

Abstract
Strength of T cell receptor (TCR) signaling, coreceptors, costimulation, antigen-presenting cell type, and cytokines all play crucial roles in determining the efficiency with which type 2 T lymphocytes (Th2, Tc2) develop from uncommitted precursors. To investigate in vivo regulatory mechanisms that control the population of type 2 T cells and disease susceptibility, we have created lines of transgenic mice in which expression of a chimeric cytokine receptor (the mouse interleukin 2 receptor β chain [IL-2Rβ] extracellular domain fused to the cytoplasmic tail of IL-4Rα) is targeted to the T lymphoid lineage using the proximal lck promoter. This chimera transduced IL-4–specific signals in response to IL-2 binding and dramatically enhanced type 2 responses (IL-4, IL-5, and immunoglobulin E production) upon in vitro TCR stimulation or in vivo antigen challenge. Thus, type 2 effector function was augmented by IL-4 signals transduced through a chimeric receptor expressed in a T cell–specific manner. This influence was sufficient for establishment of antigen-induced allergic airway hyperresponsiveness on a disease-resistant background (C57BL/6).
Keywords: interleukin 4, T cell help, signal transduction, allergic diseases
Interleukin (IL)-4 is a pleiotropic cytokine that plays an important role in the growth, differentiation, and survival of lymphocytes (1–4), as well as regulating other key hematopoietic cells such as monocytes, macrophages, and dendritic cells (5). One of the most important roles of IL-4 in immune regulation is its ability to influence the phenotype of effector T cells as they differentiate from naive precursors (1, 6–8). For instance, helper and cytolytic T lymphocytes can be divided into distinct subsets of effector cells based on their functional capabilities and the profile of cytokines they produce (9, 10). The Th1 subset of CD4+ T cells secretes IFN-γ and TNF-β, which activate cell-mediated immune responses optimally suited for immunity to intracellular pathogens (11–13); the Tc1 subset of CD8+ cells secretes similar cytokines. In contrast, the Th2 subset produces IL-5, -6, -10, and -13, in addition to IL-4, and is associated with immune responses that combat extracellular microbes, in part through help to antibody responses (12, 13).
The mechanisms by which naive T precursor cells differentiate into discrete effector cells remain a subject of intensive investigation. Factors that control differentiation into Th1 or Th2 cells include strength of signaling through the TCR, antigen presentation (including antigen dose, route of antigen administration, and type of APCs), involvement of costimulatory pathways such as those activated by CD28 ligation, and engagement of CD4 coreceptor molecule (14–17, reviewed in 18). In addition to these regulatory influences, the cytokines to which APCs and T cells are exposed during differentiation of the naive T cell are also a potent influence. Thus, IL-12 and IFN-γ favor Th1 differentiation and inhibit the emergence of Th2 cells (11–13, 19, 20). In contrast, the presence of IL-4 early during priming stimulates Th2 development and represses the production of Th1 cells and their characteristic cytokine products (21, 22). Data from in vitro systems indicate that there are conditions under which IL-4 can promote the differentiation of purified small, resting T cells into Th2 cells (6). Importantly, however, the ability of these cytokines to influence the development of CD4+ T cells in vivo has been studied almost exclusively in systems in which both APCs and naive precursor T cells are affected (23, 24). Thus, the relative contributions of T cell–autonomous signaling by IL-4 and its effects on APCs during immune responses in vivo remain to be established.
In this regard, previous work has established that, although IFN-γ may have some direct effects on T cell development (25), inhibition of IFN-γ receptor signaling in the T lineage had little effect on Th1 development (26). In contrast, targeting such inhibition to macrophages dramatically decreased Th1 development (26). Absence of interferon regulatory factor 1 (IRF-1)1 from APCs led to a similar diminution in Th1 development and a deficit of Th1 function in vivo (27, 28). However, it is unclear to what extent IL-4 acts on APCs in promoting Th2 development in vivo. In light of the potent ability of IL-4 directly to induce the CD28 ligands B7-1 and -2 (29) and to increase the rate of synthesis of class II MHC proteins (30, 31), a critical role of IL-4 in Th2 development could be mediated through changes in the strength of TCR and CD28 signaling (14, 16, 18). Moreover, under some circumstances IL-4–deficient T cells develop cytokine production characteristics consistent with the Th2 phenotype (32, 33). Taken together, these findings raise the question whether the role of IL-4 in promoting Th2 development under in vivo conditions would be promoted if IL-4 signaling were initially restricted to activated naive T cells and did not affect APCs. In addition, an apparent gain-of-function mutation in the human IL-4Rα chain is associated with atopic diseases (34), thus raising the question whether altered patterns of IL-4R signaling in T cells could potentiate allergic disease on an otherwise resistant genetic background.
To investigate such questions concerning in vivo regulation of the balance of types 1 and 2 T cells, we have created lines of mice in which the T lineage expresses a chimeric cytokine receptor. Specific portions of the human IL-4Rα cytoplasmic tail are competent to transduce signals characteristic of IL-4 when fused to human IL-2Rβ and a portion of its intracellular domain (35). To permit binding of mouse IL-2 and avoid potential confounding influences from cytoplasmic portions of IL-2Rβ, we have generated a translational fusion between the extracellular and transmembrane domain of the mouse IL-2Rβ chain and the complete intracellular domain of mouse IL-4Rα. This chimeric receptor transduced signals characteristic of IL-4 (signal transducer and activator of transcription [Stat]6 activation) in response to IL-2 binding. Such signaling was enhanced by activation of thymocytes or T cells, presumably due to the known role of IL-2Rα/CD25 in mouse IL-2 binding and function (36–38). Cells from these transgenic (Tg) mice exhibited enhanced Th2 responses (IL-4 and IL-5 production as well as help for IgE production). This influence on the development of effector T cells was sufficient to overcome the resistance of C57BL/6 mice to OVA-induced allergic airway disease. Thus, it is likely that in mice expressing the chimeric receptor transgene, the presence of IL-2 upon antigen challenge favors differentiation of naive T cells into Th2 effector cells by activation, and that this initially cell-autonomous pathway can augment Th2 effector function mediated by IL-4 signals in T cells.
Materials and Methods
Reagents.
Cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, sodium bicarbonate, 2 mM l-glutamine, nonessential amino acids, penicillin/streptomycin (all from GIBCO BRL, Gaithersburg, MD), and 5 × 10−5 M 2-mercaptoethanol (Sigma Chemical Co., St. Louis, MO), as described previously (39). Unmodified and fluorochrome- or biotin-conjugated antibodies against IL-2Rβ/CD122 (unconjugated or FITC-labeled), CD4 (FITC, r-PE, and biotin), B220 (r-PE), CD3 (unconjugated and r-PE), I-Ab,d (FITC, r-PE, and biotin), CD11b (FITC and biotin), CD11c (r-PE), IL-4 (r-PE), IFN-γ (FITC), goat anti–rat Ig (biotin), and CD28 (unconjugated) were obtained from PharMingen (San Diego, CA). Biotin-conjugated anti–mouse CD122 was generated from azide-free preparations of anti-CD122 (PharMingen) using biotin-amidocaproate N-hydroxysuccinimide ester (Sigma Chemical Co.). Streptavidin-PE and streptavidin-PerCP were obtained from BioSource International (Camarillo, CA) and Becton Dickinson & Co. (Mountain View, CA), respectively. Antibodies against Stat5 and Stat6 for electrophoretic mobility supershift assays were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Purified or biotinylated monoclonal antibodies for cytokine ELISA (IL-4, IL-5, and IFN-γ) were obtained from PharMingen.
Plasmid Construction and Generation of Tg Mice.
The chimeric IL-2R/4R cDNA was constructed by reverse transcriptase–PCR. In brief, sequences encoding the extracellular and transmembrane regions of murine IL-2R cDNA were amplified using PFU polymerase. The sense oligonucleotide primer corresponded to the 5′-flanking sequence and signal peptide (86–113 bp based on reference 40: 5′-CCTGAATTCCTCTCAGCTGTGATGGCTA-3′) with additional nucleotides encoding an EcoRI site. The antisense oligonucleotide primer corresponded to 908–927 bp, including a naturally occurring KpnI site at the downstream end (5′-CCAAGGTACCGGCACTTGAC-3′). The complete intracytoplasmic domain of IL-4R cDNA was amplified from cDNAs using primers based on a published mouse IL-4Rα cDNA sequence (41), except that a KpnI site was introduced at the upstream end to permit ligation to EcoRI, KpnI-digested IL-2Rβ amplicon. After amplification, proteinase K digestion, and cleavage with appropriate restriction endonucleases, these two PCR products were cloned by three-way ligation into the mammalian expression vector pcDNA3. After verification of expression and function in the M12 B lymphoma cell line, the cDNA was shuttled into a BamHI-digested, 5′-end–filled transgene vector. The transgene generated by subcloning the chimeric receptor cDNA into the T lineage–specific vector p1017 (lck proximal promoter followed by exons, introns, and polyadenylation signal derived from the human growth hormone gene) (42) was released as a NotI fragment encompassing the tissue-specific transcription unit, purified by standard protocols, and microinjected into the pronuclei of C57BL/6 × DBA/2 zygotes. Mice were maintained in accordance with federal and state government regulations after institutional approval. Founder lines were propagated by sequential backcrossing to C57BL/6 and, in later experiments, C57BL/ 10.D2 mice for these studies.
Northern Blot Analyses.
RNA was prepared from splenocyte suspensions stimulated under the indicated conditions according to the manufacturer's protocol after harvest and lysis in TriZol acid-phenol reagent (GIBCO BRL). These total cellular RNAs prepared by chloroform extraction and isopropanol precipitation were resolved on 1.1% agarose gels in the presence of formaldehyde, transferred to HyBond-N membranes (Amersham Pharmacia Biotech, Inc., Piscataway, NJ) and subjected to hybridization using gel-purified cDNA probes labeled by a random-primer technique. A 2.4-kb EcoRI, BamHI fragment spanning the full chimeric cDNA was used to detect the chimeric cytokine receptor (as well as endogenous IL-4Rα and IL-2Rβ bands), whereas a 255-bp cDNA fragment generated by reverse transcriptase PCR was used to detect GATA-3. The GATA-3 cDNA probe was purified after amplification (60 s each at 94, 58, and 72°C for 35 cycles) using cDNAs prepared from IL-4–stimulated splenocytes and the oligonucleotides 5′-GAAGGCATCCAGACCCGAAAC and 5′-ACCCATGGCGGTGACCATGC as primers.
Flow Cytometry and Detection of Intracellular Cytokines.
To analyze development and subsets of T lymphoid cells, suspensions of lymphoid cells were counted, prepared, stained with fluorochrome-conjugated mAbs, and analyzed as described (39). Due to the weak staining of mouse IL-2Rβ (CD122), the indicated samples were analyzed using fluorochrome-conjugated T lineage markers and indirect immunofluorescent staining of IL-2Rβ using unlabeled anti-CD122 followed by biotinylated goat anti–rat Ig, and then streptavidin–r-PE. To enhance CD122 staining in experiments involving APCs, indirect immunofluorescence was performed using biotinylated anti-CD122 followed by streptavidin–r-PE or PerCP, and larger cells were included in the initial forward and side scatter gate. For detection of cytokine production by staining intracellular pools, lymph node cells were stimulated with immobilized anti-CD3 (10 μg/ml), anti-CD28 (2 μg/ml), and recombinant mouse IL-2 (20 ng/ml) for 40 h. Monensin was added to the cell culture in the last 4 h. Cells were then stained with anti-CD4-biotin (PharMingen) and streptavidin-PerCP (Becton Dickinson & Co.), permeabilized according to the manufacturer's instructions, stained with both anti-IL-4–PE and anti-IFN-γ–FITC (PharMingen), and analyzed by flow cytometry.
Gel Mobility Shift Analyses.
Whole cell extracts were prepared from splenocyte suspensions (43, 44) stimulated under the indicated conditions using concanavalin A (Sigma Chemical Co.), purified recombinant mouse IL-4 (R & D Systems, Inc., Minneapolis, MN), mouse IL-2 (R & D Systems and PharMingen), and human IL-2 (Cetus Corp., Emeryville, CA, courtesy of the Biological Response Modifiers Program). In brief, cells were lysed at 4°C using 0.5% NP-40 supplemented with 0.15 M NaCl, 50 mM NaF, 1 mM dithiothreitol, 0.1 mM Na-vanadate, 0.4 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, and 1 μg/ml leupeptin, followed by pelleting insoluble materials. DNA binding reactions were performed using 5 μg of protein in 20 μl reactions containing 1 μg poly dI-dC competitor and the indicated 32P-labeled oligonucleotide, essentially as described (43, 44). Double-stranded oligonucleotides used contained the “N4” Stat6 binding site from residues −122 to −104 of the germline epsilon promoter in the immunoglobulin heavy chain locus, 5′-AACTTCCCAAGAACAGA and its complement (44), or the “N3” gamma-activated sequence from the IRF-1 promoter, 5′-CCTGATTTCCCCGAAATGAT and its complement (45). Identification of components in a DNA–protein complex was performed by supershift analyses using 1 μg per binding reaction of antibodies against either Stat5 or Stat6 (Santa Cruz Biotechnology). DNA–protein complexes were resolved on a 4.5% nondenaturing polyacrylamide gel in Tris-borate-EDTA buffer as described (44).
Cytokine Assays by ELISA.
Lymph node cells were stimulated with immobilized anti-CD3 (10 μg/ml) and either soluble anti-CD28 (5 μg/ml) or recombinant mouse IL-2 (20 ng/ml) in the presence or absence of neutralizing polyclonal antibody to mouse IL-4 (10 μg/ml; R & D Systems) as indicated. Similar results were obtained using a mAb against IL-4 (11B11; PharMingen). After culture for 6 d, cells were washed and restimulated with immobilized anti-CD3 plus anti-CD28 for 24 h before collection of culture supernatants for determination of cytokine production. To measure primary responses of T cells, suspensions of lymph node cells were cultured with immobilized anti-CD3 (10 μg/ml) and either anti-CD28 (2.5 μg/ml) or recombinant mouse IL-2 (20 ng/ml) for 48 h. Cytokine (IL-4, IL-5, and IFN-γ) levels from culture supernatants were determined by standard sandwich ELISA according to the manufacturer's instructions (PharMingen) and standardized to purified recombinant cytokines (PharMingen for IL-4 and IFN-γ; purified recombinant mouse IL-5 was a gift from DNAX, Palo Alto, CA).
Immunization of Mice with OVA and Antibody Measurement.
After obtaining preimmune sera, mice were injected intraperitoneally with OVA (10 μg adsorbed on 20 mg aluminum hydroxide; Sigma Chemical Co.). After 7 d, sera were collected from immunized mice, and then analyzed by isotype-specific ELISA to determine levels of OVA-specific and total antibodies. In brief, ELISA plates (Corning Glass Works, Corning, NY) were incubated overnight at 4°C with 50 μl of capture antigen solution (20 μg/ml OVA for OVA-specific antibody measurements; sheep anti–mouse IgE [Serotec Ltd., Kidlington, Oxford, UK] or anti– mouse IgG2a [Southern Biotechnology Associates, Birmingham, AL] for total isotype determinations). After discarding coating solutions, the plates were blocked with 1% BSA in PBS (2 h at room temperature) and washed. Mouse serum or standard antibodies diluted in PBS containing 0.3% BSA were added to each well. Antiserum against OVA, mouse IgE (affinity purified from mouse serum immunized with DNP; Sigma Chemical Co.) or mouse IgG2a were used as standards for OVA-specific, total IgE, or total IgG2a ELISA, respectively. Plates were then incubated for 2 h at room temperature, washed, and incubated with biotinylated detection antibodies (rat monoclonal anti–mouse IgE-biotin [BioSource International] or anti–mouse IgG2a-biotin) for 1 h, washed and incubated with avidin–alkaline phosphatase (Sigma Chemical Co.). Alkaline phosphatase activity was determined with phosphatase substrate tablets (Sigma Chemical Co.) and assessed during the linear phase of the reaction using an ELISA reader (SLT-Tecan US, Inc., Durham, NC) at 420 nm and DeltaSoft 3 analytical software. Each sample was tested in duplicate and the mean value recorded.
Allergic Airway Disease.
Mice (chimR Tg or nontransgenic [NTg], as indicated; 4-wk-old pups from the third backcross to C57BL/6) received a priming injection of OVA (10 μg in aluminum hydroxide, intraperitoneally) on day 0. These mice underwent a series of eight daily exposures (40 min each) to an aerosol generated from OVA (chicken OVA, grade V, Sigma Chemical Co.; 1% in low-endotoxin, sterile PBS), starting at day 14. The day after their eighth inhalation treatment, lung mechanics were measured in these sensitized mice and in controls that were not exposed to OVA using mechanical ventilation in a body plethysmography chamber. After cannulation of the internal jugular vein, intravenous methacholine (a bronchoconstrictor) was administered in the indicated escalating doses (46, 47), with intervals between doses to allow return of pulmonary parameters to baseline. Each resistance value represents the average of 10 measurements obtained during the peak of a response. After the final dose, cells were collected by bronchoalveolar lavage, counted, and analyzed by Wright staining.
Results
Targeted Expression of a Chimeric Cytokine Receptor in T Lineage Cells of Tg Mice.
Earlier in vivo studies have shown that systemic IL-4 has the capacity to inhibit the development of Th1 cells while promoting the Th2 phenotype (23, 24). However, it is not clear during such IL-4 exposure in vivo what are the relative contributions of IL-4 signaling within the T cell versus those in APCs. Moreover, T cells differentiated under strong polarizing conditions in vitro retained their characteristics after transfer in vivo (48), but the extent to which different levels of IL-4 signaling within T cells affect Th1 or Th2 development before exogenous antigen challenge is unknown. To investigate these questions, and to determine if enhanced IL-4 signaling after activation of naive T cells could create susceptibility to allergic diseases in vivo (34), we created a chimeric cytokine receptor that could bypass the function of endogenous IL-4 receptors.2 Because mouse IL-2 binds poorly to human IL-2 receptors, this chimeric receptor was designed as a translational fusion of the ligand-binding and transmembrane domains from the mouse IL-2Rβ chain and the intracellular domain of the mouse IL-4Rα (Fig. 1 A).
Figure 1.

Targeted expression of the chimeric IL-2Rβ/IL-4Rα transgene to T-lineage cells. (A) Structure of chimeric cytokine receptor cDNA. The chimeric IL-2R/4R receptor was composed of extracellular domain and transmembrane domain of mouse IL-2Rβ, linked to the complete intracellular domain of IL-4Rα. Tyrosine residues and two acidic domains (ID-1 and I4R) in the IL-4Rα intracytoplasmic tail are indicated. (B and C) Expression of chimeric receptor transcripts in T-lineage cells from Tg mice. RNA was prepared from NTg and Tg mice (right) (second backcross generation to B6) using the indicated lymphoid organs (B and C) or purified T or B cells (C), and from M12 B lymphoma cells (parental, −, or transfected, +, with the chimeric receptor cDNA) as indicated. Splenic T and B cells were separated by nylon wool column, and B cells were further purified by magnetic activated cell sorting (MACS) using streptavidin-microbeads after labeling with a biotinylated anti-B220 mAb. Purity determined by flow cytometry was ∼80% CD3+ (T cells) or 95% B220+ (B cells). RNA was resolved on a formaldehyde-agarose gel, transferred to HyBond filters, and probed with 32P-labeled DNA comprising the IL-2Rβ and IL-4Rα sequences present in the Tg cDNA. (C) Arrows denote the position of RNAs encoded by the transgene, two of which comigrated with endogenous RNAs hybridizing to the IL-2Rβ + IL-4Rα probe as indicated; the positions of these RNAs are also marked by open circles. An asterisk marks the band of intermediate mobility unique to transgene-positive animals (lane 2).
The IL-2 and IL-4 receptors share a number of architectural principles. IL-2Rβ and IL-4Rα are in the same subfamily of hematopoietin receptors, each binds to the Janus kinase Jak1, and the receptors share a γc chain, leading to Jak3 recruitment (49). Despite these features, they activate different Stat transcription factors and appear to differ in their respective potency for activation of mitogen-activated protein kinase and insulin receptor substrate pathways (49– 51). Accordingly, transfer of specific domains of human IL-4Rα onto a 140-amino acid cytoplasmic signaling domain from human IL-2Rβ was sufficient for IL-2 induction of CD23 on B cells, a response characteristic of IL-4 but not IL-2 (35). An additional important consideration is that, unlike the human IL-2/IL-2R system, mouse IL-2 is reported to bind mouse IL-2Rβ and activate resting T cells poorly unless IL-2Rα is expressed (36–38). Consistent with this possibility, M12 B lymphoma cells stably transfected with our chimeric receptor cDNA demonstrated that this receptor transduced IL-4–specific signals (Stat6 activation, induction of CD23 and the Ig germ line ε promoter), but only when the IL-2Rα chain, a protein virtually absent from resting naive T cells and from most thymocytes, was also transfected.2 This dependency of the mouse IL-2Rβ chain on IL-2Rα expression implied that the chimeric receptor molecule might not bind IL-2 efficiently until a cell was induced to express IL-2Rα chain, thus providing a mechanism to protect thymocyte and T cell development from abnormalities induced by IL-4 (42). Since IL-2 is a ligand that will be present at high concentration early after activation of naive T cells, these features suggested that a mIL-2Rβ/mIL-4Rα chimera might activate IL-4–specific signals in the absence of IL-4, but would signal inefficiently before T cell activation.
In light of these findings, we created Tg mice in which expression of this chimeric receptor was targeted to the T lymphoid lineage using the T lineage–specific regulatory sequences of the lck proximal promoter, thus facilitating an investigation of the regulatory roles of IL-4 signaling in mouse T lineage cells. Different copy-number integrations on the X chromosome were bred from founders; the phenotype in each of these lines is similar to the others (data not shown). Transgene-positive progeny expressed transgene-encoded transcripts in thymocytes and unfractionated splenocytes (Fig. 1 B). When peripheral lymphoid cells were fractionated into T and B cells and subjected to Northern blot analyses, there was no difference in intensity of a band representing endogenous RNA in Tg and NTg samples and no band at the position of the chimeric transcript could be detected in the B220+ cells purified from Tg mice (Fig. 1 C; a transgene-specific RNA species migrates at a position [*] between two bands containing endogenous RNAs [o]). In FACS® analyses, nearly all thymocytes and CD4+ cells from male Tg mice expressed the IL-2Rβ epitope (CD122) at their cell surface, whereas expression of endogenous IL-2Rβ chains was observed only on a smaller fraction of those cells in NTg littermates (Fig. 2 A). Of note, the level of CD122 staining on Tg thymocytes and T cells was similar to the staining intensity of positive cells among wild-type samples; the main difference was in the frequency of positive cells. Since the staining intensity of CD122 on Tg T cells (chimeric receptors plus endogenous IL-2Rβ) appeared at most fourfold greater than on wild-type cells (endogenous IL-2Rβ alone), these findings suggest that the number of chimeric receptors is only a few times higher than a cytokine receptor such as endogenous IL-2Rβ (Fig. 2 B). Consistent with the Northern blot data, direct immunofluorescent antibody staining indicated that among cells competent to express the IL-2Rα chain, CD3+ cells expressed the IL-2Rβ epitope at levels higher in Tg mice than in their NTg littermates, whereas Tg B220+ cells did not (Fig. 2 B). Moreover, the level of CD122 on I-A+B220− cells and populations labeled with cell-surface epitopes that mark macrophages (CD11b+ CD11c+) and dendritic cells (CD11b−CD11c+) was no different in Tg animals and wild-type controls, whereas increased CD122 expression was readily detected in the T cell-enriched (I-A−B220−) gate of Tg samples (Fig. 2 C). We conclude that expression of the chimeric cytokine receptor transgene in lymphoid cells is restricted to the T lineage.
Figure 2.


Cell surface expression of chimeric receptor molecules on thymocytes and peripheral T cells from Tg mice. (A) Representative fluorescence histograms from thymocytes and lymph node cells of NTg or Tg mice were sequentially stained with rat anti–mouse IL-2Rβ, biotinylated goat anti–rat Ig, and then streptavidin-PE (three-layer staining). Lymph node cells were costained with anti-CD4. Heterozygous females exhibited the effects of Lyonization, with high-level expression of IL-2Rβ protein on only 50% of thymocytes. However, the alterations (cytokine production; allergic airway disease) reported below were observed in both males and females. The data presented were from second backcross generation mice; similar results were observed using N1- and N3-generation mice. (B) Lymph node cells were costained with anti–mouse IL-2Rβ-FITC and either anti-CD3-PE or anti-B220-PE as indicated. The profiles represent the fluorescence intensity of cells from the lymphocyte gate (FSC and SSC) and CD4+, CD3+, or B220+ gate, as indicated. (C) Splenocytes (a–f ), or metrizamide gradient-enriched APCs (g–j), from wild-type (NTg) and chimeric receptor transgene-positive (Tg) mice (N3 generation) were subjected to three-color indirect immunofluorescent staining as indicated, and then analyzed by flow cytometry. Uniform compensations and a uniform forward and side-scatter gate were applied to all samples. CD122 staining in the T cell-enriched gate (a and b; I-A and B220 negative) demonstrated expression of the transgene. Higher levels of autofluorescence were present in the APC-enriched populations (c–j) due to differences between the larger APCs and lymphoid cells, thereby leading to broader CD122 histograms. In all APC-gated samples, the mean fluorescence intensity (MFI) for CD122 in Tg samples was less than the MFI of the wild-type controls. The same results were obtained using anti-CD122 directly conjugated with fluorescein and appropriate staining combinations for APC markers.
Intact Development and Deployment of the T Lineage.
Overexpression of IL-4 in thymocytes using the lck proximal promoter caused a dramatic decrease in thymic cellularity and led to a failure of export of CD8 single positive cells to populate peripheral sites (42). Accordingly, it was possible that the chimeric cytokine receptor transgene would lead to similar derangements. Alternatively, if it were functionally inert in resting T cells and most thymocytes (which do not express IL-2Rα protein/CD25), T lineage development and deployment should be normal. This latter possibility would be consistent with reports that, unlike human IL-2Rβ, mouse IL-2Rβ cannot signal proliferation of resting thymocytes or CD4+ T cells (36–38). Indeed, the size and balance of thymic populations in Tg mice were no different from thymic profiles of their NTg littermates. Moreover, although there was a subtle variation in the CD4/CD8 ratio (Tg, 2.5 ± 0.4 vs. NTg, 1.7 ± 0.1), CD4+ and CD8+ T cells populated the spleen and lymph nodes in normal numbers (Fig. 3). Although lck may be expressed in the natural T cell compartment of NK1.1-positive TCR-α/β–bearing T cells (52), there was no difference between Tg mice and wild-type (NTg) littermates in the number of these cells (data not shown). Thus, T cell development appears normal despite the potential for the chimeric cytokine receptor to deliver IL-4–like signals.
Figure 3.

Normal development in the T lineage. Suspensions of (A) thymocytes and (B) splenocytes were subjected to staining for CD4 and CD8, as indicated. Fluorescence histograms from a representative Tg male, heterozygous female, and NTg littermate are shown. The absolute numbers of thymocytes (NTg, 176 × 106; Tg, 180 × 106), splenocytes (NTg, 74 × 106; Tg, 83 × 106), T cells, and their subsets were not significantly different in Tg and NTg animals.
IL-4–independent Induction of Stat6 DNA Binding Activity in Tg T Cells.
The finding that T cell development was normal provided evidence, albeit indirect, that signaling by the chimeric receptor may be IL-2Rα dependent. To determine directly if the transgene mediates Stat6 activation under conditions that bypass potential influences from endogenous IL-4 receptors, we performed mobility shift assays using thymocytes and spleen cells that were freshly isolated or first exposed to activating stimuli (Fig. 4 and data not shown). Using an oligonucleotide probe whose spacing of the consensus repeat TTC/GAA permits stable binding by Stat6 but not Stat1–Stat5, binding activity was induced in resting spleen cells with addition of exogenous IL-4 (Fig. 4 A, lanes 5 and 10), whereas mouse IL-2 at 10 ng/ml (∼0.67 nM, near the K d of an IL-2R βγ dimer, 1 nM) did not activate Stat6 in resting thymocytes or splenocytes (lanes 2 and 7) from Tg mice or controls. Higher concentrations of mouse IL-2 (100 ng/ml) or human IL-2 (106 Cetus U/ml; ∼300 ng/ml) were sufficient to generate nuclear Stat6 binding activity (lanes 3, 4, 8, and 9), whose relatively low level may reflect inefficient activation through βγ dimers at concentrations well above their K d, as well as the presence of B cells, which lack chimeric receptor expression (Fig. 2). In contrast to resting cells (Fig. 4 B, lanes 1–6), activation (“priming”) and production of IL-2 in the cultures triggered efficient Stat6 induction in Tg (lanes 10– 12) but not NTg cells (lanes 7–9). Similar results were obtained when neutralizing antibodies against IL-4 were included during the activation phase of the culture (Fig. 4 B, lanes 13–18). The presence of Stat6 in the mobility shift complex was confirmed by supershifting with antiserum specific for Stat6, whereas anti-Stat5 failed to create a slower mobility complex when using this Stat6-specific oligonucleotide probe (Fig. 4 C). IL-2–dependent Stat6 induction in Tg cells was also observed for activated thymocytes, confirming its association with the T lineage of chimeric receptor Tg mice (Fig. 4 D). To determine the effect of the transgene on induction of an IL-4 target gene in T cells, we performed Northern blot analyses of the levels of GATA-3 mRNA, a transcription factor whose levels increase in IL-4–treated T cells and during Th2 differentiation (53). In contrast to the results with wild-type mice, activation of lymphoid cells from chimeric receptor Tg mice led to substantial induction of GATA-3 without the addition of exogenous IL-4 (Fig. 4 E). These data do not provide quantitation of the biochemical efficiency of the IL-2Rβ/ IL-4Rα chimera. Since the flow cytometric data suggest that there is a modest (less than fourfold) excess of chimeric receptors relative to wild-type CD122, our chimeric receptor may induce Stat6 less efficiently than endogenous IL-4Rα chains. Notwithstanding this possibility, we can conclude that upon activation in the absence of exogenous IL-4, T cells from Tg mice exhibit increased signaling characteristic of IL-4 compared with their NTg counterparts.
Figure 4.
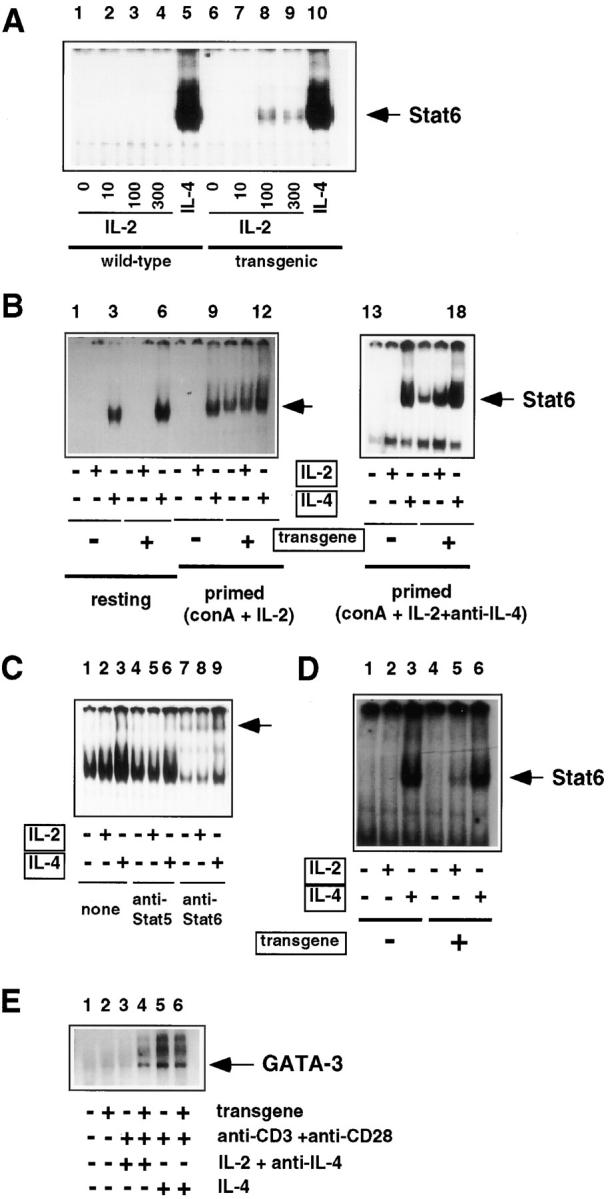
The chimeric receptor induces Stat6 in activated lymphoid cells. (A) Induction of Stat6 by IL-2. Single cell suspensions of resting splenocytes from wild-type (NTg) or chimeric receptor transgene-positive mice (N3 generation) were cultured in complete media supplemented (30 min at 37°C) with the indicated concentration of purified recombinant IL-2 (lanes 2–4 and 7–9), purified recombinant mouse IL-4 (lanes 5 and 10), or without added lymphokine (lanes 1 and 6). Extracts from these cells were then subjected to mobility shift assays using an oligonucleotide from the mouse Ig germ line ε promoter (44), which exhibits specificity for Stat6 due to an N4 spacing between the TTC and GAA. (B) Activation-dependent enhancement of IL-4–independent induction of Stat6 binding activity. Resting or activated RBC-depleted splenocytes (N3 generation) were stimulated (30 min at 37°C) with either IL-2 or IL-4 as indicated. Where indicated, cells were activated during a 15-h culture in the presence of Con A (2.5 μg/ml) and IL-2 (10 ng/ml), in the presence or absence of neutralizing anti–IL-4 antibodies, and then washed before stimulation with IL-2 or -4. Whole cell extracts were subjected to mobility shift assays as in A. (C) Stat6 but not Stat5 present in the IL-4– independent mobility shift complexes. Extracts from activated splenocytes, generated as in A, and then treated with IL-2 (lanes 2, 5 and 8) or IL-4 (lanes 3, 6, and 9) as indicated, were incubated with antiserum against Stat5 (lanes 4–6) or Stat6 (lanes 7–9) as indicated. DNA–protein complexes were then resolved by nondenaturing gel electrophoresis. (D) Induction of Stat6 in activated thymocytes. Thymocyte suspensions from Tg mice and wild-type littermates were cultured 15 h in PMA (50 ng/ml) and ionomycin (1 μg/ml) in the presence of mouse IL-2 (10 ng/ml) and neutralizing antibodies against mouse IL-4 (10 μg/ml). These activated cells were then washed and placed in culture with no added cytokine (lanes 1 and 4), mouse IL-2 (10 ng/ml; lanes 2 and 5), or mouse IL-4 (10 ng/ml; lanes 3 and 6). (E) Induction of GATA-3 mRNA in Tg lymphocytes. Portions of lymphoid cell suspensions (pooled spleen and lymph node) from Tg (N2 generation in this experiment) and NTg mice were placed in culture (lanes 3–6) or used to prepare RNA without culture (lanes 1 and 2). RNAs were prepared after culturing cells 48 h in the presence of immobilized antibodies against CD3 (10 μg/ml), soluble anti-CD28 (5 μg/ml), and either mouse IL-4 (10 ng/ml; lanes 5 and 6) or the combination of human IL-2 (10 ng/ml) and neutralizing antibodies against mouse IL-4 (10 μg/ml) (lanes 3 and 4), as indicated. Northern blot analyses of equally loaded lanes of RNA (as assessed by hybridization with an rRNA oligonucleotide; data not shown) were probed for GATA-3 as described in Materials and Methods.
Development of Type 2 T Effector Cells from Precursors.
In light of this evidence that the chimeric receptor is functional in T cells, we tested whether the transgene could influence the development of Th1 or Th2 cells during in vitro stimulation (Fig. 5). Cells from Tg mice produced over 10-fold more IL-4 than those from their NTg littermates when lymph node cells were stimulated through the TCR and costimulatory CD28 molecule for 6 d, washed, and restimulated. When effector cells developed in the presence of neutralizing antibodies against IL-4 during the primary culture, this difference between Tg and NTg mice was more dramatic (33-fold), reflecting a greater inhibitory effect of anti–IL-4 on the NTg samples. Consistent with reports that IL-4–deficient mice can generate Th2-like cells (32, 33), some IL-4 production was still observed under these conditions, perhaps reflecting the important role of costimulation through CD28. Indeed, although the output of IL-4 was diminished if anti-CD28 was omitted from the differentiation phase of these cultures, lymph node cells from chimeric receptor Tg mice exhibited a far greater competence to generate IL-4–producing effector cells when compared with their wild-type littermates. Similar, albeit less dramatic, results were obtained when IL-5 production by these cultures was used as an indicator of effector T cell development. Despite the potentiation of IL-4 production conferred by this diversion of IL-2 binding into signaling pathways characteristic of IL-4, IFN-γ production was not suppressed. However, addition of exogenous IL-4 to similar cultures has led to variable inhibition of IFN-γ production (data not shown). Taken together, these results indicate that development of the Th2 phenotype in T cells from chimeric receptor Tg mice is potentiated, and indeed can be largely independent of endogenous IL-4. Of note, under these conditions the original difference between Tg and NTg cells in levels of IL-4 signaling is confined to the T cells.
Figure 5.

Enhanced IL-4–independent Th2 development during in vitro T helper cell differentiation. Lymph node cells from a pool of three NTg or Tg littermates (N3 generation) were stimulated with immobilized anti-CD3 (10 μg/ml) and either anti-CD28 (2 μg/ml) or recombinant mouse IL-2 (20 ng/ml) in the presence or absence of polyclonal antibody to mouse IL-4 (10 μg/ml), as indicated. After 6 d of culture, cells were washed and restimulated with immobilized anti-CD3 plus anti-CD28 for 24 h. Supernatants were collected and analyzed by ELISA as described in Materials and Methods. These figures are representative of more than three (for IL-4 and IFN-γ) or two (for IL-5) independent experiments. Additional experiments using lymph node cells from individual mice of each subline showed no differences in the penetrance of the observed effects on cytokine (IL-4, IFN-γ) production.
Increased Type 2 Cytokine Response and Help to Antibody Production.
The above data demonstrate that expression of the transgene is able to influence effector T cell development in vitro. Given that the transgene amplified IL-4–specific outcomes during in vitro differentiation of T cells, we tested its effects on in vivo processes. First, we investigated this issue by measuring IL-4 and IFN-γ production after short-term polyclonal stimulation of lymph node cells in vitro, since the cytokines produced under these conditions are derived predominantly from CD4+ T cells expressing an antigen-experienced/memory phenotype (13, 54–56). Lymph node cells were stimulated 40 h with anti-CD3 and either anti-CD28 or exogenous IL-2, or both. Culture supernatants were then subjected to cytokine ELISA (Fig. 6 A), and cells were stained with fluorescein-conjugated antibodies to determine the frequency of IL-4– or IFN-γ–producing cells (Fig. 6 B). A fourfold increase in IL-4 produced by Tg cells was observed compared with those from NTg littermates. Consistent with the in vitro differentiation assays (Fig. 5), Tg cells produced similar or at most slightly reduced amounts of IFN-γ compared with NTg controls (Fig. 6 A). In concert with these data, lymph node cells from chimeric receptor Tg mice included 3% IL-4– producing CD4+ T cells, whereas at most 0.3% of CD4+ NTg cells exhibited detectable IL-4 (Fig. 6 B; the background of nonspecific staining in these assays was ∼0.3% in the CD4+ gate). Display of IFN-γ–producing cells revealed only a modest difference between NTg and Tg mice and demonstrated that the increase in IL-4 production reflected an increase in the frequency of cells that produce IL-4 but not IFN-γ, and thus correspond to a Th2 rather than a Th0 (56) phenotype. Taken together, these data suggest that the T cell–autonomous signals transduced by the chimeric cytokine receptor may function in vivo selectively to bias effector T cell development in favor of Th2 cells.
Figure 6.

Enhanced Th2 cytokines in early primary responses. (A) Lymph node cells, as in Fig. 5, were stimulated 48 h as indicated and supernatants were collected. IL-4 and IFN-γ levels in supernatants were measured by ELISA. These figures are representative of three independent experiments; additional experiments using preparations of lymph node cells from individual mice of each subline showed no differences in the penetrance of the observed effects on cytokine (IL-4, IFN-γ) production. (B) Lymph node cells, as in Fig. 5, were stimulated with immobilized anti-CD3 (10 μg/ml), anti-CD28 (2 μg/ml), and recombinant mouse IL-2 (20 ng/ml) for 40 h. Monensin was added to the cell culture in the last 4 h. Cells were stained with anti-CD4-biotin and streptavidin-PerCP, permeabilized, and stained with both anti-IL-4–PE and anti-IFN-γ-FITC, followed by FACS® analysis. These figures are representative of histograms in two independent experiments.
To measure directly the effect of the transgene on an indicator of type 2 help in vivo, we immunized mice with OVA in aluminum hydroxide. Generation of antigen-specific IgE requires T cell help and IL-4 (12, 13, 57). It may normally require days to generate antigen-specific effector cells that are efficient producers of IL-4, and for the IL-4 to lead to antibody class switching to the epsilon heavy chain exons so as to secrete IgE (58, 59). Therefore, we chose day 7 after immunization as a time point for comparison of total and antigen-specific IgE levels in chimeric receptor Tg mice to those in their wild-type littermates (Fig. 7). Both antigen-specific (Fig. 7 A) and total (B) IgE production were elevated in the Tg mice (average of 33- and 11-fold, respectively). Moreover, total IgE levels in Tg mice were elevated even before immunization, suggesting that an increase in type 2 help may be engendered by the chimeric receptor transgene during spontaneous evolution of the lymphoid repertoire in vivo. In contrast, levels of IgG2a, which is dependent on IFN-γ and thus favored by type 1 help, were little different in Tg mice from NTg littermates before immunization, and after immunization were indistinguishable from NTg controls (Fig. 7 C). These results provide in vivo evidence that T cell–specific expression of the transgene confers a selective enhancement of type 2 help, and suggest the possibility that this effect might be uncoupled from inhibition of type 1 help in vivo. Moreover, our data suggest that T cell activation that occurred spontaneously may have induced a bias toward provision of type 2 help in Tg mice before antigen challenge. Importantly, when Tg mice were rechallenged with OVA 5 wk after their first immunization, their IgE anti-OVA recall response remained dramatically greater than that of NTg controls (Fig. 8). These results indicate that the transgene-imposed bias in the helper arm of a response to a specific antigen is sustained in a recall response.
Figure 7.

Serum levels of IgE and IgG2a in mice immunized with OVA. Tg (n = 6) and NTg (n = 6) mice (N3 generation) in two independent experiments were injected intraperitoneally with 10 μg OVA adsorbed in aluminum hydroxide (20 mg). 7 d after injection, sera were collected and levels of OVA-specific and total antibodies were measured by isotyping ELISA, as described in Materials and Methods. (A) OVA-specific IgE. (B) Total IgE. (C) Total IgG2a. Each symbol represents each individual mouse and solid lines (–) indicate the mean value for each group. Immunization under these conditions elicited minimal OVA-specific IgG2a responses.
Figure 8.

Recall responses of antibody production in mice after second immunization. Tg (n = 6) and NTg (n = 6) mice in two independent experiments were injected intraperitoneally with 100 μg OVA adsorbed to 20 mg aluminum hydroxide. Primary responses measured at 7 d were similar to those obtained with 10 μg OVA (Fig. 7). The indicated antibody levels (mean ± SEM) were quantified by ELISA using sera obtained before, and 7 d after, reimmunization with OVA (100 μg in aluminum hydroxide). (A) OVA-specific IgE. (B) Total IgE. (C) Total IgG2a.
OVA-inducible Airway Hyperresponsiveness Due to Chimeric Cytokine Receptor Expression.
In light of the findings that one parameter of type 2 help in response to a specific antigen was increased in chimeric cytokine receptor Tg mice, we tested whether this effect could be sufficient to enhance an allergic response (Fig. 9). After intraperitoneal sensitization with OVA, followed by daily OVA inhalations, BALB/c mice acquire airway hyperresponsiveness to methacholine (46, 47, 60, 61). This T cell–dependent hyperresponsiveness is accompanied by eosinophilic airway infiltration and evidence of type 2 help (60, 61). In contrast to the BALB/c background, C57BL/6 mice develop an eosinophilic infiltrate, but less IL-4 production and minimal airway hyperresponsiveness, measurable as increased lung resistance (61, 62). Consistent with previous reports, the lung resistance of OVA-sensitized NTg littermates was no different than that of unsensitized mice (Fig. 9 A). In contrast, lung resistance after bronchoconstrictor challenge (Fig. 9 A) and airway eosinophilia (B) were increased in chimeric receptor Tg mice relative to littermate controls that had been OVA sensitized at the same time. We conclude that the chimeric cytokine receptor served as a dominant monogenic trait capable of intensifying an allergic disease process, presumably through enhancement of the development of type 2 T cells.
Figure 9.

The chimeric cytokine receptor transgene is sufficient to create sensitivity to allergic airway disease on a disease-resistant background. (A) Antigen sensitization of Tg, but not wild-type, mice leads to an increased bronchoconstrictor response. Mice backcrossed to C57BL/6 (N3) were sensitized to OVA by a single intraperitoneal injection (10 μg in aluminum hydroxide), followed by a series of antigen inhalations as described in Materials and Methods. Control mice were sham sensitized using PBS. These mice then underwent measurements of lung resistance to airflow after each of a series of methacholine injections at standard doses (47). The data represent the mean (± SEM) values derived from eight sensitized chimeric receptor Tg mice and an equal number of littermates in independent experiments. Significance values (comparison of sensitized Tg and NTg animals) are indicated above the graph at those methacholine doses for which differences were observed. (B) Mean values for the number and distribution of cells recovered after bronchoalveolar lavage of the lungs of the mice described in A.
Discussion
In vitro findings have identified conditions under which IL-4 can induce Th2 differentiation among highly purified T cells. In vivo experiments investigating the contributions of cytokines to regulating the populations of type 1 (IFN-γ–producing) and type 2 (IL-4–producing) effector T cells have involved perturbations that affect APCs as well as activated T cells. In light of the influences exerted by antigen dose, APC type, and costimulation intensity (13–18), it is not clear what conditions precisely mimic T cell–APC interactions in vivo. Thus, the ability of IL-4 to promote Th2 development when the initial perturbation is confined to the T lineage, and whether this would be sufficient to enhance allergic disease susceptibility, have not been established. We have employed an in vivo strategy that used a chimeric cytokine receptor to bypass endogenous IL-4 receptors by broadening the signaling specificity of IL-2. This approach was combined with the use of a T lineage-specific promoter to target expression of this receptor as a T cell–specific transgene, so that the initial in vivo perturbations of signaling are segregated from APC populations (26, 42, and Fig. 2). This diversion of IL-2 binding into IL-4 signaling pathways enhanced type 2 responses. Although a precise quantitation of the magnitude of activation of IL-4 signaling by the chimeric receptor in comparison to endogenous IL-4 receptors cannot be performed, our findings indicate that T cell–autonomous IL-4 signaling in vivo is sufficient to potentiate Th2 development on a C57BL/6 (Th1-oriented) background. Moreover, in vitro evidence suggests that Stat6 induction by IL-2 treatment of activated, Tg T cells is no greater than the response of wild-type cells to IL-4 (Fig. 4). As such, these findings complement and extend in vitro evidence suggesting that the genetic background of a T cell is a more important factor in the polarization of effector function than is the source of the APCs (63).
One current model for the regulated choice of polarized effector cytokine production divides the process into two phases. In the first phase, the naive uncommitted precursor is induced to secrete IL-2 and to activate at least low-level transcription of IL-4 and IFN-γ (64). The magnitude of IL-4 gene activation in this phase would depend primarily on the strength of TCR signaling, costimulation, and CD4 engagement (14–18, 56, 65). The initial release of IL-4, or IFN-γ and resultant IL-12, would then result in an amplification phase during which Stat6, or Stat1 and Stat4, would potentiate the differentiation process. In contrast to their wild-type counterparts, T cells bearing the chimeric receptor transgene could transduce IL-4–specific signals in the earlier phase due to the presence of IL-2, leading to enhanced Th2 development. The observed increase in IgE levels of chimeric receptor mice before immunization with an antigen suggests that these early-phase IL-4 signals may be sufficient to increase the development of effector Th2 cells as the antigen-experienced T cell effector and memory repertoires evolve in vivo. Since GATA-3 expression in Tg T cells has been reported to be sufficient to enhance Th2 development (53), the finding that GATA-3 expression is potentiated after activation of chimeric cytokine receptor-bearing T cells may provide the mechanistic basis for the T cell–autonomous increase in Th2 cells. However, other transcription factors may also be involved in this effect.
In contrast to the increase in IgE, the finding that IgG2a levels in our Tg mice are essentially normal suggests that the alteration in signaling caused by chimeric receptor expression in T cells may not be sufficient to suppress type 1 help in vivo. Moreover, no significant decrease in IFN-γ production by chimeric receptor transgene-positive cells was observed, even under conditions where Th2 development was >30-fold more potent than that of NTg counterparts. Although these data were less conclusive because of the variable ability of exogenous IL-4 to suppress in vitro IFN-γ production in our samples, they suggest that when IL-4 signaling during the early activation phase of effector T cell development is confined to the T cells, enhancement of type 2 help may be uncoupled from inhibition of type 1 help. Consistent with this possibility, transfer experiments suggest that exposure of APCs to IL-4 in vivo creates an APC population that does not support Th1 development (15). Thus, it is possible that IL-4 action on APCs may play a role in the inhibition of Th1 development, a mechanism analogous to the role of APCs in IFN-γ–induced Th1 development but in contrast to the T cell– autonomous promotion of Th2 development by IL-4. Gene knockout experiments have shown that the IL-4– induced transcription factor Stat6 is involved in inhibition of the emergence of Th1 cells as well as in Th2 development (66, 67). A potential explanation of the normal IFN-γ production and IgG2a is that this inhibitory function of Stat6 is mediated by its activity in APCs. Since IFN-γ acts on APCs to enhance IL-12 release and promote Th1 development (11, 68), one possibility for target gene regulation in the APCs is that activated Stat6 inhibits induction of interferon-responsive factors such as the transcription factor IRF-1. In this regard, an intact IRF-1 gene in the APC population is required for normal Th1 development (27, 28). Moreover, IL-4 can inhibit IRF-1 promoter induction (69) by a Stat6-dependent mechanism (Goenka, S., J. Youn, L.-y. Yu-Lee, U. Schindler, and M. Boothby, manuscript submitted for publication). Thus, one mechanism that could mediate a role for APCs in IL-4–induced repression of Th1 development is inhibition of IRF-1. A mechanism that may be T cell–autonomous has been reported to reinforce inhibition of Th1 development by IL-4 in an in vitro system. In this model, IL-4 inhibits expression on T cells of an IL-12 receptor chain (IL-12Rβ2) needed for normal signaling (70). These results were obtained using cells from BALB/c-background mice, and the extinction of IL-12Rβ2 mRNA required ∼5 d, a time point at which many cells may already have become committed to IFN-γ production (64, 70). However, our data reflect a C57BL/6 background. Thus, one possible explanation is that Th1 regulation used different mechanisms in cells derived from a BALB/c (Th2-oriented) background as compared with C57BL/6-derived cells. Also, IL-4 signaling by the chimeric receptor may primarily occur early during effector cell differentiation, when IL-2 levels are maximal, so that the mechanism inhibiting Th1 development upon initial T cell activation may differ from the process in which cells committed to a Th2 phenotype are rendered unresponsive to IL-12.
An important question underlying these studies is the degree to which genetic influences that bias the effector T cell repertoire account for differences in susceptibility to allergic diseases (34, 71). Although it is clear that the efficiency of generating type 2 help can be correlated with susceptibility of C57BL (B6 or B10.D2) and BALB/c mice to immunologic diseases (61, 72, 73), it also is clear that susceptibility arises from complex, polygenic characteristics and not only from Th1/Th2 regulation (32, 33, 74–76). For instance, in certain instances BALB/c mice are unable to heal Leishmania major infection despite inactivation of the IL-4 gene (32). A reciprocal question relates to the resistance of C57BL/6 mice to airway hyperresponsiveness in allergic airway disease (61). In particular, it is not clear if the T cell contribution to allergic disease is exclusively a reflection of Th2 development, or if other T cells may be important (for a review, see reference 77). While previous studies have shown that transfer of 5 × 106 activated, antigen-specific Th2 cells is sufficient to create susceptibility in BALB/c mice (78), other data indicate that far fewer antigen-specific T cells normally arise after immune challenge (79). Moreover, data from other disease models show that normal resistance is overcome when such large numbers of cells are used in transfers (80). The present data indicate that the introduction of a bias toward Th2 development on a C57BL/6 background is sufficient to potentiate a bronchoconstrictor response when normal numbers of T cells are present, the response evolves exclusively from antigen challenge, and effector cytokines are produced only by their natural promoters in physiologically appropriate cell types. Moreover, our findings are consistent with the possibility that a sufficient enhancement of IL-4Rα signaling in T cells could promote allergic disease susceptibility in humans (34). These data suggest that one useful application of the chimeric cytokine receptor transgene will be in simplifying the analysis of disease susceptibility traits. It can be predicted that of the many loci that contribute to differences between C57BL (B6 or B10.D2) and BALB/c mice in disease susceptibility, only some will be involved in regulation of the balance between Th1 and Th2 cells. Use of this dominant monogenic trait (the transgene) could facilitate identification of those loci that regulate functions other than effector T cell phenotype. However, it should be noted that the magnitude of such airway hyperresponsiveness was still considerably less than that typically obtained in BALB/c mice (Aronica, M.A., and J.R. Sheller, unpublished observations). Thus, the susceptibility of the BALB/c strain to allergic airway disease probably reflects genetic contributions in addition to its tendency to develop robust Th2 responses.
Acknowledgments
We gratefully acknowledge technical assistance from W. Armistead and D. Mitchell, helpful discussions and a critical reading of the manuscript by M. Rincón, G. Miller, T. Aune, and L. Van Kaer, invaluable advice from and discussions with D. Gabrilovich, and assistance with flow cytometry from J. Price (VA Medical Center and Vanderbilt University Cancer Center Flow Cytometry Core), and D. MacFarland (Howard Hughes Medical Institute Flow Cytometry Core).
This study was supported by the National Institutes of Health (NIH) Cancer Center grant CA-68485, core facilities of the Vanderbilt Cancer Center and Diabetes Research and Training Center (DRTC; P60 DK20593), an American Heart Association Scientist Development Award (J. Chen), a Glaxo-Wellcome Pulmonary Fellowship (M.A. Aronica), NIH grant GM-15431 (J. Sheller), a Leukemia Society of America Scholar's Award (M. Boothby), NIH grant R 01 GM-42550, and the Mark Collie Pilot Project Fund of the DRTC (M. Boothby).
Abbreviations used in this paper
- IRF
interferon regulatory factor
- NTg
nontransgenic
- Stat
signal transducer and activator of transcription;Tg, transgenic
Footnotes
Youn, J., et al., unpublished observations.
References
- 1.Paul WE. Interleukin-4: a prototypic immunoregulatory lymphokine. Blood. 1991;77:1859–1870. [PubMed] [Google Scholar]
- 2.Jansen JH, Fibbe WE, Willemze R, Kluin-Nelemans JC. Interleukin-4: a regulatory protein. Blut. 1990;60:269–274. doi: 10.1007/BF01736226. [DOI] [PubMed] [Google Scholar]
- 3.Foote LC, Howard RG, Marshak-Rothstein A, Rothstein TL. IL-4 induces Fas resistance in B cells. J Immunol. 1996;157:2749–2753. [PubMed] [Google Scholar]
- 4.Vella A, Teague TK, Ihle J, Kappler J, Marrack P. Interleukin 4 (IL-4) or IL-7 prevents the death of resting T cells: Stat6 is probably not required for the effect of IL-4. J Exp Med. 1997;186:325–330. doi: 10.1084/jem.186.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rennick D, Yang G, Muller-Sieburg C, Smith C, Arai N, Takabe Y, Gemmell L. Interleukin 4 (B-cell stimulatory factor 1) can enhance or antagonize the factor-dependent growth of hemopoietic progenitor cells. Proc Natl Acad Sci USA. 1987;84:6889–6893. doi: 10.1073/pnas.84.19.6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Gros G, Ben-Sasson SZ, Seder R, Finkleman FD, Paul WE. Generation of interleukin (IL)-4-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4–producing cells. J Exp Med. 1990;172:921–929. doi: 10.1084/jem.172.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swain SL, Weinberg AD, English M, Huston G. IL-4 directs the development of Th2 like helper effectors. J Immunol. 1990;145:3796–3806. [PubMed] [Google Scholar]
- 8.Abehsira-Amar O, Gibert M, Joliy M, Theze J, Jankovic D. IL-4 plays a dominant role in the differential development of Th0 into Th1 and Th2 cells. J Immunol. 1992;148:3820–3829. [PubMed] [Google Scholar]
- 9.Kim J, Woods A, Becker-Dunn E, Bottomly K. Distinct functional phenotypes of clones Ia-restricted helper T cells. J Exp Med. 1985;162:188–201. doi: 10.1084/jem.162.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 11.Hsieh C-S, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 12.Mosmann TR, Coffman RL. Th1 and Th2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 13.Swain SL, Bradley LM, Croft M, Tonkowagy S, Atkins G, Weinberg AD, Duncan DD, Hedrick SM, Dutton RW, Huston G. Helper T-cell subsets: phenotype, function and the role lymphokines in regulating their development. Immunol Rev. 1991;123:115–144. doi: 10.1111/j.1600-065x.1991.tb00608.x. [DOI] [PubMed] [Google Scholar]
- 14.Seder RA, Germain RN, Linsley PS, Paul WE. CD28-mediated costimulation of interleukin 2 (IL-2) production plays a critical role in T cell priming for IL-4 and interferon γ production. J Exp Med. 1994;179:299–304. doi: 10.1084/jem.179.1.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cua DJ, Coffman RL, Stohlman SA. Exposure to T helper 2 cytokines in vivobefore encounter with antigen selects for T helper subsets via alterations in antigen-presenting cell function. J Immunol. 1996;157:2830–2836. [PubMed] [Google Scholar]
- 16.Rulifson IC, Sperling AI, Fields PE, Fitch FW, Bluestone JA. CD28 costimulation promotes the production of Th2 cytokines. J Immunol. 1997;158:658–665. [PubMed] [Google Scholar]
- 17.Fowell DJ, Magram J, Turck CW, Killeen N, Locksley RM. Impaired Th2 subset development in the absence of CD4. Immunity. 1997;6:559–569. doi: 10.1016/s1074-7613(00)80344-1. [DOI] [PubMed] [Google Scholar]
- 18.Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol. 1997;15:297–322. doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- 19.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu C-Y, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFNγ production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 20.Wenner CA, Guler ML, Macatonia SE, O'Garra A, Murphy KM. Roles of IFN-γ and IFN-α in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 21.Seder RA, Gazzinelli R, Sher A, Paul WE. IL-12 acts directly on CD4+T cells to enhance priming for IFNγ production and diminishes IL-4 inhibition of such priming. Proc Natl Acad Sci USA. 1993;90:10188–10192. doi: 10.1073/pnas.90.21.10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 23.Burstein HJ, Tepper RI, Leder P, Abbas AK. Humoral immune functions in IL-4 transgenic mice. J Immunol. 1991;147:2950–2956. [PubMed] [Google Scholar]
- 24.Yoshimoto K, Swain SL, Bradley LM. Enhanced development of Th2-like primary CD4 effectors in response to sustained exposure to limited rIL-4 in vivo. . J Immunol. 1996;156:3267–3274. [PubMed] [Google Scholar]
- 25.Bradley LM, Dalton DK, Croft M. A direct role for IFN-γ in regulation of Th1 cell development. J Immunol. 1996;157:1350–1358. [PubMed] [Google Scholar]
- 26.Dighe AS, Campbell D, Hsieh C-S, Clarke S, Greaves DR, Gordon S, Murphy KM, Schreiber RD. Tissue-specific targeting of cytokine unresponsiveness in transgenic mice. Immunity. 1995;3:657–666. doi: 10.1016/1074-7613(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 27.Lohoff M, Ferrick D, Mittrucker H-W, Duncan GS, Bischof S, Rollinghoff M, Mak TW. Interferon regulatory factor-1 is required for a T helper 1 immune response in vivo. . Immunity. 1997;6:681–689. doi: 10.1016/s1074-7613(00)80444-6. [DOI] [PubMed] [Google Scholar]
- 28.Taki S, Sato T, Ogasawara K, Fukuda T, Sato M, Hida S, Suzuki G, Mitsuyama M, Shin E-H, Kojima S, et al. Multistage regulation of Th-1 type immune responses by the transcription factor IRF-1. Immunity. 1997;6:673–679. doi: 10.1016/s1074-7613(00)80443-4. [DOI] [PubMed] [Google Scholar]
- 29.Stack RM, Lenschow DJ, Gray GS, Bluestone JA, Fitch FW. IL-4 treatment of small splenic B cells induces the costimulatory molecules B7-1 and B7-2. J Immunol. 1994;152:5723–5733. [PubMed] [Google Scholar]
- 30.Mond JJ, Carman J, Sarma C, Ohara J, Finkelman FD. Interferon-γ suppresses B cell stimulation factor (BSF-1) induction of class II MHC determinants on B cells. J Immunol. 1986;137:3534–3537. [PubMed] [Google Scholar]
- 31.Boothby M, Gravallese E, Liou H-C, Glimcher LH. A DNA binding protein regulated by IL-4 and by differentiation in B cells. Science. 1988;242:1559–1562. doi: 10.1126/science.3144043. [DOI] [PubMed] [Google Scholar]
- 32.Noben-Trauth N, Kropf P, Muller I. Susceptibility to Leishmania majorinfection in interleukin-4-deficient mice. Science. 1996;271:987–990. doi: 10.1126/science.271.5251.987. [DOI] [PubMed] [Google Scholar]
- 33.Kropf P, Etges R, Schopf L, Chung C, Sypek J, Muller I. Characterization of T cell-mediated responses in nonhealing and healing Leishmania majorinfections in the absence of endogenous IL-4. J Immunol. 1997;159:3434–3443. [PubMed] [Google Scholar]
- 34.Hershey GKK, Friedrich MF, Esswein LA, Thomas ML, Chatila TA. The association of atopy with a gain-of-function mutation in the alpha subunit of the interleukin-4 receptor. N Engl J Med. 1997;337:1720–1725. doi: 10.1056/NEJM199712113372403. [DOI] [PubMed] [Google Scholar]
- 35.Wang HY, Paul WE, Keegan AD. IL-4 function can be transferred to the IL-2 receptor by tyrosine containing sequences found in the IL-4 receptor α chain. Immunity. 1996;4:113–121. doi: 10.1016/s1074-7613(00)80676-7. [DOI] [PubMed] [Google Scholar]
- 36.Nemoto T, Takeshita T, Ishii N, Kondo M, Higuchi M, Satomi S, Nakamura M, Mori S, Sugamura K. Differences in the interleukin-2 (IL-2) receptor system in human and mouse: α chain is required for formation of the functional mouse IL-2 receptor. Eur J Immunol. 1995;25:3001–3005. doi: 10.1002/eji.1830251102. [DOI] [PubMed] [Google Scholar]
- 37.Hattori M, Okazaki H, Ishida Y, Onuma M, Kano S, Honjo T, Minato N. Expression of murine IL-2 receptor β-chain on thymic and splenic lymphocyte subpopulations as revealed by the IL-2-induced proliferative response in human IL-2 receptor α-chain transgenic mice. J Immunol. 1990;144:3809–3815. [PubMed] [Google Scholar]
- 38.Asano M, Ishida Y, Sabe H, Kondo M, Sugamura K, Honjo T. IL-2 can support growth of CD8+ T cells but not CD4+ T cells of human IL-2 receptor β-chain transgenic mice. J Immunol. 1994;153:5373–5381. [PubMed] [Google Scholar]
- 39.Boothby M, Mora AL, Scherer DC, Brockman J, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of NF-κB. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kono T, Doi T, Yamada G, Hatakeyama M, Minamoto S, Tsudo M, Miyasaka M, Miyata T, Taniguchi T. Murine interleukin 2 receptor β chain: dysregulated gene expression in lymphoma line EL-4 caused by a promoter insertion. Proc Natl Acad Sci USA. 1990;87:1806–1810. doi: 10.1073/pnas.87.5.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mosley B, Beckmann MP, March CJ, Idzerda RL, Gimpel SD, VandenBos T, Friend D, Alpert A, Anderson D, Jackson J, et al. The murine interleukin 4 receptor: molecular cloning and characterization of secreted and membrane bound forms. Cell. 1989;59:335–348. doi: 10.1016/0092-8674(89)90295-x. [DOI] [PubMed] [Google Scholar]
- 42.Lewis DB, Yu CC, Forbush KA, Carpenter J, Sato TA, Grossman A, Liggitt DH, Perlmutter RM. Interleukin 4 expressed in situ selectively alters thymocyte development. J Exp Med. 1989;173:89–100. doi: 10.1084/jem.173.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryan JJ, McReynolds LJ, Keegan A, Wang L-H, Garfein E, Rothman P, Nelms K, Paul WE. Growth and gene expression are predominantly controlled by distinct regions of the human IL-4 receptor. Immunity. 1996;4:123–132. doi: 10.1016/s1074-7613(00)80677-9. [DOI] [PubMed] [Google Scholar]
- 44.Wang D-Z, Cherrington AL, Famakin-Mosuro BM, Boothby M. Independent pathways to de-repression of the mouse immunoglobulin heavy chain epsilon germ-line promoter. Int Immunol. 1996;8:977–989. doi: 10.1093/intimm/8.7.977. [DOI] [PubMed] [Google Scholar]
- 45.Schindler C, Shuai K, Prezioso VR, Darnell JE., Jr Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science. 1992;257:809–813. doi: 10.1126/science.1496401. [DOI] [PubMed] [Google Scholar]
- 46.Tu Y-P, Larsen GL, Irvin GL. Utility of murine systems to study asthma pathogenesis. Eur Respir Rev. 1995;5:224–230. [Google Scholar]
- 47.Irvin CG, Tu Y-P, Sheller JR, Funk CD. 5-Lipoxygenase products are necessary for ovalbumin-induced airway responsiveness in mice. Am J Physiol. 1997;272:L1053–L1058. doi: 10.1152/ajplung.1997.272.6.L1053. [DOI] [PubMed] [Google Scholar]
- 48.Swain SL. Generation and in vivopersistence of polarized Th1 and Th2 memory cells. Immunity. 1994;1:543–552. doi: 10.1016/1074-7613(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 49.O'Shea JJ. Jaks, STATs, cytokine signal transduction, and immunoregulation: are we there yet? . Immunity. 1997;7:1–11. doi: 10.1016/s1074-7613(00)80505-1. [DOI] [PubMed] [Google Scholar]
- 50.Keegan AD, Nelms K, White M, Wang L-M, Pierce JH, Paul WE. An IL-4 receptor region containing an insulin receptor motif is important for IL-4-mediated IRS-1 phosphorylation and cell growth. Cell. 1994;76:811–820. doi: 10.1016/0092-8674(94)90356-5. [DOI] [PubMed] [Google Scholar]
- 51.Welham MJ, Duronio V, Schrader JW. Interleukin-4-dependent proliferation dissociates p44erk-1, p42erk-2, and p21rasactivation from cell growth. J Biol Chem. 1994;269:5865–5873. [PubMed] [Google Scholar]
- 52.Bendelac A, Rivera MN, Park S-H, Roark JH. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu Rev Immunol. 1997;15:535–562. doi: 10.1146/annurev.immunol.15.1.535. [DOI] [PubMed] [Google Scholar]
- 53.Zheng W-p, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 54.Bradley LM, Duncan DD, Yoshimoto K, Swain SL. Memory effectors: a potent, IL-4-secreting helper T cell population that develops in vivoafter restimulation with antigen. J Immunol. 1993;150:3119–3130. [PubMed] [Google Scholar]
- 55.Natesan M, Razi-Wolf Z, Reiser H. Costimulation of IL-4 production by murine B7-1 and B7-2 molecules. J Immunol. 1996;156:2783–2791. [PubMed] [Google Scholar]
- 56.Seder, R.A., and W.E. Paul. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12: 635–673. [DOI] [PubMed]
- 57.Finkelman FD, Holmes J, Katona IM, Urban JF, Jr, Beckmann MP, Park LS, Schooley KA, Coffman RL, Mosmann TR, Paul WE. Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol. 1990;8:303–333. doi: 10.1146/annurev.iy.08.040190.001511. [DOI] [PubMed] [Google Scholar]
- 58.Mandler R, Finkelman FD, Levine AD, Snapper CM. IL-4 induction of IgE class switching by lipopolysaccharide-activated murine B cells occurs predominantly through sequential switching. J Immunol. 1993;150:407–418. [PubMed] [Google Scholar]
- 59.Snapper CM, Marcu KB, Zelazowski P. The immunoglobulin class switch: beyond “accessibility.” . Immunity. 1997;6:217–223. doi: 10.1016/s1074-7613(00)80324-6. [DOI] [PubMed] [Google Scholar]
- 60.Drazen JM, Arm JP, Austen KF. Sorting out the cytokines of asthma. J Exp Med. 1996;183:1–5. doi: 10.1084/jem.183.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Lamm WJE, Albert RK, Chi EY, Henderson WR, Lewis DB. Influence of the route of allergen administration and genetic background on the murine allergic pulmonary response. Am J Respir Crit Care Med. 1997;155:661–669. doi: 10.1164/ajrccm.155.2.9032210. [DOI] [PubMed] [Google Scholar]
- 62.Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a mouse model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsieh CS, Macatonia SE, O'Garra A, Murphy KM. T cell genetic background determines default T helper phenotype development in vitro. J Exp Med. 1995;181:713–721. doi: 10.1084/jem.181.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakamura T, Kamogawa Y, Bottomly K, Flavell RA. Polarization of IL-4- and IFN-γ-producing CD4+ T cells following activation of naive CD4+T cells. J Immunol. 1997;158:1085–1094. [PubMed] [Google Scholar]
- 65.Murphy KM, Murphy TL, Szabo SJ, Jacobson NG, Guler ML, Gorham JD, Gubler U. Regulation of IL-12 receptor expression in early T-helper responses implies two phases of Th1 differentiation: capacitance and development. Chem Immunol. 1997;68:54–69. doi: 10.1159/000058694. [DOI] [PubMed] [Google Scholar]
- 66.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S-i, Nakanishi K, Yoshida N, Kishimoto T, Kira S. Essential role of Stat6 in IL-4 signaling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 67.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, Chu C, Quelle FW, Nosaka T, Vignali DAA et. al. Lack of IL-4-induced Th2 response and IgE class switching in mice with a disrupted Stat6 gene. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 68.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-γ. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 69.Ohmori Y, Hamilton TA. IL-4-induced Stat6 suppresses IFN-γ-stimulated Stat1-dependent transcription in mouse macrophages. J Immunol. 1997;159:5474–5482. [PubMed] [Google Scholar]
- 70.Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R β2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Postma DS, Bleecker ER, Amelung PJ, Holroyd KJ, Xu J, Panhuysen CIM, Meyers DA, Levitt RC. Genetic susceptibility to asthma—bronchial hyperresponsiveness coinherited with a major gene for atopy. N Engl J Med. 1995;333:894–900. doi: 10.1056/NEJM199510053331402. [DOI] [PubMed] [Google Scholar]
- 72.Scott B, Liblau R, Degermann S, Marconi LA, Ogata L, Caton AJ, McDevitt HO, Lo D. A role for non-MHC genetic polymorphism in susceptibility to spontaneous autoimmunity. Immunity. 1994;1:73–82. doi: 10.1016/1074-7613(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 73.Reiner SL, Locksley RM. The regulation of immunity to Leishmania major. . Annu Rev Immunol. 1995;13:151–177. doi: 10.1146/annurev.iy.13.040195.001055. [DOI] [PubMed] [Google Scholar]
- 74.DeSanctis GT, Merchant M, Beier DR, Dredge RD, Grobholz JK, Martin TR, Lander ES, Drazen JM. Quantitative locus analysis of airway hyper-responsiveness in A/J and C57BL/6 mice. Nat Genet. 1995;11:150–154. doi: 10.1038/ng1095-150. [DOI] [PubMed] [Google Scholar]
- 75.DeSanctis GT, Itoh A, Green FH, Qin S, Kimura T, Grobholz JK, Martin TR, Maki T, Drazen JM. T-lymphocytes regulate genetically determined airway hyperresponsiveness in mice. Nat Med. 1997;3:460–462. doi: 10.1038/nm0497-460. [DOI] [PubMed] [Google Scholar]
- 76.Beebe AM, Mauze S, Schork NJ, Coffman RL. Serial backcross mapping of multiple loci associated with resistance to Leishmania majorin mice. Immunity. 1997;6:551–557. doi: 10.1016/s1074-7613(00)80343-x. [DOI] [PubMed] [Google Scholar]
- 77.Holtzman MJ, Sampath D, Castro M, Look DC, Jayaraman S. The one-two of T helper cells: does interferon-γ knock out the Th2 hypothesis for asthma? . Am J Respir Cell Mol Biol. 1996;14:316–318. doi: 10.1165/ajrcmb.14.4.8600934. [DOI] [PubMed] [Google Scholar]
- 78.Cohn L, Homer RJ, Marinov A, Rankin J, Bottomly K. Induction of airway mucus production by T helper 2 (Th2) cells: a critical role for interleukin 4 in cell recruitment but not mucus production. J Exp Med. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McHeyzer-Williams MG, Davis MM. Antigen-specific development of primary and memory T cells in vivo. . Science. 1995;268:106–111. doi: 10.1126/science.7535476. [DOI] [PubMed] [Google Scholar]
- 80.Kurts C, Carbone FR, Barnden M, Blanas E, Allison J, Heath WR, Miller JFAP. CD4+ T cell help impairs CD8+T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J Exp Med. 1997;186:2057–2062. doi: 10.1084/jem.186.12.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]