (CsA, 20 μg/ml), PD 098059 (PD, 50 μM), SB 203580 (SB, 20 μM), wortmannin (W, 100 nM), and rapamycin (R, 20 ng/ml). None of the inhibitors alone induced DNA fragmentation at the concentrations used. The result is the average of duplicates ± SD. Experiments were repeated twice with identical results. (B) MKK1 but not MKK3 prevented Fas- mediated apoptosis in Jurkat cells. Jurkat cells were transfected with pcDNA3, active MKK1 (29), or active MKK3b (30), together with 5 μg of pGreen Lantern-1 (GIBCO BRL) by the DEAE-dextran method. 30 h later, half of the transfected cells were treated with CH11 (black bars) and the other half were untreated (white bars). Viability of cells was determined after another 15 h by the cells expressing green fluorescence (reference 34). (C) Transfection of MKK3b led to activation of p38 MAPK. Jurkat cells were transfected with pcDNA3 (Vec) or active MKK3b (MKK3), and cell extracts were prepared 40 h later. The content of phosphorylated p38 MAPK (pp38) and p38 MAPK (p38) were determined by immunoblots with antiphosphorylated T180/Y182 p38 MAPK antibody (New England Biolabs, Inc., Beverly, MA) and anti-p38 MAPK antibody C-20 (Santa Cruz Biotechnology, Inc.), respectively.
MAPK Activation Suppressed FADD-induced Apoptosis but not Caspase-3–induced Cell Death.
We next attempted to identify the stage along the Fas-initiated apoptotic pathway that was antagonized by MKK. Jurkat cells were transfected with FADD and caspase-3 (CPP32) together with the vector or active MKK1 (Fig. 2 A). The cell death induced by FADD was significantly prevented by cotransfection with active MKK1. In contrast, overexpression of active MKK1 did not prevent cell death triggered by caspase-3 (Fig. 2 A). The inhibition by MKK was at the early stage of the Fas-mediated apoptotic pathway. Because only a small fraction of Jurkat cells (<10%) were transfected with the gene of interest in the transient assay, we further examined the preventive effect of MAPK in 293T cells. Tests using green fluorescence protein indicated a transfection efficient >50%. The high efficiency of transfection was demonstrated by the MAPK activation in 293T cells transfected with active MKK1 (Fig. 2 C), in which prominent MAPK activity was detected 6 h after transfection. The activation of MAPK then subsided, but was still higher than that of the untransfected cells 12 and 24 h after transfection. Transfection of FADD and caspase-3 also led to extensive cell death of 293T cells as measured by PI staining (Fig. 2 B). The coexpression of active MKK1 in 293T cells suppressed the cell death induced by FADD. Similar to that observed in Jurkat cells, overexpression of active MKK1 did not rescue the apoptosis induced by caspase-3. MAPK activation interfered with the Fas-initiated apoptotic cascade at the stage before the activation of caspase-3.
Figure 2.

MAPK activation suppressed FADD-induced apoptosis but not caspase-3–induced cell death. (A) Jurkat cells were transfected by the DEAE-dextran method with pcDNA3 (Control), FADD (1 μg), or caspase-3 (CPP32, 3 μg), together with the vector (Vec) or active MKK1 (3 μg). 3 μg of pGreen Lantern-1 was included in each transfection and pcDNA3 was used to make a total dose of 10 μg. Viability was determined 30 h after transfection as described in the legend to Fig. 1. Data are the average of two independent transfections. 100% viability represents cells transfected with pGreen Lantern-1 and pcDNA3 only. (B) 293T cells were transfected by the calcium phosphate method with the same plasmids used in A. The apoptosis of the transfected cells was determined by staining with 20 μg/ml PI and analyzed by FACScan® (Becton Dickinson). The fraction of cells with sub-G1 DNA content was used as percent cell death. (C) Active MKK1 induced MAPK activation. 293T cells were transfected with active MKK1, and cell extracts were prepared at the indicated time points. MAPK was precipitated by anti-ERK2 antibody, and phosphorylation of myelin basic protein (MBP) was determined by immunocomplex kinase assay.
MAPK Activation Induced FLIP Expression.
The antagonism of FADD-triggered cell death by active MKK1 resembled the inhibitory activity of FLIP. Both the short form and long form of human FLIP (hFLIPS and hFLIPL) interact with FADD and FLICE, and suppress FADD- induced cell death (16–18, 20). We used primers to detect the expression of both long form and short form (hFLIPL/S) or long form only (hFLIPL) by RT-PCR. FLIP expression in Jurkat cells was induced by T cell activation from stimulation with Con A (Fig. 3 A). The expression profiles of hFLIPL/S and hFLIPL were similar. The optimum induction of FLIP mRNA was 4 h after Con A activation. The induction of hFLIPL/S was suppressed in the presence of MKK inhibitor PD 098059, suggesting that MKK was required for FLIP expression. We further checked if activation of MKK1 would lead to induction of FLIP. The endogenous FLIP mRNA in 293T cells was weak but clearly detectable. Transfection of 293T cells with active MKK1 significantly increased the expression of hFLIPL/S (Fig. 3 B). The induction was visible 6 h after transfection and peaked at 24 h. Vector alone did not lead to any increase in hFLIPL/S mRNA 24 h after transfection. Both the endogenous and the induced expression of FLIP were largely suppressed in the presence of PD 098059 (Fig. 3 B), supporting their dependence on MKK.
Figure 3.
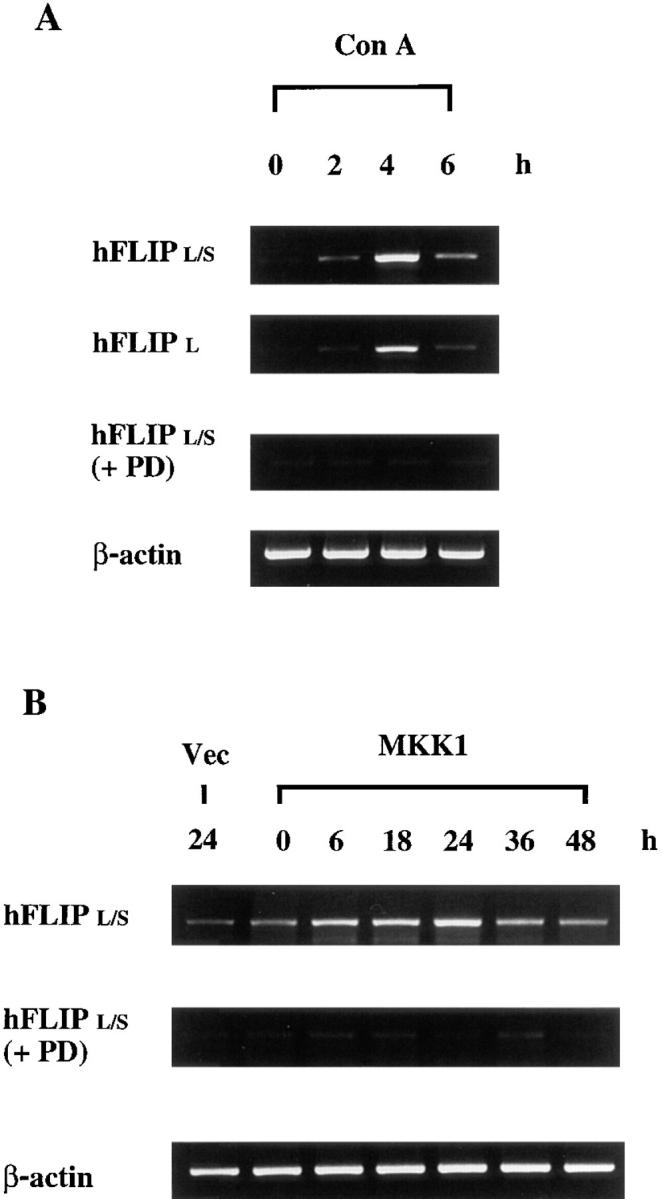
FLIP expression was induced by activated MKK. (A) Jurkat T cells were activated with Con A in the absence or presence of PD 098059, and total RNA was isolated 2, 4, and 6 h after activation. (B) 293T cells were transfected with active MKK1 or pcDNA3 (Vec) in the absence or presence of PD 098059, and RNA was prepared at the indicated time points. 2 μg of total RNA was used for cDNA synthesis by using oligo-dT as primer. 1/10 of the cDNA synthesized was then amplified by using primers specific for hFLIPL/S (long and short forms), hFLIPL (long form only), and actin. 20% of the PCR products were resolved on an agarose gel for comparison.
Antagonism of Fas-mediated Apoptosis in Normal T cells by Con A Activation Was Mediated by MAPK and FLIP.
Because signaling in lymphoma and in embryonic kidney cells may be distinct from those in normal T cells, we further examined whether activation of normal T cells antagonized Fas-initiated apoptosis. MRL +/+ thymocytes, known to express a high level of Fas (38), were stimulated with immobilized anti–mouse Fas antibody Jo2. Significant cell death of +/+ thymocytes was detected 4 h after Fas engagement (Fig. 4 A). The apoptosis observed was Fas-specific because Jo2 did not promote any death in lpr/lpr thymocytes. Con A by itself promoted immature T cell death. However, costimulation with Con A largely prevented Fas-mediated apoptosis in thymocytes (Fig. 4 A). The inhibitory effect of Con A was abrogated in the presence of PD 098059 (10 μM). Similar to that observed in Jurkat cells, Con A activation led to FLIP expression in mouse thymocytes (Fig. 4 B). The FLIP induction in thymocytes was significantly earlier than Jurkat cells after Con A activation. The sensitivity to PD 098059 indicated that Con A–induced FLIP expression in thymocytes was also dependent on MKK.
Figure 4.


Antagonism of Fas-induced apoptosis in normal thymocytes by Con A activation was mediated by MKK. (A) Freshly isolated thymocytes from 8-wk-old MRL +/+ and MRL lpr/lpr mice were stimulated with immobilized anti-Fas antibody Jo2 (coated at 5 μg/ml) and/or Con A (4 μg/ml) in the absence or presence of PD 098059 (PD, 10 μM) for 4 h. Cell death was determined by Annexin V (Clontech, Palo Alto, CA) staining. Data shown are thymocytes from +/+ mice except Jo2 (lpr). The result is the average of duplicates ± SD. Experiments were repeated twice with identical results. (B) Induction of FLIP was MKK-dependent in thymocytes. +/+ thymocytes were activated with Con A in the absence or presence of 10 μM PD 098059, and total RNA was isolated 1, 2, and 3 h after activation. FLIP transcript was determined as described in the legend to Fig. 3 except primers for mFLIP were used during PCR analysis.
In addition to thymocytes, we used mature T lymphocytes isolated from spleen. Fas expression in splenic T cells was induced by TPA/A23187, and T cells were then cultured in the presence of IL-2. Treatment of preactivated splenic T cells with Jo2 led to extensive cell death (Fig. 5 A). Fas-mediated apoptosis in activated splenic T cells was prevented by Con A stimulation in a PD 098059–sensitive manner (Fig. 5 A). MKK was also essential for the induction of FLIP in Con A–treated splenic T cells (Fig. 5 B). Therefore, MKK and FLIP antagonized Fas-induced apoptosis in normal T cells as well as in T lymphomas.
Figure 5.


Inhibition of Fas-induced apoptosis in preactivated splenic T cells by Con A activation was mediated by MKK. (A) +/+ splenic T cells were purified and activated with TPA/A23187 for 24 h. The cells were then washed and incubated in IL-2 (10 U/ml) for another 3 d. The viable cells were recovered by Percoll (Amersham Pharmacia Biotech, Uppsala, Sweden) and stimulated with anti-Fas antibody Jo2 (coated at 5 μg/ml) and/or Con A (4 μg/ml) in the absence or presence of PD 098059 (PD, 20 μM) for 16 h. For Con A stimulation, 4 × 104 glutaraldehyde-fixed TA3 (reference 27) were added in each well. DNA content was determined by staining with 20 μg/ml PI as described in the legend to Fig. 1. The result is the average of duplicates ± SD. Experiments were repeated twice with identical results. (B) Activated splenic T cells were stimulated with Con A–fixed TA3 in the absence or presence of 20 μM PD 098059, and total RNA was isolated 2 and 4 h after activation. FLIP transcript was determined as described in the legend to Fig. 4.
Antagonism of FLIP Induction by Antisense Construct Abrogated the Protective Effect of MKK1.
We next examined whether the antiapoptotic effect of MKK was indeed mediated by the induced expression of FLIP. The FLIP cDNA was obtained using RT-PCR based on the published FLIP sequence (16, 19). The isolated FLIP effectively prevented FADD-induced cell death in 293T cells (Fig. 6 A). An antisense construct of FLIP was then prepared which contained the 5′ half of FLIP cDNA (−74 to +615 nt) in antisense orientation. The expression of HA-FLIP protein level was suppressed by 50% when cotransfected with an equal amount of the FLIP antisense into 293T cells (not shown). The effectiveness of the FLIP antisense construct was further confirmed by inhibition of the FLIP-mediated antiapoptotic effect (Fig. 6 A). The FLIP antisense construct alone did not induce cell death in 293T cells. The coexpression of the FLIP antisense construct with active MKK1 in Jurkat cells abrogated the suppression of Fas-induced apoptosis (Fig. 6 B). The FLIP antisense construct by itself did not affect the viability of Jurkat cells. Similarly, FADD-triggered cell death of 293T cells was no longer prevented by MKK1 when FLIP antisense was cotransfected (Fig. 6 C). Blockage of FLIP expression by the antisense construct eliminated the antagonistic effect of MKK1 in Jurkat and 293T cells. This was also the case for normal T lymphocytes. Even though the transfection efficiency in preactivated splenic T lymphocytes was much lower than Jurkat cells, transient transfection of active MKK1 conferred resistance of preactivated splenic T lymphocytes to Jo2-induced apoptosis (Fig. 6 D). The presence of FLIP antisense removed the protective effect of MKK1 in normal T cells. Therefore, the interference of Fas- and FADD-induced apoptosis by MKK was likely mediated by FLIP in different types of cells.
Figure 6.

Antagonism of Fas/FADD-induced apoptosis was abrogated by inhibition of FLIP induction. (A) Expression of FLIP inhibited FADD-induced apoptosis, which was abrogated by the FLIP antisense. 293T cells were transfected with 2 μg of FADD, 2 μg of HA-pcDNA3-FLIP (FLIP), with or without 5 μg of pcDNA3-antisense-FLIP (FLIP-anti). The fraction of cells with sub-G1 DNA content was determined 24 h later and used as percent cell death. (B) Coadministration of the FLIP antisense blocked the preventive effect of MKK1 on Fas-induced apoptosis in Jurkat cells. Jurkat T cells were transfected with pGreen Lantern-1 and/or MKK1, in the presence or absence of 5 μg pcDNA3-antisense-FLIP. 30 h later, half of the transfected cells were treated with CH11 (black bars) and the other half were untreated (white bars). Viability of cells was determined after another 15 h by the cells expressing green fluorescence in a 5-mm2 area (reference 34). Numbers are averages of five independent measurements. (C) Coadministration of FLIP antisense reversed the antagonist effect of MKK1 on FADD-induced death. 293T cells were transfected with FADD, MKK1, and/or pcDNA3-antisense-FLIP. Percent cell death represents the fraction of cells with sub-G1 DNA content. (D) FLIP antisense abrogated the protective effect of MKK1 on Fas-initiated apoptosis in normal T cells. Preactivated splenic T cells were transfected with pGreen Lantern-1, vector (Control), or MKK1, in the presence or absence of 5 μg pcDNA3-antisense-FLIP by the DEAE-dextran method. Viability was determined as described in B except Jo2 was used to induce cell death.
Discussion
In this study, we illustrated that MAPK activation effectively prevents Fas- and FADD-initiated cell death. In addition, the death-inhibitory activity of MAPK is likely due to the induced expression of FLIP. MKK inhibitor PD 098059 suppressed the induction of FLIP in T cells by Con A (Figs. 3 A, 4 B, and 5 B), and the protective effect of Con A was abrogated (Figs. 1 A, 4 A, and 5 A). FLIP mRNA induction reached its optimum 4 h after Con A stimulation, but decreased thereafter. The transient induction of FLIP mRNA may seem incompatible with the prolonged suppression of Fas- and FADD-triggered cell death 24 h after induction. The prolonged antiapoptotic effect of FLIP may be due to its location, as demonstrated by both FLIP and viral FLIP being incorporated in the Fas-containing death-inducing signaling complex (16, 21). Transient expression of FLIP may generate enough FLIP protein to suppress the activity of the death-inducing signaling complex. This was further supported by the fact that the antisense construct of FLIP, which inhibited the expression of FLIP, eliminated most of the antagonistic effect of MKK1 (Fig. 6, B–D). Induction of FLIP may thus account for most of the antiapoptotic activity of active MKK1. Notably, the central role of FLIP in the regulation of Fas-induced apoptosis in T cells is supported by increasing evidence. Toso, a recently identified specific inhibitor of Fas-mediated apoptosis, acts through induction of FLIP expression (39). Part of the mechanism by which IL-2 enhances Fas-mediated T cell apoptosis is mediated by an inhibition of FLIP expression (40).
Recent studies indicate that apoptotic signal transmitted by Fas engagement is enhanced by TCR signals in T hybridomas and in mature T cells (41, 42). Similarly, we have observed that anti-CD3 alone failed to protect Fas-induced apoptosis of activated splenic T cells (not shown). Our observation that Con A activation led to inhibition of Fas-initiated cell death in T cells (Figs. 1, 4, and 5) suggests that costimulation signals are required for effective antagonism of FADD-mediated apoptosis. This is consistent with a report on the protection against Fas-induced apoptosis by superantigen and CD28 in mature human T cells (24).
FADD is the apoptotic mediator of several DRs, including Fas, TNFR1, DR3, and DR5 (2, 3, 9–14). Hence, FADD is activated by death signals through the binding of the Fas ligand, TNF, and TNF-related apoptosis–inducing ligand (TRAIL), whereas MAPK is activated by engagement of tyrosine kinase receptor, G protein–coupled receptor, and protooncogenes (43, 44). The activation of MAPK is essential for cell proliferation and mitogenesis, yet its role in antiapoptosis is less definite. MAPK has been shown to inhibit apoptosis induced by nerve growth factor deprivation in PC12 cells (45). MAPK has also been demonstrated to be involved in the inhibition of ceramide-induced apoptosis by sphingosine 1-phosphate (46) and in the suppression of cardiac myocyte apoptosis by cardiotrophin 1 (47). Because of the diverse biological targets of MAPK, the mechanism of MAPK in the antagonism of apoptosis remains mostly elusive. This study reveals a novel mechanism by which MAPK antagonizes FADD-mediated death through the induction of a FADD inhibitor. Therefore, the antagonism of FADD-initiated apoptosis by MKK represents an interesting cross-talk between two distinct signaling pathways, one apoptotic and the other mitogenic. Given the range of physiological functions and pathological consequences mediated by DRs, the antagonism of FADD by MKK illustrates how the early stage of DR-induced apoptosis can be interrupted. Interestingly, the Fas-activated apoptotic process is accompanied by an early cleavage of Raf-1, the activator of MKK (48). In this manner, the antiapoptotic MKK pathway is inhibited during the progression of the death pathway. Further molecular characterization of the antagonism between FADD and MKK will help efforts to place DR-mediated apoptosis under the appropriate control.
Acknowledgments
We thank Dr. Vishva Dixit for CPP32 and pcDNA3-AU1-FADD, Dr. Natalie Ahn for active MKK1, and Dr. Jiahuai Han for active MKK3b. We also thank Mr. Douglas Platt for editorial correction of the manuscript.
Abbreviations used in this paper
- DR
death receptor
- FADD
Fas-associated death domain protein
- FLICE
FADD-like ICE
- FLIP
FLICE-inhibitory protein
- ICE
IL-1β–converting enzyme
- MAPK
mitogen-activated protein kinase
- MKK
MAPK kinase
- nt
nucleotide(s)
- PI
propidium iodide
- RT
reverse transcription
Footnotes
This project was supported by grant DOH86-HR-508 from the Department of Health, grant NSC 86-2316-B001-012 M30 from the National Science Council, and a grant from Academia Sinica.
References
- 1.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 2.Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 3.Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- 4.Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, et al. Flice, a novel FADD-homologue ICE/ CED-3-like protease, is recruited to the CD95(Fas/APO-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 5.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/ FADD-interacting protease in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85:803–815. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 6.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO J. 1997;16:2794–2804. doi: 10.1093/emboj/16.10.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like protease during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 8.Hirata H, Takahashi A, Kobayashi S, Yonehara S, Sawai H, Okazaki T, Yamamoto K, Sasada M. Caspases are activated in a branched protease cascade and control distinct downstream process in Fas-induced apoptosis. J Exp Med. 1998;187:587–600. doi: 10.1084/jem.187.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chinnaiyan AM, Tepper CG, Seldin MF, O'Rourke K, Kischkel FC, Hellbardt S, Krammer PH, Peter ME, Dixit VM. FADD/MORT1 is a common mediator of CD95 (Fas/Apo-1) and tumor necrosis factor-induced apoptosis. J Biol Chem. 1996;271:4961–4965. doi: 10.1074/jbc.271.9.4961. [DOI] [PubMed] [Google Scholar]
- 10.Chinnaiyan AM, O'Rourke K, Yu G-L, Lyons RH, Garg M, Duan DR, Xing L, Gentz R, Ni J, Dixit VM. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–992. doi: 10.1126/science.274.5289.990. [DOI] [PubMed] [Google Scholar]
- 11.Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, Timour MS, Gerhart MJ, Schooley KA, Smith CA, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–5397. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-κB pathway. Immunity. 1997;7:821–830. doi: 10.1016/s1074-7613(00)80400-8. [DOI] [PubMed] [Google Scholar]
- 13.Schneider P, Thome M, Burns K, Bodmer J-L, Hofmann K, Kataoka T, Holler N, Tschopp J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-κB. Immunity. 1997;7:831–836. doi: 10.1016/s1074-7613(00)80401-x. [DOI] [PubMed] [Google Scholar]
- 14.Yeh W-C, de la Pompa JL, McCurrach ME, Shu H-B, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Cado D, Chen A, Kabra NH, Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392:296–300. doi: 10.1038/32681. [DOI] [PubMed] [Google Scholar]
- 16.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer J-L, Schroter M, Burns K, Mattmann C, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 17.Goltsev YV, Kovalenko AV, Arnold E, Varfolomeev EE, Brodianskii VM, Wallach D. CASH, a novel caspase homologue with death effector domains. J Biol Chem. 1997;272:19641–19644. doi: 10.1074/jbc.272.32.19641. [DOI] [PubMed] [Google Scholar]
- 18.Hu S, Vincenz C, Ni J, Gentz R, Dixit VM. I-FLICE, a novel inhibitor tumor necrosis factor receptor-1 and CD-95-induced apoptosis. J Biol Chem. 1997;272:17255–17257. doi: 10.1074/jbc.272.28.17255. [DOI] [PubMed] [Google Scholar]
- 19.Shu H-B, Halpin DR, Goeddel DV. Casper is a FADD- and caspase-related inducer of apoptosis. Immunity. 1997;6:751–763. doi: 10.1016/s1074-7613(00)80450-1. [DOI] [PubMed] [Google Scholar]
- 20.Srinivasula SM, Ahmad M, Ottlie S, Bullrich F, Banks S, Wang Y, Fernandes-Alnemri T, Croce CM, Litwack G, Tomaselli KJ, et al. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J Biol Chem. 1997;272:18542–18545. doi: 10.1074/jbc.272.30.18542. [DOI] [PubMed] [Google Scholar]
- 21.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer J-L, Schroter M, et al. Viral FLICE-inhibitory proteins (FLIPS) prevent apoptosis induced by death receptor. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 22.Klas C, Debatin K-M, Jonker RR, Krammer PH. Activation interferes with APO-1 pathway in mature human T cells. Int Immunol. 1993;6:625–630. doi: 10.1093/intimm/5.6.625. [DOI] [PubMed] [Google Scholar]
- 23.Holmstrom TH, Chow SC, Elo I, Coffey ET, Orrenius S, Sistonen L, Eriksson JE. Suppression of Fas/APO-1-mediated apoptosis by mitogen-activated kinase signaling. J Immunol. 1998;160:2626–2636. [PubMed] [Google Scholar]
- 24.McLeod JD, Walker LSK, Patel YI, Boulougouris G, Sansom DM. Activation of human T cells with superantigen (staphylococcal enterotoxin B) and CD28 confers resistance to apoptosis via CD95. J Immunol. 1998;160:2072–2079. [PubMed] [Google Scholar]
- 25.Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with receptor of tumor necrosis factor. J Exp Med. 1989;169:1747–1756. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsueh Y-P, Lai M-Z. JNK but not MAP kinase is sensitive to cAMP in T lymphocytes. J Biol Chem. 1995;270:18094–18098. doi: 10.1074/jbc.270.30.18094. [DOI] [PubMed] [Google Scholar]
- 27.Lee M-R, Liou M-L, Liou M-L, Yang Y-F, Lai M-Z. cAMP analogs prevent activation-induced apoptosis of T cell hybridoma. J Immunol. 1993;151:5208–5217. [PubMed] [Google Scholar]
- 28.Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of CED-3, is a crmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 29.Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande GF, Woude, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38β) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- 31.Lai M-Z, Jang Y-J, Chen L-K, Gefter ML. Restricted V-(D)-J junctional regions in the T cell response to λ repressor: identification of residues critical for antigen recognition. J Immunol. 1990;144:4851–4856. [PubMed] [Google Scholar]
- 32.Hsueh Y-P, Liang H-E, Ng S-Y, Lai M-Z. CD28 costimulation activates CREB in T lymphocytes. J Immunol. 1997;158:85–93. [PubMed] [Google Scholar]
- 33.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 34.Hsu S-C, Wu C-C, Luh T-Y, Chou C-K, Han S-H, Lai M-Z. Apoptotic signal of Fas is not mediated by ceramide. Blood. 1998;91:2658–2663. [PubMed] [Google Scholar]
- 35.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 36.del Carmen Ruiz-Ruiz, M., M. Izquierdo, G. de Murcia, and A. Lopez-Rivas. Activation of protein kinase C attenuates early signals in Fas-mediated apoptosis. Eur J Immunol. 1997;27:1442–1450. doi: 10.1002/eji.1830270622. [DOI] [PubMed] [Google Scholar]
- 37.Hauser P, Papoff G, Eramo A, Reif K, Cantrell DA, Ruberti G. Protection of CD95-mediated apoptosis by activation of phosphatidylinositide 3-kinase and protein kinase B. Eur J Immunol. 1998;28:57–69. doi: 10.1002/(SICI)1521-4141(199801)28:01<57::AID-IMMU57>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 38.Nishimura Y, Ishii A, Kobayashi Y, Yamasaki Y, Yonehara S. Expression and function of mouse Fas antigen on immature and mature T cells. J Immunol. 1995;154:4395–4403. [PubMed] [Google Scholar]
- 39.Hitoshi Y, Lorens J, Kitada S, Fisher J, LaBarge M, Ring HZ, Francke U, Reed JC, Kinoshita S, Nolan GP. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity. 1998;8:461–471. doi: 10.1016/s1074-7613(00)80551-8. [DOI] [PubMed] [Google Scholar]
- 40.Rafaeli Y, Parijs LV, London CA, Tschopp J, Abbas AK. Biochemical mechanism of IL-2 regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–623. doi: 10.1016/s1074-7613(00)80566-x. [DOI] [PubMed] [Google Scholar]
- 41.Wong B, Arron J, Choi Y. T cell receptor signals enhance susceptibility to Fas-mediated apoptosis. J Exp Med. 1997;186:1939–1944. doi: 10.1084/jem.186.11.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hornung F, Zheng L, Lenardo MJ. Maintenance of clonotype specificity in CD95/Apo-1/Fas-mediated apoptosis of mature T lymphocytes. J Immunol. 1997;159:3816–3822. [PubMed] [Google Scholar]
- 43.Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci USA. 1993;90:5889–5892. doi: 10.1073/pnas.90.13.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 45.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinase on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 46.Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind JS, Spiegel S. Suppression of ceramide-mediated cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 47.Sheng Z, Knowlton K, Chen J, Hoshijima M, Brown JH, Chien KR. Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via a mitogen-activated protein kinase-dependent pathway. Divergence from downstream CT-1 signals for myocardial cell hypertrophy. J Biol Chem. 1997;272:5783–5791. doi: 10.1074/jbc.272.9.5783. [DOI] [PubMed] [Google Scholar]
- 48.Widmann C, Gibson S, Johnson GL. Caspase-dependent cleavage of signaling protein during apoptosis. J Biol Chem. 1998;273:7141–7147. doi: 10.1074/jbc.273.12.7141. [DOI] [PubMed] [Google Scholar]