

Abstract
To evaluate the role of natural immunoglobulin (Ig)M in the immediate response against microbial infection, we tested mutant mice that are deficient in secreted (s)IgM in an acute peritonitis model induced by cecal ligation and puncture (CLP). 20% of wild-type mice died within 32 h of CLP, whereas 70% of sIgM-deficient mice died within the same time period. The increased susceptibility was associated with a reduced level of tumor necrosis factor (TNF)-α, a decreased neutrophil recruitment and an increased bacterial load in the peritoneum, and elevated levels of endotoxin and proinflammatory cytokines in the circulation. Resistance to CLP by sIgM-deficient mice was restored by reconstitution with polyclonal IgM from normal mouse serum. Reconstitution with a monoclonal IgM specific to phosphatidylcholine, a conserved cell membrane component, has a modest effect but a monoclonal IgM specific to phosphocholine is not protective. These findings demonstrate a critical role of natural IgM in the immediate defense against severe bacterial infection.
Keywords: natural antibody, immunoglobulin M, complement, bacterial infection, immediate defense
The spontaneously occurring immunoglobulins in human cord blood, in “antigen-free” mice, and in normal individuals in the absence of apparent antigen stimulation are referred to as natural antibodies (for reviews, see references 1–3). Most of these antibodies are of IgM class produced by B-1 cells. B-1 cells differ from conventional B cells in that they are generated predominantly during fetal and neonatal development (4–6). Because of the preferential usage of JH-proximal VH gene segments and the lack of terminal deoxynucleotidyl transferase activity in precursor B cells during early ontogeny (7–9), the repertoire of natural antibodies is much more restricted than those produced by conventional B cells. A large proportion of the natural antibodies are polyreactive to phylogenetically conserved structures such as nucleic acids, heat shock proteins, carbohydrates, and phospholipids (4–6, 10). For example, 5–15% of murine B-1 cells express IgM specific to phosphatidylcholine (PtC), a common membrane component exposed after treatment of red blood cells with proteolytic enzyme bromelain (11, 12).
The physiologic functions of natural antibodies have long been a subject of interest. Among many postulated functions, natural IgM, together with factors of the innate immunity, is thought to provide a first line of defense against microbial infection (1–3). In addition to its natural presence, IgM is a pentamer and could potentially bind to 10 antigenic determinants per molecule. The polyreactivities enable it to react with a broad spectrum of antigens simultaneously. Furthermore, IgM is a potent complement activator. Activation of complement can directly result in the lysis of invading bacteria or opsonization of infectious particles for efficient phagocytosis by macrophages and polymorphonuclear leukocytes. However, due to the lack of suitable animal models, the putative function of natural IgM has not been critically examined under physiological conditions.
We have previously constructed a mutant mouse strain in which B cells are specifically deficient in secreted (s)IgM but still express membrane-bound IgM and secrete other Ig isotypes (13). To determine the physiological role of natural IgM in bacterial infection, we examined the susceptibility of sIgM-deficient mice in an acute septic peritonitis model induced by cecal ligation and puncture (CLP [14]). We show that sIgM-deficient mice are much more susceptible than wild-type mice as indicated by an inability to clear bacteria from peritoneum, a systemic release of proinflammatory cytokines, and a high mortality rate. Resistance to CLP by sIgM-deficient mice was restored by reconstitution with polyclonal IgM from normal mouse serum and to a less extent by monoclonal IgM specific to PtC but not to phosphocholine (PC). Our findings demonstrate a critical role of natural IgM in the immediate response against acute systemic bacterial infection.
Materials and Methods
Mice.
Mice deficient in sIgM were described previously (13). Mutant mice either in the mixed C57BL/6 × 129 background or pure 129 background were maintained in specific pathogen–free facilities and used at 6–8 wk of age. Studies were performed according to institutional guidelines for animal use and care.
CLP.
The surgical procedure was performed as described (15). In brief, mice were anesthetized by avertin (0.2 ml of 2.5% solution per 10 g body wt), and a 0.5-cm midline incision was made in the peritoneum. The distal two thirds of the cecum was ligated with a silk suture, and the cecum was punctured once with an 18.5-gauge needle, and then gently squeezed to ensure that the holes were completely open. The cecum was returned to the peritoneal cavity, and the body wall was stitched and the incision was closed with 9-mm stainless steel wound clips. Sham controls were operated on in the same manner but without ligation and puncture. In some experiments, mice were killed 3 h after CLP. Peritoneal lavage was harvested after injection of 3 ml of PBS with 2% FCS and used for further assays (see below). In other experiments, mice were bled through the tail vein at 1.5, 3, 6, and 12 h after CLP and sera were used for further assays. Statistical analysis for survival was performed using the Stata program (Stata Corp., College Station, TX).
IgM Purification.
Polyclonal IgM was isolated from normal mouse serum (Sigma Chemical Co., St. Louis, MO) using an anti-IgM affinity column. Sera were precipitated by ammonium sulfate (50% saturation), and the precipitate was dissolved in PBS and dialyzed. Recovered solution was filtered through a 0.45-μm filter and applied to a 5-ml protein G–sepharose column (Sigma Chemical Co.) to remove IgG. The flow-through was reconstituted with NaCl to a final concentration of 0.5 M and applied to a 4-ml anti-IgM sepharose column (Zymed Laboratories, Inc., South San Francisco, CA). Bound protein was eluted with 0.1 M glycine/0.15 M NaCl, pH 2.5, and neutralized with 1 M Tris, pH 8.0. Protein content in each fraction was determined by spectrophotometry, and positive fractions were pooled, dialyzed, and concentrated (Amicon, Inc., Beverly, MA). 2C8 hybridoma secreting PtC-specific IgM was provided by Dr. Leonore A. Herzenberg (Stanford University, Stanford, CA), and 5E11 hybridoma secreting PC-specific IgM was obtained from Dr. J. Latham Claflin (University of Michigan Medical School, Ann Arbor, MI [12, 16]). Anti-PtC and anti-PC IgM was isolated with anti-IgM affinity column from ascites produced in recombination activating gene (RAG)-2–deficient mice. Purity of IgM was analyzed by SDS-PAGE followed by Coomassie stain. Western blot and ELISA were used to assess IgG contamination and degradation of IgM. All batches of purified IgM had no detectable degradation and <1% IgG contamination (data not shown). Endotoxin levels in purified IgM was <200 EU/ml as determined by Limulus amebocyte lysate assay (Associates of Cape Cod, Woods Hole, MA). In IgM reconstitution experiments, sIgM-deficient mice were given 0.5 mg i.v. of polyclonal or monoclonal IgM in 0.5 ml PBS 4 h before CLP.
Assays.
Peritoneal lavage fluid was assayed for Escherichia coli, neutrophils, and the levels of endotoxin (LPS), TNF-α, and IL-6. Serum was assayed for the levels of endotoxin, TNF-α, and IL-6. E. coli colony-forming units (CFU) were determined by overnight culture of serial dilutions of peritoneal lavage fluid on Luria broth–agar plates at 37°C. Colonies of E. coli were identified in a background of heterogeneous colonies by morphology and confirmed by culture on MacConkey II agar and by assay with the Enteric Identification System (Organon Teknika, Durham, NC). Neutrophils in the peritoneal lavage were analyzed by flow cytometry using PE-conjugated anti-granulocyte antibody and FITC-conjugated anti-CD19 antibody (PharMingen, San Diego, CA). 104 live cells were collected for each sample, and neutrophils were identified as GR+CD19−. TNF-α and IL-6 were measured by ELISA using commercial kits (Endogen, Inc., Cambridge, MA; and Intergen Co., Purchase, NY). ELISA was performed according to the manufacturer's specification. Concentrations of TNF-α and IL-6 in individual samples were determined by comparison with a standard curve derived from the cytokine standard supplied with the kits. Peritoneal and serum LPS were measured using Limulus amebocyte lysate assay. LPS concentrations of individual samples were calculated using a standard curve derived from the LPS standard provided with the kit.
Results and Discussion
To examine the role of natural IgM in the immediate response to microbial infection, we determined the susceptibility of sIgM-deficient mice to acute septic peritonitis induced by CLP that releases endogenous bacteria from the cecum into the peritoneal cavity. 32 h after CLP, 70% of sIgM-deficient mice died compared with 20% of the wild-type mice (Fig. 1), indicating that the absence of natural IgM rendered mutant mice much more susceptible to the acute bacterial infection (P < 0.0001). The increased susceptibility of mutant mice to CLP was due to the absence of sIgM, because reconstitution of mutant mice with a single dose of 0.5 mg i.v. of total IgM isolated from normal mouse serum 4 h before CLP restored their survival to the same level as wild-type mice (Fig. 1). Similarly, sIgM-deficient mice were also more sensitive to challenge by individual species of pathogenic bacteria such as group B Streptococcus. The lethal dose to 50% (LD50) animals challenged was 10-fold lower for sIgM-deficient mice than for wild-type mice (our unpublished observations). sIgM-deficient mice also showed increased incidence of spontaneous bacterial infection by opportunistic bacteria, including Pasteurella pneumotropica, in specific pathogen–free facilities (our unpublished observations). These data show that natural IgM is required for the protection against bacterial infection.
Figure 1.

Natural IgM confers resistance to CLP. sIgM-deficient (−/−) and wild-type (+/+) mice at 6-8 wk of age were subject to CLP. IgM-reconstituted sIgM-deficient mice (−/− IgM) were given as a single dose of 0.5 mg i.v. of total IgM affinity- purified from normal mouse serum 4 h before CLP. Mice were monitored for survival within the first 32 h.
Resistance to CLP is dependent on complement, mast cells, and TNF-α (17–20). The susceptibility of sIgM-deficient mice to CLP is similar to mice deficient in complement component C3 or C4 or mast cells (17–19). The requirement of natural IgM for resistance to CLP is probably based on its ability to bind to bacteria and activate complement. To test this possibility, we determined the immediate response by assaying the levels of TNF-α, IL-6, and LPS, neutrophil infiltration, and bacterial load in the peritoneal lavage 3 h after CLP. As in C3-deficient mice, the levels of TNF-α and IL-6 in mutant mice were approximately half those in wild-type mice (Table 1). Without CLP, both sIgM-deficient and wild-type mice had very few neutrophils in the peritoneum (<1%) and similar numbers of cells in the lavage after CLP (data not shown). Again, as in C3-deficient mice, the percentage of neutrophils in the peritoneal lavage recovered from sIgM-deficient mice was significantly reduced compared with that from wild-type mice (55 vs. 82%; Table 1, and data not shown). Furthermore, 10 times more E. coli were recovered from the peritoneal lavage of sIgM-deficient mice than from wild-type mice (Table 1). Associated with the higher bacterial load, approximately twice the amount of endotoxin (LPS) was detected in sIgM-deficient mice as in wild-type mice. Reconstitution of sIgM-deficient mice with total IgM restored the levels of TNF-α and neutrophils and reduced E. coli load in the peritoneal lavage (Table 1), consistent with the increased survival. These data show that the effects resulting from the absence of sIgM on the induction of TNF-α, neutrophil infiltration, and bacterial load in the peritoneum are very similar to those seen in the absence of C3, indicating that natural IgM functions through the complement pathway.
Table 1.
Analyses of Peritoneal Lavage 3 h after CLP
| +/+ | −/− | −/− IgM | −/− PtC-IgM | −/− PC-IgM | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TNF-α (pg/ml) | 1,507 | 892 | 2,068 | 1,643 | 304 | |||||
| IL-6 (pg/ml) | 2,734 | 1,401 | ND | ND | ND | |||||
| Neutrophils (%) | 81.7 | 54.8 | 83.5 | 69.6 | 22.1 | |||||
| E. coli (CFU) | 34.9 × 103 | 350 × 103 | 2.3 × 103 | 62.5 × 103 | 510 × 103 | |||||
| LPS (EU/ml) | 36.7 | 62.9 | 23.8 | 51.2 | 64.0 |
sIgM-deficient mice (−/−), wild-type mice (+/+), and IgM-reconstituted sIgM-deficient mice were subject to CLP. Peritoneal lavage was carried out by injection of 3 ml i.p. of PBS with 2% FCS 3 h after CLP. IgM-reconstituted mice were given a single dose of 0.5 mg i.v. of purified polyclonal IgM from normal mouse serum or monoclonal IgM specific to PtC or PC 4 h before CLP. Peritoneal lavages of sIgM-deficient and wild-type mice were each pooled from seven mice, and the levels of TNF-α, IL-6, and LPS, E. coli counts, and neutrophils were assayed (see Materials and Methods). The levels of TNF-α and LPS, E. coli counts, and neutrophils were assayed in peritoneal lavage from individual IgM-reconstituted mice. The average of four mice is shown. Similar results were obtained in a separate experiment.
IgM is the most potent complement activator among the five classes of Igs. A single bound IgM molecule is sufficient to activate complement to lyse a red blood cell (21). Binding of natural IgM to bacteria immediately after infection likely results in the activation of complement through the classical pathway. Since serum from sIgM-deficient mice lysed antibody-opsonized red blood cells just as efficiently as serum from wild-type mice in a hemolytic assay (data not shown), the increased susceptibility of sIgM-deficient mice to CLP is probably associated with the absence of IgM-mediated complement activation. C3- or C4-deficient mice appear to be even more sensitive to CLP than sIgM-deficient mice as indicated by 100% mortality within 24 h (17). This may be because complement can also be activated through the alternative and lectin pathways, and complement is important in the efficient clearance of bacteria. In addition, sIgM-deficient mice have relatively normal levels of IgGs (13), some of which are probably natural antibodies. Although the IgG proteins may contribute to the survival of sIgM-deficient mice, they are clearly not sufficient to compensate fully the absence of natural IgM.
Uncontrolled bacterial infection in the peritoneum leads to a fatal systemic infection and inflammation. To determine the systemic inflammatory response to CLP in the absence of natural IgM, we collected serum from both sIgM-deficient and wild-type mice at 1.5, 3, 6, and 12 h after CLP and assayed for the levels of LPS, TNF-α, and IL-6. We divided the sera into four groups based on the genotype of the mice and whether the mice died or survived at 32 h after CLP. As shown in Fig. 2, the levels of serum LPS were similar to those of sham controls 3 h after CLP but significantly higher at 6 and 12 h after CLP. Among the four groups of mice, sIgM-deficient mice that died had a significantly higher level of LPS by 6 h after CLP, consistent with previous findings indicating that natural IgM is involved in the clearance of LPS (22). Similarly, sIgM-deficient mice that died had a significantly elevated level of TNF-α 3 h after CLP (Fig. 2). Although the initial local release of TNF-α is beneficial to the containment of bacterial infection, systemic release of TNF-α is usually associated with wide-spread inflammation. The level of proinflammatory cytokine IL-6 was also elevated in the serum 6 and 12 h after CLP in sIgM-deficient mice as well as in wild-type mice that died (Fig. 2). IL-6 is a major cytokine that induces the production of acute phase proteins, such as C-reactive protein, that are thought to have similar functions as natural antibodies (23–25). The similar levels of serum IL-6 in both sIgM-deficient and wild-type mice suggest that acute phase proteins were similarly elevated in both types of mice. Thus, natural IgM appears to have some unique functions in the immediate response against bacterial infection that cannot be replaced by acute phase proteins. In the absence of natural IgM, local bacterial infection leads to wide-spread inflammation and high mortality.
Figure 2.
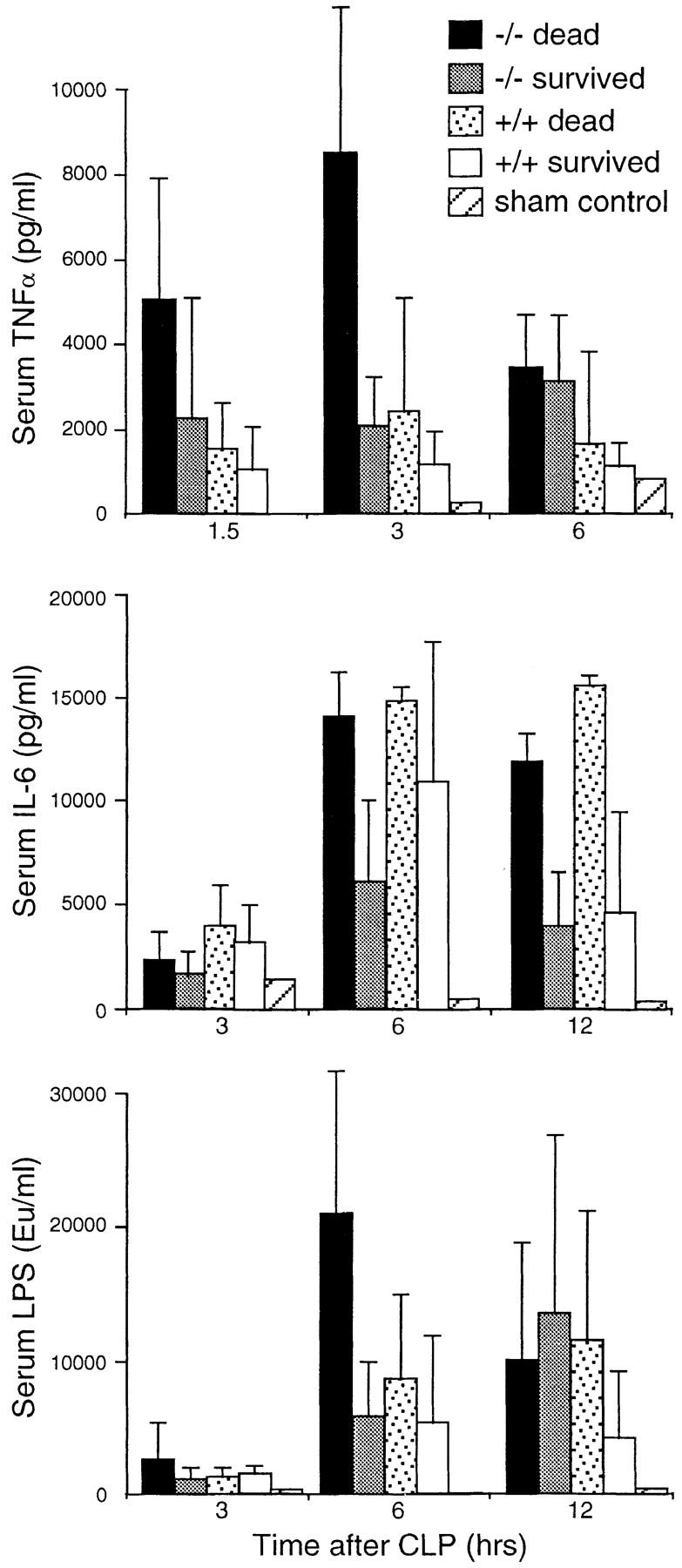
Comparison of the levels of LPS, TNF-α, and IL-6 in the serum of sIgM-deficient and wild-type mice at different time points after CLP. Sera were collected at 1.5, 3, 6, and 12 h after CLP and divided into four groups based on the genotype of the mice and whether the mice died or survived at 32 h after CLP. Sham control was operated on without ligation and puncture. Concentrations of LPS, TNF-α, and IL-6 were determined by ELISA. Error bars indicate SD. For TNF-α assay, the numbers of mice used in each category are as follows: −/− dead, 12; −/− survived, 10; +/+ dead, 10; and +/+ survived, 16. Eight mice were used in each category for IL-6 assay. Six mice were used in each category for LPS assay. Numbers of sham controls for TNF-α, IL-6, and LPS were 2, 1, and 1, respectively.
Reconstitution of sIgM-deficient mice with polyclonal IgM from normal mouse serum restored their resistance to CLP (Fig. 1). Therefore, it was of obvious interest to test whether the resistance can be restored by reconstitution with monoclonal IgM specific to PtC or PC. PtC is a common membrane component and anti-PtC is the most widely expressed specificity by natural IgM (11, 12). PC is chemically related to PtC and is found as a determinant of the pneumococcal cell wall (26). Anti-PC IgM has been shown to confer protection against certain types of Streptococcus pneumoniae (27, 28). sIgM-deficient mice were given a single dose of 0.5 mg i.v. of anti-PtC or PC IgM 4 h before CLP. 3 h after CLP, mice were killed and peritoneal lavage was harvested to assay for the levels of TNF-α and LPS, neutrophil infiltration, and bacterial load. As in reconstitution with polyclonal IgM, reconstitution with anti-PtC IgM resulted in elevated levels of TNF-α and neutrophils and a reduced E. coli load and LPS in the peritoneal lavage (Table 1). In contrast, reconstitution with anti-PC IgM had no obvious effect. The difference between the two monoclonal IgM preparations was probably due to their differences in specificity, because both were >98% pure, had minimal endotoxin contamination, and reconstituted to a similar level as assayed by ELISA of serum taken 3 h after CLP (data not shown). The resistance conferred by anti-PtC IgM was clearly evident but it appeared not to be as effective as polyclonal IgM, probably because polyclonal IgM bind bacteria through many different antigenic determinants. That anti-PtC IgM had a clear effect suggests that it is involved in the immediate defense against bacterial infection under physiological conditions.
Activation of complement is essential for the initial containment of systemic bacterial infection as demonstrated by the susceptibility of complement-deficient humans, guinea pigs, and mice to bacterial infection (17, 29–31). C5b–C9 membrane attack complex can lyse bacteria directly. C3a and C5a peptides function as chemoattractants to recruit leukocytes to the site of infection. Opsonization of bacteria by C3b and iC3b promotes efficient phagocytosis by neutrophils. In addition, complement is required for the efficient activation of mast cells to release TNF-α for the initiation of a local inflammatory response (17). However, the optimal clearance of bacteria by complement requires the presence of natural antibodies. Gram-positive bacteria are generally resistant to complement-mediated lysis because the thick peptidoglycan layer in their cell wall prevents insertion of the membrane-attack complex (32). Although gram-negative bacteria are effectively lysed by complement, these bacteria contain LPS in their cell walls which when released in substantial amount can be fatal within hours (33, 34). Furthermore, sialic acids of the pneumococcal capsular polysaccharides can inhibit the activation of the alternative complement pathway (35). The capsular polysaccharides also inhibit phagocytosis by macrophages and neutrophils (36). Effective responses to bacterial infection require antibodies to opsonize bacteria for efficient phagocytosis and to neutralize the released endotoxin (22). What is the source of antibodies before the onset of a specific humoral response to a particular infectious agent? Natural IgM is ideally suited for these purposes because of its polyreactivities and the exquisite ability to activate complement, and because normally it is already present. The susceptibility of sIgM-deficient mice to CLP and restoration of resistance by reconstitution with polyclonal IgM and to a lesser extent by anti-PtC IgM clearly demonstrate a critical role of natural IgM in the immediate defense against bacterial infection.
Acknowledgments
We thank Drs. Leonore A. Herzenberg and J. Latham Claflin for anti-PtC and PC hybridomas, respectively; Drs. Yong Wang and Jay Austen for helping with hemolytic assay; Dr. Herman Eisen for critical reading of the manuscript; and members of the laboratory for help and discussion.
This work was supported in part by National Institutes of Health grants AI41762 (to J. Chen), AI36389, and AI39246 (to M.C. Carroll), and by a pre-doctoral fellowship from Boehringer Ingelheim (to M. Boes).
References
- 1.Tlaskalová-Hogenová H, Mandel L, Stepánková R, Bártová J, Barot R, Leclerc M, Kováru F, Trebichavsky I. Autoimmunity: from physiology to pathology. Natural antibodies, mucosal immunity and development of B cell repertoire. Folia Biol (Praha) 1992;38:202–215. [PubMed] [Google Scholar]
- 2.Avrameas S. Natural autoantibodies: from ‘horor autotoxicus' to ‘gnothi seauton.' . Immunol Today. 1991;12:154–159. doi: 10.1016/S0167-5699(05)80045-3. [DOI] [PubMed] [Google Scholar]
- 3.Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7:812–818. doi: 10.1016/0952-7915(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 4.Kantor AB, Herzenberg LA. Origin of murine B cell lineages. Annu Rev Immunol. 1993;11:501–538. doi: 10.1146/annurev.iy.11.040193.002441. [DOI] [PubMed] [Google Scholar]
- 5.Hardy RR, Hayakawa K. CD5 B cells, a fetal B cell lineage. Adv Immunol. 1994;55:297–339. doi: 10.1016/s0065-2776(08)60512-x. [DOI] [PubMed] [Google Scholar]
- 6.Casali, P., M.T. Kasaian, and G. Haughton. 1994. B-1 (CD5) cells. In Autoimmunity, Physiology and Disease. A. Coutinho and M.D. Kazatchkine, editors. Wiley-Liss, Inc., New York. 57–88.
- 7.Yancopoulos GD, Desiderio SV, Paskind M, Kearney JF, Baltimore D, Alt FW. Preferential utilization of the most JH-proximal VHgene segments in pre-B-cell lines. Nature. 1984;311:727–733. doi: 10.1038/311727a0. [DOI] [PubMed] [Google Scholar]
- 8.Feeney AJ. Lack of N regions in fetal and neonatal mouse immunoglobulin V-D-J junctional sequences. J Exp Med. 1990;172:1377–1390. doi: 10.1084/jem.172.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu H, Förster I, Rajewsky K. Sequence homologies, N sequence insertion and JH gene utilization in VHDJHjoining: implications for the joining mechanism and the ontogenetic timing of Ly1 B cell and B-CLL progenitor generation. EMBO (Eur Mol Biol Organ) J. 1990;9:2133–2140. doi: 10.1002/j.1460-2075.1990.tb07382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmberg, D., and J. Kearney. 1994. Selection of B cell repertoire and natural autoantibodies. In Autoimmunity, Physiology and Disease. A. Coutinho and M.D. Kazatchkine, editors. Wiley-Liss, Inc., New York. 89–106.
- 11.Mercolino TJ, Arnold LW, Hawkins LA, Haughton G. Normal mouse peritoneum contains a large population of Ly1+(CD5) B cells that recognize phosphatidyl choline. J Exp Med. 1988;168:687–698. doi: 10.1084/jem.168.2.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hardy RR, Carmack CE, Shinton SA, Riblet RJ, Hayakawa K. A single VH gene is utilized predominantly in anti-BrMRBC hybridomas derived from purified Ly-1 B cells: definition of the VH11 family. J Immunol. 1989;142:3642–3651. [PubMed] [Google Scholar]
- 13.Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J Immunol. 1998;160:4776–4787. [PubMed] [Google Scholar]
- 14.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock—a review of laboratory models and a proposal. J Surg Res. 1980;29:189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 15.Echtenacher B, Falk W, Männel DN, Krammer PH. Requirement of endogenous tumor necrosis factor/ cachectin for recovery from experimental peritonitis. J Immunol. 1990;145:3762–3766. [PubMed] [Google Scholar]
- 16.Claflin JL, Berry J. Genetics of the phosphocholine-specific antibody response to Streptococcus pneumoniae. . J Immunol. 1988;141:4012–4019. [PubMed] [Google Scholar]
- 17.Prodeus AP, Zhou X, Maurer M, Galli SJ, Carroll MC. Impaired mast cell-dependent natural immunity in complement C3-deficient mice. Nature. 1997;390:172–175. doi: 10.1038/36586. [DOI] [PubMed] [Google Scholar]
- 18.Echtenacher B, Männel DN, Hultner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–77. doi: 10.1038/381075a0. [DOI] [PubMed] [Google Scholar]
- 19.Malaviya R, Ikeda T, Ross E, Abraham SN. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-α. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- 20.Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330:662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 21.Cooper NR. The classical complement pathway: activation and regulation of the first complement component. Adv Immunol. 1985;37:151–216. doi: 10.1016/s0065-2776(08)60340-5. [DOI] [PubMed] [Google Scholar]
- 22.Reid RR, Prodeus AP, Khan W, Hsu T, Rosen FS, Carroll MC. Endotoxin shock in antibody-deficient mice. J Immunol. 1997;159:970–975. [PubMed] [Google Scholar]
- 23.Hirano T, Akira S, Taga T, Kishimoto T. Biological and clinical aspects of interleukin 6. Immunol Today. 1990;11:443–449. doi: 10.1016/0167-5699(90)90173-7. [DOI] [PubMed] [Google Scholar]
- 24.Baumann H, Gauldie J. The acute phase response. Immunol Today. 1994;15:75–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 25.Steel DM, Whitehead AS. The major acute phase reactants: C-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol Today. 1994;15:81–89. doi: 10.1016/0167-5699(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 26.Brundish DE, Baddiley J. Pneumococcal C-substance, a ribitol teichoic acid containing choline phosphate. Biochem J. 1968;110:573–582. doi: 10.1042/bj1100573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Briles DE, Nahm M, Schroer K, Davie J, Baker P, Kearney J, Barletta R. Antiphosphocholine antibodies found in normal mouse serum are protective against intravenous infection with type 3 Streptococcus pneumoniae. . J Exp Med. 1981;153:694–705. doi: 10.1084/jem.153.3.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yother J, Forman C, Gray BM, Briles DE. Protection of mice from infection with Streptococcus pneumoniaeby anti-phosphocholine antibody. Infect Immun. 1981;36:184–188. doi: 10.1128/iai.36.1.184-188.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alper CA. Increased susceptibility to infection associated with abnormalities of complement-mediated functions and of the third complement component (C3) N Engl J Med. 1970;282:350–354. doi: 10.1056/nejm197002122820701. [DOI] [PubMed] [Google Scholar]
- 30.Quezado ZMN, Hoffman WD, Winkelstein JA, Yatsiv I, Koev CA, Cork LC, Elin RJ, Eickacker PQ, Natanson C. The third component of complement protects against Escherichia coliendotoxin-induced shock and multiple organ failure. J Exp Med. 1994;179:569–575. doi: 10.1084/jem.179.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci USA. 1995;92:11490–11494. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marques MB, Kasper DL, Pangburn MK, Wessels MR. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B Streptococci. Infect Immun. 1992;60:3986–3993. doi: 10.1128/iai.60.10.3986-3993.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrison DC, Ryan JL. Endotoxins and disease mechanisms. Annu Rev Med. 1987;38:417–432. doi: 10.1146/annurev.me.38.020187.002221. [DOI] [PubMed] [Google Scholar]
- 34.Glauser MP, Zanetti G, Baumgartner J-D, Cohen J. Septic shock: pathogenesis. Lancet. 1991;338:732–736. doi: 10.1016/0140-6736(91)91452-z. [DOI] [PubMed] [Google Scholar]
- 35.Edwards MS, Kasper DL, Jennings HJ, Baker CJ, Nicholson-Weller A. Capsular sialic acid prevents activation of the alternative complement pathway by type III, group B streptococci. J Immunol. 1982;128:1278–1283. [PubMed] [Google Scholar]
- 36.Wessels MR, Paoletti LC, Kasper DL, DiFabio JL, Michon F, Holme K, Jennings HL. Immunogenicity in animals of a polysaccharide–protein conjugate vaccine against type III group B Streptococcus. . J Clin Invest. 1990;86:1428–1433. doi: 10.1172/JCI114858. [DOI] [PMC free article] [PubMed] [Google Scholar]