

Abstract
The mechanisms underlying the survival of intracellular parasites such as mycobacteria in host macrophages remain poorly understood. In mice, mutations at the Nramp1 gene (for natural resistance-associated macrophage protein), cause susceptibility to mycobacterial infections. Nramp1 encodes an integral membrane protein that is recruited to the phagosome membrane in infected macrophages. In this study, we used microfluorescence ratio imaging of macrophages from wild-type and Nramp1 mutant mice to analyze the effect of loss of Nramp1 function on the properties of phagosomes containing inert particles or live mycobacteria. The pH of phagosomes containing live Mycobacterium bovis was significantly more acidic in Nramp1- expressing macrophages than in mutant cells (pH 5.5 ± 0.06 versus pH 6.6 ± 0.05, respectively; P <0.005). The enhanced acidification could not be accounted for by differences in proton consumption during dismutation of superoxide, phagosomal buffering power, counterion conductance, or in the rate of proton “leak”, as these were found to be comparable in wild-type and Nramp1-deficient macrophages. Rather, after ingestion of live mycobacteria, Nramp1-expressing cells exhibited increased concanamycin-sensitive H+ pumping across the phagosomal membrane. This was associated with an enhanced ability of phagosomes to fuse with vacuolar-type ATPase–containing late endosomes and/or lysosomes. This effect was restricted to live M. bovis and was not seen in phagosomes containing dead M. bovis or latex beads. These data support the notion that Nramp1 affects intracellular mycobacterial replication by modulating phagosomal pH, suggesting that Nramp1 plays a central role in this process.
Keywords: mycobacterium tuberculosis, phagosome, phagocytosis, macrophage, proton pump
Infectious diseases are a major reemerging health problem, accounting for 33% of world mortality (1.7 × 107 deaths per year). The most deadly diseases of this group include acute respiratory infections (4.4 × 106), tuberculosis (3.1 × 106), diarrhoeal diseases (3.1 × 106), and malaria (2.1 × 106) (references 1–4). The widespread emergence of multidrug resistance in virtually all infectious diseases has increased the severity of the problem (5, 6). One example is the striking return of tuberculosis and other mycobacterial diseases caused by the increased incidence of these infections in AIDS patients (500-fold greater than in the normal population) and by the emergence of highly virulent and multidrug-resistant strains of Mycobacterium tuberculosis (reference 7). The host defense mechanisms against these infections and the processes underlying long-term persistence and replication of mycobacteria and other such intracellular parasites in mononuclear phagocytes remain unclear and need to be better understood (8).
Microorganisms are normally internalized by macrophages and sequestered into membrane-bound vacuoles termed phagosomes. In most instances, the phagosome subsequently matures into an effective microbicidal organelle through fusion with early endosomes, late endosomes, and lysosomes, thereby becoming acidic and acquiring lytic enzymes (9, 10). M. tuberculosis survives intracellularly by prematurely arresting the process of phagosomal maturation (11, 12). Mycobacterial phagosomes retain the ability to fuse with early and recycling endosomes, but are virtually unable to fuse with late endosomes or compartments containing lysosomal enzymes (13). Although it is generally accepted that this defect is observed only with viable mycobacteria, the bacterial factors responsible for the maturational arrest and the underlying molecular targets of this action remain largely unexplained.
Innate resistance or susceptibility to mycobacterial infections has been detected in human populations (14, 15) and in animal models such as the laboratory mouse (16, 17). In a few instances, the genetic determinants of resistance or susceptibility have been identified. In the mouse, the Bcg/ Ity/Lsh locus on chromosome 1 confers natural resistance to infection with a group of seemingly unrelated intracellular parasites including several mycobacterial species (M. bovis, M. lepraemurium, M. smegmatis, M. avium, and M. intracellulare), Salmonella typhimurium, and Leishmania donovani (17–22). Susceptibility in vivo is expressed as an uncontrolled microbial replication in the spleen and liver early during infection, which is caused by an inability of the tissue macrophages to restrict intracellular proliferation (20, 22–25). The Bcg locus has been identified by positional cloning (Nramp1 [natural resistance–associated macrophage protein];1 reference 20), and has been shown to encode an integral membrane phosphoglycoprotein of 110 kD that is expressed almost exclusively in macrophages (26). In inbred strains of mice, susceptibility to infection is associated with a single amino acid substitution in Nramp1 (G169R), which causes rapid degradation of the protein (21). In vivo typing of animals showing either a loss of function null allele (knockout) or a gain of function Nramp1 transgene (Nramp1G169 in C57BL/6J mice) have established that Nramp1 and Bcg are indeed allelic (27, 28). In humans, NRAMP1 mRNA is expressed in both granulocytes and mononuclear phagocytes, and polymorphic variants at or near NRAMP1 have been found associated with increased susceptibility to leprosy (29). The pleiotropic effect of mutations at Nramp1 on resistance to infections with antigenically unrelated microbes suggests that this protein plays a key role in basic antimicrobial defense mechanisms of phagocytes. Immunolocalization studies have shown that Nramp1 is expressed in the late endosomal/early lysosomal (lysosomal-associated membrane protein [LAMP] 1–positive) compartment of the macrophage, and is recruited to the membrane of the phagosome through fusion events during the maturation process that follows phagocytosis (30). Therefore, Nramp1 is likely to confer resistance to mycobacterial infection by directly altering the phagosomal milieu.
The generation of an acidic interior is generally believed to be essential to the microbicidal activity of phagosomes (9, 31). Acidification of the phagosomal lumen, which is initiated and maintained primarily by the action of vacuolar-type proton ATPases (V-ATPases; references 9, 32), can exert a direct toxic effect on internalized bacteria. In addition, it is required for the activation of some lysosomal hydrolases, which typically have low pH optima (33, 34). Importantly, phagosomes containing mycobacteria fail to acidify normally, at least in part because of exclusion of V-ATPases (9, 11, 35), caused by alterations in fusogenic properties of the mycobacterial phagosomes.
These results together with the established phagosomal location of Nramp1 prompted us to investigate the possibility that Nramp1 may control mycobacterial replication through effects on phagosomal maturation and acidification. To this end, we compared the properties of phagosomes induced by M. bovis in macrophages from either normal, Nramp1positive mice or from animals bearing a null mutation at the Nramp1 locus (27, 28). The results indicate that Nramp1 plays a key role in the events leading to phagosomal acidification, and ultimately, inhibition of mycobacterial replication.
Materials and Methods
Materials, Solutions, and Antibodies.
Nigericin, 2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein (BCECF) acetoxymethyl ester, Oregon green, N-hydroxysuccinimidyl 5-(and 6-)-carboxyfluorescein (NHS-CF), FITC-conjugated dextran, FITC-conjugated human holotransferrin, and Texas red–conjugated dextran were from Molecular Probes, Inc. (Eugene, OR). Streptolysin O (SLO) was obtained from Dr. S. Bhakdi (Institute for Medical Microbiology, Johannes Gutenberg University, Mainz, Germany). Concanamycin was obtained from Kamiya Biochemical Company (Thousand Oaks, CA). Diphenylene iodonium was synthesized in our laboratory as described (36). Valinomycin was from Calbiochem-Novabiochem Corp. (La Jolla, CA). All other chemicals were of reagent grade and were obtained from Sigma Chemical Co. (St. Louis, MO), Fisher Scientific Co. (Pittsburgh, PA), or Pharmacia Biotech (Piscataway, NJ).
Polyclonal antibodies to the 39-kD subunit of the V-ATPase were raised by injecting rabbits with a glutathione-S-transferase fusion protein encoding the entire subunit and were then affinity purified as described (9). mAbs to LAMP-2 were obtained from the Developmental Studies Hybridoma Bank, maintained by both the University of Iowa (Ames, Iowa) and Johns Hopkins University School of Medicine (Baltimore, MD; reference 37). Cy3-conjugated donkey anti–mouse, anti–rabbit, and anti–rat IgG and FITC-conjugated donkey anti–rat IgG were obtained from Jackson ImmunoResearch Labs. (West Grove, PA).
PBS consisted of (mM): 140 NaCl, 10 KCl, 8 sodium phosphate, 2 potassium phosphate, pH 7.4. Powdered medium RPMI 1640 (HCO3 −-free, Hepes buffered) was from Sigma Chemical Co. The K+-rich medium contained (mM): 140 KCl, 5 glucose, and 15 Hepes, pH 7.4. In all cases the osmolarity was adjusted to 290 ± 5 mosM with the major salt. Permeabilization buffer consisted of (mM): 140 potassium glutamate, 5 NaCl, 10 glucose, 10 Hepes-K, and 0.1 EGTA, pH 7.35.
Cell and Bacterial Culture.
Mice expressing wild-type Nramp1 (strain 129/sv) were obtained from Taconic Farms (Germantown, NY). Knockout mice with a null mutation at the Nramp1 locus were generated on a 129/sv genetic background, as described (27, 28). Resident peritoneal macrophages were obtained from Nramp1 mutant (Nramp1 −/−) and wild-type mice as described (38). In brief, the peritoneal cavities of 6–8-wk-old mice were lavaged with 10 ml of ice-cold PBS. The cells, comprising ∼30% macrophages as determined by Wright staining, were washed three times in cold PBS, then resuspended in Hepes-buffered medium RPMI 1640 with 10% FCS. Cells (106/ml) were then incubated with bacteria as described below and plated on glass coverslips. Nonadherent cells (predominantly lymphocytes) were removed by washing with fresh medium.
M. bovis strain bacillus Calmette-Guérin, substrain Montreal (BCG) was obtained from the Armand Frappier Institute (Laval, Quebec, Canada) and maintained as described (39). In brief, one pelicule cultured on Sauton medium for 21 d was homogenized to 100 mg/ml and frozen at −80°C. Before infecting macrophages, freshly cultured bacteria were passaged for 1 wk in Dubos liquid medium at 37°C. Where indicated, BCG were killed by boiling for 5 min. For measurement of phagosomal pH, BCG were washed in PBS and 0.05% Tween 20 to promote disaggregation, then labeled by incubation in NHS-CF (0.1 mg/ml) and Oregon green (1 mg/ml) on ice for 30 min. Unbound dye was removed by repeated washing. The labeling procedure had no effect on bacterial viability, which was determined as follows: BCG were incubated with acridine-orange (100 ng/ml) for 15 min on ice and washed twice in PBS. Under these conditions, dead bacteria emit red fluorescence and are detectable under rhodamine optics. Heat-killed bacteria were used for comparison.
Measurement of Phagosomal pH.
Measurements of phagosomal pH (pHp) were obtained through the combined application of video microscopy and fluorescence ratio imaging. Suspended peritoneal macrophages from Nramp1 mutant (Nramp1 −/−) and wild-type mice were exposed to fluoresceinated M. bovis . The labeled bacteria were suspended in RPMI, added to cells at a ratio of ∼10 particles/cell and incubated for 1 h at 37°C. Peritoneal macrophages and bacteria were then sedimented together in a microcentrifuge tube for 10 s and then plated on glass coverslips to allow for adherence to occur. After 1 h at 37°C, coverslips were washed in RPMI containing 10% serum. Where indicated, phagocytosis was allowed to occur in Na+-free, K+-rich medium (see above).
For pH measurements, cells were washed with PBS and placed in a thermostatted Leiden holder on the stage of a Zeiss IM-35 microscope (Carl Zeiss Inc., Thornwood, NY) equipped with a ×63, 1.4 numerical aperture oil-immersion objective. A Sutter filter wheel was used to alternately position the two excitation filters (500BP10 and 440BP10 nm) in front of a xenon lamp. To minimize dye bleaching and photodynamic damage, neutral density filters were used to reduce the intensity of the excitation light reaching the cells. The excitation light was directed to the cells via a 510-nm dichroic mirror. Data were recorded every 60 s by irradiating the cells for 250 ms at each of the excitation wavelengths. Image acquisition was controlled by Metafluor software (Universal Imaging Corp., West Chester, PA), operating on a Pentium Dell computer (Dell Inc., Toronto, Ontario, Canada). The fluorescent light was directed onto a 535BP25-nm emission filter placed in front of a cooled CCD camera (Princeton Instruments Inc., Princeton, NJ). The sample was continuously illuminated at 620 nm by placing a red filter in front of the transmitted incandescent source. By placing an additional dichroic mirror in the light path, the red light was directed to a video camera and optical disk recorder (Panasonic TQ-2028F), allowing continuous visualization of cell morphology by Nomarski microscopy. Calibration of the fluorescence ratio versus pH was performed in situ for each experiment by equilibrating the cells in isotonic K+-rich medium buffered to varying pH values (between 6.0 and 7.45) in the presence of the K+/H+ ionophores nigericin (5 μM) and monensin (2 μM) as described (40). Calibration curves were constructed by plotting the extracellular pH, which is assumed to be identical to the internal pH (41), against the corresponding fluorescence ratio.
Determination of Phagosomal Buffering Power.
The buffering power of the phagosome was determined fluorimetrically in cells that had internalized fluoresceinated mycobacteria. After stabilization of phagosomal pH, defined concentrations of NH4Cl were added and the resulting change in pHp was determined. The buffering capacity of the compartment studied was calculated as detailed elsewhere (42) and is expressed as mmol/pH unit/liter phagosomal volume (mM/pH).
Determination of Phagosomal V-ATPase Activity.
To measure V-ATPase activity, cells having internalized fluoresceinated mycobacteria as above were bathed in permeabilization buffer and incubated with SLO (0.1 mg/ml) for 5 min. That plasma membrane permeabilization occurred under these conditions was demonstrated by the onset of cellular swelling and the uptake of trypan blue. Intracellular compartments were not permeabilized by the SLO treatment, as FITC-dextran–labeled endosomes remained intact under similar conditions and phagosomes retained the ability to acidify. Phagosomal V-ATPase activity was then reinitiated by perfusing the SLO-treated cells with permeabilization buffer containing 5 mM ATP plus Mg2+.
Measurement of Early (Recycling) Endosomal pH.
To measure the pH of the early/recycling endosomal compartment (pHe), macrophages from knockout mice were treated with FITC-labeled human holotransferrin (20 μg/ml in RPMI) for 15 min at 37°C. Cells were then washed in PBS to remove free transferrin and placed in a thermostatted Leiden holder for single-cell imaging as described above. Cells were irradiated at each of the two excitation wavelengths (500BP10 and 440BP10 nm) for 500 ms and, where indicated, were treated with 100 nM concanamycin. Calibration of pHe was achieved using monensin and nigericin as described above.
Immunofluorescence.
Immunofluorescence studies were performed on cells from wild-type and Nramp1 −/− mice that had internalized labeled mycobacteria or latex particles. Mycobacteria were labeled with NHS-CF (0.01 mg/ml) as described above. Latex particles were opsonized by incubating in FCS for 1 h at 37°C, washed in PBS to remove unbound opsonin, and added to cells at a density of 10 beads per cell, as described (38). Where indicated, cells were preincubated with FITC dextran (1 mg/ml) for 15 min at 37°C to label early endosomes. Cells were fixed for 3 h with 4% paraformaldehyde in PBS at room temperature and washed in PBS containing 100 mM glycine for 10 min. The cells were then permeabilized in 0.1% Triton X-100 in PBS for 20 min at 22°C, washed in ice-cold PBS, and blocked with 5% donkey serum in PBS for 1 h at room temperature. For localization of the 39-kD subunit of the V-ATPase, coverslips were then incubated with a 1:100 dilution of the affinity-purified antibody and a 1:1,000 dilution of Cy3-labeled anti-rabbit IgG. The specificity of the anti–39-kD subunit antibody was confirmed by preincubating it with a fivefold molar excess of purified fusion protein for 1 h at room temperature, which prevented binding to the cells (not shown). For localization of LAMP-2, coverslips were fixed, permeabilized, and blocked as above then incubated with a 1:4 dilution of anti–LAMP-2 antibody (hybridoma culture supernatant) and a 1:600 dilution of Cy3-labeled anti-rat IgG.
After staining, cells were mounted using Slow Fade (Molecular Probes, Inc.) and sealed with nail polish. Fluorescence was analyzed with a laser confocal microscope (model TCS4D; Leica Instruments GmbH, Heidelberg, Germany) with a ×63 objective. Composites of confocal images were assembled and labeled using Photoshop and Illustrator software (Adobe, Mountain View, CA). All experiments were performed at least four times. Representative confocal images are displayed where appropriate.
Results
Determination of the pH of Phagosomes Containing M. bovis.
Single cell spectroscopy was used to measure the pH of phagosomes induced by ingestion of M. bovis in macrophages isolated from Nramp1-expressing (wild-type) and Nramp1 −/− mutant mice. Mycobacteria were covalently labeled with fluorescent, pH-sensitive dyes that emitted signals detectable by ratio imaging. Two dyes with different H+ affinity (fluorescein, pKa = 6.4, and Oregon green, pKa = 4.7) were used in combination, extending the range of reliable pH measurement from ∼4.0 to 7.5. The procedure used to label the bacteria had no effect on their viability, as previously determined by Oh et al. (43). Phagocytosis was induced by incubating wild-type or Nramp1 −/− macrophages with labeled bacteria for 1 h at 37°C. Fig. 1 illustrates a representative experiment performed in wild-type macrophages. Fig. 1 A shows a differential interference contrast (Nomarski) microscopy image of macrophages interacting with live M. bovis. As displayed in the accompanying fluorescence ratio image (Fig. 1 B), several of these bacteria (e.g., filled white arrows) are in an acidic environment (note pseudocolor calibration scale on the right), while others (e.g., open arrow) are exposed to near neutral pH. It is likely that the latter are exposed to the extracellular milieu, whereas the acidic bacteria are located within phagosomes. However, Nomarski microscopy could not reliably discriminate between extra- and intracellular bacteria, nor could all the adherent extracellular bacteria be removed by washing. Therefore, we routinely used three functional criteria to verify that the acidic particles had indeed been internalized. Bacteria within phagosomes were identified not only by their acidic initial pH, but also by the occurrence of a rapid alkalinization upon addition of NH4Cl. Preferential permeation of NH3 across the plasma and phagosomal membranes, followed by its protonation in the phagosomal lumen, result in selective alkalinization of intraorganellar bacteria with little effect on extracellular particles (Fig. 1, C and D). Similarly, addition of the K+/H+ exchange ionophore nigericin is expected to alter pHp, but not that of extracellular bacteria. Conversely, abrupt changes in the pH of the bathing solution were predicted to affect acutely the pH of extracellular bacteria, but not that of intraphagosomal organisms (data not shown). By using a combination of these criteria, we could effectively identify internalized mycobacteria and were able to assess the effects of Nramp1 on the extent and mechanisms of phagosomal acidification.
Figure 1.

Measurement of pHp. Peritoneal macrophages isolated from wild-type Nramp1-expressing mice (strain 129/sv) were allowed to interact with live, fluoresceinated M. bovis, some of which became internalized. The cells were then plated on glass coverslips, mounted in thermoregulated chambers, and visualized using differential interference contrast optics (A), while the fluorescence was measured with alternating excitation at 440 and 490 nm. The fluorescence ratio (B and C) was used for the measurement of the pH in the vicinity of the bacterial surface. After acquiring baseline pH measurements (B), the cells were exposed to 20 mM NH4 + and recording was resumed (C). A pseudocolor pH scale is shown to the right. Internalized bacteria, identified by their resting acidic pH and responsiveness to NH4 + (filled white arrowheads) and an adherent extracellular bacterium (open arrowhead) are indicated. A typical time course of pH determination in internalized (filled red circles) and adherent bacteria (open circles) are shown in D . The addition of 20 mM NH4 + is indicated. Representative of 37 individual experiments.
Phagosomal pH in Wild-type and Nramp1− /− Mutant Macrophages.
We used the method described in Fig. 1 to compare the pH of phagosomes induced by live and heat-killed M. bovis in resident peritoneal macrophages obtained from wild-type (Nramp1 +/+) and mutant (Nramp1 −/−) mice. As shown in Fig. 2 A and summarized in Fig. 2 B, the pH of phagosomes containing live M. bovis was significantly more acidic in normal macrophages than in Nramp1 mutant macrophages (pH 5.5 ± 0.06 versus pH 6.6 ± 0.05, respectively; P <0.005). The values obtained in the Nramp1 mutants are consistent with previous reports of the phagosomal pH in macrophages from mice that are naturally susceptible to the mycobacteria and bear a mutant G169D allele at Nramp1 (11, 19, 43). Of note, the phagosomal pH in both cell types became indistinguishable after addition of nigericin (not shown), indicating that the optical properties of the two systems are similar and validating the calibration procedure. Importantly, the attenuated phagosomal acidification seen in the Nramp1 −/− mice was specific for live mycobacteria, as phagosomes containing either heat-killed bacteria or opsonized latex particles acidified normally (Fig. 2 C), i.e., to an extent comparable to that observed in phagosomes of Nramp1-expressing cells containing either live (Fig. 2 B) or dead bacteria, or latex beads (not shown). This confirms that the phagosomal acidification mechanism(s) are fully competent in both cell types, and that they differ only in their responsiveness to live mycobacteria.
Figure 2.

Determination of pHp in wild-type and Nramp1 −/− macrophages. Phagosomal pH was measured by ratio imaging as described for Fig. 1 (see Materials and Methods) in peritoneal macrophages obtained from wild-type (Nramp +/+) mice and Nramp1 mutant mice (Nramp1 −/−). Phagosome formation was induced using either live, fluoresceinated M. bovis (A, B, and leftmost bar in C), dead M. bovis (middle bar in C), or opsonized latex beads (rightmost bar in C). (A) frequency histogram comparing the pH in wild-type (Nramp +/+) mice and Nramp1 mutant mice (Nramp1 −/−); (B) summary of results comparing Nramp +/+ and Nramp1 −/− cells; data are means ± SE of 37 individual determinations; (C) summary of results comparing different phagocytic particles in Nramp1 −/− cells. Data are means ± SE of 24 determinations.
To confirm that genetic ablation of the Nramp1 gene was directly responsible for the defect in phagosomal acidification, we also measured pHp in macrophages obtained from C57/BL6J mice, a naturally susceptible mouse strain with a mutant G169D allele at Nramp1 (20, 28, 44). In macrophages from these mice the pHp was significantly higher in phagosomes containing live M. bovis than in those induced by dead bacteria (6.5 ± 0.05 versus 5.6 ± 0.06, respectively; P <0.05). These data provide strong evidence that Nramp1 plays a direct role in the control of phagosomal acidification after internalization of mycobacteria.
Mechanism of Mycobacterial Phagosome Acidification in Wild-type and Nramp1− /− Macrophages.
Having demonstrated differences in phagosomal pH between Nramp1-expressing and Nramp1 −/− macrophages, we next investigated the mechanistic basis for the differential acidification. V-ATPases are believed to be responsible for the acidification of normal phagosomes. In susceptible host cells, such ATPases are largely absent from mycobacterial phagosomes (11, 12), seemingly accounting for their elevated pH. However, although higher than that of other phagosomes, the pH of mycobacterial phagosomes is nevertheless nearly one unit more acidic than that of the surrounding cytosol (11, 43). Therefore, we considered the possibility that the intermediate acidification observed was the consequence of a Donnan equilibrium, generated by nondiffusible anionic charges within the phagosome, possibly on the bacterial wall. This prediction was tested by adding the H+-selective ionophore carbonyl cyanide-m-chlorophenyl hydrazone (CCCP) to macrophages that had internalized live M. bovis. Increasing the passive H+ conductance would be expected to have little effect or even accentuate a pH gradient induced by a Donnan potential. Contrary to this prediction, the acidification noted in mycobacterial phagosomes from Nramp1 −/− macrophages was rapidly dissipated by the protonophore (Fig. 3 A), implying that intraphagosomal H+ were not at electrochemical equilibrium. Similarly, CCCP alkalinized the pH of mycobacterial phagosomes from Nramp1-expressing macrophages (Fig. 3 A). These findings indicate that a Donnan equilibrium is not an important contributor to phagosomal acidification and that H+ are actively accumulated in the lumen of phagosomes from both normal and Nramp −/− mice, though to different extents.
Figure 3.


Effects of protonophores and V-ATPase inhibitors on pHp. pHp was measured by ratio imaging as described for Fig. 1 (see Materials and Methods) in peritoneal macrophages obtained from wild-type mice (open symbols) and Nramp1 −/− mice (filled symbols). Phagosome formation was induced using live, fluoresceinated M. bovis. After acquiring baseline measurements, 10 μM CCCP (A) or 100 nM concanamycin (B, CCM) was added to both samples. Data are representative of five determinations.
Fig. 3 B shows that, in both types of macrophages, the active accumulation of H+ is sensitive to concanamycin. At the concentration used (100 nM), this inhibitor selectively blocks V-ATPases (45). Therefore, active H+ pumps appear to be functional in the phagosomal membrane of both wild-type and Nramp1 −/− mice. This observation agrees with recent observations made in macrophages from C57BL/6J mice (Nramp1D169), a strain that is naturally susceptible to mycobacteria (9).
Steady state pHp is dictated by the net result of proton pumping, efflux and metabolic production or consumption, in combination with the endogenous buffering power. Alterations in one or more of these parameters must be responsible for the differences in pHp noted between wild-type and Nramp1 −/− macrophages. The contribution of each of these parameters was analyzed.
Phagosomal Buffering Power.
In principle, the observed differences in pH between phagosomes from wild-type and Nramp1 −/− mice could be attributable to differences in their buffering capacity. To test this possibility, the phagosomal buffering power was measured by pulsing cells that had internalized live M. bovis with small aliquots of NH4Cl (1–2 mM). The change in pHp resulting from entry and protonation of NH3 was used to compute the buffering power. Because buffering capacity can vary with pH, care was taken to make the comparisons in the same range of pHp. This is particularly important because the initial pHp of the two types of macrophages is different. As shown in Table 1, the buffering capacity of mycobacterial phagosomes in the 6.5 to 6.8 range was similar in both wild-type and Nramp1 −/− macrophages. Therefore, other parameters must be responsible for their differential steady state pHp.
Table 1.
Physiological Characteristics of Phagosomes from Nramp1 Wild-type (Nramp1+ /+) and Nramp1− /− Mice
| Nramp1 +/+ | Nramp1 −/− | |||
|---|---|---|---|---|
| Buffering power* (mmol/pH/liter) | 18 ± 2 | 16 ± 4 | ||
| H+ consumption by ROI‡ (ΔpHp) | 0.2 ± 0.05 | 0.3 ± 0.1 | ||
| Net H+ efflux§ (pH/min) | 0.2 ± 0.03 | 0.2 ± 0.02 | ||
| Counterion conductance‖ (ΔpHp) | 0.3 ± 0.03 | 0.2 ± 0.04 |
Macrophages from wild-type and Nramp1 −/− mice that had internalized live M. bovis were pulsed with small aliquots of NH4Cl (1–2 mM). The change in pHp resulting from entry and protonation of NH3 was used to compute the buffering power, which was compared in the 6.5– 6.8 range in both cell types.
Macrophages from both strains that had internalized live M. bovis were treated with the flavoprotein inhibitor diphenylene iodonium (3 μM), and steady state pHp was reached. Results are expressed as pHp after addition minus pHp before addition.
Net phagosomal H+ efflux was induced in macrophages from wild-type and Nramp1 −/− mice that had internalized live M. bovis by addition of concanamycin (100 nM). Because the buffering power of both types of phagosomes is similar, the fluxes are expressed as the rates of change of pH per unit time, and were made at comparable pHp.
Cells from both strains were allowed to internalize live M. bovis in a K+-rich medium. After steady state pHp was reached, cells were treated with valinomycin (10 μM), a conductive K+ ionophore. Results are expressed as pHp after addition minus pHp before addition.
Effects of the Electrogenic Na+ Pump on the Protonmotive Force.
Because the process of H+ pumping by the V-ATPase is rheogenic, it is affected by changes in the transmembrane potential. In some systems, the rate or extent of acidification of endosomes has been shown to be modulated by the parallel activity of the Na+ pump, an electrogenic ATPase (46, 47). Therefore, we considered the possibility that enhanced Na+ pump activity contributed to the reduced acidification of Nramp1 −/− phagosomes. To test this hypothesis, ouabain was added to the medium before phagocytosis and was present throughout. High (millimolar) concentrations of the drug were used to ensure inhibition of the pump, which in rodents has low affinity for the glycoside. Ouabain had no significant effect on pHp (not shown). Therefore, it is unlikely that differential electrogenic effects of the Na+/K+-ATPase contribute to the difference in pHp noted between wild-type and Nramp1 −/− macrophages.
H+ Consumption by Superoxide Dismutation.
It has been proposed that superoxide radicals, formed within the phagosome by the NADPH oxidase, consume H+ during the dismutation reaction, forming hydrogen peroxide and water (48). Therefore, we considered the possibility that Nramp1 affected pHp indirectly by altering the rate of superoxide production or dismutation. Macrophages from wild-type and Nramp1 −/− mice that had internalized live M. bovis were treated with the flavoprotein inhibitor diphenylene iodonium, at concentrations known to produce complete inhibition of the NADPH oxidase. As shown in Table 1, the inhibitor had no effect on steady state pHp in macrophages from either strain. This finding suggests that because macrophages generate superoxide at relatively low rates, unlike neutrophils (49), the dismutation reaction does not contribute significantly to net H+ consumption in the phagosome. Hence, superoxide dismutation cannot account for the differential behavior of the two types of macrophages.
Phagosomal H+ (Equivalent) “Leak”.
Steady state pHp is established by the differential between the rates of H+ influx (pumping) and efflux (leakage of H+ and H+-equivalent species). Because both wild-type and Nramp1 −/− macrophages seem to share the same pumping mechanism, we considered the possibility that leaks of different magnitude might account for their different pHp. Net H+ efflux was induced by inhibiting the V-ATPases with concanamycin, as in Fig. 3. Because the buffering power of both types of phagosomes is similar, the fluxes are expressed as the rates of change of pH per unit of time. The measurements were made at comparable pHp to ensure that any differences in the fluxes reflected the permeability to H+ and not its driving force. As shown in Table 1, proton leakage rates were found to be similar in both wild-type and Nramp1 −/− mice. Therefore, the difference in the steady state pHp between the two types of mice is probably due to differential influx (pumping).
ATP-dependent H+ Pumping.
At steady state, H+ pumping is obscured by H+ efflux (leakage) of equal magnitude and opposite direction. The leak is highest at acidic pHp, when the force driving efflux is greater. Conversely, the activity of the pump diminishes as the lumen becomes acidic. Therefore, to assess the ability of phagosomes to pump H+ encumbered minimally by leakage, we measured the rate of initial acidification, near neutral pHp. For this purpose, we first dissipated the H+ gradient by depletion of cytosolic ATP, using SLO to permeabilize the plasma membrane. This antibiotic forms large pores in the plasmalemma, without permeabilizing internal organelles, including the phagosome (50, 51). As shown in Fig. 4 A, upon addition of SLO to cells suspended in medium devoid of ATP and Mg2+, the phagosomal acidification dissipates within minutes. Importantly, this pH change was not observed when the medium contained ATP and Mg2+ (data not shown), indicating that it is not a consequence of direct phagosomal permeabilization by SLO. Instead, neutralization of pHp appears to be due largely to washout of ATP and Mg2+, the substrates required by the V-ATPase, since acidification was restored by their readdition. Accordingly, the reacidification was virtually eliminated by simultaneous addition of concanamycin (Fig. 4 A).
Figure 4.
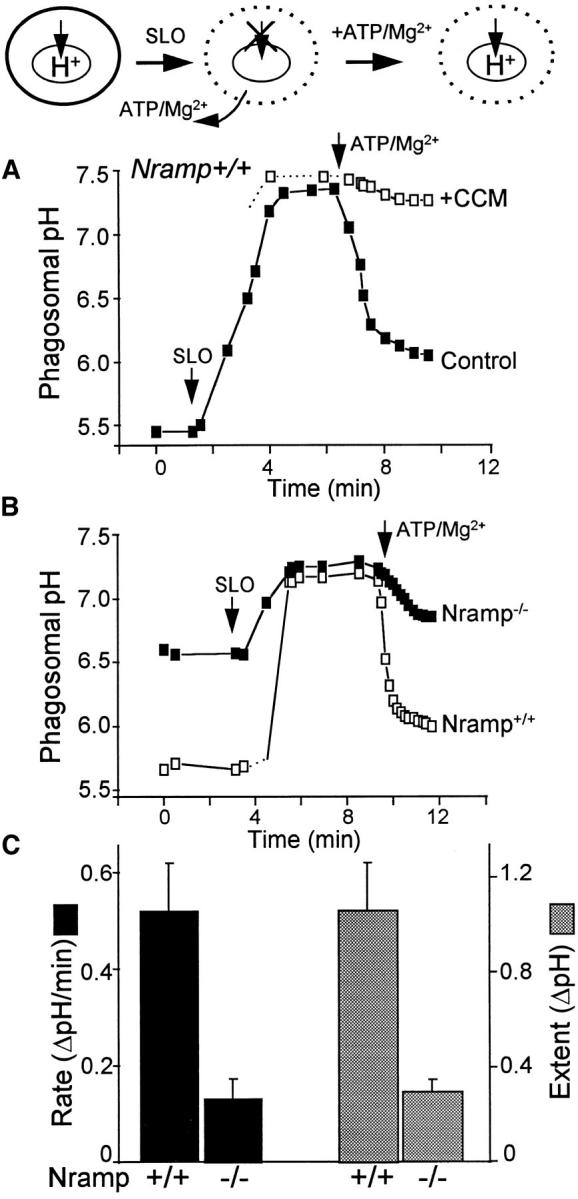
pHp determinations in SLO-permeabilized cells. The experimental protocol is diagramatically illustrated at the top. In brief, intact cells that normally have acidic phagosomes (left) were permeabilized using 0.1 μg/ml SLO in media devoid of ATP/Mg2+, resulting in inhibition of H+ pumping due to depletion of substrate (middle). Readdition of ATP/ Mg2+ induces resumption of pumping (right). Phagosomal pH was measured by ratio imaging as described for Fig. 1 (see Materials and Methods) in peritoneal macrophages obtained from wild-type (Nramp +/+) mice and from Nramp1 mutant mice (Nramp −/−). (A) Measurements in Nramp1 +/+ cells. Where indicated, cells were treated with SLO, and ATP and Mg were reintroduced in the absence (filled symbols) or presence of 100 nM concanamycin (CCM; open symbols). (B) Nramp1 +/+ cells (open symbols) or Nramp1 −/− cells (filled symbols) were treated with SLO. ATP and Mg were reintroduced where indicated. A and B are each representative of five determinations. (C) Summary of results from five experiments like that shown in B. The initial rate (black bars) and extent (stippled bars) of ATP/ Mg-induced acidification were measured. Data are means ± SE of five separate experiments.
As shown in Fig. 4 B, the initial rate and extent of acidification recorded upon reintroduction of ATP and Mg2+ were significantly greater in M. bovis phagosomes of wild-type mice than in their Nramp1 −/− counterparts. The results of five similar experiments are summarized in Fig. 4 C. From these observations, we conclude that elimination of Nramp1 results in a decreased ability to pump H+ into mycobacterial phagosomes. This is most simply explained by a reduction in the number of V-ATPases, but other possible explanations exist (see below).
Counterion Permeability.
To maintain electroneutrality in the course of net translocation of H+ by the V-ATPase, stoichiometric movement of counterions is required. It has been postulated that the rate of acidification can be limited not by the ability of the ATPases to pump H+, but by the passive permeability of the membrane to counterions (52). Accordingly, the difference in the pumping rates noted between wild-type and Nramp1 −/− mice could be attributed to reduced counterion permeability in the case of the latter. Two lines of evidence argue against this possibility. First, addition of CCCP to the Nramp1 −/− macrophages rapidly increased pHp (Fig. 3). Because in the steady state H+ pumping and leakage are identical, dissipation of the acidification by CCCP indicates that the rate of leakage greatly exceeded pumping. The excess leakage of H+ induced by the protonophore must have been supported by a flux of counterions that was greater than that needed to operate the pump in the steady state, implying that the resting conductance is greater than necessary to support the basal rate of pumping, i.e., that counterion permeability is not rate limiting.
One might argue that the conductive pathway may display rectification, i.e., that the counterion permeability is greater in one direction than in the other. A second type of experiment was devised to consider this possibility. Cells from both strains were allowed to internalize live M. bovis in a K+-rich medium. After steady state pHp was reached, cells were treated with valinomycin, a conductive K+ ionophore. Under these conditions, cations are readily able to exit the phagosomal lumen, providing an adequate counterion for H+ pumping. As shown in Table 1, addition of valinomycin had minimal effects on pHp in both strains of mice. These findings confirm that counterion conductance is not limiting to phagosomal acidification and therefore cannot account for the differential acidification seen in wild-type and Nramp1 −/− macrophages.
Phago-lysosomal Fusion in Wild-type and Nramp1−/− Macrophages.
In general, phagosomes are believed to acquire H+ pumps after fusion with late endosomes/lysosomes, which are a rich source of V-ATPases (9, 10, 32, 53). In permissive cells, live mycobacteria are known to impair phagosome fusion to V-ATPase–containing vesicles (11), likely accounting for the inability of the phagosomes to acidify normally. Therefore, it is conceivable that expression of Nramp1 restores the ability of the phagosomes to interact with late endosomes and lysosomes. This possibility was assessed by quantifying by immunofluorescence the insertion of LAMP-2, a marker of the membranes of late endosomes and lysosomes, into the phagosomal membrane of wild-type and Nramp1 −/− macrophages. Phagosomes induced by live M. bovis or latex particles were compared in both types of cells. As illustrated in Fig. 5, C, F, and H, both types of macrophages accumulated LAMP-2 on the membrane of phagosomes formed by internalization of latex beads. In contrast, recruitment of LAMP-2 to the membrane of M. bovis phagosomes was significantly impaired in Nramp1 −/− macrophages (Fig. 5, D, E, and G; overall 7% positive) when compared with wild-type cells analyzed under similar conditions (Fig. 5, A, B, and G; overall 26% positive). Therefore, increased fusion of mycobacterial phagosomes with late endosomes/lysosomes in wild-type macrophages may explain their increased V-ATPase activity and greater acidification.
Figure 5.

Delivery of LAMP-2 to phagosomes in wild-type and Nramp1 −/− cells. Macrophages obtained from Nramp1 +/+ (A–C) or Nramp1 −/− (D–F) mice were allowed to internalize live, fluoresceinated M. bovis or opsonized latex beads as specified. The cells were then fixed, permeabilized, and incubated with antibodies to LAMP-2, followed by Cy3-labeled secondary antibodies. Representative confocal fluorescence images are shown in A–F. The fraction of phagosomes stained by anti–LAMP-2 antibodies in M. bovis phagosomes is summarized in G (means ± SE of seven determinations), and the fraction of phagosomes stained in latex bead phagosomes is shown in H (seven determinations).
Delivery of V-ATPases to Mycobacterial Phagosomes from Nramp1−/− Macrophages.
Although increased fusion to late endosomal/lysosomal compartments provides an explanation for the increased V-ATPase activity in wild-type compared with Nramp1 −/− cells, it fails to account for the presence of V-ATPases and partial acidification seen in Nramp1-negative phagosomes containing live mycobacteria. It has previously been demonstrated that, although mycobacterial phagosomes of permissive cells do not fuse with late endosomes and lysosomes (11, 12, 54), they retain the capacity to interact with early and recycling endosomes (9, 13, 55). Importantly, in other systems these compartments can accumulate H+ in a concanamycin-sensitive manner (9, 53, 56), and may thus represent a source of V-ATPases for mycobacterial phagosomes in Nramp1 −/− mice. Therefore, we investigated whether fusion with early endosomes was responsible for delivery of V-ATPases to mycobacterial phagosomes in Nramp1 −/− mice. The results of these studies are summarized in Fig. 6. Macrophages from Nramp1 −/− mice were allowed to internalize fixable Texas red–dextran (a fluid phase marker) for 15 min, a period sufficient to direct this dye to the early endosomal compartment (57; Fig. 6 A). These macrophages were then fixed, and the subcellular distribution of the V-ATPase was detected with an antibody raised to its 39-kD subunit. The V-ATPases demonstrated a punctate, vesicular pattern suggestive of endo-membrane localization (Fig. 6 B). As shown in Fig. 6 C (superimposed images from A and B), there was significant, yet incomplete, colocalization of the 39-kD subunit with the fluid phase marker, suggesting the presence of V-ATPases in the early endosomal compartment. To assess the propensity of M. bovis-containing phagosomes to fuse with early endosomes, macrophages that had internalized mycobacteria were treated with Texas red–dextran as above (Fig. 6, D and E). In 48% of instances, the mycobacterial phagosome was found to be outlined by the endosomal marker, confirming that phagosome-endosome fusion had occurred in these cells (Fig. 6 F).
Figure 6.


Localization of V-ATPases in early endosomes and in phagosomes. (A–C) Macrophages obtained from Nramp1 −/− mice were allowed to internalize Texas red–labeled fixable (1 mg/ml) dextran for 15 min at 37°C. The cells were then fixed, permeabilized, and incubated with an affinity-purified polyclonal antibody against the 39-kD subunit of the V-ATPase, followed by an FITC-labeled secondary antibody. (A) Localization of Texas red–labeled dextran; (B) localization of the V-ATPase subunit; (C) areas of overlap between the dextran and the ATPase. (D–F) Macrophages obtained from Nramp1 −/− mice were allowed to internalize live, fluoresceinated M. bovis and were subsequently incubated with Texas red–labeled dextran for 15 min at 37°C. The cells were then fixed and visualized by Nomarski (D) and confocal immunofluorescence microscopy. (E) Localization of fluoresceinated bacteria; (F) distribution of Texas red–labeled dextran. Images are representative of at least five experiments of each kind. (G) Macrophages obtained from Nramp1 −/− mice were allowed to internalize FITC-labeled human holotransferrin (20 μg/ml), and pHe was measured using single-cell imaging. Where indicated, concanamycin (100 nM) was added. Data represent means ± SEM of three separate experiments.
Even though most of the material internalized by fluid phase endocytosis recycles back to the surface (58), and despite the use of comparatively short pulses, a small fraction of the fluorescence may have reached late endosomes. Therefore, it was essential to confirm that early/recycling endosomes of Nramp1 −/− mutant mice expressed functional V-ATPases. Therefore, macrophages from these mice were treated with FITC-labeled transferrin, which enters the cells via clathrin-dependent endocytosis and accumulates in the early/recycling endosomal compartment without ever reaching late endosomes (59, 60). The pH of this compartment was determined to be more acidic than the cytoplasm (6.6 ± 0.02, Fig. 6 G), in agreement with values determined in other cell types (60, 61). Upon addition of concanamycin, the acidification was rapidly dissipated (Fig. 6 G), confirming that the accumulation of H+ within early/ recycling endosomes is mediated by V-ATPases. These data suggest that mycobacterial phagosomes from Nramp1 −/− mice can acquire V-ATPases as a result of fusion with early endosomes.
Discussion
This study determined that phagosomes containing mycobacteria (M. bovis) in wild-type mice are significantly more acidic than those formed in macrophages from Nramp1 −/− mutant mice (Fig. 2). This difference in pH could conceivably account for host resistance to mycobacterial infection by several mechanisms. First, the enhanced acidification is expected to have a direct bacteriostatic effect, since mycobacteria grow optimally in the pH range 6.5–7.9 (43). In addition, the acidic milieu can activate microbicidal enzymes delivered to the phagosome during fusion with lysosomes (31). Also, the low pHp favors the protonation of nitrite to nitrous acid. Dismutation of the latter promotes the formation of reactive nitrogen species that are effective bactericidal agents (62). The latter can promote the formation of reactive species and is therefore a more effective bactericidal agent. Finally, the increased acidification would promote the dismutation of superoxide to hydrogen peroxide, which in turn functions as the substrate for myeloperoxidase delivered by phago-lysosomal fusion. Of note, the defect in phagosomal acidification noted in Nramp1 −/− mice was also detected in macrophages from C57BL/6 mice, a strain susceptible to M. bovis infection and that bears a naturally occurring mutant G169D allele at the Nramp1 locus that prevents expression of the protein (27, 28). Together, these data suggest that the role of Nramp1 in restricting the replication of intracellular pathogens such as mycobacteria is at least in part by alteration of pHp.
Several possible mechanisms could account for the differences in pHp between normal and Nramp1 −/− macrophages, and these were each investigated in turn. It has previously been speculated that the generation of intraphagosomal superoxide could consume a significant amount of H+ through dismutation during the course of hydrogen peroxide production. However, in macrophages this reaction is unlikely to contribute significantly to the higher pHp of Nramp1 −/− phagosomes, as inhibition of the NADPH oxidase had no discernible effect on pHp (Table 1). Likewise, phagosomal buffering power, counterion conductance and the rate of H+ (equivalent) leak were found to be comparable between the two types of cells, and are hence unlikely to explain the observed difference in the extent of acidification (Table 1). Rather, the lower pHp of wild-type macrophages was most likely due to increased rates of active proton accumulation in mycobacterial phagosomes. Support for this notion was obtained using cells treated with SLO (Fig. 4): selective permeabilization of the plasma membrane by this toxin resulted in reversible inhibition of V-ATPase activity, caused by depletion of cytosolic ATP/Mg. Accordingly, readdition of ATP/Mg2+ induced rapid phagosomal acidification at a rate significantly greater in wild-type mice than in the Nramp1 −/− counterparts. These data strongly support the conclusion that mycobacterial phagosomes in Nramp1-expressing macrophages are more acidic due to increased activity of V-ATPases.
The results of several laboratories, including our own (9, 11, 12), demonstrate a paucity of ATPases in mycobacterial phagosomes, as evaluated by immunological means. On the other hand, the functional determinations presented here indicate that concanamycin-sensitive proton pumping is in fact detectable in such phagosomes. To reconcile these observations, we considered a model whereby V-ATPases could induce acidification of the phagosomal lumen while being physically absent from the phagosomal membrane (9). Proton equivalents could be delivered to the phagosomal lumen by acid carrier vesicles that are derived from endosomes (or other acidic organelles), yet are themselves devoid of V-ATPases. Though unable to interact with lysosomes, the mycobacterial phagosome could nevertheless fuse with such vesicles and become partially acidic. Continued vesicular traffic to the phagosome would explain the sustained, concanamycin-sensitive acidification in a compartment that lacks ATPases. Although attractive, this model appears untenable, in view of the data obtained using SLO-permeabilized cells. Fusion of vesicular compartments within cells is known to require various cytosolic cofactors, including GTP, NSF, and SNAPs, which are expected to wash out of cells perforated by SLO. Nevertheless, in such cells phagosomal acidification was effectively reactivated by the mere readdition of Mg2+ and ATP, implying that vesicular fusion was not required for this process.
A more likely explanation for the observed acidification is therefore that V-ATPases are in fact present and functional on the phagosomal membrane itself, though at a comparatively low density that is difficult to detect by chemical or immunological means. What then is the likely source of such phagosomal ATPases? Recent reports by the laboratories of Russell (13) and Horowitz (55) demonstrated that mycobacterial phagosomes can fuse and remain in dynamic equilibrium with (components of) the early and recycling endosomal compartments of susceptible macrophages. Such compartments could in principle deliver the proton pumps to the phagosome, as suggested by our functional and immunofluorescence data. As shown in Fig. 6, early/recycling endosomes of Nramp1 −/− macrophages acidify in a concanamycin-sensitive manner, confirming that they possess V-ATPases. Partial colocalization with the 39-kD subunit of the pump supports this notion. Moreover, in confirmation of the earlier results (13, 16), early endosomes were found to be capable of fusing with phagosomes containing live mycobacteria (Fig. 6).
The enhanced V-ATPase activity and lower pHp observed in cells from mice expressing Nramp1 likely reflects the additional ability of these phagosomes to fuse readily with late endosomes and/or lysosomes (63; Fig. 5 G), a process that is seemingly impaired by the bacteria in the Nramp1 −/− mice. The observations can be most simply explained by postulating that the arrest in phagosomal maturation induced by internalized live mycobacteria in susceptible mice is circumvented by Nramp1 (9, 11, 54, 55). What is the mechanism whereby Nramp1 facilitates phago-lysosomal interaction? Nramp1, which resides in the membrane of lysosomes in quiescent cells, may itself act as a fusogen, in a manner analogous to SNARE or rab proteins, thus promoting interaction with the maturing phagosome (38, 64–66). No direct evidence in support of this mechanism has been provided and there is no homology between the structure of Nramp1 and those of proteins involved in vesicular docking and fusion. Rather, recent findings indicate that members of the Nramp family function as transporters for divalent cations (67). The Nramp2 protein is closely related to Nramp1, sharing a high degree of primary sequence homology (77% similarity) and very similar structural features (68). Recent electrophysiological studies in Xenopus oocytes demonstrated that Nramp2 functions as a divalent cation transporter, translocating Fe2+, Zn2+, Mn2+, Cd2+, and several other cations (69). Nramp2-mediated transport is pH dependent, and the divalent cations seem to be cotransported with H+ (69). In parallel, it was reported that mice bearing a mutation at the Nramp2 locus show severe microcytic anemia, leading to the conclusion that Nramp2 is the major transferrin-independent Fe2+ uptake system of the intestine (70). In addition, the yeast Saccharomyces cerevisiae has three Nramp proteins (Smf1, Smf2, Smf3) that share strong homology to the mammalian family members (47% similarity), one of which (Smf1) has been demonstrated to be a Mn2+ transporter (67). We have recently shown that, despite their large evolutionary distance, Nramp2 can complement a double smf1/2 yeast mutant and restore growth on EGTA and at alkaline pH (71). This conservation of function among distant Nramp family members strongly suggests that Nramp1 may also be a divalent cation transporter, acting at the lysosomal and/or phagosomal membrane. In this event, the transported substrate could conceivably affect phago-lysosome fusion. Interestingly, in vitro studies have recently shown that endosome– endosome fusion is dependent upon the availability of Zn2+ (72). Moreover, calcium is known to be essential for phago-lysosomal fusion in neutrophils (73), though not in macrophages (74). Inhibition of mycobacterial replication may also result from the Nramp1-mediated elimination of metals that are essential for their growth or for protection against cellular microbicidal agents. Regardless of the mechanism, it is important to point out that the observed effects of Nramp1 on phagosomal maturation are limited to phagosomes containing live mycobacteria, since the pH and fusogenic activity of phagosomes containing dead bacteria or latex beads appear to be similar in wild-type and Nramp1 −/− macrophages (Fig. 5). Therefore, it is reasonable to propose that Nramp1 antagonizes the effect of a factor produced by live mycobacteria, which interferes with phagosomal maturation.
In summary, this study shows that the unrestricted replication of intracellular pathogens in vivo caused by elimination of Nramp1 function in a Nramp1 −/− host is concomitant to impaired acidification of phagosomes containing live mycobacteria. The presence of the Nramp1 protein appears to bypass the arrest in phagosomal maturation typically observed in susceptible strains by enhancing the capacity of the mycobacterial phagosome to fuse to late endosomes and/or lysosomes, leading to increased V-ATPase activity at the phagosomal membrane, and consequently enhanced phagosomal acidification. Studies are now ongoing to identify the putative substrates translocated by Nramp1 that may modulate phagosomal maturation.
Abbreviations used in this paper
- BCG
Mycobacterium bovis strain bacillus Calmette-Guérin, substrain Montreal
- CCCP
carbonyl cyanide-m-chlorophenyl hydrazone
- LAMP
lysosomal-associated membrane protein
- NHS-CF
N-hydroxysuccinimidyl 5-(and 6-)-carboxyfluorescein
- Nramp
natural resistance-associated macrophage protein
- pHe
endosomal pH
- pHp
phagosomal pH
- SLO
streptolysin O
- V-ATPases
vacuolar-type proton ATPases
Footnotes
This research was funded by operating grants awarded to P. Gros from the National Institute of Allergy and Infectious Diseases (RO1 AI-35247-0352) and to S. Grinstein and O.D. Rotstein by the Medical Research Council of Canada and the National Sanatorium Association. S. Grinstein is cross-appointed to the Department of Biochemistry of the University of Toronto. P. Gros and S. Grinstein are International Scholars of the Howard Hughes Medical Institute. D.J. Hackam is the recipient of an Ethicon-Society of University Surgeons Surgical Research Award and a Medical Research Council of Canada Fellowship.
References
- 1.Young P. White House to expand response to infectious diseases. Am Soc Microbiol News. 1996;62:450–451. [Google Scholar]
- 2.Berkelman RL, Bryan RT, Osterholm MT, Leduc JW, Hughes JM. Infectious disease surveillance: a crumbling foundation. Science. 1996;264:368–370. doi: 10.1126/science.8153621. [DOI] [PubMed] [Google Scholar]
- 3.Bloom BR. Tuberculosis: back to a frightening future. Nature. 1992;358:538–540. doi: 10.1038/358538b0. [DOI] [PubMed] [Google Scholar]
- 4.Kaye K, Frieden TR. Tuberculosis control: the relevance of classic principles in an era of acquired immunodeficiency syndrome and multidrug resistance. Epidemiologic Rev. 1996;18:52–63. doi: 10.1093/oxfordjournals.epirev.a017916. [DOI] [PubMed] [Google Scholar]
- 5.Davies J. Inactivation of antibiotics and the dissemination of resistance genes. Science. 1994;264:375–382. doi: 10.1126/science.8153624. [DOI] [PubMed] [Google Scholar]
- 6.Neu HC. The crisis in antibiotic resistance. Science. 1992;257:1064–1072. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- 7.Heubner RE, Castro KG. The changing face of tuberculosis. Annu Rev Med. 1995;46:47–55. doi: 10.1146/annurev.med.46.1.47. [DOI] [PubMed] [Google Scholar]
- 8.Young DB, Duncan K. Prospects for new interventions in the treatment and prevention of Mycobacterial disease. Annu Rev Microbiol. 1997;49:641–673. doi: 10.1146/annurev.mi.49.100195.003233. [DOI] [PubMed] [Google Scholar]
- 9.Hackam DJ, Rotstein OD, Zhang WJ, Demaurex N, Woodside M, Tsai O, Grinstein S. Regulation of phagosomal acidification. Differential targeting of Na+/H+ exchangers, Na+/K+-ATPases, and vacuolar type H+-ATPases. J Biol Chem. 1997;272:29810–29820. doi: 10.1074/jbc.272.47.29810. [DOI] [PubMed] [Google Scholar]
- 10.Desjardins M, Huber LA, Parton RG, Griffiths G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J Cell Biol. 1994;124:677–688. doi: 10.1083/jcb.124.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, Allen RD, Gluck SL, Heuser J, Russell DG. Lack of acidification in mycobacteriumphagosomes produced by exclusion of the vesicular proton-ATPase. Science. 1994;263:678–681. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- 12.Russell DG, Dant J, Sturgill-Koszycki S. Mycobacterium avium- and Mycobacterium tuberculosis-containing vacuoles are dynamic, fusion-competent vesicles that are accessible to glycosphingolipids from the host cell plasmalemma. J Immunol. 1996;156:4764–4773. [PubMed] [Google Scholar]
- 13.Sturgill-Koszycki S, Shiable UE, Russell DG. Mycobacterium-containing phagosomes are accessible to early endosomes and reflect a transitional state in normal phagosome biogenesis. EMBO (Eur Mol Biol Organ) J. 1996;15:6960–6968. [PMC free article] [PubMed] [Google Scholar]
- 14.Stead WW. Genetics and resistance to tuberculosis: could resistance be enhanced by genetic engineering? . Ann Int Med. 1992;116:937–941. doi: 10.7326/0003-4819-116-11-937. [DOI] [PubMed] [Google Scholar]
- 15.Schurr E, Morgan K, Gros P, Skamene E. Genetics of leprosy. Am J Trop Med Hyg. 1991;44:4–11. doi: 10.4269/ajtmh.1991.44.4. [DOI] [PubMed] [Google Scholar]
- 16.McLeod R, Buschman E, Arbuckle LD, Skamene E. Immunogenetics in the analysis of resistance to intracellular pathogens. Curr Opin Immunol. 1995;7:539–552. doi: 10.1016/0952-7915(95)80100-6. [DOI] [PubMed] [Google Scholar]
- 17.Malo D, Skamene E. Genetic control of host resistance to infection. Trends Genet. 1994;10:365–371. doi: 10.1016/0168-9525(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 18.Appelberg R, Sarmento AM. The role of macrophage activation and of Bcg-encoded macrophage functions in the control of Mycobacterium aviuminfection in mice. Clin Exp Immunol. 1990;80:324–331. doi: 10.1111/j.1365-2249.1990.tb03288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gros P, Skamene E, Forget A. Genetic control of natural resistance to Mycobacterium bovis(BCG) in mice. J Immunol. 1981;127:2417–2421. [PubMed] [Google Scholar]
- 20.Vidal SM, Malo D, Vogan K, Skamene E, Gros P. Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. . Cell. 1993;73:469–485. doi: 10.1016/0092-8674(93)90135-d. [DOI] [PubMed] [Google Scholar]
- 21.Malo D, Vogan K, Vidal S, Hu J, Cellier M, Schurr E, Fuks A, Bumstead N, Morgan K, Gros P. Haplotype mapping and sequence analysis of the mouse Nrampgene predict susceptibility to infection with intracellular parasites. Genomics. 1994;23:51–61. doi: 10.1006/geno.1994.1458. [DOI] [PubMed] [Google Scholar]
- 22.Skamene E, Schurr E, Gros P. Infection genomics: Nramp1 as a major determinant of natural resistance to intracellular infections. Annu Rev Med. 1998;49:275–287. doi: 10.1146/annurev.med.49.1.275. [DOI] [PubMed] [Google Scholar]
- 23.Goto Y, Buschman E, Skamene E. Regulation of host resistance to Mycobacterium tuberculosis by the Bcggene. Immunogenetics. 1989;30:218–221. doi: 10.1007/BF02421210. [DOI] [PubMed] [Google Scholar]
- 24.Stach JL, Gros P, Forget A, Skamene E. Phenotypic expression of genetically-controlled natural resistance to Mycobacterium bovis(BCG) J Immunol. 1984;132:218–221. [PubMed] [Google Scholar]
- 25.Denis M, Forget A, Pelletier M, Gervais F, Skamene E. Killing of Mycobacterium smegmatisby macrophages from genetically susceptible and resistant mice. J Leukocyte Biol. 1990;47:25–30. doi: 10.1002/jlb.47.1.25. [DOI] [PubMed] [Google Scholar]
- 26.Vidal SM, Pinner E, Lepage P, Gauther S, Gros P. Natural resistance to intracellular infections: Nramp1 encodes a membrane phosphoglycoprotein absent in macrophages from susceptible (Nramp1 D169) mouse strains. J Immunol. 1996;157:3559–3568. [PubMed] [Google Scholar]
- 27.Vidal S, Tremblay ML, Govoni G, Gauthier G, Sebastiani G, Malo D, Skamene E, Olivier M, Jothy S, Gros P. The Ity/Lsh/Bcg locus: natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1gene. J Exp Med. 1995;182:655–666. doi: 10.1084/jem.182.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Govoni G, Vidal S, Gauthier G, Skamene E, Malo D, Gros P. The Bcg/Ity/Lsh locus: genetic transfer of resistance to infections in C57BL/6J mice transgenic for the Nramp1(Gly169) allele. Infect Immun. 1996;64:2923–2929. doi: 10.1128/iai.64.8.2923-2929.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abel L, Sanchez FO, Oberti J, Thuc NV, Van Hoa L, Lap VD, Skamene E, Lagrange PH, Schurr E. Susceptibility to leprosy is linked to the human NRAMP1 gene. J Infect Dis. 1998;177:133–145. doi: 10.1086/513830. [DOI] [PubMed] [Google Scholar]
- 30.Gruenheid S, Pinner E, Desjardins M, Gros P. Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J Exp Med. 1997;185:717–730. doi: 10.1084/jem.185.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenberg, S., and S.C. Silverstein. 1993. Phagocytosis. In Fundamental Immunology. W.E. Paul, editor. Raven Press, New York. 941–964.
- 32.Lukacs GL, Rotstein OD, Grinstein S. Phagosomal acidification is mediated by a vacuolar-type H+-ATPase in murine macrophages. J Biol Chem. 1990;265:21099–21107. [PubMed] [Google Scholar]
- 33.Verkruyse LA, Hormann SL. Lysosomal targeting of palmitoyl-protein thioesterase. J Biol Chem. 1996;271:15831–15836. doi: 10.1074/jbc.271.26.15831. [DOI] [PubMed] [Google Scholar]
- 34.Mortensen UH, Breddam K. A conserved glutamic acid bridge in serine carboxypeptidases, belonging to the alpha/beta hydrolase fold, acts as a pH-dependent protein-stabilizing element. Protein Sci. 1994;3:838–842. doi: 10.1002/pro.5560030513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosisphagosome and evidence that phagosomal maturation is inhibited. J Exp Med. 1995;181:257–270. doi: 10.1084/jem.181.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collette J, McGreer D, Crawford R, Chubb F, Sandin RD. Synthesis of some cyclic iodonium salts. J Am Chem Soc. 1956;78:3819–3820. [Google Scholar]
- 37.Chen JW, Murphy TL, Willingham MC, Pastan I, August JT. Identification of two lysosomal membrane glycoproteins. J Cell Biol. 1985;101:85–95. doi: 10.1083/jcb.101.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hackam DJ, Rotstein OD, Bennett MK, Klip A, Grinstein S, Manolson MF. Characterization and subcellular localization of target membrane soluble NSF attachment protein receptors (t-SNAREs) in macrophages. Syntaxins 2,3 and 4 are present on phagosomal membranes. J Immunol. 1996;156:4377–4383. [PubMed] [Google Scholar]
- 39.Forget A, Skamene E, Gros P, Miailhe AC, Turcotte R. Differences in response among inbred mouse strains to infection with small doses of Mycobacterium bovisBCG. Infect Immun. 1981;32:42–47. doi: 10.1128/iai.32.1.42-47.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hackam DJ, Rotstein OD, Zhang WJ, Schreiber AD, Grinstein S. Rho is required for the initiation of calcium transients and phagocytosis by Fc receptors in macrophages. J Exp Med. 1997;186:955–966. doi: 10.1084/jem.186.6.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grinstein S, Furuya W. Characterization of the amiloride sensitive Na+/H+antiport of human neutrophils. Am J Physiol. 1986;250:283–291. doi: 10.1152/ajpcell.1986.250.2.C283. [DOI] [PubMed] [Google Scholar]
- 42.Roos A, Boron WF. Intracellular pH. Physiol Rev. 1981;61:296–434. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- 43.Oh YK, Straubinger RM. Intracellular fate of Mycobacterium avium: use of dual label spectrofluorimetry to investigate the influence of bacterial viability and opsonization on phagosomal pH and phagosome-lysosome interaction. Infect Immun. 1996;64:319–325. doi: 10.1128/iai.64.1.319-325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teixeira HC, Munk ME, Kaufman SH. Frequencies of IFN gamma- and IL-4 producing cells during Mycobacterium bovisBCG infection in two genetically susceptible mouse strains: role of alpha/beta T cells and NK1.1 cells. Immunol Lett. 1995;46:15–19. doi: 10.1016/0165-2478(95)00009-t. [DOI] [PubMed] [Google Scholar]
- 45.Muroi M, Takasu A, Yamasaki M, Takatsuki A. Folimycin (concanamycin A), an inhibitor of V-type H(+)- ATPase, blocks cell surface expression of virus-envelope glycoproteins. Biochem Biophys Res Commun. 1993;193:999–1005. doi: 10.1006/bbrc.1993.1724. [DOI] [PubMed] [Google Scholar]
- 46.Cain CC, Sipe DM, Murphy RF. Regulation of endocytic pH by the Na+, K+-ATPase in living cells. Proc Natl Acad Sci USA. 1989;86:544–548. doi: 10.1073/pnas.86.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuchs R, Schmid S, Mellman I. A possible role for Na+, K+-ATPase in regulating ATP-dependent endosome acidification. Proc Natl Acad Sci USA. 1989;86:539–543. doi: 10.1073/pnas.86.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Segal AW, Geisow M, Garcia R, Harper A, Miller R. The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature. 1981;290:406–409. doi: 10.1038/290406a0. [DOI] [PubMed] [Google Scholar]
- 49.Rotrosen, D. 1992. The respiratory burst oxidase. In Inflammation: Basic Principles and Clinical Correlates. J.I. Gallin, I.M. Goldstein, and R. Synderman, editors. Raven Press, New York. 589–602.
- 50.Galli T, Chilcote T, Mundigl O, Binz T, Niemann H, De Camilli P. Tetanus toxin-mediated cleavage of cellubrevin impairs exocytosis of transferrin receptor containing vesicles in CHO cells. J Cell Biol. 1994;125:1015–1024. doi: 10.1083/jcb.125.5.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Funato K, Beron W, Yang CZ, Mukhopadhyay A, Stahl PD. Reconstitution of phagosome-lysosome fusion in streptolysin-O permeabilized cells. J Biol Chem. 1997;272:16147–16151. doi: 10.1074/jbc.272.26.16147. [DOI] [PubMed] [Google Scholar]
- 52.Nanda A, Grinstein S. Protein kinase C activates an H+ (equivalent) conductance in the plasma membrane of human neutrophils. Proc Natl Acad Sci USA. 1991;88:10816–10820. doi: 10.1073/pnas.88.23.10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forgac M. Structure and function of vacuolar class of ATP-driven proton pumps. Physiol Rev. 1989;69:765–796. doi: 10.1152/physrev.1989.69.3.765. [DOI] [PubMed] [Google Scholar]
- 54.Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem. 1997;272:13326–13331. doi: 10.1074/jbc.272.20.13326. [DOI] [PubMed] [Google Scholar]
- 55.Clemens DL, Horwitz MA. The Mycobacterium tuberculosisphagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J Exp Med. 1996;184:1349–1355. doi: 10.1084/jem.184.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gluck SL. The vacuolar H+-ATPases: versatile proton pumps participating in constitutive and specialized functions of eukaryotic cells. Int Rev Cytol. 1993;137:105–137. [PubMed] [Google Scholar]
- 57.Mellman I. Membranes and sorting. Curr Opin Cell Biol. 1996;8:497–498. doi: 10.1016/s0955-0674(96)80026-3. [DOI] [PubMed] [Google Scholar]
- 58.Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 59.Dautry-Varsat A, Ciechanover AA, Lodish HF. pH and the recycling of transferrin during receptor mediated endocytosis. Proc Natl Acad Sci USA. 1983;80:2258–2262. doi: 10.1073/pnas.80.8.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamashiro DJ, Tycko B, Fluss SR, Maxfield FR. Segregation of transferrin to a mildly acidic (pH 6.5) para-Golgi compartment in the recycling pathway. Cell. 1984;37:789–800. doi: 10.1016/0092-8674(84)90414-8. [DOI] [PubMed] [Google Scholar]
- 61.Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis. Physiol Rev. 1997;77:759–804. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- 62.Stuehr DJ, Nathan CF. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Chastellier C, Frehel C, Offredo C, Skamene E. Implication of phagosome-lysosome fusion in restriction of Mycobacterium aviumgrowth in bone marrow macrophages from genetically resistant mice. Infect Immun. 1993;61:3775–3784. doi: 10.1128/iai.61.9.3775-3784.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sudhof TC, De Camilli P, Niemann H, Jahn R. Membrane fusion machinery: insights from synaptic proteins. Cell. 1993;75:1–4. [PubMed] [Google Scholar]
- 65.Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 66.Novick P, Brennwald B. Friends and family: the role of the Rab GTPases in vesicular traffic. Cell. 1993;75:597–601. doi: 10.1016/0092-8674(93)90478-9. [DOI] [PubMed] [Google Scholar]
- 67.Supek F, Supekova L, Nelson H, Nelson N. A yeast manganese transporter related to the macrophage protein involved in conferring resistance to mycobacteria. Proc Natl Acad Sci USA. 1996;93:5105–5110. doi: 10.1073/pnas.93.10.5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gruenheid S, Cellier M, Vidal S, Gros P. Identification and characterization of a second mouse Nramp gene. Genomics. 1995;25:514–525. doi: 10.1016/0888-7543(95)80053-o. [DOI] [PubMed] [Google Scholar]
- 69.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 70.Fleming MD, Trenor CC, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC. Microcytic anemia mice have a mutation in Nramp2, a candidate iron transporter. Nat Genet. 1997;16:383–386. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- 71.Pinner E, Gruenheid S, Raymond M, Gros P. Functional complementation of the yeast divalent cation transporter family SMF by NRAMP2,a member of the mammalian natural resistance associated macrophage protein family. J Biol Chem. 1997;272:28933–28938. doi: 10.1074/jbc.272.46.28933. [DOI] [PubMed] [Google Scholar]
- 72.Aballay A, Sarrouf MN, Colombo MI, Stahl PD, Mayorga LS. Zn2+depletion blocks endosome fusion. Biochem J. 1995;312:919–923. doi: 10.1042/bj3120919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stendahl O, Krause K-H, Krischer J, Jerstrom P, Theler J-M, Clark RA, Carpentier J-L, Lew DP. Redistribution of intracellular Ca2+stores during phagocytosis in human neutrophils. Science. 1994;265:1439–1441. doi: 10.1126/science.8073285. [DOI] [PubMed] [Google Scholar]
- 74.Zimmerli S, Majeed M, Gustavsson M, Stendahl O, Sanan DA, Ernst JD. Phagosome–lysosome fusion is a calcium-independent event in macrophages. J Cell Biol. 1996;132:49–61. doi: 10.1083/jcb.132.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]