

Abstract
The induction of optimal systemic antitumor immunity involves the priming of both CD4+ and CD8+ T cells specific for tumor-associated antigens. The role of CD4+ T helper cells (Th) in this response has been largely attributed to providing regulatory signals required for the priming of major histocompatibility complex class I restricted CD8+ cytolytic T lymphocytes, which are thought to serve as the dominant effector cell mediating tumor killing. However, analysis of the effector phase of tumor rejection induced by vaccination with irradiated tumor cells transduced to secrete granulocyte/macrophage colony-stimulating factor indicates a far broader role for CD4+ T cells in orchestrating the host response to tumor. This form of immunization leads to the simultaneous induction of Th1 and Th2 responses, both of which are required for maximal systemic antitumor immunity. Cytokines produced by these CD4+ T cells activate eosinophils as well as macrophages that produce both superoxide and nitric oxide. Both of these cell types then collaborate within the site of tumor challenge to cause its destruction.
Keywords: cancer, vaccine, T helper cell, macrophage, eosinophil
The exquisite specificity of antigen recognition by the T cell arm of the immune response provides an important basis for cancer immunotherapy. The ability to discriminate tumor cells from normal tissues is critical for enabling effective tumor destruction while minimizing toxicity. Indeed, the isolation of tumor-specific T cells from cancer patients has fueled the search for tumor-associated antigens. The molecular identification of several such antigens, particularly in human melanoma, has enabled the pursuit of vaccine strategies that specifically target defined tumor antigens (1–3).
In the setting where relevant tumor rejection antigens have yet to be defined, vaccination with modified whole tumor cells as the antigen source has been explored as a means to prime systemic antitumor immunity. The host response to this form of vaccination has been shown to be significantly enhanced when the immunizing tumor cells are transduced with genes encoding cytokines or other molecules involved in the regulation of immune responses (4). For both tumor cell–based and defined antigen vaccine strategies, T cell subset depletion studies have usually demonstrated the requirement for both CD4+ and CD8+ T cells for systemic tumor rejection to occur.
Given that most nonhematopoietic tumors express MHC class I molecules, which serve as the restricting element for CD8+ T cell recognition, but do not express MHC class II molecules, which are required for CD4+ T cell recognition, it has been assumed that the predominant tumoricidal effector mechanism is killing by CD8+ CTL. The requirement for CD4+ T cells in these responses has been attributed to providing help during priming to achieve full activation and effector function of tumor-specific CTL. Indeed, MHC class I restricted CD8+ T cells that specifically lyse tumor cells in vitro are frequently measured to document vaccine efficacy and to serve as a surrogate end point in clinical tumor vaccine trials.
However, several lines of evidence suggest a broader role for CD4+ T cells in mediating other significant antitumor effector functions. First, studies in which mice were vaccinated several weeks before a live tumor challenge demonstrated that although immunologically intact mice were able to reject tumor, the depletion of CD4+ T cells with a mAb just before the tumor challenge resulted in the complete loss of tumor rejection (5). In these experiments, T cell help for CTL priming was fully available at the time of vaccination. This outcome suggests that CD4+ T cells play a more direct role in the effector phase of tumor rejection. Second, we have demonstrated previously that vaccination against tumor cells completely lacking MHC class I expression resulted in tumor rejection that was comparable to that seen against a challenge with an MHC class I positive variant of the same tumor (6). Depletion of either CD4+ T cells or NK cells, but not CD8+ T cells, resulted in the inability to reject the MHC class I negative variant, suggesting that CD4+ T cells may provide help as well as a measure of antigen specificity to effector cells of the immune response that do not themselves have the capacity for antigen-specific recognition.
To dissect the mechanism of CD4+ T cell effector function during tumor rejection, we studied immune responses induced by a genetically modified whole cell vaccine. A comparison of genetically modified whole cell vaccines, using the B16 melanoma (7) transduced with a large set of individual cytokine genes, demonstrated that GM-CSF– transduced vaccines generated potent and long-lasting systemic antitumor immunity that was dependent on both CD4+ and CD8+ T cells (5). Induction of immunity against the poorly immunogenic B16 tumor appears to be due to the ability of the locally produced GM-CSF to activate bone marrow–derived APCs during the priming phase to process and present tumor antigens to both CD4+ and CD8+ T cells (8–10). In the following studies, we were able to demonstrate that rather than simply providing help for CD8+ T cells, CD4+ T cells express both Th1 and Th2 cytokines and recruit other antitumor effector cells in addition to CD8+ T cells.
Materials and Methods
In Vivo Vaccination: Tumor Challenge Experiments.
6–8-wk-old female C57BL/6 mice were obtained from the National Cancer Institute (Bethesda, MD) and housed in the Johns Hopkins Oncology Center Animal Facility. In the same facility, CD4−/−, CD8−/−, γ-IFN−/−, IL-4−/−, IL-5−/−, X-CGD, and inducible nitric oxide synthase (iNOS−/−)1 mice that had been backcrossed to the C57BL/6 background for greater than seven generations were bred, and female offspring were used for experiments starting at 6–8 wk of age. CD4−/− and CD8−/− mice were gifts from Dr. Tak Mak (Ontario Cancer Institute, University of Toronto, Toronto, Canada). IL-4−/−, γ-IFN−/−, and iNOS−/− mice were obtained from The Jackson Laboratories (Bar Harbor, ME). IL-5−/− and X-CGD mice were gifts of Dr. Eric Pearlman (Case Western Reserve, Cleveland, OH) and Dr. Mary Dinauer (Indiana University School of Medicine, Indianapolis, IN), respectively. In brief, mice were injected subcutaneously in the left flank with 106 irradiated (50 Gy) B16 tumor cells transduced with the GM-CSF gene (B16-GM-CSF), resulting in the production of 420 ng GM-CSF/106 cells/24 h (5). 2 wk later, mice were challenged in the right flank with 105 live nontransduced B16 cells (B16-WT). B16-GM-CSF and B16 wild-type cells were provided by Dr. Glenn Dranoff (Dana Farber Cancer Institute, Boston, MA). Before injection, cells were harvested while in log phase growth from in vitro cell culture by trypsinization and were washed three times in serum-free 1× HBSS. All injections were in 0.1-ml vol. All experimental groups were matched for both age and sex. Data from the immunization challenge experiments are presented as Kaplan-Meier plots, representing the percentage of animals without detectable tumor. Mice were monitored twice weekly for tumor growth. All individual experiments included a minimum of 10 mice/group, and data from at least two repeat experiments are pooled in the Kaplan-Meier analyses.
Quantitative RT-PCR.
TRIZOL (GIBCO BRL, Gaithersburg, MD) was used to extract total RNA from lymphocytes draining the site of s.c. vaccination with irradiated B16-GM-CSF and from the site of s.c. challenge with live B16-WT cells. The SUPERSCRIPT Preamplification System (GIBCO BRL) was used to reverse transcribe total RNA to cDNA. The cDNA and cytokine competitor (a gift from Dr. Richard Locksley, University of California San Francisco, San Francisco, CA) were then PCR amplified for γ-IFN (forward: 5′-CATTGAAAGCCTAGAAAGTCTG-3′; reverse: 5′-CTCATGAATGCATCCTTTTTCG-3′), yielding 267- and 320-bp fragments, respectively. The cDNA and cytokine competitor were also PCR-amplified for IL-4 (forward: 5′-CATCGGCATTTTGAACGAGGTCA-3′; reverse: 5′-CTTATCGATGAATCCAGGCATCG-3′), yielding 240- and 360-bp fragments, respectively. The following cycling conditions were used: 94°C for 3 min, then 35 cycles at 94°C for 40 sec, 60°C for 20 sec, and 72°C for 40 sec, followed by a final extension at 72°C for 10 min. All reactions used PCR Gem 100 (Perkin-Elmer Corp., Norwalk, CT) for hot start and 32P-labeled primers for later determination of final product yield. The products were then resolved on a 6% polyacrylamide gel, dried on a Bio-Rad gel dryer (Bio-Rad Lab., Hercules, CA), and the final product yields were quantified on a PhosphorImager scanner (Molecular Dynamics, Sunnyvale, CA). The cytokine cDNA was held constant and the competitor concentration was varied until the yield of the two products were at least within threefold of each other. The initial concentration of the cytokine in the PCR reaction was then calculated by the following formula: initial cytokine level = (cytokine product signal/competitor product signal) × (starting concentration of competitor). The number of copies per microgram of total RNA was then calculated.
Immunohistochemistry for iNOS.
The biopsy of the tumor challenge site was removed and flash frozen in O.C.T. compound (Sakura Finetek USA, Inc., Torrance, CA) and dry-ice cooled isopentane. 5-μm tissue sections were fixed for 10 min in acetone and washed with TBS. Sections were quenched with a solution of 0.05% H2O2, 1% normal goat serum, and 0.5% milk for 30 min and then blocked with TBS/milk/1% normal goat serum for 10 min. The sections were then incubated with either α-iNOS antibody (11) or Mac-3 antibody (American Type Culture Collection, Rockville, MD) for 60 min. After washing, sections were incubated with biotinylated secondary antibody, goat α-rabbit IgG Fc for the α-iNOS antibody (Jackson ImmunoResearch Labs, Inc., West Grove, PA) and rabbit α-rat IgG (Vector Labs, Inc., Burlingame, CA) for the Mac-3 antibody, for 30 min. After washing, sections were incubated with ExtAvidin alkaline phosphatase (Sigma Chemical Co., St. Louis, MO) for 60 min. Slides were washed, and then a Fast Red substrate was used for detection.
Results
Antitumor Immunity Requires CD4+-dependent Effector Cells in Addition to CD8+ T Cells.
Wild-type, CD4 knockout (CD4−/−) (12) and CD8 knockout (CD8−/−) mice (13) were vaccinated subcutaneously in the left flank with 106 irradiated B16-GM-CSF (B16 cells transduced with GM-CSF) and challenged 2 wk later in the right flank with 105 wild-type (nontransduced) B16 melanoma cells. This dose of wild-type B16 cells is roughly 100-fold greater than the minimum dose that forms tumors in nonvaccinated C57BL/6 wild-type mice, but is rejected in the majority of animals previously vaccinated with B16-GM-CSF. In contrast to vaccinated wild-type mice, immunization of CD4−/− mice failed to prime a systemic immune response capable of rejecting this tumor challenge (Fig. 1). Interestingly, although similarly immunized CD8−/− mice (13) are impaired in their ability to reject this tumor challenge, a significant fraction of CD8−/− mice mounted a successful tumor rejection. In several experiments over a range of tumor challenge doses, the fraction of immunized CD8−/− mice that rejected a tumor challenge was roughly half of that observed in immunized wild-type mice (data not shown). In contrast, no measurable antitumor responses were ever demonstrated in CD4−/− mice. These observations directly demonstrate the existence of other CD4+ T cell–dependent effector mechanisms besides MHC class I restricted CD8+ CTL.
Figure 1.

The contribution of CD4+ and CD8+ T cell subsets to the systemic response to B16-GM-CSF vaccination. Wild-type, CD4−/−, and CD8−/− mice were injected subcutaneously in the left flank with 106 irradiated (50 Gy) B16-GM-CSF cells. Mice were challenged 2 wk later in the right flank with 105 live B16-WT cells, and examined twice weekly for the development of tumor. These data represent the results of three different experiments in which each experimental group consisted of 10 mice.
Vaccination with B16-GM-CSF Induces the Expression of Both Th1 and Th2 Cytokines.
The best characterized function of CD4+ T cells is the elaboration of cytokines that regulate downstream effector function of both cellular and humoral immunity. The division of CD4+ T cell or Th function into Th1 and Th2 phenotypes originally was based on the observation that murine T helper clones produced certain cytokines in a mutually exclusive fashion (14–16). Subsequent studies indicated that Th1 cytokines, such as γ-IFN, as well as cytokines that promote Th1 differentiation, such as IL-12, inhibit Th2 development (17– 19), whereas Th2 cytokines, such as IL-4 and IL-10, inhibit Th1 development (20–22). Taken together, these findings suggested that productive in vivo Th responses would develop uniquely along either a Th1 or a Th2 pathway and that the ensuing immunologic effector mechanisms would reflect the distinct pattern of cytokine production associated with the particular pathway of Th differentiation.
We used the B16 vaccination-challenge system to evaluate the expression of γ-IFN, the prototypical Th1 cytokine, and IL-4, the prototypical Th2 cytokine at the site of tumor challenge. Competitive quantitative PCR analysis of γ-IFN and IL-4 mRNA at the site of tumor challenge demonstrated that both of these messages were significantly increased, relative to naive mice (Fig. 2 A). The increase in γ-IFN and IL-4 mRNA was similar in vaccinated CD8−/− mice to that observed in wild-type mice. In contrast, γ-IFN and IL-4 mRNA were not increased at the tumor challenge site in vaccinated CD4−/− mice, indicating that both γ-IFN and IL-4 production are dependent upon activated CD4+ T cells. These results contrast with the findings in other studies on immune responses to parasites and intracellular bacteria that demonstrated that the production of one or the other cytokine ultimately dominates the effector phase of the response, leading to a polarized pattern of Th cytokine secretion that is associated with the ability to clear the infection (23–29).
Figure 2.


The role of cytokines in the systemic immune response to vaccination with B16-GM-CSF. (A) Mice were vaccinated with 106 irradiated (50 Gy) B16-GM-CSF tumor cells and challenged 2 wk later in the opposite flank with 105 live B16-WT tumor cells. 4 d after live tumor challenge, mice were killed and total RNA was extracted from a biopsy of the tumor challenge site. At least four mice were used for each group. γ-IFN and IL-4 mRNA levels were then determined by a competitive quantitative RT-PCR assay (14). Results are shown as number of copies of γ-IFN or IL-4 message per microgram of total RNA. (B) Wild-type, γ-IFN−/−, IL-4−/−, and IL-5−/− mice were vaccinated with irradiated B16-GM-CSF cells and challenged with B16-WT cells 2 wk later in the opposite flank, as in Fig. 1. These data represent the results of three different experiments in which each experimental group consisted of 10 mice.
Vaccination with B16-GM-CSF Requires Both Th1 and Th2 Cytokines for Maximal Systemic Tumor Immunity.
To determine whether the Th1 cytokine, γ-IFN, or the Th2 cytokine, IL-4, was required for the induction of systemic antitumor immunity by B16-GM-CSF, vaccination-challenge experiments were performed in γ-IFN−/− and IL-4−/− mice (30, 31). As demonstrated in Fig. 2 B, protective immunity against B16 melanoma challenge was significantly decreased in both sets of cytokine gene knockout mice. In the case of γ-IFN−/− mice, protection against tumor challenge was completely eliminated, whereas in IL-4−/− mice, protection was reduced by ∼50% relative to vaccinated wild-type mice. Thus, in accordance with the PCR results in Fig. 2 A, and in contrast to the immune response in previous infectious models, induction of maximal systemic antitumor immunity was dependent on both the Th1 and the Th2 components of the immune response.
Eosinophils Are a Th2 Effector Cell.
To further define potential downstream effector mechanisms mediating tumor rejection, tissue from the tumor challenge site was examined. As seen in Fig. 3, eosinophils are one of the most prominent infiltrating cell types at the site of tumor challenge in vaccinated mice (in addition to macrophages and lymphocytes). Eosinophils represent a candidate Th2-dependent antitumor effector cell. Their differentiation from myeloid progenitors requires the Th2 cytokine IL-5 (32), and their recruitment from the blood into tissues is partially dependent on IL-4 (33). Indeed, the dense eosinophil infiltrate at the tumor challenge site is reminiscent of that seen in IL-4 transduced tumor cells after in vivo injection (34, 35). Eosinophils are completely absent from the tumor challenge site in vaccinated CD4−/− mice and are ∼80% reduced in vaccinated IL-4−/− mice, indicating that CD4+ T cell–derived IL-4 is critical for their recruitment. In contrast, the challenge site of vaccinated CD8−/− and γ-IFN−/− mice demonstrated an eosinophil infiltrate similar to that of wild-type mice.
Figure 3.
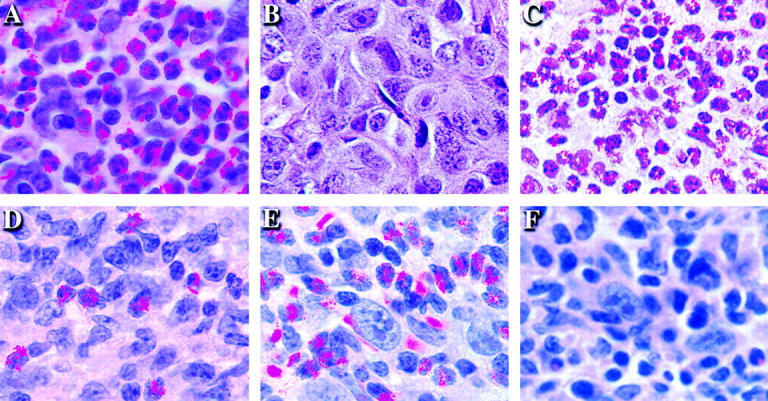
The dependence of eosinophil recruitment to the tumor challenge site on CD4+ T cells and cytokines. Mice were vaccinated with 106 irradiated (50 Gy) B16-GM-CSF cells and challenged 2 wk later on the opposite flank with 105 live B16-WT cells. After 4 d, the site of live wild-type tumor challenge was removed and sections were stained with hematoxylin and eosin. (A) Wild-type, (B) CD4−/−, (C) CD8−/−, (D) IL-4−/−, (E) γ-IFN−/−, and (F) IL-5−/− C57BL/6 mice.
Several lines of evidence support the notion that the infiltrating eosinophils are important Th2 dependent antitumor effectors, rather than simply inactive bystanders. First, protection against tumor challenge was significantly diminished in vaccinated IL-5−/− mice (Fig. 2 B) (36). Because IL-5 is critical for differentiation of bone marrow progenitors into eosinophils, mature eosinophils fail to develop in IL-5−/− mice. Second and most importantly, the loss of systemic antitumor immunity was associated with the absence of eosinophils at the tumor challenge sites in IL-5−/− mice (Fig. 3). Third, earlier studies of both mouse and human tissues revealed eosinophil degranulation at the tumor site, as demonstrated by the presence of eosinophil granule specific major basic protein in the interstitial space (37). This degranulation is a characteristic feature of activated eosinophils (38).
Tumor-specific CD8+ T Cells Are Not Sufficient for Maximal Tumor Immunity.
A number of potential antitumor effector mechanisms could depend on the Th1 component. The majority of studies on antitumor immunity have focused on CTL generation as the critical Th1-dependent effector mechanism. We wished to examine whether the priming of tumor-specific CTL was impaired in γ-IFN knockout mice, which might account for the marked dependence of successful tumor rejection on the ability to produce this cytokine. Interestingly, induction of CTL specific for an immunodominant MHC class I restricted B16 antigen derived from the tyrosinase related protein-2 protein (39) was identical in wild-type, IL-4−/−, and γ-IFN−/− mice (Fig. 4). Although CTL induction was normal in the cytokine gene knockout mice, tyrosinase related protein-2–specific CTL could not be detected in B16-GM-CSF vaccinated CD4−/− mice. This finding is consistent with recent reports that have identified CD154/CD40 interactions between CD4+ T cells and APCs as mediators of T cell help for CTL priming via the cross-priming pathway (40–42). The fact that there was not a loss of antitumor CTL activity associated with the corresponding loss of in vivo tumor protection in the cytokine gene knockout mice does not mean that CTL are unimportant as antitumor effectors, but it does indicate that CTL alone are insufficient to eliminate tumor challenges.
Figure 4.

The role of CD4+ T cells and cytokines in priming tumor-specific CTL. Wild-type, CD4−/−, γ-IFN−/−, and IL-4−/− mice were vaccinated with 106 irradiated (50 Gy) B16-GM-CSF tumor cells. After 2 wk, splenocytes were isolated and cultured for 7 d with 5 μg/ml TRP-2 peptide and 10 U/ml IL-2. On day 7, live T cells were incubated with 51Cr-labeled MC57G targets at the indicated effector to target ratios in the presence of 1 μg/ml TRP-2 peptide or irrelevant peptide. The percent peptide-specific lysis is calculated as the difference between the percent specific lysis due to the TRP-2 peptide and the percent specific lysis due to the irrelevant peptide.
Antitumor Immunity Requires Production of Nitric Oxide (NO) and Superoxide From Tumoricidal Macrophages.
Another potential Th1 effector mechanism is NO production by iNOS in activated macrophages. NO production by macrophages has been demonstrated to have tumoricidal activity both in vitro and in vivo (43–54). Indeed, immunohistochemical staining at the challenge site of B16-GM-CSF vaccinated wild-type mice showed an abundant infiltration of macrophages (Fig. 5 B). This infiltrate was still evident in the cytokine gene knockout mice, but was absent in CD4−/− mice (Fig. 5 C). Infiltrating macrophages stained strongly positive for iNOS in vaccinated wild-type mice (Fig. 5 H). In contrast, despite the presence of large numbers of macrophages in B16-GM-CSF vaccinated γ-IFN−/− mice (Fig. 5 E), iNOS was virtually absent at the challenge site in γ-IFN−/− mice (Fig. 5 K). iNOS expression was present at the challenge site of IL-4−/− mice (Fig. 5 L), although levels were reduced relative to wild-type mice. The reduction in iNOS levels in IL-4−/− mice was somewhat unexpected, given that IL-4 inhibits iNOS expression in macrophages (55–57). One possible explanation for this reduction in IL-4−/− mice could be the compensatory hyperexpression of other cytokines, such as IL-10, that indirectly inhibit iNOS expression through the inhibition of TNF-α expression, a costimulator for iNOS induction (21, 22, 58–60). It is also possible that the decreased iNOS expression is secondary to the decreased tissue eosinophilia, as eosinophils themselves are known to express TNF-α (61, 62). Furthermore, eosinophils also express eosinophil peroxidase and macrophage inflammatory protein-1, both of which induce TNF-α release from macrophages (63, 64). Therefore, it is possible that eosinophils function as a positive modulator for iNOS release by macrophages. Furthermore, the dramatic absence of iNOS expression in γ-IFN−/− mice, which fail to reject tumor in response to vaccination, suggests that NO production by tumor infiltrating macrophages may represent an important in vivo antitumor effector mechanism.
Figure 5.

Macrophages and iNOS expression at the site of wild-type tumor challenge. Mice were vaccinated with 106 irradiated (50 Gy) B16-GM-CSF tumor and challenged 2 wk later with 105 live B16-WT tumor. After 4 d, mice were killed and the live wild-type tumor challenge site was sectioned and stained for macrophages (A–F) and iNOS (G–L) (24). (A) Control for macrophage staining: no primary antibody; (B) macrophages at site of tumor challenge in wild-type mice; (C) CD4−/− mice; (D) CD8−/− mice; (E) γ-IFN−/− mice; and (F) IL-4−/− mice; (G) control for iNOS staining: no primary antibody; (H) iNOS at site of tumor challenge in wild-type mice; (I) CD4−/− mice; (J) CD8−/− mice; (K) γ-IFN−/− mice; and (L) IL-4−/− mice.
Direct functional evidence for the role of iNOS in tumor rejection was sought in vaccination-challenge experiments in iNOS−/− mice. In accordance with the previous immunohistochemical staining, vaccinated iNOS−/− mice show a substantial decrease in protection against challenge with tumor (Table 1), thereby demonstrating the critical role for NO in antitumor killing.
Table 1.
The Role of iNOS and NADPH Oxidase in Mediating Tumor Rejection in Response to B16-GM-CSF Vaccination
| WT no vaccine | NOS−/− no vaccine | WT B16-GM-CSF | NOS−/− B16-GM-CSF | |||||
|---|---|---|---|---|---|---|---|---|
| Exp. 1 | 0/5 (0%) | 0/5 (0%) | 10/10 (100%) | 6/10 (60%) | ||||
| Exp. 2 | 0/5 (0%) | 0/5 (0%) | 9/10 (90%) | 5/10 (50%) | ||||
| WT no vaccine | X-CGD no vaccine | WT B16-GM-CSF | X-CGD B16-GM-CSF | |||||
| Exp. 1 | 0/5 (0%) | 0/5 (0%) | 8/10 (80%) | 3/10 (30%) | ||||
| Exp. 2 | 0/5 (0%) | 0/5 (0%) | 8/10 (80%) | 3/10 (30%) | ||||
| Exp. 3 | 0/5 (0%) | 0/5 (0%) | 9/10 (90%) | 4/10 (40%) |
Wild-type (WT), iNOS−/−, and X-CGD C57BL/6 mice were injected subcutaneously in the left flank with 106 irradiated (50 Gy) BI6-GM-CSF cells. Mice were challenged 2 wk later in the right flank with 105 live B16-WT cells. Results are presented as number of tumor free survivors after 8 wk over the total number of mice. P = 0.0084 by two-sided Fisher's exact test comparing vaccinated wild-type and NOS−/− mice. Fisher's exact test (rather than the χ2 test) was used to test the difference in the proportion of tumor-free survivors comparing vaccinated wild-type versus NOS−/− mice because of the small numbers of expected and observed vaccinated wild-type mice that developed tumors. The test suggests a significant difference between the proportion of tumor-free survivors in the wild-type and NOS−/− mice (P = 0.0084), with an odds ratio of 15.54 (95% confidence interval: 1.73–139.65 using Woolf's procedure). P = 0.001 by χ2 test comparing vaccinated wild-type and X-CGD mice.
Activated phagocytes can also mediate killing through the production of superoxide, which is produced by the NADPH oxidase–enzyme complex. Mutations in the CYBB gene which encodes the 91-kD glycoprotein component of the oxidase (gp91phox) account for the X-linked form of chronic granulomatous disease, an immunodeficiency syndrome characterized by recurrent infections with catalase-positive microorganisms. Phagocytes from chronic granulomatous disease patients or genetic knockout mice of gp91phox (X-CGD mice) (65) mount a severely defective respiratory burst with little or no generation of superoxide or hydrogen peroxide. X-CGD mice immunized with B16-GM-CSF showed a substantial decrease in protection against challenge with B16 tumor (Table 1), thereby implicating superoxide in the antitumor response. The loss of tumor protection in both the iNOS−/− and X-CGD mice indicates that both NO and superoxide involved in phagocytic killing play a major role in the antitumor effect induced by B16-GM-CSF.
Discussion
Taken together, the experiments presented here demonstrate that effective antitumor immunity is critically dependent upon the CD4+ T cell, which is responsible for orchestrating multiple immunologic effector arms, dependent on both Th1 and Th2 cytokines. We have demonstrated previously that the priming phase of the immune response to GM-CSF–secreting tumor cells involves the recruitment of bone marrow–derived APCs which process and present tumor antigens to both CD4+ and CD8+ T cells (10). In this study, we have examined the effector mechanisms required for the successful rejection of tumor found at distant sites. As the challenge tumor used in these studies is MHC class II negative, the effector phase of the response also requires processing of tumor antigens by infiltrating APCs for presentation to CD4+ T cells. These observations underscore the critical role of bone marrow–derived APCs in two phases of the antitumor immune response—priming of de novo T cell responses (thought to be mediated largely by activated dendritic cells) and amplification of the effector phase through the processing and presentation of tumor antigen to memory CD4+ T cells. Through the local release of cytokines, these cells direct the effector response, recruiting and activating tumoricidal macrophages, eosinophils, and the other populations seen histologically. This cooperative interaction between APC and memory CD4+ T cell is the hallmark of a classic delayed type hypersensitivity response.
The requirement for CD4+ T cells in the effector phase of an immune response to tumors that do not themselves express MHC class II is consistent with earlier results in adoptive transfer systems. Infusion of tumor-specific CD4+ T cells was shown to be capable of eradicating the MHC class II negative FBL-3 murine leukemia line (66). Class II positive macrophages were found to be required to present processed tumor antigens to CD4+ T cells (67). Furthermore, several vaccine strategies that directly enhance the priming of tumor-specific CD4+ T cells have been shown to augment the systemic rejection of MHC class II negative tumors (68–71).
Previous studies in cancer immunology have focused largely on Th1 effector mechanisms, and particularly on CTL generation. Interestingly, CTL may play a smaller role in eradication of the B16 challenge tumor than in other cancer vaccine systems because of the expression of Fas ligand by B16 (as well as by many human melanomas), which may protect the tumor from direct CTL-mediated killing (72). Instead, it appears that both macrophages and eosinophils play a major role in the immune response against B16 and probably act together to achieve more efficient tumor killing. This is supported by previous studies showing that (a) eosinophil peroxidase can synergize with macrophage reactive oxygen intermediates to kill tumor cells (73) and (b) peroxidases can catalyze the oxidation of nitrite to generate further cytotoxic radicals (74). However, neither macrophages nor eosinophils have an intrinsic capacity for tumor specificity. Instead, the tumor specificity of these effectors is based on their activation by neighboring tumor-specific Th cells.
To date, the major effort in tumor antigen identification has focused almost exclusively on antigens recognized by tumor-specific, MHC class I restricted CTL. Several antigen-specific vaccine strategies incorporating class I restricted tumor antigens identified through these efforts are being explored currently in animal models and early phase clinical trials. In the absence of known MHC class II restricted tumor antigens, such strategies rely on the effects of adjuvants or incorporate surrogate class II antigens from infectious pathogens to provide help for CTL induction. The studies presented here suggest that although Th responses generated by this type of vaccination may enhance CTL induction during priming, they are unlikely to participate in the effector phase that requires tumor-specific CD4+ T cell help. Ultimately, the optimal tumor antigen–specific vaccine will ideally incorporate a panel of dominant tumor antigens recognized by both CD4+ and CD8+ T cells.
The finding that GM-CSF transduced whole cell vaccines simultaneously induce Th1 and Th2 differentiation contrasts with previous findings of in vivo immune responses to both intracellular bacteria and parasites. In resistant mouse strains, such as C3H or C57BL/6, infection with the intracellular parasite Leishmania major exclusively induces a Th1 response, characterized by high γ-IFN and low IL-4 production (23–25). Furthermore, eradication of L. major has been shown to be dependent upon γ-IFN– dependent stimulation of iNOS in macrophages (75–84). Conversely, in susceptible mouse strains, such as BALB/c, infection with L. major induces exclusively a Th2 immune response, characterized by high IL-4 and low γ-IFN production (23–25, 28, 85). Based upon this altered pattern of Th differentiation and the direct inhibition of macrophage activation by IL-4 and IL-10 (22, 56, 60, 86–88), BALB/c mice fail to mount effective immunity against L. major challenge. In contrast, protective immunity against helminth infections, such as Schistosoma mansoni and Nippostrogylus brasiliensis, is associated with Th2 development and the induction of IL-4 (26, 27, 89).
In each of these models, the definitive demonstration that particular Th1 or Th2 cytokines are critical in directing appropriate immune responses comes from the finding that elimination of a given cytokine, either by in vivo antibody depletion or by genetic knockout, results in a loss of protection against the particular pathogen. For example, γ-IFN blockade renders formerly resistant mice susceptible to L. major infection (90, 91). In addition, administration of anti–IL-4 antibody to BALB/c mice renders them resistant to L. major infection (92). Conversely, IL-4 blockade or IL-12 administration renders resistant mice susceptible to the helminth infections Trichuris muris, Heligmosomoides polygyrus, and N. brasiliensis infections (29, 93, 94).
In these models, a Th0 response, characterized by a mixture of Th1 and Th2 cytokines, is often seen initially. However, the immune response in these models typically commits along Th1 or Th2 lines by 1–2 wk after infectious challenge (90, 92, 95–97). It is still not known whether the dual Th1/Th2 phenotype in this antitumor response is produced by one or two different populations of Th cells. However, because the RT-PCR cytokine data at the challenge site were obtained 18 d after vaccination, the mice most likely had already differentiated past the Th0 stage. Furthermore, the finding that complete rejection of challenge tumors introduced 2 wk after vaccination requires both IL-4 and γ-IFN–dependent responses suggests that the dual Th1/Th2 response persists for an extended period of time.
It also remains to be determined whether the dual Th1/ Th2 effector response seen here is a general characteristic of other cell-based tumor vaccines, or is unique to the mechanism by which paracrine GM-CSF production initiates immune responses. Interestingly, a similar Th1/Th2 pattern was seen in response to a defined tumor antigen, i.e., a B cell lymphoma idiotype protein after immunization with GM-CSF producing lymphoma cells (98). Further characterization of the Th response made by antigen-specific T cells in response to tumor vaccination is being performed currently using TCR transgenic mice specific for a defined tumor antigen. It is likely that one of the reasons for the superiority of paracrine GM-CSF vaccines relative to other cytokine-transduced vaccines relates to the central role of GM-CSF in inducing bone marrow progenitors to differentiate into dendritic cells. Currently, we are attempting to determine whether dendritic cells that differentiate under GM-CSF control are uniquely capable of inducing and maintaining Th1 and Th2 differentiation simultaneously.
These findings of induction and maintenance of simultaneous Th1 and Th2 responses are somewhat surprising, given that Th1-associated cytokines such as IL-12 and γ-IFN inhibit Th2 differentiation and Th2-associated cytokines such as IL-4 and IL-10 inhibit Th1 differentiation. Currently, we do not know whether the dual Th1/Th2 response is based on the development of Th cells that simultaneously produce Th1 and Th2 cytokines, or a mixture of separate Th1 and Th2 populations. Nonetheless, the finding that Th1 and Th2 effector mechanisms can actually collaborate with each other in directing an effective antitumor response rather than antagonizing each other has important implications for the development of cancer immunotherapies in general. Ultimately, the most effective cancer immunotherapies may indeed be those that can simultaneously marshal multiple Th1 and Th2 effector mechanisms that can cooperate in most effectively killing both the primary cancer and metastatic tumor deposits.
Acknowledgments
We would like to thank Dr. Tak Mak for providing the CD4−/− and CD8−/− mice, Dr. Eric Pearlman for the IL-5−/− mice, and Dr. Mary Dinauer for the X-CGD mice.
This work was supported by Public Health Service grant AI/AG37934-01 and gifts from the Topercer family. H. Levitsky is a Scholar of the Leukemia Society of America.
Footnotes
Abbreviations used in this paper: iNOS, inducible nitric oxide synthase; NO, nitric oxide.
References
- 1.Pardoll DM. Tumour antigens. A new look for the 1990s. Nature. 1994;369:357. doi: 10.1038/369357a0. [DOI] [PubMed] [Google Scholar]
- 2.Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawakami Y, Rosenberg SA. Human tumor antigens recognized by T-cells. Immunol Res. 1997;16:313–339. doi: 10.1007/BF02786397. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll DM. Cancer vaccines. Nat Med. 1998;4:525–531. doi: 10.1038/nm0598supp-525. [DOI] [PubMed] [Google Scholar]
- 5.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony–stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levitsky HI, Lazenby A, Hayashi RJ, Pardoll DM. In vivo priming of two distinct antitumor effector populations: the role of MHC class I expression. J Exp Med. 1994;179:1215–1224. doi: 10.1084/jem.179.4.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fidler IJ. Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res. 1975;35:218–224. [PubMed] [Google Scholar]
- 8.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inaba K, Steinman RM, Pack MW, Aya H, Inaba M, Sudo T, Wolpe S, Schuler G. Identification of proliferating dendritic cell precursors in mouse blood. J Exp Med. 1992;175:1157–1167. doi: 10.1084/jem.175.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang AY, Golumbek P, Ahmadzadeh M, Jaffee E, Pardoll D, Levitsky H. Role of bone marrow– derived cells in presenting MHC class I–restricted tumor antigens. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- 11.Lowenstein CJ, Hill SL, Lafond-Walker A, Wu J, Allen G, Landavere M, Rose NR, Herskowitz A. Nitric oxide inhibits viral replication in murine myocarditis. J Clin Invest. 1996;97:1837–1843. doi: 10.1172/JCI118613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rahemtulla A, Fung-Leung WP, Schilham MW, Kundig TM, Sambhara SR, Narendran A, Arabian A, Wakeham A, Paige CJ, Zinkernagel RM, et al. Normal development and function of CD8+cells but markedly decreased helper cell activity in mice lacking CD4. Nature. 1991;353:180–184. doi: 10.1038/353180a0. [DOI] [PubMed] [Google Scholar]
- 13.Fung-Leung WP, Schilham MW, Rahemtulla A, Kundig TM, Vollenweider M, Potter J, van Ewijk W, Mak TW. CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell. 1991;65:443–449. doi: 10.1016/0092-8674(91)90462-8. [DOI] [PubMed] [Google Scholar]
- 14.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 15.Cher DJ, Mosmann TR. Two types of murine helper T cell clone. II. Delayed-type hypersensitivity is mediated by TH1 clones. J Immunol. 1987;138:3688–3694. [PubMed] [Google Scholar]
- 16.Cherwinski HM, Schumacher JH, Brown KD, Mosmann TR. Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J Exp Med. 1987;166:1229–1244. doi: 10.1084/jem.166.5.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Botran R, Sanders VM, Mosmann TR, Vitetta ES. Lymphokine-mediated regulation of the proliferative response of clones of T helper 1 and T helper 2 cells. J Exp Med. 1988;168:543–558. doi: 10.1084/jem.168.2.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gajewski TF, Fitch FW. Anti-proliferative effect of IFN-γ in immune regulation. I. IFN-γ inhibits the proliferation of Th2 but not Th1 murine helper T lymphocyte clones. J Immunol. 1988;140:4245–4252. [PubMed] [Google Scholar]
- 19.Gajewski TF, Goldwasser E, Fitch FW. Anti-proliferative effect of IFN-γ in immune regulation. II. IFN-γ inhibits the proliferation of murine bone marrow cells stimulated with IL-3, IL-4, or granulocyte–macrophage colony-stimulating factor. J Immunol. 1988;141:2635–2642. [PubMed] [Google Scholar]
- 20.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–2095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O'Garra A. IL-10 acts on the antigen presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 1991;146:3444–3451. [PubMed] [Google Scholar]
- 22.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 23.Locksley RM, Heinzel FP, Sadick MD, Holaday BJ, Gardner KD., Jr Murine cutaneous leishmaniasis: susceptibility correlates with differential expansion of helper T-cell subsets. Ann Inst Pasteur Immunol. 1987;138:744–749. doi: 10.1016/s0769-2625(87)80030-2. [DOI] [PubMed] [Google Scholar]
- 24.Scott P, Natovitz P, Coffman RL, Pearce E, Sher A. Immunoregulation of cutaneous leishmaniasis. T cell lines that transfer protective immunity or exacerbation belong to different T helper subsets and respond to distinct parasite antigens. J Exp Med. 1988;168:1675–1684. doi: 10.1084/jem.168.5.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinzel FP, Sadick MD, Holaday BJ, Coffman RL, Locksley RM. Reciprocal expression of interferon γ or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J Exp Med. 1989;169:59–72. doi: 10.1084/jem.169.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Street NE, Schumacher JH, Fong TA, Bass H, Fiorentino DF, Leverah JA, Mosmann TR. Heterogeneity of mouse helper T cells. Evidence from bulk cultures and limiting dilution cloning for precursors of Th1 and Th2 cells. J Immunol. 1990;144:1629–1639. [PubMed] [Google Scholar]
- 27.Pearce EJ, Caspar P, Grzych JM, Lewis FA, Sher A. Downregulation of Th1 cytokine production accompanies induction of Th2 responses by a parasitic helminth, Schistosoma mansoni. . J Exp Med. 1991;173:159–166. doi: 10.1084/jem.173.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heinzel FP, Sadick MD, Mutha SS, Locksley RM. Production of interferon γ, interleukin 2, interleukin 4, and interleukin 10 by CD4+lymphocytes in vivo during healing and progressive murine leishmaniasis. Proc Natl Acad Sci USA. 1991;88:7011–7015. doi: 10.1073/pnas.88.16.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urban JF, Jr, Katona IM, Paul WE, Finkelman FD. Interleukin 4 is important in protective immunity to a gastrointestinal nematode infection in mice. Proc Natl Acad Sci USA. 1991;88:5513–5517. doi: 10.1073/pnas.88.13.5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 31.Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Kohler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362:245–248. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 32.Campbell HD, Tucker WQ, Hort Y, Martinson ME, Mayo G, Clutterbuck EJ, Sanderson CJ, Young IG. Molecular cloning, nucleotide sequence, and expression of the gene encoding human eosinophil differentiation factor (interleukin 5) Proc Natl Acad Sci USA. 1987;84:6629–6633. doi: 10.1073/pnas.84.19.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schleimer RP, Sterbinsky SA, Kaiser J, Bickel CA, Klunk DA, Tomioka K, Newman W, Luscinskas FW, Gimbrone MA, Jr, McIntyre BW, et al. IL-4 induces adherence of human eosinophils and basophils but not neutrophils to endothelium. Association with expression of VCAM-1. J Immunol. 1992;148:1086–1092. [PubMed] [Google Scholar]
- 34.Tepper RI, Coffman RL, Leder P. An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science. 1992;257:548–551. doi: 10.1126/science.1636093. [DOI] [PubMed] [Google Scholar]
- 35.Golumbek PT, Lazenby AJ, Levitsky HI, Jaffee LM, Karasuyama H, Baker M, Pardoll DM. Treatment of established renal cancer by tumor cells engineered to secrete interleukin-4. Science. 1991;254:713–716. doi: 10.1126/science.1948050. [DOI] [PubMed] [Google Scholar]
- 36.Kopf M, Brombacher F, Hodgkin PD, Ramsay AJ, Milbourne EA, Dai WJ, Ovington KS, Behm CA, Kohler G, Young IG, Matthaei KI. IL-5–deficient mice have a developmental defect in CD5+B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity. 1996;4:15–24. doi: 10.1016/s1074-7613(00)80294-0. [DOI] [PubMed] [Google Scholar]
- 37.Simons JW, Jaffee EM, Weber CE, Levitsky HI, Nelson WG, Carducci MA, Lazenby AJ, Cohen LK, Finn CC, Clift SM, et al. Bioactivity of autologous irradiated renal cell carcinoma vaccines generated by ex vivo granulocyte–macrophage colony-stimulating factor gene transfer. Cancer Res. 1997;57:1537–1546. [PMC free article] [PubMed] [Google Scholar]
- 38.Kato M, Abraham RT, Okada S, Kita H. Ligation of the β2 integrin triggers activation and degranulation of human eosinophils. Am J Respir Cell Mol Biol. 1998;18:675–686. doi: 10.1165/ajrcmb.18.5.2885. [DOI] [PubMed] [Google Scholar]
- 39.Bloom MB, Perry-Lalley D, Robbins PF, Li Y, el-Gamil M, Rosenberg SA, Yang JC. Identification of tyrosinase-related protein 2 as a tumor rejection antigen for the B16 melanoma. J Exp Med. 1997;185:453–459. doi: 10.1084/jem.185.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 41.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 42.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 43.Hibbs JB, Jr, Taintor RR, Chapman HA, Jr, Weinberg JB. Macrophage tumor killing: influence of the local environment. Science. 1977;197:279–282. doi: 10.1126/science.327547. [DOI] [PubMed] [Google Scholar]
- 44.Stuehr DJ, Nathan CF. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li LM, Kilbourn RG, Adams J, Fidler IJ. Role of nitric oxide in lysis of tumor cells by cytokine-activated endothelial cells. Cancer Res. 1991;51:2531–2535. [PubMed] [Google Scholar]
- 46.Kwon NS, Stuehr DJ, Nathan CF. Inhibition of tumor cell ribonucleotide reductase by macrophage- derived nitric oxide. J Exp Med. 1991;174:761–767. doi: 10.1084/jem.174.4.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yim CY, Bastian NR, Smith JC, Hibbs JB, Samlowski WE. Macrophage nitric oxide synthesis delays progression of ultraviolet light-induced murine skin cancers. Cancer Res. 1993;53:5507–5511. [PubMed] [Google Scholar]
- 48.Xie K, Huang S, Dong Z, Gutman M, Fidler IJ. Direct correlation between expression of endogenous inducible nitric oxide synthase and regression of M5076 reticulum cell sarcoma hepatic metastases in mice treated with liposomes containing lipopeptide CGP 31362. Cancer Res. 1995;55:3123–3131. [PubMed] [Google Scholar]
- 49.Xie K, Huang S, Dong Z, Juang SH, Gutman M, Xie QW, Nathan C, Fidler IJ. Transfection with the inducible nitric oxide synthase gene suppresses tumorigenicity and abrogates metastasis by K-1735 murine melanoma cells. J Exp Med. 1995;181:1333–1343. doi: 10.1084/jem.181.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bastian NR, Yim CY, Hibbs JB, Jr, Samlowski WE. Induction of iron-derived EPR signals in murine cancers by nitric oxide. Evidence for multiple intracellular targets. J Biol Chem. 1994;269:5127–5131. [PubMed] [Google Scholar]
- 51.Cui S, Reichner JS, Mateo RB, Albina JE. Activated murine macrophages induce apoptosis in tumor cells through nitric oxide-dependent or -independent mechanisms. Cancer Res. 1994;54:2462–2467. [PubMed] [Google Scholar]
- 52.Dong Z, Staroselsky AH, Qi X, Xie K, Fidler IJ. Inverse correlation between expression of inducible nitric oxide synthase activity and production of metastasis in K-1735 murine melanoma cells. Cancer Res. 1994;54:789–793. [PubMed] [Google Scholar]
- 53.Tanguay S, Bucana CD, Wilson MR, Fidler IJ, von Eschenbach AC, Killion JJ. In vivo modulation of macrophage tumoricidal activity by oral administration of the liposome-encapsulated macrophage activator CGP 19835A. Cancer Res. 1994;54:5882–5888. [PubMed] [Google Scholar]
- 54.Yim CY, McGregor JR, Kwon OD, Bastian NR, Rees M, Mori M, Hibbs JB, Jr, Samlowski WE. Nitric oxide synthesis contributes to IL-2–induced antitumor responses against intraperitoneal Meth A tumor. J Immunol. 1995;155:4382–4390. [PubMed] [Google Scholar]
- 55.Liew FY, Millott S, Li Y, Lelchuk R, Chan WL, Ziltener H. Macrophage activation by interferon γ from host-protective T cells is inhibited by interleukin (IL)3 and IL4 produced by disease-promoting T cells in leishmaniasis. Eur J Immunol. 1989;19:1227–1232. doi: 10.1002/eji.1830190712. [DOI] [PubMed] [Google Scholar]
- 56.Oswald IP, Wynn TA, Sher A, James SL. Interleukin 10 inhibits macrophage microbicidal activity by blocking the endogenous production of tumor necrosis factor α required as a costimulatory factor for interferon γ–induced activation. Proc Natl Acad Sci USA. 1992;89:8676–8680. doi: 10.1073/pnas.89.18.8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bogdan C, Vodovotz Y, Paik J, Xie QW, Nathan C. Mechanism of suppression of nitric oxide synthase expression by interleukin 4 in primary mouse macrophages. J Leukocyte Biol. 1994;55:227–233. doi: 10.1002/jlb.55.2.227. [DOI] [PubMed] [Google Scholar]
- 58.Gazzinelli RT, Oswald IP, Hieny S, James SL, Sher A. The microbicidal activity of interferon γ–treated macrophages against Trypanosoma cruzi involves an l-arginine–dependent, nitrogen oxide–mediated mechanism inhibitable by interleukin 10 and transforming growth factor β. Eur J Immunol. 1992;22:2501–2506. doi: 10.1002/eji.1830221006. [DOI] [PubMed] [Google Scholar]
- 59.Gazzinelli RT, Oswald IP, James SL, Sher A. IL-10 inhibits parasite killing and nitrogen oxide production by IFN-γ–activated macrophages. J Immunol. 1992;148:1792–1796. [PubMed] [Google Scholar]
- 60.Oswald IP, Gazzinelli RT, Sher A, James SL. IL-10 synergizes with IL-4 and transforming growth factor β to inhibit macrophage cytotoxic activity. J Immunol. 1992;148:3578–3582. [PubMed] [Google Scholar]
- 61.Costa JJ, Matossian K, Resnick MB, Beil WJ, Wong DT, Gordon JR, Dvorak AM, Weller PF, Galli SJ. Human eosinophils can express the cytokines tumor necrosis factor α and macrophage inflammatory protein-1 α. J Clin Invest. 1993;91:2673–2684. doi: 10.1172/JCI116506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beil WJ, Weller PF, Tzizik DM, Galli SJ, Dvorak AM. Ultrastructural immunogold localization of tumor necrosis factor-α to the matrix compartment of eosinophil secondary granules in patients with idiopathic hypereosinophilic syndrome. J Histochem Cytochem. 1993;41:1611–1615. doi: 10.1177/41.11.8409368. [DOI] [PubMed] [Google Scholar]
- 63.Spessotto P, Dri P, Bulla R, Zabucchi G, Patriarca P. Human eosinophil peroxidase enhances tumor necrosis factor and hydrogen peroxide release by human monocyte– derived macrophages. Eur J Immunol. 1995;25:1366–1373. doi: 10.1002/eji.1830250535. [DOI] [PubMed] [Google Scholar]
- 64.Fahey TJ, III, Tracey KJ, Tekamp-Olson P, Cousens LS, Jones WG, Shires GT, Cerami A, Sherry B. Macrophage inflammatory protein 1 modulates macrophage function. J Immunol. 1992;148:2764–2769. [PubMed] [Google Scholar]
- 65.Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. . J Exp Med. 1997;185:207–218. doi: 10.1084/jem.185.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Greenberg PD, Kern DE, Cheever MA. Therapy of disseminated murine leukemia with cyclophosphamide and immune Lyt-1+,2- T cells. Tumor eradication does not require participation of cytotoxic T cells. J Exp Med. 1985;161:1122–1134. doi: 10.1084/jem.161.5.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kern DE, Klarnet JP, Jensen MC, Greenberg PD. Requirement for recognition of class II molecules and processed tumor antigen for optimal generation of syngeneic tumor-specific class I–restricted CTL. J Immunol. 1986;136:4303–4310. [PubMed] [Google Scholar]
- 68.Ostrand-Rosenberg S, Roby CA, Clements VK. Abrogation of tumorigenicity by MHC class II antigen expression requires the cytoplasmic domain of the class II molecule. J Immunol. 1991;147:2419–2422. [PubMed] [Google Scholar]
- 69.Clements VK, Baskar S, Armstrong TD, Ostrand-Rosenberg S. Invariant chain alters the malignant phenotype of MHC class II+tumor cells. J Immunol. 1992;149:2391–2396. [PubMed] [Google Scholar]
- 70.Baskar S, Ostrand-Rosenberg S, Nabavi N, Nadler LM, Freeman GJ, Glimcher LH. Constitutive expression of B7 restores immunogenicity of tumor cells expressing truncated major histocompatibility complex class II molecules. Proc Natl Acad Sci USA. 1993;90:5687–5690. doi: 10.1073/pnas.90.12.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu TC, Guarnieri FG, Staveley OCKF, Viscidi RP, Levitsky HI, Hedrick L, Cho KR, August JT, Pardoll DM. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci USA. 1995;92:11671–11675. doi: 10.1073/pnas.92.25.11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hahne M, Rimoldi D, Schroter M, Romero P, Schreier M, French LE, Schneider P, Bornand T, Fontana A, Lienard D, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–1366. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 73.Nathan CF, Klebanoff SJ. Augmentation of spontaneous macrophage-mediated cytolysis by eosinophil peroxidase. J Exp Med. 1982;155:1291–1308. doi: 10.1084/jem.155.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van der Vliet A, Eiserich JP, Halliwell B, Cross CE. Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite. A potential additional mechanism of nitric oxide–dependent toxicity. J Biol Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 75.Green SJ, Meltzer MS, Hibbs JB, Jr, Nacy CA. Activated macrophages destroy intracellular Leishmania major amastigotes by an l-arginine–dependent killing mechanism. J Immunol. 1990;144:278–283. [PubMed] [Google Scholar]
- 76.Liew FY, Li Y, Millott S. Tumor necrosis factor α synergizes with IFN-γ in mediating killing of Leishmania majorthrough the induction of nitric oxide. J Immunol. 1990;145:4306–4310. [PubMed] [Google Scholar]
- 77.Liew FY, Li Y, Millott S. Tumour necrosis factor (TNF-α) in leishmaniasis. II. TNF-α–induced macrophage leishmanicidal activity is mediated by nitric oxide from l-arginine. Immunology. 1990;71:556–559. [PMC free article] [PubMed] [Google Scholar]
- 78.Liew FY, Millott S, Parkinson C, Palmer RM, Moncada S. Macrophage killing of Leishmania parasite in vivo is mediated by nitric oxide from l-arginine. J Immunol. 1990;144:4794–4797. [PubMed] [Google Scholar]
- 79.Liew FY, Li Y, Moss D, Parkinson C, Rogers MV, Moncada S. Resistance to Leishmania majorinfection correlates with the induction of nitric oxide synthase in murine macrophages. Eur J Immunol. 1991;21:3009–3014. doi: 10.1002/eji.1830211216. [DOI] [PubMed] [Google Scholar]
- 80.Mauel J, Ransijn A, Buchmuller-Rouiller Y. Killing of Leishmania parasites in activated murine macrophages is based on an l-arginine–dependent process that produces nitrogen derivatives. J Leukocyte Biol. 1991;49:73–82. doi: 10.1002/jlb.49.1.73. [DOI] [PubMed] [Google Scholar]
- 81.Evans TG, Thai L, Granger DL, Hibbs JB. Effect of in vivo inhibition of nitric oxide production in murine leishmaniasis. J Immunol. 1993;151:907–915. [PubMed] [Google Scholar]
- 82.Assreuy J, Cunha FQ, Epperlein M, Noronha-Dutra A, O'Donnell CA, Liew FY, Moncada S. Production of nitric oxide and superoxide by activated macrophages and killing of Leishmania major. . Eur J Immunol. 1994;24:672–676. doi: 10.1002/eji.1830240328. [DOI] [PubMed] [Google Scholar]
- 83.Stenger S, Thuring H, Rollinghoff M, Bogdan C. Tissue expression of inducible nitric oxide synthase is closely associated with resistance to Leishmania major. . J Exp Med. 1994;180:783–793. doi: 10.1084/jem.180.3.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 85.Morris L, Troutt AB, McLeod KS, Kelso A, Handman E, Aebischer T. Interleukin-4 but not gamma interferon production correlates with the severity of murine cutaneous leishmaniasis. Infect Immun. 1993;61:3459–3465. doi: 10.1128/iai.61.8.3459-3465.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liew FY, Li Y, Severn A, Millott S, Schmidt J, Salter M, Moncada S. A possible novel pathway of regulation by murine T helper type-2 (Th2) cells of a Th1 cell activity via the modulation of the induction of nitric oxide synthase on macrophages. Eur J Immunol. 1991;21:2489–2494. doi: 10.1002/eji.1830211027. [DOI] [PubMed] [Google Scholar]
- 87.al-Ramadi BK, Meissler JJ, Jr, Huang D, Eisenstein TK. Immunosuppression induced by nitric oxide and its inhibition by interleukin-4. Eur J Immunol. 1992;22:2249–2254. doi: 10.1002/eji.1830220911. [DOI] [PubMed] [Google Scholar]
- 88.Ding L, Linsley PS, Huang LY, Germain RN, Shevach EM. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the upregulation of B7 expression. J Immunol. 1993;151:1224–1234. [PubMed] [Google Scholar]
- 89.Urban JF, Jr, Madden KB, Svetic A, Cheever A, Trotta PP, Gause WC, Katona IM, Finkelman FD. The importance of Th2 cytokines in protective immunity to nematodes. Immunol Rev. 1992;127:205–220. doi: 10.1111/j.1600-065x.1992.tb01415.x. [DOI] [PubMed] [Google Scholar]
- 90.Belosevic M, Finbloom DS, Van Der Meide PH, Slayter MV, Nacy CA. Administration of monoclonal anti–IFN-γ antibodies in vivo abrogates natural resistance of C3H/HeN mice to infection with Leishmania major. . J Immunol. 1989;143:266–274. [PubMed] [Google Scholar]
- 91.Wang ZE, Reiner SL, Zheng S, Dalton DK, Locksley RM. CD4+ effector cells default to the Th2 pathway in interferon γ–deficient mice infected with Leishmania major. . J Exp Med. 1994;179:1367–1371. doi: 10.1084/jem.179.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sadick MD, Heinzel FP, Holaday BJ, Pu RT, Dawkins RS, Locksley RM. Cure of murine leishmaniasis with anti-interleukin 4 monoclonal antibody. Evidence for a T cell–dependent, interferon γ–independent mechanism. J Exp Med. 1990;171:115–127. doi: 10.1084/jem.171.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Finkelman FD, Madden KB, Morris SC, Holmes JM, Boiani N, Katona IM, Maliszewski CR. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine– anti-cytokine antibody complexes. J Immunol. 1993;151:1235–1244. [PubMed] [Google Scholar]
- 94.Finkelman FD, Madden KB, Cheever AW, Katona IM, Morris SC, Gately MK, Hubbard BR, Gause WC, Urban JF., Jr Effects of interleukin 12 on immune responses and host protection in mice infected with intestinal nematode parasites. J Exp Med. 1994;179:1563–1572. doi: 10.1084/jem.179.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chatelain R, Varkila K, Coffman RL. IL-4 induces a Th2 response in Leishmania major–infected mice. J Immunol. 1992;148:1182–1187. [PubMed] [Google Scholar]
- 96.Reed SG, Scott P. T-cell and cytokine responses in leishmaniasis. Curr Opin Immunol. 1993;5:524–531. doi: 10.1016/0952-7915(93)90033-o. [DOI] [PubMed] [Google Scholar]
- 97.Reiner SL, Zheng S, Wang ZE, Stowring L, Locksley RM. Leishmania promastigotes evade interleukin 12 (IL-12) induction by macrophages and stimulate a broad range of cytokines from CD4+T cells during initiation of infection. J Exp Med. 1994;179:447–456. doi: 10.1084/jem.179.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levitsky HI, Montgomery J, Ahmadzadeh M, Staveley-O'Carroll K, Guarnieri F, Longo DL, Kwak LW. Immunization with granulocyte–macrophage colony-stimulating factor–transduced, but not B7-1–transduced, lymphoma cells primes idiotype-specific T cells and generates potent systemic antitumor immunity. J Immunol. 1996;156:3858–3865. [PubMed] [Google Scholar]