

Abstract
Many tumor-associated antigens are nonmutated, poorly immunogenic tissue differentiation antigens. Their weak immunogenicity may be due to “self”-tolerance. To induce autoreactive T cells, we studied immune responses to gp100/pmel 17, an antigen naturally expressed by both normal melanocytes and melanoma cells. Although a recombinant vaccinia virus (rVV) encoding the mouse homologue of gp100 was nonimmunogenic, immunization of normal C57BL/6 mice with the rVV encoding the human gp100 elicited a specific CD8+ T cell response. These lymphocytes were cross-reactive with mgp100 in vitro and treated established B16 melanoma upon adoptive transfer. To understand the mechanism of the greater immunogenicity of the human version of gp100, we characterized a 9-amino acid (AA) epitope, restricted by H-2Db, that was recognized by the T cells. The ability to induce specific T cells with human but not mouse gp100 resulted from differences within the major histocompatibility complex (MHC) class I–restricted epitope and not from differences elsewhere in the molecule, as was evidenced by experiments in which mice were immunized with rVV containing minigenes encoding these epitopes. Although the human (hgp10025–33) and mouse (mgp10025–33) epitopes were homologous, differences in the three NH2-terminal AAs resulted in a 2-log increase in the ability of the human peptide to stabilize “empty” Db on RMA-S cells and a 3-log increase in its ability to trigger interferon γ release by T cells. Thus, the fortuitous existence of a peptide homologue with significantly greater avidity for MHC class I resulted in the generation of self-reactive T cells. High-affinity, altered peptide ligands might be useful in the rational design of recombinant and synthetic vaccines that target tissue differentiation antigens expressed by tumors.
Keywords: melanoma, tumor-associated antigen, gp100, xenoimmunization, CD8+ T lymphocyte
The recent cloning of tumor antigens recognized by T cells has caused considerable interest in the development of antigen-specific cancer vaccines (1–3). Some antigens are especially attractive candidates for use in vaccines due to their shared nature between individuals, including the melanocyte differentiation antigens (MDA)1 gp100, melanoma antigen recognized by T lymphocytes (MART)-1, and tyrosinase (2), as well as several proteins in the MAGE family (1). However, as indicated by results from clinical trials thus far, inducing therapeutic T cells to these antigens has been difficult. One reason for the apparent hyporesponsiveness of the human immune system to many tumor antigens may be that they are normal, nonmutated “self”- proteins, expressed on normal tissues as well as on tumor cells. An incomplete understanding of the processes of central and peripheral tolerance has hampered the development of successful cancer vaccines targeting these autoantigens, limiting the use of the growing number of candidate tumor antigens.
The absence of an immune response to a defined autoantigen can be due to negative selection of self-antigen– specific T cells during maturation in the thymus, termed “central” tolerance (4). A low level of autoreactivity is required for positive selection in the thymus (5, 6).
T cells with low reactivity to autoantigens thus persist. Mature T lymphocytes with reactivity to self-antigens may remain in a functionally tolerant state, termed “ignorance”, if they do not traffic to antigen-bearing cells, or if the target antigen is not processed and presented to a level that can trigger the specific TCR. Mature self-reactive T cells that encounter antigen on normal tissues in the absence of an activating costimulatory microenvironment can be functionally eliminated by anergization or physically by deletion, thus effecting extrathymic or peripheral tolerance (7, 8).
The mechanisms of breaking tolerance to self-antigens may be relevant for the induction of immune responses to tissue differentiation antigens expressed by tumors. To study requirements for activation of self-reactive, tumor-specific T cells specific for a naturally expressed antigen, we targeted gp100, a normal, nonmutated MDA. In humans, gp100 is expressed both by normal melanocytes and the majority of malignant melanomas tested (9). CD8+ T lymphocytes with reactivity to gp100 have been detected in patients with metastatic melanoma. The mouse homologue for gp100, also known as pmel 17, has been cloned previously (10, 11), and like its human counterpart is normally expressed in melanocytes in an unmanipulated C57BL/6 mouse as well as in mouse melanomas. We sought to determine the requirements to break T cell tolerance to a naturally expressed self-antigen. Furthermore, we evaluated the functional characteristics of autoreactive T cells in the recognition and destruction of a spontaneous mouse melanoma, B16, in vivo.
Materials and Methods
Animals and Cell Lines.
Female C57BL/6 (H-2b) mice, 6–10 wk old, obtained from Frederick Cancer Research Center (Frederick, MD) and maintained in a barrier facility, were used for all experiments. EL-4 thymoma (H-2b) and the derived β-galactosidase (β-gal)–transfected clone E22 have been described previously (12). B16 (H-2b), hereafter named B16.WT, is a spontaneous murine melanoma expressing gp100, MART-1, tyrosinase, tyrosinase-related protein (TRP)-1, and TRP-2 by FACS® and Western blot analysis (data not shown). B16.B7-1 is a hyperpigmented clone of B16.WT that was stably transfected using a Moloney mouse leukemia virus encoding the gene for B7-1 driven by a LTR promoter. JB/MS is a pigmented, chemically induced melanoma expressing gp100, provided by Dr. Vincent Hearing (National Cancer Institute, NIH, Bethesda, MD). 293Kb and 293KbDb are highly transfectable human renal 293 cells stably transfected with Kb and Kb plus Db, respectively. RMA/S (H-2b) is a cell line deficient in endogenous peptide loading (13). EL-4, B16.WT, RMA/S, MCA205, and MC38, a C57BL/6-derived colon carcinoma, were maintained in complete medium (CM; RPMI 1640 with 10% heat-inactivated fetal bovine serum [FBS; Biofluids, Rockville, MD], 0.03% l-glutamine, 100 μg/ml streptomycin, 100 μg/ml penicillin, and 50 μg/ml gentamycin sulfate [NIH Media Center, Bethesda, MD]). B16.B7-1 and E22 were maintained in CM with 400 μg/ml of bioactive G418. 293Kb and 293KbDb were maintained in DMEM with 10% heat-inactivated FBS, 0.03% l-glutamine, 100 μg/ml streptomycin, 100 μg/ml penicillin, 50 μg/ml gentamycin sulfate, and 400 μg/ml of bioactive G418.
Peptides.
All synthetic peptides were synthesized using regular F-MOC chemistry. The following synthetic peptides were all synthesized by Peptide Technologies (Washington, D.C.) to a purity >99% by HPLC and amino acid (AA) analysis: KVPRNQDWL, spanning AAs 25–33 of human (h)gp100; EGSRNQDWL, spanning AAs 25–33 of mouse (m)gp100; ASNENMETM, spanning AAs 366–374 of the nucleoprotein of influenza A (NP); DAPIYTNV, spanning AAs 96–103 of Escherichia coli β-gal (12); and SIINFEKL, spanning AAs 257–264 of chicken OVA. The NP, hgp100, and mgp100 peptides are restricted by H-2Db, and the β-gal and OVA peptides by H-2Kb.
Recombinant Viruses.
All recombinant vaccinia viruses (rVVs) used in this study were generated by insertion of the foreign genes by homologous recombination and subsequent purification of recombinant progeny as described by Earl et al. (14). rVVs encoding the human and mouse gp100 epitopes as minigenes were constructed using a recombination plasmid, pKT1401, containing an early promoter driving the signal sequence of the adenovirus E3/19k protein followed by an additional Ala using code GCA, resulting in a putative signal peptide cleavage site, and fused to oligonucleotides encoding the epitope sequences for mgp100, hgp100, or β-galactosidase as described above. pKT1401 was recombined into the VV locus encoding the small subunit of viral ribonucleotide reductase. Correct integration was checked by PCR-based viral genome analysis using primers flanking the viral locus encoding the small subunit of ribonucleotide reductase (12). rVVmgp100, rVVmMART-1, rVVmTyr, rVVmTRP-1, and rVVmTRP-2 were based on the plasmid pSC65, in which the completely synthetic early/late promoter pSE/liter drives expression of the antigen and the early/late promoter p7.5E/liter drives expression of the LacZ gene (15). The rVVSE/Lβ-gal has been described previously (15, 16). Cloning of the genes for mgp100 and mMART-1/Melan-A has been described previously (11), and the cDNAs for hgp100, mTyr, mTRP-1, and mTRP-2 were gifts from Dr. Y. Kawakami (Surgery Branch, NIH; reference 9), Dr. H. Yamamoto (Tohoku University, Sendai, Japan; reference 17), Dr. S. Shibahara (Friedrich Miescher Institut, Basel, Switzerland; reference 18), and Dr. V. Hearing (Laboratory of Cell Biology, NCI, NIH; reference 19), respectively. The rVVhgp100, recombinant fowlpox virus (rFPV)hgp100, rFPVmgp100, and rFPVβ-gal were provided by Therion Biologics Corp. (Boston, MA). Plasmid DNA constructs (pDNA) were based on the pCDNA3 backbone, and encoded hgp100, mgp100, or β-galactosidase under the control of the CMV promoter. Expression of rVV, rFPV, and pDNA were confirmed by immunostaining (20) as well as by Western blot of transfected and infected cells using antisera (21, 22) provided by Dr. V. Hearing. Recombinant adenoviruses were provided by Dr. Bruce Roberts (Genzyme Corp., Framingham, MA), and encoded the genes for hgp100, mgp100, or β-galactosidase under the control of the CMV promoter (11).
Generation of gp100-reactive T Cell Line.
Mice were vaccinated twice at 3-wk intervals by a hand-held helium-driven device (Geniva Inc., Middletown, WI) with 1 μg of hgp100 DNA (23), and 3 wk after the second vaccination splenocytes were cultured with rVVhgp100-infected dendritic cells (DCs), generated as previously described (16) in CM with 30 IU/ml rhIL-2 (a gift from Chiron Corp., Emeryville, CA) for 7 d, and were subsequently restimulated every 7–10 d with 2 × 105 B16.B7-1 (60,000 rad) and 5 × 106 irradiated splenocytes (1,500 rad) in CM with 30 IU/ml rhIL-2. T cell clones were derived by limiting dilution using 0.3 T cells per well cocultured with 4 × 105 irradiated splenocytes and 2 × 104 irradiated B16.B7-1 cells in U-bottomed 96-well plates. The generation of the β-gal reactive cell line has been described previously (12). All T cells were used between 5 and 10 d after restimulation. For peptide-induced T cells, mice were vaccinated as specified in figure legends. Spleens were harvested on day 21 after the last vaccination, separated into single cell suspension, and cultured in T-25 flasks in 15 ml CM at 5 × 107 cells per flask. Peptide was added to a final concentration of 1 μM. After 6 d of culture, cells were washed in CM and used in a cytokine release assay.
Cytokine Release Assay.
To determine cytokine release, 5 × 104 target cells per well were added to effector T cells in round-bottomed 96-well plates at an E/T ratio of 1:1 for established T cell lines and 5:1 for secondary cultures, and were incubated for 24 h in CM. Supernatants were collected and tested using the mIFN-γ ELISA kit (Endogen, Cambridge, MA) or the mGM-CSF ELISA kit (R&D Systems, Minneapolis, MN) according to manufacturer's protocol. rVV-, rFPV- and rAd-infected targets were infected at multiplicities of infection of >10, >10, and >100, respectively, incubated for 2 h, and then washed twice in CM. Peptide-pulsing was performed at 1 μM in CM for 2 h at 37°C and followed by two washes in CM. Transfected targets were prepared using Lipofectamine (GIBCO BRL, Gaithersburg, MD) following manufacturer's guidelines.
MHC Class I Stabilization Assay.
RMA/S cells were incubated at room temperature for 24 h, pulsed with peptide at the indicated concentrations for 45 h at room temperature, and then incubated at 37°C for 4 h to allow turnover of “empty” MHC class I molecules (13). Cells were then stained with FITC-conjugated Db-specific mAb KH95 (PharMingen, San Diego, CA) for 1 h at 4°C and staining was asserted using a FACScan® (Becton Dickinson, Sunnyvale, CA).
Adoptive Transfer Treatment.
For adoptive transfer experiments, mice were injected with 2 × 105 B16 intravenously on day 0 followed by intravenous injection of the indicated number of T cells on day 3. rhIL-2 (3 × 105 IU) was given twice daily intraperitoneally for 5 d starting immediately after adoptive transfer.
Alternatively, mice were injected intravenously with the indicated number of T cells, followed 5 d later by 2 × 105 B16 intravenously. Mice were killed on day 17 after tumor injection and pulmonary nodules were enumerated. All in vivo experiments were performed in a blinded, randomized fashion.
Results
gp100 Is Recognized by Mouse T Cells.
To generate gp100-specific T cells, mice were immunized with mgp100 encoded in plasmid DNA or rVV, two vectors with proven efficacy in the generation of specific T cells (3, 23, 24). Splenocytes from immunized mice were restimulated in vitro using syngeneic DCs infected with appropriate rVV, rFPV, or rAd, and subsequent restimulations were performed with irradiated splenocytes and irradiated B16.B7-1, a clone of B16 retrovirally transduced to express the costimulatory molecule CD80 (B7-1). None of the cultures exhibited gp100-specific reactivity when tested by IFN-γ release against gp100-positive targets (data not shown). Since xenogeneic antigen can, in some instances, induce immune reactivity where the autologous antigen failed (25–27), mice were immunized with pDNA encoding hgp100.
A CD4−CD8+ lytic T lymphocyte line was generated by gene-gun administration of pDNA encoding hgp100 followed by restimulation of splenocytes ex vivo with DCs that had been infected with rVVhgp100. After two subsequent in vitro restimulations with B16.B7-1 melanoma, the T cells recognized B16 melanoma, as well as the immortalized normal melanocyte line Melan-A, with a high degree of specificity (Fig. 1). The EL-4 thymoma and the MC38 sarcoma were not recognized. However, recognition could be conferred upon 293 KbDb cells after infection with rVV encoding either mgp100 or hgp100, but not after infection with rVV containing any of the other known MDAs, tyrosinase, TRP-1, or TRP-2.
Figure 1.

B16 melanoma-reactive T cells specifically recognize nonmutated gp100. Splenocytes from hgp100-immunized mice were cultured as described in Materials and Methods, and cocultured for 24 h with various targets shown on the ordinate, including B16 melanoma, the immortalized normal melanocyte line Melan-A, and human 293 kidney cells expressing the mouse restriction elements H-2Kb and H-2Db infected with rVV encoding mouse melanocyte differentiation antigens. Supernatants were assayed for IFN-γ by ELISA. Specific IFN-γ release was detected against targets expressing mgp100 or hgp100.
Antitumor Activity of gp100-specific T Cells.
We sought to evaluate the relevance of mgp100 as a tumor rejection antigen. To exclude the potential influence of T cells with a specificity other than gp100, the bulk T cell line was cloned by limiting dilution. 12 clones were evaluated and each had reactivities identical to the bulk T cell line shown in Fig. 1. The gp100-reactive T cell clone (clone no. 9) was tested in vitro (Fig. 2 A) and in vivo (Fig. 2 B). Mice bearing 3-d-old B16 pulmonary nodules were infused with clone no. 9 followed by rIL-2, resulting in a dramatic tumor destruction (P <0.0001, 4 × 106 β-gal–specific T cells + rhIL-2 versus 4 × 106 clone no. 9 T cells + rhIL-2), whereas treatment was ineffective using control β-gal–reactive T cells or rIL-2 alone (P >0.5 versus no treatment) (Fig. 2 B). Separate gp100-reactive T cells clones were comparable in their ability to reject B16 in vivo (data not shown). These results suggest that gp100 functions as a true tumor rejection antigen in established murine melanoma.
Figure 2.
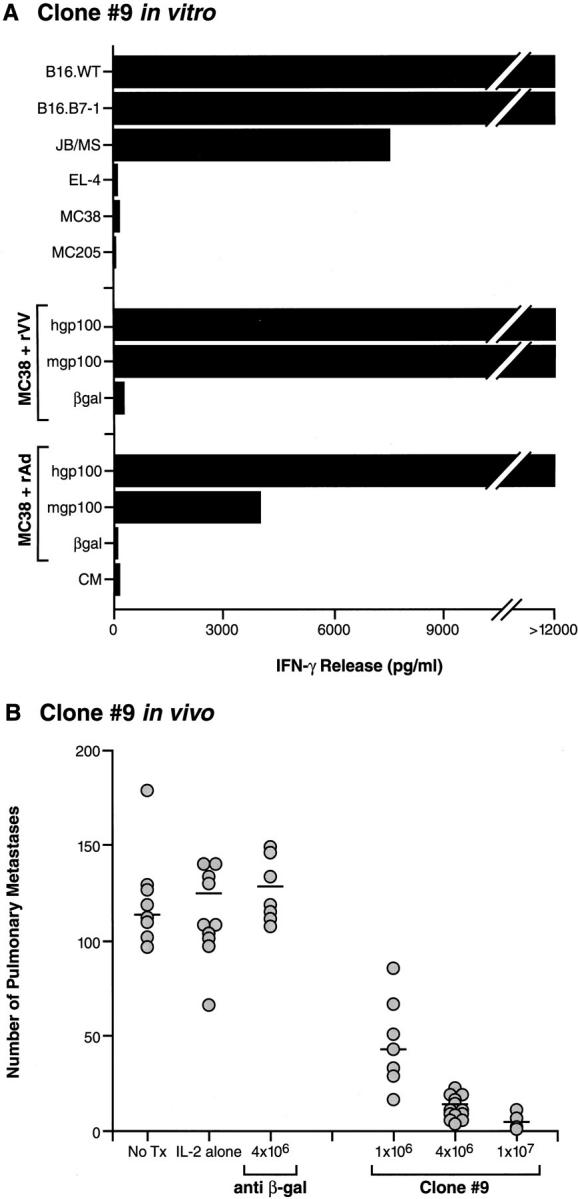
gp100 is a tumor rejection antigen. (A) A clone of bulk gp100-reactive T cells, clone no. 9, was established by limiting dilution at 0.3 cells/well. Upon coculture, clone no. 9 specifically recognized B16.WT, B16.B7-1, and JB/MS melanomas, but not control H-2b tumors EL-4, MCA205, and MC38. Clone no. 9 also recognized MC38 sarcoma infected with either rVV or rAd encoding hgp100 or mgp100, but not control rVV or rAd. (B ) Specific in vivo reactivity of gp100-specific T cell clone no. 9 was evaluated by injecting mice with 2 × 105 B16.WT cells intravenously. 3 d later, the specified numbers of clone no. 9 T cells were adoptively transferred. 3 × 105 IU rhIL-2 was then given intraperitoneally two times daily for 5 d. Mice were killed 18 d after tumor inoculation and pulmonary nodules were enumerated. Transfer of T cell clone no. 9, but not β-gal–specific T cells or rhIL-2 alone, dramatically reduced the number of pulmonary nodules in a dose-dependent manner, indicating that gp100 can function as a tumor rejection antigen in vivo. Additional independent clones yielded similar results in repeat experiments.
Identification of a Cross-reactive, MHC Class I–restricted Epitope in gp100.
We sought to understand why T cells could only be induced by the xenogeneic, human form of gp100 and not the self sequence. To determine whether the difference in immunogenicity resided in the actual peptides from gp100 that were recognized by T cells, or in differences in sequences outside of those recognized, we characterized the epitopes recognized by the T cell clone. To identify the MHC class I molecule that restricted gp100 recognition, we used the human renal cell carcinoma line, 293, stably transfected with the mouse restriction elements, Kb and Db. Both lines were transiently transfected with plasmids encoding either mouse or human gp100, or with a control plasmid encoding NP. Only 293 KbDb cells (and not 293 Kb) cells expressing mgp100 or hgp100 triggered the release of IFN-γ from gp100-reactive T cells, suggesting that Db was the dominant and perhaps only restriction element for gp100 recognition (Fig. 3 A).
Figure 3.

B16-specific T cells recognize a Db-restricted 9-AA peptide from gp100. In experiment 1, human 293 kidney cells stably transfected with Kb and Db were transfected with human and mouse gp100 pDNA and cocultured for 24 h with gp100-specific T cells. Supernatants were assayed for IFN-γ by ELISA. Specific IFN-γ release was detected when T cells were cocultured with transfected 293 KbDb, but not 293 Kb, indicating that gp100 recognition was predominantly Db-restricted. In experiment 2, human 293 kidney cells stably transfected with Kb and Db were transfected with 3′ exonuclease truncated constructs of hgp100 pDNA and cocultured for 24 h with gp100-reactive T cells. Supernatants were assayed for IFN-γ by ELISA. Specific IFN-γ release was detected with each truncated construct, including the 300-bp cDNA fragment, indicating that a specific immunogenic peptide was located in the 100 NH2-terminal AAs of the gp100 molecule. In experiment 3, peptides corresponding to the binding motif for Db were identified in the first 100 residues of the hgp100 molecule and synthesized together with the corresponding sequences from mgp100. Individual peptides were added to gp100-reactive T cells, and supernatants were assayed for GM-CSF by ELISA. Specific GM-CSF release was detected with peptide pair gp10025–33, suggesting it was the 9-AA epitope responsible for the gp100-reactivity of the T cells.
At 626 AAs in length, many possible epitopes in gp100 could be recognized by gp100-reactive T cells. To reduce the number of candidate peptides, a set of progressively shorter versions of the gene encoding hgp100 was used (28). These shortened forms were generated by 3′ exonuclease digestion or PCR amplification of the original hgp100 cDNA. Fragments were then transfected into 293 KbDb cells that were used as targets for T cell recognition (Fig. 3 B). Even the shortest fragment of the hgp100 cDNA, with a length of 300 bp by gel electrophoresis, conferred a high degree of recognition upon the 293 transfectants, implying that a major epitope was located in the first 100 AAs of the molecule.
To identify the sequence of the epitope, 9-AA-long peptides were evaluated for their potential to bind to Db using a computer-generated epitope forecast, based on previously published peptide binding data, that is designed to predict binding affinity for a variety of human and mouse MHC class I alleles (29, 30; and http://bimas.dcrt.nih.gov/molbio/hla_bind). Based on these predictions, sets of synthetic peptides were made from both hgp100 and mgp100, which although similar are not identical. When the peptides were added to cultures of gp100-reactive T cells, one peptide pair, gp10025–33, was recognized with a high degree of specificity in a GM-CSF release assay (Fig. 3 C).
T Cell Reactivity to gp100 Can Be Induced by Xenoimmunization.
With the identification of a defined peptide epitope, we explored the immunological mechanism underlying the disparate ability of mgp100 and hgp100 to induce gp100-specific T cells. Mice were vaccinated with rVVmgp100, encoding mgp100, or rVVhgp100, encoding hgp100, and splenocytes were isolated after 3 wk and stimulated for 6 d in vitro with synthetic peptides corresponding to hgp10025–33, mgp10025–33, or OVA257–264, and then tested in a GM-CSF release assay (Table 1, first two columns). Splenocytes from mice vaccinated with rVVmgp100 and restimulated with mgp10025–33 or hgp10025–33 peptide failed to develop peptide reactivity. Conversely, splenocytes from mice vaccinated with hgp100 and restimulated with either mgp10025–33 or hgp10025–33 peptide developed reactivity to both peptides. Similar results were obtained using splenocytes from mice vaccinated with recombinant fowlpoxvirus or plasmid DNA (data not shown). These observations indicated that T cells recognizing mgp100 could be induced exclusively by xenoimmunization with the hgp100 molecule.
Table 1.
IFN-γ Release by Peptide-stimulated Splenocytes from gp100-immunized Mice
| Immunization | rVVhgp100 |
rVVmgp100 | rVVEShgp10025–33 | rVVESmgp10025–33 | Naive | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In vitrostimulation | ||||||||||||||||||||||||||||
| hgp | mgp | OVA | hgp | mgp | OVA | hgp | mgp | OVA | hgp | mgp | OVA | hgp | mgp | |||||||||||||||
| Peptide targets | ||||||||||||||||||||||||||||
| None | 264 | 584 | 236 | 137 | 0 | 51 | 279 | 428 | 5,297 | 862 | 1,082 | 1,324 | 215 | 456 | ||||||||||||||
| hgp | 15,120 | 20,751 | 250 | 137 | 1 | 0 | 15,945 | 11,197 | 1,267 | 1,295 | 1,580 | 819 | 229 | 428 | ||||||||||||||
| mgp | 2,567 | 19,307 | 257 | 94 | 58 | 37 | 940 | 11,268 | 1,438 | 911 | 847 | 1,629 | 215 | 527 | ||||||||||||||
| OVA | 165 | 343 | 144 | 137 | 37 | 208 | 172 | 449 | 300 | 648 | 698 | 2,141 | 257 | 478 | ||||||||||||||
Mice were vaccinated with indicated rVVs, and splenocytes were isolated 3 wk later and restimulated for 6 d with 1 μg/ml peptide. Subsequent stimulation of cultured cells with hgp10025–33 or mgp10025–33 peptide resulted in IFN-γ release only by splenocytes from mice immunized with rVVhgp100 or rVVEShgp10025–33. hgp, hgp100 AA 25–33 (KVPRNQDWL); mgp, mgp100 AA 25–33 (EGSRNQDWL); OVA, ovalbumin, AA 257–264 (SIINFEKL). Four repeat experiments yielded similar results; numbers indicate IFN-γ (pg/ml) secreted by 2 × 105 CTLs in 24 h; numbers in bold indicate secretion >3-fold over control peptide.
Increased Immunogenicity of hgp100 Is Intrinsic to the MHC Class I–restricted Epitope.
Several mechanisms could account for the apparent immunological unresponsiveness to mgp100 and the ability of hgp100 to break it. Nonhomologous regions of the full-length hgp100 (which is 76% identical to mgp100 at the AA level; reference 11) could result in intramolecular epitope-spreading (31, 32) or facilitate antibody-mediated antigen capture by APCs (26, 33– 35). Alternatively, sequence differences in the relevant epitopes or their flanking sequences could result in differential proteolytic cleavage or transporter associated with antigen processing (TAP)–mediated transport across the endoplasmic reticulum (ER) membrane (36). To explore these possibilities, we constructed a series of rVV-containing minigenes encoding the relevant 9-AA T cell epitopes preceded by the E3/19K adenoviral ER-insertion signal sequence (ES), previously shown to result in TAP-independent transport of antigenic peptides (37), and followed by a double stop codon. These constructs eliminated differences in flanking sequences and other nonhomologous regions of the molecule.
Mice were vaccinated with rVVESmgp100, rVVEShgp10025–33, or rVVESβ-gal96–103, encoding the 9-AA mgp10025–33, hgp10025–33, and β-gal96–103 peptides, respectively (38). Splenocytes were cultured for 6 d with mgp10025–33 or hgp10025–33 peptides then tested for specificity in a cytokine release assay (Table 1). Reactivities were similar to those obtained with rVV encoding full-length gp100 molecules: immunization with the mouse minigene gp100 construct failed to induce T cells, whereas immunization with the hgp100 construct induced T cells that were cross-reactive with both mgp10025–33 and hgp10025–33 peptides. These results were not consistent with a major role for differential peptide processing and transport, antibody-facilitated antigen presentation, or intramolecular immunodominance in the differential in vivo immunogenicity of mgp100 and hgp100.
MHC Class I–Peptide Interactions and the Immunogenicity of gp100.
To evaluate the relative avidity of the gp100-specific T cells for the mgp10025–33 and hgp10025–33 epitopes, we pulsed the peptides onto EL-4 cells (H-2b) (Fig. 4). A striking difference in the relative T cell activities to the mgp10025–33 and hgp10025–33 peptides was observed. The mgp10025–33 peptide was recognized at concentrations as low as 10−9 M, with half-maximal recognition occurring at about 10−8 M, whereas the hgp10025–33 peptide was recognized at concentrations as low as 10−11 M. Alanine-substitutions at positions 1, 2, or 3 in either the mouse or human versions of the synthetic peptides did not substantially alter recognition by gp100-reactive T cells (Fig. 5 A). Indeed, a triple-substituted peptide, AAARNQDWL, fully retained the ability to sensitize EL-4 for recognition. (Fig. 5 B). The first three AAs could even be deleted and a substantial degree of recognition remained (Fig. 5 B). However, substitution of any of the AAs at positions 4 through 9 abrogated recognition of both the hgp10025–33 and mgp10025–33 peptide variants. To determine the involvement of MHC class I binding affinity, an MHC class I stabilization assay was done on RMA/S cells, which lack activity of the TAP transporters (13). FACS® analysis revealed 50% stabilization of Db by hgp10025–33 peptide at a concentration ∼100-fold lower than for mgp10025–33 (Fig. 6). This indicated that the apparent avidity of gp100-reactive T cells for the hgp10025–33 peptide could be largely attributed to its greater ability to stabilize the restricting MHC class I molecule, H-2Db.
Figure 4.

Recognition of mgp10025–33 and hgp10025–33 peptides at limiting concentrations. EL-4 thymoma cells were incubated with the mgp10025–33 peptide EGSRNQDWL and hgp10025–33 peptide KVPRNQDWL at the concentrations shown on the abscissa for 2 h at 37°C, washed twice, and cocultured for 24 h with gp100-reactive T cells. Supernatants were assayed for IFN-γ by ELISA, which was expressed as percentage of the maximal release (at 1 μM peptide, as shown on the abscissa). Half-maximal recognition of hgp10025–33 was reached at a concentration ∼1,000-fold lower than that needed for mgp10025–33. Data shown is an average of two independent experiments.
Figure 5.

Fine specificity of gp100-reactive T cells: EL-4 thymoma cells were incubated with either alanine-substituted peptide variants (A) or NH2-terminally deleted peptide variants (B) for 2 h at 37°C, washed twice, and cocultured for 24 h with gp100-reactive T cells. Supernatants were assayed for GM-CSF by ELISA.
Figure 6.

The TAP-deficient cell line RMA-S, expressing “empty” MHC class I molecules, was incubated with the concentrations shown on the abscissa with the mgp10025–33 and hgp10025–33 peptides, as well as control peptides, for 45 min at 25°C, then incubated for 4 h at 37°C. Cells were then stained with FITC-conjugated H-2Db–specific mAb KH95 for 1 h at 4°C, and staining was assessed using flow cytometric analysis and expressed as percentage of the maximal stabilization at 100 μM. The hgp10025–33 peptide stabilized H-2Db molecules at a 100-fold lower concentration than did the mgp10025–33 peptide.
Self-reactive T Cells Induced with Altered Antigen Function In Vivo.
One major potential use for self-reactive T cells is their application in cancer treatment. To evaluate the usefulness of altered antigen in the development of a synthetic cancer vaccine capable of eliciting therapeutic T cells, mice were vaccinated once with rVVhgp100 and restimulated for three rounds with splenocytes pulsed with 1 μM mgp10025–33 peptide. CD8+ T cells were tested for gp100 recognition by IFN-γ release (Fig. 7 A). T cells strongly recognized both mgp10025–33 and hgp10025–33 peptides, but not control OVA257–264 peptide, and recognized as well melanoma B16.WT, B16.B7-1, and JB/MS, a chemically induced C57BL/6 melanoma. Control H-2b, gp100− tumor lines were not recognized.
Figure 7.

rVVhgp100-induced T cells are tumor specific and can treat established melanoma. (A) Mice were vaccinated with 107 rVVhgp100 intravenously 3 wk before splenocytes were isolated and cultured with 1 μM of mgp10025–33 peptide for 6 d, after which the cells underwent two more cycles of restimulation with mgp10025–33-pulsed splenocytes. The resulting CD4−CD8+ T cells were cocultured for 24 h with various tumor targets, as well as MC38 sarcoma infected with indicated rVV or MC38 sarcoma pulsed with 1 μM of peptide, at an E/T ratio of 1:1, and IFN-γ release was measured by ELISA. rVVhgp100-induced T cells were mgp10025–33– and hgp10025–33–specific, and specifically recognized mgp100+ melanomas, as well as MC38 sarcoma infected with rVVhgp100 or rVVmgp100 but not rVVβ-gal, and MC38 sarcoma pulsed with hgp10025–33 or mgp10025–33 peptide, but not OVA257-264 peptide. (B) Mice were injected intravenously with 2 × 105 B16.WT and treated 3 d later with 5 × 106 of the T cells that are shown in A, or with 5 × 106 β-gal–specific T cells, followed by 3 × 105 IU rhIL-2 intraperitoneally twice daily for 5 d. 18 d after tumor inoculation, the number of pulmonary nodules was specifically reduced by mgp 10025–33–specific T cells. (C) Mice were injected intravenously with 4 × 106 of the T cells that are shown in A, or 4 × 106 β-gal–specific T cells. 5 d later, mice were challenged intravenously with 2 × 105 B16.WT. 18 d after tumor inoculation, the number of pulmonary nodules was dramatically lower in mice receiving mgp 10025–33–specific T cells, indicating in vivo survival of functional, autoreactive T cells. Two repeat experiments yielded similar results.
The function of T lymphocytes generated using purely recombinant and synthetic forms of gp100 were evaluated by adoptive transfer of T cells to mice bearing tumors established for 3 d in vivo. Mice receiving mgp10025–33–specific T cells experienced a significant reduction in tumor burden compared with mice receiving control T cells (Fig. 7 B). The survival of these cells was evaluated in vivo by transfer 5 d before tumor challenge, with T cells retaining their antitumor activity for this period (Fig. 7 C). This indicated that tumor-specific T cells induced with an antigen-based cancer vaccine can survive in vivo and mediate efficient tumor destruction.
Discussion
The nonhomologous sequences flanking a MHC-restricted epitope could influence the immunogenicity of the epitope through a variety of mechanisms. The full-length hgp100 and mgp100 molecules are 76% identical at the AA level (11). Xenoimmunization could induce antibodies to nonhomologous determinants on the xenoantigen. When expressed on the surface of B cells that produce them, these antibodies could capture the xenoantigen and make it available for B cell processing and presentation on MHC class II to activate CD4+ T cells (35). This mechanism has been postulated to play a role in the initiation of human autoimmune diseases such as SLE, which is largely mediated by CD4+ T cells and autoreactive antibodies (25, 34). Recent data suggest that B cells may cross-present antigen on MHC class I after capture (39). However, there is a vigorous debate over the ability of B cells to activate “virgin” T cells (i.e., T cells that have not previously been activated by antigen) (33, 40). DCs can also capture immune complexes containing xenoantigen through Fc receptors and present it through MHC class I and class II pathways, inducing de novo activation of autoreactive T cells (41). In this scenario, the completely autologous mgp100 would not induce such antibodies and thus would fail to be captured and presented. The involvement of extra-epitope sequences in the immunogenicity of the human gp100 molecules is not consistent with the results shown in Table 1 where the immunogenicity of human full-length and minimal determinant constructs are compared. In fact, the two constructs elicit comparable CD8+ T cell responses to the 25–33 epitope.
Another mechanism by which xenoimmunization could enhance immunogenicity is by the processing of a given epitope. For example, the hgp10025–33 peptide might be processed more efficiently than the mgp10025–33 peptide, since the two differ in the three NH2-terminal AAs, as well as in the AA sequences surrounding the peptides in the full-length molecule (36, 42). The differences between the processing of the human and mouse sequences is minimized and possibly eliminated by the use of minigene constructs preceded by ER-insertion signal sequences that bypass proteasome-mediated peptide liberation as well as TAP-mediated peptide transport (Table 1).
Indeed, the increased immunogenicity of human gp100 appeared to reside completely within the 9-mer peptide. The hgp10025–33 peptide differed from its mouse counterpart in three NH2-terminal AAs. In fact, these three differences were rather dramatic, with a positively charged lysine (K) replacing a negatively charged glutamic acid (E), a medium-sized valine (V) replacing a small glycine (G), and proline (P), which is a cyclic residue and known to reduce the number of possible conformations due to impaired hydrogen bonding, replacing serine (S), which may not induce such a structural distortion. Despite the presence in both peptides of optimal anchor residues at both dominant anchor positions, 5 (N) and 9 (L), the difference in the three NH2-terminal residues resulted in a dramatically increased affinity of the hgp10025–33 peptide for the mouse MHC class I allele, H-2Db, compared with the mgp10025–33 peptide (Fig. 6). Clearly, the ability of the peptide to stabilize Db molecules on the surface of RMA-S cells could reflect the amount of peptide presented on the surface of APCs, which in turn could determine the activation of T cell precursors. In retrospect, it was fortuitous that the interspecies differences in the gp100 sequences created such a high binder, whereas the similarities preserved sufficient T cell receptor contact residues to allow cross-recognition.
The efficacy of the mechanism described here through which xenoimmunization induced autoreactive T cells required that a number of criteria be satisfied. There must be an MHC-binding epitope in the autologous protein which is naturally processed and presented that can be recognized by the available T cell repertoire. Most importantly, a homologous epitope from the xenogeneic protein must also be naturally processed and presented, but must be presented in the context of MHC at a higher density on the surface of the APCs. The chance that such an epitope will be found in any give xenoantigen may be small. However, an understanding of this mechanism clearly points the way to the rational design of immunogens based on the enhancement of the stability of peptide–MHC complexes.
The data presented here suggest that the main difference between the mgp10025–33 peptide and its hgp10025–33 analogue resides in binding to MHC class I. In human in vitro studies, modification of the second AA of the hgp100 peptide epitope hgp100209–217 to methionine has been shown to significantly increase its affinity to HLA-A2, leading to dramatically increased ability to raise gp100-specific T cells from patient PBLs in vitro (43). The higher affinity peptide is also more effective in vaccinating patients in vivo, increasing gp100-specific T cell precursor levels, and possibly resulting in higher treatment response rates (43a). Similarly, other groups have reported increased immunogenicity of peptide epitopes with enhanced MHC class I binding (26, 44, 45).
Taken together, these data suggest that there is indeed unresponsiveness to gp100 in mice. However, the unresponsiveness is relative and can be broken by using a peptide homologue with higher affinity for MHC class I. One mechanism through which a peptide with higher MHC class I binding can break tolerance is based on the assumption that T cell tolerance exists to a level of antigen rather than to the identity of the antigen (44, 46, 47). In the case of gp100, CD8+ T cell precursors with the ability to recognize a certain amount of peptide in the context of MHC class I are inactivated, through either thymic and/or peripheral deletion or anergization. The remaining T cells have TCRs with an affinity that is too low to be triggered by the levels of gp100 peptide present on melanocytes or APCs in lymph nodes draining the skin. Therefore, these T cells are never activated, deleted, or anergized. Instead, they remain “ignorant” of MHC class I with mgp10025–33 peptide. A vaccination with autologous mgp100 will not trigger these T cells unless the vaccine is able to significantly raise the amount of peptide–MHC class I complex on professional APCs to a level high enough to surpass the TCR threshold. Only then do the T cells become activated. We are currently evaluating the clinical efficacy of antitumor effects of gp100-based cancer vaccines that contain epitopes with enhanced stability of peptide–MHC complexes.
Acknowledgments
The authors wish to thank Deborah R. Surman and Mr. Paul J. Spiess for expert technical assistance; Ms. Martha Blalock for assistance with graphics; Drs. Dennis Panicali and Linda Gritz (Therion Biologics) for providing rVVs; and Dr. Bruce Roberts for providing recombinant adenoviruses.
Abbreviations used in this paper
- AA
amino acid
- β-gal
β-galactosidase
- CM
complete medium
- DC
dendritic cell
- ER
endoplasmic reticulum
- FPV
fowlpox virus
- h
human
- m
mouse
- MART
melanoma antigen recognized by T lymphocytes
- MDA
melanocyte differentiation
- NP
the nucleoprotein from influenza A
- pDNA
plasmid DNA
- rVV
recombinant vaccinia virus
- TAP
the transporter associated with antigen processing
- TRP
tyrosinase-related protein
Footnotes
Correspondence to Nicholas P. Restifo, Senior Investigator, National Cancer Institute, Bldg. 10, Rm. 2B42, Bethesda, MD 20892-1502. Phone: 301-496-4904; Fax: 301-496-0011; E-mail: restifo@nih.gov
References
- 1.Boon T, Coulie PG, Van den Eynde B. Tumor antigens recognized by T cells. Immunol Today. 1997;18:267–268. doi: 10.1016/s0167-5699(97)80020-5. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today. 1997;18:175–182. doi: 10.1016/s0167-5699(97)84664-6. [DOI] [PubMed] [Google Scholar]
- 3.Restifo NP. The new vaccines: building viruses that elicit antitumor immunity. Curr Opin Immunol. 1996;8:658–663. doi: 10.1016/s0952-7915(96)80082-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kappler JW, Roehm N, Marrack P. T cell tolerance by clonal elimination in the thymus. Cell. 1987;49:273–280. doi: 10.1016/0092-8674(87)90568-x. [DOI] [PubMed] [Google Scholar]
- 5.Sant'Angelo DB, Waterbury PG, Cohen BE, Martin WD, Van Kaer L, Hayday AC, Janeway CA., Jr The imprint of intrathymic self-peptides on the mature T cell receptor repertoire. Immunity. 1997;7:517–524. doi: 10.1016/s1074-7613(00)80373-8. [DOI] [PubMed] [Google Scholar]
- 6.Hu Q, Bazemore WC, Girao C, Opferman JT, Sun J, Shabanowitz J, Hunt DF, Ashton-Rickardt PG. Specific recognition of thymic self-peptides induces the positive selection of cytotoxic T lymphocytes. Immunity. 1997;7:221–231. doi: 10.1016/s1074-7613(00)80525-7. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz RH. T cell clonal anergy. Curr Opin Immunol. 1997;9:351–357. doi: 10.1016/s0952-7915(97)80081-7. [DOI] [PubMed] [Google Scholar]
- 8.Wallace PM, Rodgers JN, Leytze GM, Johnson JS, Linsley PS. Induction and reversal of long-lived specific unresponsiveness to a T-dependent antigen following CTLA4Ig treatment. J Immunol. 1995;154:5885–5895. [PubMed] [Google Scholar]
- 9.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwon BS, Halaban R, Ponnazhagan S, Kim K, Chintamaneni C, Bennett D, Pickard RT. Mouse silver mutation is caused by a single base insertion in the putative cytoplasmic domain of Pmel 17. Nucleic Acids Res. 1995;23:154–158. doi: 10.1093/nar/23.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhai Y, Yang JC, Spiess P, Nishimura MI, Overwijk WW, Roberts B, Restifo NP, Rosenberg SA. Cloning and characterization of the genes encoding the murine homologues of the human melanoma antigens MART1 and gp100. J Immunother. 1997;20:15–25. doi: 10.1097/00002371-199701000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overwijk WW, Surman DR, Tsung K, Restifo NP. Identification of a Kb-restricted CTL epitope of beta-galactosidase: potential use in development of immunization protocols for “self” antigens. Methods (Orlando) 1997;12:117–123. doi: 10.1006/meth.1997.0461. [DOI] [PubMed] [Google Scholar]
- 13.Ljunggren HG, Stam NJ, Ohlen C, Neefjes JJ, Hoglund P, Heemels MT, Bastin J, Schumacher TN, Townsend A, Karre K. Empty MHC class I molecules come out in the cold. Nature. 1990;346:476–480. doi: 10.1038/346476a0. [DOI] [PubMed] [Google Scholar]
- 14.Earl PL, Cooper N, Moss B. Preparation of cell cultures and vaccinia virus stocks. Curr Prot Mol Biol. 1991;16:1–117. doi: 10.1002/0471142727.mb1616s43. [DOI] [PubMed] [Google Scholar]
- 15.Chakrabarti S, Sisler JR, Moss B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques. 1997;23:1094–1097. doi: 10.2144/97236st07. [DOI] [PubMed] [Google Scholar]
- 16.Bronte V, Carroll MW, Goletz TJ, Wang M, Overwijk WW, Marincola F, Rosenberg SA, Moss B, Restifo NP. Antigen expression by dendritic cells correlates with the therapeutic effectiveness of a model recombinant poxvirus tumor vaccine. Proc Natl Acad Sci USA. 1997;94:3183–3188. doi: 10.1073/pnas.94.7.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamamoto H. Cloning and sequencing of mouse tyrosinase cDNA. Jpn J Genet. 1987;62:271–274. [Google Scholar]
- 18.Shibahara S, Tomita Y, Sakakura T, Nager C, Chaudhuri B, Muller R. Cloning and expression of cDNA encoding mouse tyrosinase. Nucleic Acids Res. 1986;14:2413–2427. doi: 10.1093/nar/14.6.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson IJ, Chambers DM, Tsukamoto K, Copeland NG, Gilbert DJ, Jenkins NA, Hearing V. A second tyrosinase-related protein, TRP-2, maps to and is mutated at the mouse slaty locus. EMBO (Eur Mol Biol Organ) J. 1992;11:527–535. doi: 10.1002/j.1460-2075.1992.tb05083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Surman, D.R., K.R. Irvine, E.P. Shulman, T.M. Allweis, S.A. Rosenberg, and N.P. Restifo. 1998. Generation of polyclonal rabbit antisera to mouse melanoma associated antigens using gene gun immunization. J. Immunol. Methods. In press. [DOI] [PMC free article] [PubMed]
- 21.Kobayashi T, Urabe K, Orlow SJ, Higashi K, Imokawa G, Kwon BS, Potterf B, Hearing VJ. The Pmel 17/ silver locus protein. Characterization and investigation of its melanogenic function. J Biol Chem. 1994;269:29198–29205. [PubMed] [Google Scholar]
- 22.Winder A, Kobayashi T, Tsukamoto K, Urabe K, Aroca P, Kameyama K, Hearing VJ. The tyrosinase gene family—interactions of melanogenic proteins to regulate melanogenesis. Cell Mol Biol Res. 1994;40:613–626. [PubMed] [Google Scholar]
- 23.Irvine KR, Rao JB, Rosenberg SA, Restifo NP. Cytokine enhancement of DNA immunization leads to effective treatment of established pulmonary metastases. J Immunol. 1996;156:238–245. [PMC free article] [PubMed] [Google Scholar]
- 24.Moss B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci USA. 1996;93:11341–11348. doi: 10.1073/pnas.93.21.11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mamula MJ. Lupus autoimmunity: from peptides to particles. Immunol Rev. 1995;144:301–314. doi: 10.1111/j.1600-065x.1995.tb00074.x. [DOI] [PubMed] [Google Scholar]
- 26.Kozhich AT, Caspi RR, Berzofsky JA, Gery I. Immunogenicity and immunopathogenicity of an autoimmune epitope are potentiated by increasing MHC binding through residue substitution. J Immunol. 1997;158:4145–4151. [PubMed] [Google Scholar]
- 27.Fong L, Ruegg CL, Brockstedt D, Engleman EG, Laus R. Induction of tissue-specific autoimmune prostatitis with prostatic acid phosphatase immunization: implications for immunotherapy of prostate cancer. J Immunol. 1997;159:3113–3117. [PubMed] [Google Scholar]
- 28.Kawakami Y, Eliyahu S, Jennings C, Sakaguchi K, Kang X, Southwood S, Robbins PF, Sette A, Appella E, Rosenberg SA. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154:3961–3968. [PubMed] [Google Scholar]
- 29.Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–175. [PubMed] [Google Scholar]
- 30.Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 31.Vanderlugt CJ, Miller SD. Epitope spreading. Curr Opin Immunol. 1996;8:831–836. doi: 10.1016/S0952-7915(96)80012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moudgil KD, Chang TT, Eradat H, Chen AM, Gupta RS, Brahn E, Sercarz EE. Diversification of T cell responses to carboxy-terminal determinants within the 65-kD heat-shock protein is involved in regulation of autoimmune arthritis. J Exp Med. 1997;185:1307–1316. doi: 10.1084/jem.185.7.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mamula MJ, Fatenejad S, Craft J. B cells process and present lupus autoantigens that initiate autoimmune T cell responses. J Immunol. 1994;152:1453–1461. [PubMed] [Google Scholar]
- 34.Mamula MJ, Lin RH, Janeway CA, Jr, Hardin JA. Breaking T cell tolerance with foreign and self co- immunogens. A study of autoimmune B and T cell epitopes of cytochrome c. J Immunol. 1992;149:789–795. [PubMed] [Google Scholar]
- 35.Simitsek PD, Campbell DG, Lanzavecchia A, Fairweather N, Watts C. Modulation of antigen processing by bound antibodies can boost or suppress class II major histocompatibility complex presentation of different T cell determinants. J Exp Med. 1995;181:1957–1963. doi: 10.1084/jem.181.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng Y, Yewdell JW, Eisenlohr LC, Bennink JR. MHC affinity, peptide liberation, T cell repertoire, and immunodominance all contribute to the paucity of MHC class I–restricted peptides recognized by antiviral CTL. J Immunol. 1997;158:1507–1515. [PubMed] [Google Scholar]
- 37.Eisenlohr LC, Bacik I, Bennink JR, Bernstein K, Yewdell JW. Expression of a membrane protease enhances presentation of endogenous antigens to MHC class I–restricted T lymphocytes. Cell. 1992;71:963–972. doi: 10.1016/0092-8674(92)90392-p. [DOI] [PubMed] [Google Scholar]
- 38.Restifo NP, Bacik I, Irvine KR, Yewdell JW, McCabe BJ, Anderson RW, Eisenlohr LC, Rosenberg SA, Bennink JR. Antigen processing in vivo and the elicitation of primary CTL responses. J Immunol. 1995;154:4414–4422. [PMC free article] [PubMed] [Google Scholar]
- 39.Ke Y, Kapp JA. Exogenous antigens gain access to the major histocompatibility complex class I processing pathway in B cells by receptor-mediated uptake. J Exp Med. 1996;184:1179–1184. doi: 10.1084/jem.184.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992;258:1156–1159. doi: 10.1126/science.1439825. [DOI] [PubMed] [Google Scholar]
- 41.Abbas AK, Haber S, Rock KL. Antigen presentation by hapten-specific B lymphocytes. II. Specificity and properties of antigen-presenting B lymphocytes, and function of immunoglobulin receptors. J Immunol. 1985;135:1661–1667. [PubMed] [Google Scholar]
- 42.Mamula MJ. The inability to process a self-peptide allows autoreactive T cells to escape tolerance. J Exp Med. 1993;177:567–571. doi: 10.1084/jem.177.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, Kawakami Y. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol. 1996;157:2539–2548. [PubMed] [Google Scholar]
- 43a.Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola F, Topalian SL, Restifo NP, Dudley ME, Schwartz SL, Spiess PJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lipford GB, Bauer S, Wagner H, Heeg K. In vivo CTL induction with point-substituted ovalbumin peptides: immunogenicity correlates with peptide-induced MHC class I stability. Vaccine. 1995;13:313–320. doi: 10.1016/0264-410x(95)93320-9. [DOI] [PubMed] [Google Scholar]
- 45.Pogue RR, Eron J, Frelinger JA, Matsui M. Amino-terminal alteration of the HLA-A*0201-restricted human immunodeficiency virus pol peptide increases complex stability and in vitro immunogenicity. Proc Natl Acad Sci USA. 1995;92:8166–8170. doi: 10.1073/pnas.92.18.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 47.Liu GY, Fairchild PJ, Smith RM, Prowle JR, Kioussis D, Wraith DC. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]