

Abstract
Hepatocellular carcinoma (HCC) is a common complication of chronic hepatitis B virus (HBV) infection. The pathogenetic mechanisms potentially responsible for HCC during chronic HBV infection are not well defined. This study demonstrates that chronic immune-mediated liver cell injury triggers the development of HCC in the absence of viral transactivation, insertional mutagenesis, and genotoxic chemicals. These results strongly suggest that the immune response to HBV is both necessary and sufficient to cause liver cancer during chronic HBV infection, and that all other procarcinogenic events associated with HCC are probably dependent on this process.
Keywords: cytotoxic T lymphocytes, hepatitis B virus, chronic hepatitis, hepatocellular carcinoma, transgenic mice
Antiviral T cells are believed to play a major role in the control of hepatitis B virus (HBV)1 infection. In support of this notion, the T cell response to HBV has been shown to be vigorous, polyclonal, and multispecific in patients with acute hepatitis who ultimately clear the virus, whereas it is relatively weak and narrowly focused in patients with chronic hepatitis, except during acute exacerbations of chronic disease (1, 2). The relative inefficiency of the T cell response in persistently infected patients is thought to cause chronic hepatitis by destroying some but not all of the infected cells, setting up a cycle of liver cell destruction and regeneration in the context of continuous intrahepatic inflammation that often terminates in hepatocellular carcinoma (HCC; references 3–7).
The mechanisms responsible for malignant transformation in chronic HBV infection are not well defined, and both viral and host factors have been implicated in the process. On the one hand, all cases of HCC occur after many years of chronic hepatitis, which could theoretically provide the mitogenic and mutagenic environment to precipitate random genetic and chromosomal damage and lead to the development of HCC. On the other hand, most tumors contain clonally integrated HBV DNA and microdeletions in the flanking cellular DNA, which could theoretically deregulate cellular growth control mechanisms (8). Furthermore, the HBV X gene product has been shown to transactivate cellular genes associated with cellular growth control (9–11) and inhibit p53 gene function in vitro (12), suggesting that deregulated X gene expression from integrated fragments of subviral DNA could play a role in hepatocarcinogenesis (13). Similarly, COOH-terminally truncated viral envelope proteins expressed from integrated subviral DNA may have transactivating activity (14, 15) and could potentially contribute to carcinogenesis in chronic HBV infection. However, like many retroviruses, HBV integration does not occur in resting hepatocytes; therefore, if HBV integration plays a role in hepatocarcinogenesis, antecedent events must occur that trigger hepatocellular turnover.
In an effort to clarify the carcinogenic potential of chronic hepatitis, we showed previously that transgenic mice that produce hepatotoxic quantities of the HBV large envelope polypeptide (16–19) display hepatocellular injury, regenerative hyperplasia, chronic inflammation, Kupffer cell hyperplasia, oxygen radical production, glutathione depletion, oxidative DNA damage, transcriptional deregulation, and aneuploidy that inexorably progress to HCC (18– 23). Although those studies demonstrated that HBV can cause HCC in the absence of insertional mutagenesis, X gene expression, or genotoxic chemicals, they did not prove that chronic immune-mediated hepatitis was a procarcinogenic stimulus in itself. Therefore, this study was undertaken to determine whether HCC can be triggered by a chronic, virus-specific immune response in HBV transgenic mice.
Materials and Methods
HBV Transgenic Mice.
Production and characterization of transgenic mouse lineage 107-5D (official designation Tg[Alb-1,HBV]Bri66; inbred B10D2, H-2d) have been described previously (17). Lineage 107-5D contains the entire HBV envelope coding region (subtype ayw) under the constitutive transcriptional control of the mouse albumin promoter (17). These mice express the HBV small, middle, and large envelope proteins in their hepatocytes (17, 18), are immunologically tolerant to hepatitis B surface antigen (HBsAg) at the T cell level (24), and display no evidence of liver disease during their lifetime, although they do develop “ground glass” hepatocytes due to overexpression of the large envelope protein (17). There is no X-RNA or X-protein expression detectable in the livers of these animals (our unpublished observations). Importantly, the mice develop a severe MHC class I–restricted necroinflammatory liver disease after the adoptive transfer of HBsAg-specific CTLs (17, 25, 26).
Thymectomy, Irradiation, Bone Marrow Reconstitution, and Spleen Cell Transfer.
18 8–10-wk-old male transgenic mice were thymectomized as described (27). 7 d later, the mice were irradiated (900 cGy) from a 137Cs source (Gammacell 40 irradiator; Atomic Energy of Canada, Ltd., Ottawa, Canada), then divided into two groups for bone marrow reconstitution (BMR) with nontransgenic or transgenic donor bone marrow that had been depleted of T cells by complement fixation using anti-Thy1.2 Ab (PharMingen, San Diego, CA). Group 1 recipients (nine mice), the primary experimental animals in this study, were reconstituted by the intravenous injection of 107 bone marrow cells collected from the femurs and tibias of syngeneic nontransgenic B10D2 (H-2d) mice. To control for the potential hepatocarcinogenic effect of irradiation, group 2 recipients (nine mice) were reconstituted with bone marrow cells derived from their immunologically tolerant inbred transgenic littermates. 1 wk after bone marrow reconstitution, the group 1 animals received 2 × 108 splenocytes from nontransgenic B10D2 (H-2d) mice that had been infected intraperitoneally 3 wk earlier with a recombinant vaccinia virus (HBs-vac) which expresses HBsAg, as described (25). At the same time, the group 2 animals received the same number of splenocytes from saline-injected, immunologically tolerant transgenic littermates. A third group of 10 transgenic mice (group 3) remained unmanipulated for the entire experiment for baseline comparison.
Disease Model.
All three groups of mice were analyzed for evidence of HBsAg-specific immunity and liver disease before and after BMR and after spleen cell transfer. Serum levels of HBsAg and anti-HBs were assessed by radioimmunoassay (AUSRIA II; Abbott Laboratories, North Chicago, IL) and enzyme-linked immunoassay (AUSAB EIA; Abbott Laboratories), respectively, as described (28). Hepatocellular injury was monitored biochemically as serum alanine aminotransferase (sALT) activity (19). Results were expressed as mean units per liter ± SEM of sALT activity, and differences between experimental and control groups were assessed for statistical significance by Student's t test. Tumor development was assessed by abdominal palpation and confirmed by autopsy, at which time the number of tumors visible at the surface of each liver was counted. Livers were then sectioned and examined for evidence of additional tumors not visible on the surface, and the diameter of each tumor was measured with a millimeter rule. Mice were killed by cervical dislocation. Tissue samples were fixed in 10% zinc-buffered formalin (Anatech Ltd., Battle Creek, MI), embedded in paraffin, sectioned (3 μm), and stained with hematoxylin and eosin as described (19). The intrahepatic distribution of HBsAg was assessed by the indirect immunoperoxidase method using 3-amino-9-ethyl carbazole (AEC; Shandon, Inc., Pittsburgh, PA) as a coloring substrate, as described previously (18). Liver tissue was also snap-frozen in liquid nitrogen and stored at −80°C for molecular analysis. 10 μg of total RNA was extracted from nontumorous liver tissue of representative mice from each group and subjected to RNase protection analysis to monitor the expression of CD3γ, CD4, CD8α, F480, and a panel of inflammatory cytokines as described previously (28).
CTL Analysis.
Donor and recipient spleen cells were stimulated in vitro every 7 d with irradiated P815 cells that stably express the HBV envelope proteins (P815preS1 cells), as described (29). 2 wk later, the cytolytic activity of the resultant T cell lines was assessed in a standard 4-h cytotoxicity assay with 51Cr-labeled P815, P815preS1 target cells, and P815 cells that had been pulsed either with different concentrations of a synthetic peptide representing residues 28–39 of HBsAg or with media, as described (25). In selected experiments, the HBsAg-specific cytolytic activity against P815 and P815-S target cells (E/T ratio 50:1) was tested in the presence of a rat mAb specific for mouse CD4 or CD8 (1 μg/ml; PharMingen), or a control Ab (purified rat IgG, 1 μg/ml; PharMingen). All experiments were performed in duplicate. Percent specific 51Cr release was derived as follows: (experimental cpm − spontaneous cpm)/(total cpm − spontaneous cpm) × 100. Spontaneous release was always <20% of the total.
Results
Anti-HBs Abs and Transaminase Profiles after Adoptive Transfer.
Two groups of 8–10-wk-old male transgenic mice were thymectomized (27), irradiated, and reconstituted with either syngeneic nontransgenic (group 1) or transgenic (group 2) T cell–depleted donor bone marrow. 1 wk after BMR, group 1 recipients received 2 × 108 splenocytes from syngeneic nontransgenic mice that had been primed with HBs-vac, as described (25). Simultaneously, group 2 animals received the same number of splenocytes from saline-injected, immunologically tolerant transgenic littermates, thereby serving as radiation controls. A third group of 10 transgenic mice (group 3) remained unmanipulated for the entire experiment.
In keeping with our previous reports (24–26), the HBs-vac–immunized nontransgenic donors displayed high titers of serum anti-HBs Abs and HBsAg-specific CTLs in their spleens (not shown). In contrast, the transgenic donors were anti-HBs–negative, and their splenocytes displayed no HBsAg-specific CTL activity, confirming that this lineage is immunologically tolerant to HBsAg. Consistent with the transfer of HBsAg-specific B cells, HBsAg disappeared from the serum of group 1 mice within 3 d after adoptive transfer (Fig. 1 A), concomitant with the appearance of anti-HBs (not shown). Furthermore, consistent with the transfer of HBsAg-specific CTLs, sALT activity increased from preinjection levels of 20–40 U/liter to ∼4,000 U/liter in group 1 recipients within 7 d after adoptive transfer, and fell progressively thereafter (Fig. 1 B). Importantly, sALT activity never returned to baseline in these animals, remaining at least two to three times above normal (except for the 270-d time point) and rising by 17 mo after BMR and spleen cell transfer (Fig. 1 B, and Table 1), at which time the animals were killed. We do not think the decrease in sALT activity in group 1 at 270 d reflects temporary resolution of disease, since the mice showed significant histological evidence of chronic hepatitis before (at 240 d) and after (at 510 d) this time point (Fig. 2). The rise in sALT in groups 1 and 2 at 510 d reflects a combination of tumor necrosis and the destruction of normal hepatocytes by the expanding tumors. In contrast to these observations, serum HBsAg levels did not fall in group 2 or group 3 mice (Fig. 1 A), in keeping with the absence of anti-HBs in their serum (not shown). Similarly, sALT activity remained in the normal range in group 2 and group 3 animals throughout most of the study, although they did rise late in the experiment in a few of the group 2 radiation controls (Fig. 1 B, and Table 1).
Figure 1.
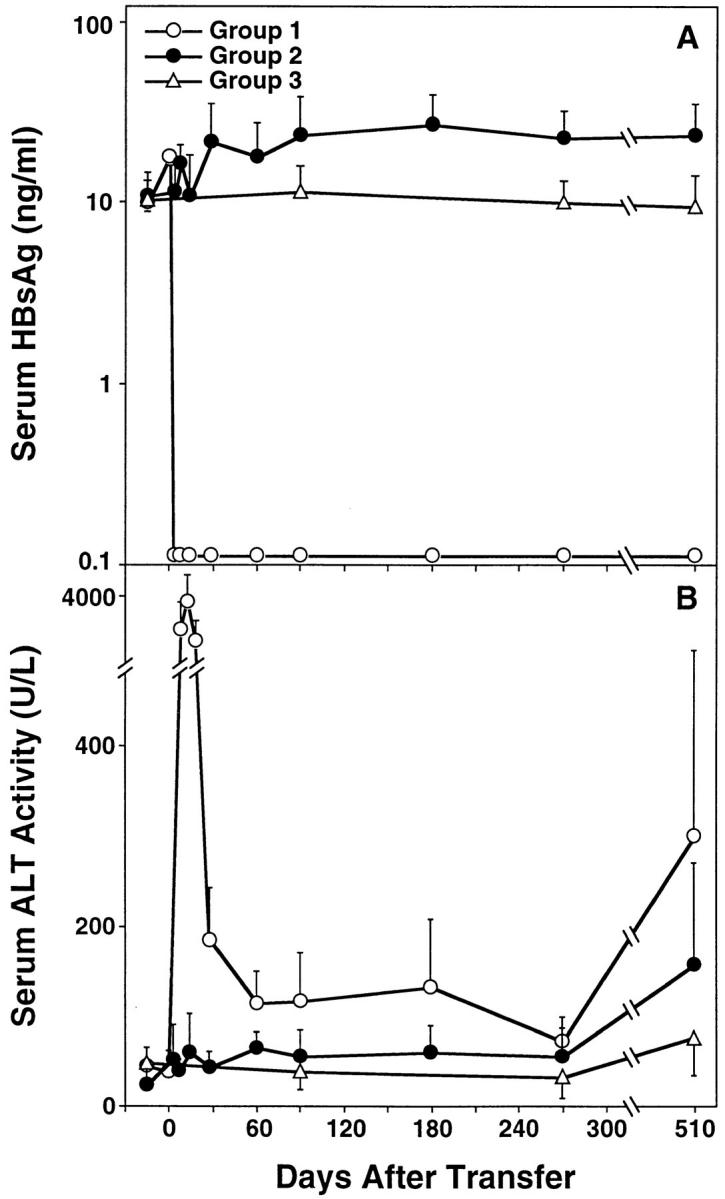
Kinetics of serum HBsAg (A) and ALT (B) in HBV transgenic mice. Group 1 mice (open circles; n = 9) were thymectomized, lethally irradiated, and reconstituted with bone marrow and spleen cells from syngeneic nontransgenic donors that had been previously immunized with recombinant vaccinia virus HBs-vac. Similarly treated group 2 transgenic control animals (filled circles; n = 9) were reconstituted with bone marrow and spleen cells from immunologically tolerant transgenic donors that had been injected with saline. All results were compared with group 3 unmanipulated age- and sex-matched transgenic mice (open triangles; n = 10). Adoptive transfer of splenocytes was performed on day 0. Results were expressed as mean units per liter ± SEM of sALT activity, and differences between experimental and control groups were assessed for statistical significance by Student's t test.
Table 1.
Biochemical, Histological, and Immunological Features of HBV Transgenic Mice as a Consequence of Chronic Immune-mediated Hepatitis
| Group | Mouse ID | Age at killing | Final HBsAg | Final αHBs | Initial sALT | Peak sALT | Final sALT | Liver tumor | No. of tumors | Largest tumor | Tumor histology | CTL (% lysis) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mo | mm | |||||||||||||||||||||||
| 1 | 283 | 18 | − | + | 12 | 2100 | 308 | + | 4–5 | 10 | HCC | 46 | ||||||||||||
| 284 | 18 | − | + | 60 | 3090 | 780 | + | 3 | 3 | Adenoma | 35 | |||||||||||||
| 285 | 18 | ND | + | 60 | 5130 | 360 | + | 4 | 12 | HCC | ND | |||||||||||||
| 180 | 19 | − | + | 48 | 3300 | 188 | + | >25 | 20 | HCC | 81 | |||||||||||||
| 183 | 19 | − | + | 66 | 2850 | 392 | + | 8 | 15 | HCC | 38 | |||||||||||||
| 243 | 19 | − | + | 18 | 6210 | 232 | + | >25 | 20 | HCC | 59 | |||||||||||||
| 251 | 19 | ND | + | 30 | 4920 | 136 | + | 1 | 12 | HCC | ND | |||||||||||||
| 254 | 19 | − | + | 60 | 4410 | 220 | + | 5 | 15 | HCC | 69 | |||||||||||||
| 177 | 20 | − | + | 60 | 2580 | 80 | + | 6 | 6 | HCC | 60 | |||||||||||||
| 2 | 27 | 17 | ND | − | 48 | 84 | 84 | − | 0 | 0 | ND | |||||||||||||
| 22 | 18 | ND | − | 30 | 72 | 72 | − | 0 | 0 | ND | ||||||||||||||
| 355 | 21 | + | − | 48 | 348 | 348 | + | 1 | 10 | Adenoma | 0 | |||||||||||||
| 359 | 21 | + | − | 30 | 184 | 184 | − | 0 | 0 | 1 | ||||||||||||||
| 387 | 21 | ND | − | 24 | 120 | 120 | − | 0 | 0 | ND | ||||||||||||||
| 388 | 21 | + | − | 24 | 116 | 116 | − | 0 | 0 | 17 | ||||||||||||||
| 8 | 21 | + | − | 24 | 88 | 88 | + | 1 | 9 | HCC | 0 | |||||||||||||
| 396 | 21 | + | − | 30 | 108 | 84 | − | 0 | 0 | 6 | ||||||||||||||
| 34 | 21 | + | − | 18 | 332 | 332 | − | 0 | 0 | 0 | ||||||||||||||
| 3 | 308 | 16 | + | − | 42 | 180 | 180 | − | 0 | 0 | 2 | |||||||||||||
| 290 | 19 | − | − | 54 | 54 | 32 | − | 0 | 0 | 2 | ||||||||||||||
| 315 | 21 | + | − | 24 | 40 | 40 | − | 0 | 0 | ND | ||||||||||||||
| 303 | 21 | + | − | 42 | 104 | 104 | − | 0 | 0 | ND | ||||||||||||||
| 351 | 21 | + | − | 72 | 80 | 80 | − | 0 | 0 | ND | ||||||||||||||
| 352 | 21 | + | − | 36 | 48 | 48 | − | 0 | 0 | ND | ||||||||||||||
| 342 | 21 | + | − | 72 | 72 | 68 | − | 0 | 0 | ND | ||||||||||||||
| 356 | 21 | + | − | 36 | 84 | 84 | − | 0 | 0 | 1 | ||||||||||||||
| 357 | 21 | + | − | 48 | 96 | 96 | − | 0 | 0 | 0 | ||||||||||||||
| 358 | 21 | + | − | 36 | 52 | 52 | − | 0 | 0 | 0 |
Figure 2.
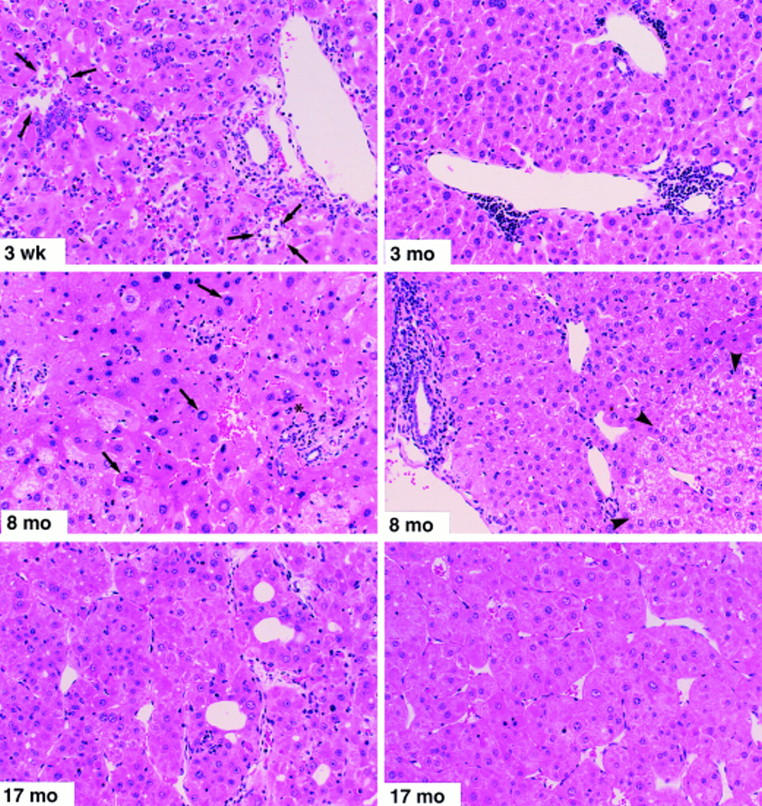
Histopathological features of prolonged chronic immune-mediated hepatitis in HBV transgenic mice (group 1). Mice were killed by cervical dislocation at the indicated time points. The 3-wk specimen illustrates portal and parenchymal inflammatory infiltrates with focal hepatocellular dropout (arrows) and hepatocellular apoptosis (asterisk). The 3-mo specimen displays lymphocytic portal infiltrates and moderate disarray of the lobular architecture. In addition, several lobular inflammatory foci were observed at this time point (not shown). The 8-mo specimen (left) illustrates marked lobular disarray, small focal lobular inflammatory infiltrates associated with degenerating hepatocytes, acidophil bodies (asterisk), and marked variation in the size and shape of many hepatocytes, some of which display nuclear pseudoinclusions (arrows). The other 8-mo specimen (right) illustrates a portal inflammatory infiltrate (at the left of the photo) with piecemeal necrosis of the limiting plate, and a glycogen-rich altered hepatic focus (arrowheads) containing numerous hepatocytes with prominent nucleoli (features traditionally associated with preneoplastic foci). Both of the 17-mo specimens, taken from large (>15 mm diameter) tumors from different animals, illustrate the classical histological features of trabecular HCC consisting of relatively well-differentiated hepatocytes with granular eosinophilic cytoplasm, arranged in nests and cords several cells thick that display increased mitotic activity, compress the adjacent hepatic parenchyma, and display areas of hemorrhage and necrosis (not shown). Hematoxylin and eosin; original magnification: ×200.
Chronic Hepatitis and HCC.
The differential sALT activity in the animals was reflected in the gross and histological appearance of their livers at various times after BMR and spleen cell transfer as illustrated in Fig. 2. Group 1 animals analyzed 3 wk after spleen cell transfer (top left) revealed a subacute inflammatory liver disease of variable severity characterized by a diffuse lymphomononuclear and polymorphonuclear inflammatory infiltrate in the parenchyma and portal areas, focal hepatocellular necrosis and dropout (arrows), and disorganization of the hepatic lobular architecture. 3 mo later (top right), the mice displayed multiple portal and intralobular inflammatory infiltrates and moderate lobular disarray indicative of chronic hepatitis. By 8 mo of age (middle), the liver disease in these animals was more severe. The left panel illustrates marked lobular disarray, small focal inflammatory infiltrates associated with degenerating hepatocytes, acidophil bodies (asterisk), and marked variation in the size and shape of the hepatocytes, some of which display nuclear pseudoinclusions (arrows). The other 8-mo specimen (right) illustrates a portal inflammatory infiltrate with piecemeal necrosis of the limiting plate, and a glycogen-rich altered hepatic focus (arrowheads) containing numerous hepatocytes with prominent nucleoli (features traditionally associated with preneoplastic foci). Eventually, when the animals were killed 17 mo later (bottom), they all displayed multiple liver tumors, most of which were >10 mm in diameter (Table 1). The tumors in all but one of these animals displayed the classical histological features of trabecular HCC (Fig. 2, bottom) characterized by trabecular cords composed of several layers of well-differentiated neoplastic hepatocytes that contained abundant granular eosinophilic cytoplasm and prominent nucleoli covered by a thin endothelial lining and were arranged in nests and cords several cells thick that displayed increased mitotic activity (not shown), compressed the adjacent hepatic parenchyma, and displayed areas of hemorrhage and necrosis (not shown).
In contrast to these findings, the radiation control mice in group 2 displayed much less severe necroinflammatory changes in their livers at the time of autopsy, consistent with their lower sALT levels. Interestingly, two of the nine mice in this group displayed solitary liver tumors up to 10 mm in diameter, one of which was classified histologically as a well-differentiated, trabecular HCC, whereas the other was a benign adenoma characterized by sheets of normal-appearing, glycogen-rich hepatocytes without portal tracts or central veins (Table 1). Not surprisingly, the livers of group 3 mice were grossly and histologically normal, except for the presence of HBsAg+ ground glass hepatocytes characteristic of this lineage (not shown). The livers of all of the animals in this study contained HBsAg+ hepatocytes at the time of autopsy; however, the frequency of such hepatocytes was much lower in group 1 animals (0.01– 15%) than in groups 2 and 3 (30–60%), presumably reflecting the destruction of antigen-positive hepatocytes by the immune response and, perhaps, the downregulation of antigen expression by inflammatory cytokines, as described previously (28, 30).
Intrahepatic T Cell and Cytokine Profiles in Reconstituted Animals.
To characterize the intrahepatic inflammatory response, total hepatic RNA from the nontumorous livers from six representative animals of each group (Fig. 3, lanes 1–18) was analyzed for the presence of T cell, macrophage, and cytokine transcripts using an RNase protection assay we have described previously (28, 30). The results were compared with total hepatic RNA from a similar group of transgenic mice killed at the peak of a CTL-induced acute hepatitis (Fig. 3, lanes 19–21) 3 d after adoptive transfer of 107 HBsAg-specific CTLs from a clone (6C2) we have shown previously to cause fulminant hepatitis in these animals (26), at which time their sALT activity was 3,213 ± 303 (mean ± SEM) U/liter, i.e., 10–20 times higher than group 1 and 2 animals, respectively. As expected, the highest level of T cell, macrophage, and cytokine mRNA expression was observed in the acute hepatitis specimens (lanes 19–21), followed, in order, by group 1 (lanes 1–6), then group 2 (lanes 7–12), and finally group 3 (lanes 13– 18) which represents the baseline for this analysis. It is noteworthy that IFN-γ mRNA was detectable only in the acutely inflamed livers, suggesting that the quality as well as the magnitude of the inflammatory infiltrate is different in these acute and chronic hepatitis models. These results demonstrate that the T cell markers and TNF-α mRNA content were greater in group 1 than group 2 at the time of autopsy. Considering the different sALT profiles in these two groups throughout the experiment (Fig. 1), these results suggest that the inflammatory liver disease began much earlier and was more severe in group 1 than in group 2, probably explaining the higher incidence of HCC in group 1 animals.
Figure 3.

Intrahepatic T cell and cytokine profiles in HBV transgenic mice. Total RNA was extracted from nontumorous liver tissue of representative mice from each group 17 mo after adoptive transfer (groups 1 and 2) and from unmanipulated age-matched controls (group 3), and subjected to RNase protection analysis to monitor the expression of CD3γ, CD4, CD8α, F480, and a panel of inflammatory cytokines. Lanes 19–21 contain total liver RNA from three HBV transgenic mice that were injected with an HBsAg-specific CTL clone which induced a severe transient acute hepatitis in transgenic recipients (references 26 and 28). The ribosomal protein light 32 (L32) RNA was used to normalize the amount of RNA loaded in each lane.
HBsAg-specific CTL Responses in Reconstituted Animals.
To assess the possible contribution of CTL response to the pathogenesis of the liver disease in all three groups of recipients, spleens harvested at the end of the experiment were examined for HBsAg-specific cytotoxic activity. As seen in Fig. 4 A, all of the group 1 mice (left) displayed strong HBsAg-specific CTL responses. In contrast, only one of the group 2 control mice (middle) showed a weak CTL response which was not detectable in any of the group 3 controls (right). The CTL activity of group 1 spleen cells was inhibited by an mAb specific for CD8 but not CD4 (Fig. 4 B, left). The antigenic fine specificity of the group 1 CTL response was examined using P815 target cells pulsed with a previously defined Ld-restricted immunodominant CTL epitope located between residues 28 and 39 of HBsAg (HBsAg 28–39; reference 29). As shown in Fig. 4 B (middle), CTLs from all but one of the group 1 animals were specific for that epitope, although their cytotoxic activity was generally weaker than that of HBs-vac–primed nontransgenic mouse splenocytes (Fig. 4 B, right). Splenocytes from the single group 1 mouse whose CTLs did not recognize peptide HBsAg 28–39 responded to a subdominant Dd-restricted CTL epitope located between amino acids 281 and 289 in HBsAg (not shown).
Figure 4.

(A) In vitro cytotoxic activity of splenocytes from group 1, 2, and 3 mice at the time of autopsy. Splenocytes from individual mice were stimulated in vitro every 7 d with irradiated P815 cells that stably express the HBV envelope proteins (P815preS1 cells). 2 wk later, the cytolytic activity of the resultant T cell lines was assessed in a standard 4-h cytotoxicity assay with 51Cr-labeled P815 cells and P815 transfectants that express the HBV small envelope polypeptide (P815-S) at the E/T ratios shown. Each line represents the cytolytic activity detectable in an individual mouse. (B) Characteristics of in vitro cytotoxic activity of splenocytes from group 1 mice and HBs-vac–primed nontransgenic mice. Left, The influence of anti-CD4 and anti-CD8 Abs on the HBsAg-specific cytolytic activity of group 1 spleen cells that had been stimulated for an additional 1 wk and tested for cytolytic activity against P815 and P815-S target cells at an E/T ratio of 50:1 in the presence of a rat mAb specific for mouse CD4 or CD8 or a control Ab. Columns with the same pattern indicate cytotoxic activity of splenocytes from the same mouse. Middle, Cytolytic activity of group 1 spleen cells against P815 cells that had been pulsed either with the indicated concentrations of a synthetic peptide representing residues 28–39 of HBsAg or with media at an E/T ratio of 50:1. Right, Cytolytic activity of similarly stimulated splenocytes from HBs-vac–primed nontransgenic B10D2 (H-2d) mice identical to those used as group 1 donors. Middle and right, Each line represents the cytolytic activity detectable in an individual mouse.
Discussion
Hepatocarcinogenesis during chronic HBV infection is a complex multifactorial process in which liver cell turnover plays an important role. In support of this notion, we have reported that hepatocellular injury triggered by overexpression of the HBV large envelope protein triggers hepatocellular regeneration, oxidative DNA damage, clonal expansion, and HCC (18–23). Although the large envelope protein is probably not expressed at toxic levels during natural HBV infection, the same downstream events (i.e., regeneration, DNA damage, etc.) could be induced in the chronically infected liver by CTL-mediated destruction of infected hepatocytes.
To test this hypothesis, we developed a model of chronic immune-mediated liver disease using transgenic mice that express nontoxic concentrations of the large, middle, and small envelope proteins in the hepatocyte. Similar to human chronic HBsAg carriers, these mice are immunologically tolerant to HBsAg and develop no evidence of liver disease except ground glass hepatocytes during their lifetime. 3-mo-old male transgenic mice (group 1) were thymectomized, lethally irradiated (900 rads), and reconstituted with bone marrow and spleen cells from syngeneic nontransgenic donors that had been previously immunized with HBsAg-vac and displayed HBsAg-specific CTLs and anti-HBs Abs. Similarly treated transgenic control animals (group 2) were reconstituted with bone marrow and spleen cells from immunologically tolerant transgenic donors. All results were compared with unmanipulated age- and sex-matched transgenic mice (group 3).
All of the group 1 mice developed acute hepatitis and cleared HBsAg from their serum. Subsequently, all of these mice developed chronic hepatitis, and their livers contained increased numbers of CD4+ and CD8+ T cells. All nine of the animals in this group developed one or several large liver tumors, all but one of which were histologically consistent with HCC. Importantly, an HBsAg-specific CTL response was detectable in the spleen of these animals. These results suggest that the mice developed a chronic inflammatory liver disease due to a relatively inefficient intrahepatic immune response that killed some of the HBsAg+ hepatocytes without downregulating HBV gene expression. This contrasts with the profound downregulation of HBV gene expression that occurs during CTL-induced acute hepatitis in these mice and related lineages due to the antiviral effects of IFN-γ and TNF-α produced by the antigen-activated CTLs (28, 30, 31). Indeed, HBsAg+ hepatocytes were present in the livers of these animals at the time of autopsy, representing a continuing supply of target cells for T cell recognition and continuing liver cell injury. Interestingly, however, the number of HBsAg+ hepatocytes was lower in group 1 animals than in the other two groups, possibly reflecting partial downregulation by intrahepatic cytokines, particularly TNF-α, which was more strongly induced in group 1 than in the other animals (Fig. 4).
In contrast to group 1, the group 2 mice did not clear HBsAg from their serum or develop acute hepatitis, presumably because the adoptively transferred transgenic spleen cells did not contain HBsAg-specific B cells or CTLs. Nonetheless, these animals developed chronic hepatitis very slowly as they aged, and the disease was less severe than that observed in group 1 animals. In keeping with these observations, HBsAg-specific CTLs were not detectable in their spleens at autopsy, and their livers contained fewer activated T cells than group 1 animals. Accordingly, only a single liver tumor was detectable in only two of the nine (22%) animals in this group, and only one of them was histologically consistent with HCC. As expected, all 13 of the control animals in group 3 displayed histologically normal livers, except for ground glass hepatocytes and minimal inflammatory infiltrates, for their lifetimes, and none produced CTLs or developed HCC.
Although the primary immunological mechanism responsible for chronic hepatitis in group 1 mice is not known at this time, in view of our previous experiments demonstrating that adoptively transferred HBsAg-specific CD4+ and CD8+ T cell clones cause acute hepatitis in this lineage (25, 26, 32), it is likely that one or both T cell subsets are involved. Whereas the initial episode of acute hepatitis was clearly related to the spleen cell transfer, the chronic hepatitis could be due to primed splenic memory cells or to naive T cells emerging from the nontransgenic bone marrow in these animals. This interesting distinction will be pursued in future experiments.
We do not understand the basis for the elevated transaminases and inflammatory liver disease appearing late in the course of this experiment in some of the group 2 animals. We suspect that this reflects a slow-onset, radiation-induced hepatitis. However, it is also possible that HBs-specific T cell precursors could have emerged from the marrow, escaped central deletion because of the absence of the thymus, and expanded slowly in the periphery without any pathological consequences until they reached sufficient numbers after several months in some of the mice to cause hepatitis. The absence of HBs-specific CTLs in the spleens of these mice suggests that CD4+ T cells may have contributed to the liver disease in these animals, as we suggested above for the group 1 animals.
The pathogenetic importance of immune-mediated hepatocellular injury in hepatocarcinogenesis in this study is strengthened by the fact that HCC occurs in the context of necrosis, inflammation, and regeneration (cirrhosis) in several human liver diseases other than HBV, including chronic hepatitis C (for a review, see reference 33), alcoholism (34), hemochromatosis (35), glycogen storage disease (36), α1-antitrypsin deficiency (37, 38), and primary biliary cirrhosis (39). Therefore, irrespective of etiology or pathogenesis, it would appear that chronic liver cell injury is a premalignant condition that initiates a cascade of events characterized by increased rates of cellular DNA synthesis and production of endogenous mutagens coupled with compromised cellular detoxification and repair functions. If these processes are sustained for a sufficiently long period of time, they would be expected to cause the multiple genetic and chromosomal changes necessary to trigger the development of HCC. This notion is supported by the close correlation between chronic inflammation and carcinogenesis both in animal models of wounding and inflammation that lead to skin cancer (40–42), and in humans, where continuous oral irritation leads to oral cancer (43), chronic inflammatory bowel disease predisposes to bowel cancer (44– 47), chronic cystitis precedes bladder cancer (48–52), and reflux esophagitis leads to esophageal cancer (53, 54).
Although these associations strongly suggest that chronic necroinflammation may be procarcinogenic in regenerative tissues, they do not constitute proof of this concept. However, these results provide definitive evidence that HBV-specific chronic immune-mediated liver cell injury is sufficient to initiate and sustain the process of hepatocarcinogenesis in this model. Furthermore, they demonstrate that the immune response is procarcinogenic despite the absence of cofactors such as viral integration, X gene expression, or genotoxic agents, which have been proposed to contribute to the development of HCC in humans. Since the immunological, virological, and histological features of this model closely resemble human chronic hepatitis, the results suggest that an ineffective immune response is the principal oncogenic factor during chronic HBV infection in humans. It is ironic that the same T cell response that can eradicate HBV from the liver when it is strong can be procarcinogenic by triggering a chronic necroinflammatory liver disease when it is unable to completely terminate the infection. If this is correct, therapeutic enhancement of the T cell response to HBV in chronically infected patients should prevent HCC.
Acknowledgments
We thank M.V. Hobbs for providing the probe sets used in the RNase protection assay; J.A. Koziol for statistical analysis; H. Kishimoto and J. Sprent for thymectomy instruction; A. Brown, R. Koch, and M. Chadwell for excellent technical assistance; J. Summers for critiquing this manuscript before submission; and J. Newmann for assistance with manuscript preparation.
This work was supported by grant R37-CA40489 from the National Institutes of Health. Y. Nakamoto was partially supported by the Uehara Memorial Foundation. This is manuscript 11378-MEM from The Scripps Research Institute.
Abbreviations used in this paper
- anti-HBs Ab
antibody to HBsAg
- BMR
bone marrow reconstitution
- HBsAg
hepatitis B surface antigen
- HBs-vac
recombinant vaccinia virus that expresses HBsAg
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- sALT
serum alanine aminotransferase
References
- 1.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 2.Tsai SL, Chen PJ, Lai MY, Yang PM, Sung JL, Huang JH, Hwang LH, Chang TH, Chen DS. Acute exacerbations of chronic type B hepatitis are accompanied by increased T cell responses to hepatitis B core and e antigens. Implications for hepatitis B e antigen seroconversion. J Clin Invest. 1992;89:87–96. doi: 10.1172/JCI115590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colombo M, de Franchis R, Del Ninno E, Sangiovanni A, De Fazio C, Tommasini M, Donato MF, Piva A, Di Carlo V, Dioguardi N. Hepatocellular carcinoma in Italian patients with cirrhosis. N Engl J Med. 1991;325:675–680. doi: 10.1056/NEJM199109053251002. [DOI] [PubMed] [Google Scholar]
- 4.Okuda K. Hepatocellular carcinoma: recent progress. Hepatology. 1992;15:948–963. doi: 10.1002/hep.1840150532. [DOI] [PubMed] [Google Scholar]
- 5.Ikeda K, Saitoh S, Koida I, Arase Y, Tsubota A, Chayama K, Kumada H, Kawanishi M. A multivariate analysis of risk factors for hepatocellular carcinogenesis: a prospective observation of 795 patients with viral and alcoholic cirrhosis. Hepatology. 1993;18:47–53. [PubMed] [Google Scholar]
- 6.Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, Nakanishi K, Fujimoto I, Inoue A, Yamazaki H, Kawashima T. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med. 1993;328:1797–1801. doi: 10.1056/NEJM199306243282501. [DOI] [PubMed] [Google Scholar]
- 7.Grisham JW. Interspecies comparison of liver carcinogenesis: implications for cancer risk assessment. Carcinogenesis (Oxf) 1997;18:59–81. doi: 10.1093/carcin/18.1.59. [DOI] [PubMed] [Google Scholar]
- 8.Matsubara K, Tokina T. Integration of hepatitis B virus DNA and its implications for hepatocarcinogenesis. Mol Biol Med. 1990;7:243–260. [PubMed] [Google Scholar]
- 9.Maguire HF, Hoeffler JP, Siddiqui A. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science. 1991;252:842–844. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- 10.Kekule AS, Lauer U, Weiss L, Luber B, Hofschneider PH. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature. 1993;361:742–745. doi: 10.1038/361742a0. [DOI] [PubMed] [Google Scholar]
- 11.Natoli G, Avantaggiati ML, Chirillo P, Costanzo A, Artini M, Balsano C, Levrero M. Induction of the DNA-binding activity of c-jun/c-fos heterodimers by the hepatitis B virus transactivator pX. Mol Cell Biol. 1994;14:989–998. doi: 10.1128/mcb.14.2.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang XW, Forrester K, Yeh H, Feitelson MA, Gu J-R, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci USA. 1994;91:2230–2234. doi: 10.1073/pnas.91.6.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T, Hurokawa K. High level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology. 1994;19:810–819. [PubMed] [Google Scholar]
- 14.Hildt E, Saher G, Bruss V, Hofschneider PH. The hepatitis B virus large surface protein (LHBs) is a transcriptional activator. Virology. 1996;225:235–239. doi: 10.1006/viro.1996.0594. [DOI] [PubMed] [Google Scholar]
- 15.Meyer M, Caselmann WH, Schluter V, Schreck R, Hofschneider PH, Baeuerle PA. Hepatitis B virus transactivator MHBst: activation of NF-kappa B, selective inhibition by antioxidants and integral membrane localization. EMBO (Eur Mol Biol Organ) J. 1992;11:2991–3001. doi: 10.1002/j.1460-2075.1992.tb05369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chisari FV, Pinkert CA, Milich DR, Filippi P, McLachlan A, Palmiter RD, Brinster RL. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science. 1985;230:1157–1160. doi: 10.1126/science.3865369. [DOI] [PubMed] [Google Scholar]
- 17.Chisari FV, Filippi P, McLachlan A, Milich DR, Riggs M, Lee S, Palmiter RD, Pinkert CA, Brinster RL. Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice. J Virol. 1986;60:880–887. doi: 10.1128/jvi.60.3.880-887.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chisari FV, Filippi P, Buras J, McLachlan A, Popper H, Pinkert CA, Palmiter RD, Brinster RL. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc Natl Acad Sci USA. 1987;84:6909–6913. doi: 10.1073/pnas.84.19.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989;59:1145–1156. doi: 10.1016/0092-8674(89)90770-8. [DOI] [PubMed] [Google Scholar]
- 20.Dunsford HA, Sell S, Chisari FV. Hepatocarcinogenesis due to chronic liver cell injury in hepatitis B virus transgenic mice. Cancer Res. 1990;50:3400–3407. [PubMed] [Google Scholar]
- 21.Pasquinelli C, Bhavani K, Chisari FV. Multiple oncogenes and tumor suppressor genes are structurally and functionally intact during hepatocarcinogenesis in hepatitis B virus transgenic mice. Cancer Res. 1992;52:2823–2829. [PubMed] [Google Scholar]
- 22.Hagen TM, Wehr C, Huang S-N, Curnutte J, Fowler P, Martinez V, Ames BN, Chisari FV. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci USA. 1994;91:12808–12812. doi: 10.1073/pnas.91.26.12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang S-N, Chisari FV. Strong, sustained hepatocellular proliferation precedes hepatocarcinogenesis in hepatitis B surface antigen transgenic mice. Hepatology. 1995;21:620–626. [PubMed] [Google Scholar]
- 24.Wirth S, Guidotti LG, Ando K, Schlicht HJ, Chisari FV. Breaking tolerance leads to autoantibody production but not autoimmune liver disease in HBV envelope transgenic mice. J Immunol. 1995;154:2504–2515. [PubMed] [Google Scholar]
- 25.Moriyama T, Guilhot S, Klopchin K, Moss B, Pinkert CA, Palmiter RD, Brinster RL, Kanagawa O, Chisari FV. Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science. 1990;248:361–364. doi: 10.1126/science.1691527. [DOI] [PubMed] [Google Scholar]
- 26.Ando K, Moriyama T, Guidotti LG, Wirth S, Schreiber RD, Schlicht HJ, Huang S, Chisari FV. Mechanisms of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. J Exp Med. 1993;178:1541–1554. doi: 10.1084/jem.178.5.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, and W. Strober. 1994. Current Protocols in Immunology. John Wiley & Sons, Inc., New York.
- 28.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 29.Ando K, Guidotti LG, Wirth S, Ishikawa T, Missale G, Moriyama T, Schreiber RD, Schlicht HJ, Huang S, Chisari FV. Class I restricted cytotoxic T lymphocytes are directly cytopathic for their target cells in vivo. J Immunol. 1994;152:3245–3253. [PubMed] [Google Scholar]
- 30.Guidotti LG, Ando K, Hobbs MV, Ishikawa T, Runkel RD, Schreiber RD, Chisari FV. Cytotoxic T lymphocytes inhibit hepatitis B virus gene expression by a noncytolytic mechanism in transgenic mice. Proc Natl Acad Sci USA. 1994;91:3764–3768. doi: 10.1073/pnas.91.9.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilles PN, Fey G, Chisari FV. Tumor necrosis factor-alpha negatively regulates hepatitis B virus gene expression in transgenic mice. J Virol. 1992;66:3955–3960. doi: 10.1128/jvi.66.6.3955-3960.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franco A, Guidotti LG, Hobbs MV, Pasquetto V, Chisari FV. Pathogenetic effector function of CD4-positive T helper 1 cells in hepatitis B virus transgenic mice. J Immunol. 1997;159:2001–2008. [PubMed] [Google Scholar]
- 33.Alter, H.J. 1988. Transfusion-associated non-A, non-B hepatitis: the first decade. In Viral Hepatitis and Liver Disease. A.J. Zuckerman, editor. Alan R. Liss, New York. 534–542.
- 34.Lieber CS, Garro A, Leo MA, Mak KM, Worner T. Alcohol and cancer. Hepatology. 1986;6:1005–1019. doi: 10.1002/hep.1840060533. [DOI] [PubMed] [Google Scholar]
- 35.Niederau C, Fischer R, Sonnenberg A, Stremmel W, Trampisch HJ, Strohmeyer G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313:1256–1262. doi: 10.1056/NEJM198511143132004. [DOI] [PubMed] [Google Scholar]
- 36.Limmer J, Fleig WE, Leupold D, Bittner R, Ditschunest H, Berger H-G. Hepatocellular carcinoma in type 1 glycogen storage disease. Hepatology. 1988;8:531–537. doi: 10.1002/hep.1840080317. [DOI] [PubMed] [Google Scholar]
- 37.Carlson J, Eriksson S. Chronic “cryptogenic” liver disease and malignant hepatoma in intermediate alpha-1-antitrypsin deficiency identified by a pi 2-specific monoclonal antibody. Scand J Gastroenterol. 1985;20:835–841. doi: 10.3109/00365528509088831. [DOI] [PubMed] [Google Scholar]
- 38.Eriksson S, Carlson J, Velez RN. Risk of cirrhosis and primary liver cancer in alpha-1-antitrypsin deficiency. N Engl J Med. 1986;314:736–740. doi: 10.1056/NEJM198603203141202. [DOI] [PubMed] [Google Scholar]
- 39.Melia WM, Wilkinson ML, Portmann BC, Johnson PJ, Williams R. Hepatocellular carcinoma in the non-cirrhotic liver: a comparison with that complicating cirrhosis. Q J Med. 1984;53:391–400. [PubMed] [Google Scholar]
- 40.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science. 1985;230:676–678. doi: 10.1126/science.2996144. [DOI] [PubMed] [Google Scholar]
- 41.Sieweke MH, Stoker AW, Bissell MJ. Evaluation of the cocarcinogenic effect of wounding in Rous sarcoma virus tumorigenesis. Cancer Res. 1989;49:6419–6424. [PubMed] [Google Scholar]
- 42.Lacey M, Alpert S, Hanahan D. Bovine papillomavirus genome elicits skin tumours in transgenic mice. Nature. 1986;322:609–612. doi: 10.1038/322609a0. [DOI] [PubMed] [Google Scholar]
- 43.Konstantinidis A, Smulow JB, Sonnenschein C. Tumorigenesis at a predetermined oral site after one intraperitoneal injection of N-nitroso-N-methylurea. Science. 1982;216:1235–1237. doi: 10.1126/science.7079755. [DOI] [PubMed] [Google Scholar]
- 44.Korelitz BI. Carcinoma of the intestinal tract in Crohn's disease: results of a survey conducted by the National Foundation for Ileitis and colitis. Am J Gastroenterol. 1983;78:44–46. [PubMed] [Google Scholar]
- 45.Collins RH, Jr, Feldman M, Fordtran JS. Colon cancer, dysplasia, and surveillance in patients with ulcerative colitis. A critical review. N Engl J Med. 1987;316:1654–1658. doi: 10.1056/NEJM198706253162609. [DOI] [PubMed] [Google Scholar]
- 46.D'Argenio G, Cosenza V, Delle M, Cave, Iovino P, Delle N, Valle, Lombardi G, Mazzacca G. Butyrate enemas in experimental colitis and protection against large bowel cancer in a rat model. Gastroenterology. 1996;110:1727–1734. doi: 10.1053/gast.1996.v110.pm8964397. [DOI] [PubMed] [Google Scholar]
- 47.Johnson LD, Ausman LM, Sehgal PK, King NW., Jr A prospective study of the epidemiology of colitis and colon cancer in cotton-top tamarins (Saguinus oedipus) . Gastroenterology. 1996;110:102–115. doi: 10.1053/gast.1996.v110.pm8536845. [DOI] [PubMed] [Google Scholar]
- 48.Locke JR, Hill DE, Walzer Y. Incidence of squamous cell carcinoma in patients with long-term catheter drainage. J Urol. 1985;133:1034–1035. doi: 10.1016/s0022-5347(17)49366-9. [DOI] [PubMed] [Google Scholar]
- 49.Kantor AF, Hartge P, Hoover RN, Fraumeni JF., Jr Epidemiological characteristics of squamous cell carcinoma and adenocarcinoma of the bladder. Cancer Res. 1988;48:3853–3855. [PubMed] [Google Scholar]
- 50.Yamamoto M, Wu HH, Momose H, Rademaker A, Oyasu R. Marked enhancement of rat urinary bladder carcinogenesis by heat-killed Escherichia coli. . Cancer Res. 1992;52:5329–5333. [PubMed] [Google Scholar]
- 51.Kawai K, Yamamoto M, Kameyama S, Kawamata H, Rademaker A, Oyasu R. Enhancement of rat urinary bladder tumorigenesis by lipopolysaccharide-induced inflammation. Cancer Res. 1993;53:5172–5175. [PubMed] [Google Scholar]
- 52.Kawai K, Kawamata H, Kemeyama S, Rademaker A, Oyasu R. Persistence of carcinogen-altered cell population in rat urothelium which can be promoted to tumors by chronic inflammatory stimulus. Cancer Res. 1994;54:2630–2632. [PubMed] [Google Scholar]
- 53.Dahms BB, Rothstein FC. Barrett's esophagus in children: a consequence of chronic gastroesophageal reflux. Gastroenterology. 1984;86:318–323. [PubMed] [Google Scholar]
- 54.Cameron AJ, Ott BJ, Payne WS. The incidence of adenocarcinoma in columnar-lined (Barrett's) esophagus. N Engl J Med. 1985;313:857–859. doi: 10.1056/NEJM198510033131404. [DOI] [PubMed] [Google Scholar]