

Abstract
DCs (dendritic cells) function as sentinels of the immune system. They traffic from the blood to the tissues where, while immature, they capture antigens. They then leave the tissues and move to the draining lymphoid organs where, converted into mature DC, they prime naive T cells. This suggestive link between DC traffic pattern and functions led us to investigate the chemokine responsiveness of DCs during their development and maturation. DCs were differentiated either from CD34+ hematopoietic progenitor cells (HPCs) cultured with granulocyte/macrophage colony–stimulating factor (GM-CSF) plus tumor necrosis factor (TNF)-α or from monocytes cultured with GM-CSF plus interleukin 4. Immature DCs derived from CD34+ HPCs migrate most vigorously in response to macrophage inflammatory protein (MIP)-3α, but also to MIP-1α and RANTES (regulated on activation, normal T cell expressed and secreted). Upon maturation, induced by either TNF-α, lipopolysaccharide, or CD40L, DCs lose their response to these three chemokines when they acquire a sustained responsiveness to a single other chemokine, MIP-3β. CC chemokine receptor (CCR)6 and CCR7 are the only known receptors for MIP-3α and MIP-3β, respectively. The observation that CCR6 mRNA expression decreases progressively as DCs mature, whereas CCR7 mRNA expression is sharply upregulated, provides a likely explanation for the changes in chemokine responsiveness. Similarly, MIP-3β responsiveness and CCR7 expression are induced upon maturation of monocyte- derived DCs. Furthermore, the chemotactic response to MIP-3β is also acquired by CD11c+ DCs isolated from blood after spontaneous maturation. Finally, detection by in situ hybridization of MIP-3α mRNA only within inflamed epithelial crypts of tonsils, and of MIP-3β mRNA specifically in T cell–rich areas, suggests a role for MIP-3α/CCR6 in recruitment of immature DCs at site of injury and for MIP-3β/CCR7 in accumulation of antigen-loaded mature DCs in T cell–rich areas.
Keywords: dendritic cells, chemokines, migration, maturation, regulation, in vivo expression
Dendritic cells (DCs)1 are professional APCs that are found in all lymphoid and nonlymphoid organs (1). DCs are bone marrow–derived and likely migrate as precursors through blood stream to tissues, where they become resident cells such as Langerhans cells in the epidermis. Moreover, after pathogen invasion, DCs are probably recruited at the site of inflammation (2), as illustrated in the rat, where intratracheal antigen instillation induces accumulation of DCs in the airway epithelium (3). In the periphery, DCs such as Langerhans cells capture the antigens (4–8), then migrate via the lymphatics and home to the T cell–rich area of lymph nodes, where they are called interdigitating cells (IDCs) (9–14). At this site, they present the processed antigens to naive T cells and generate an antigen-specific primary T cell response (15–18). During their migration from peripheral tissues to lymphoid organs, DCs undergo a maturation process encompassing dramatic changes in phenotype and functions (11, 19, 20). In particular, immature DCs such as fresh Langerhans cells capture and process soluble proteins efficiently and are effective at activating specific memory and effector T cells. Conversely, mature DCs such as IDCs of lymphoid organs are poor in antigen capture and processing but markedly efficient in naive T cell priming (4–7, 21, 22). This last difference correlates with the levels of expression of MHC class II and accessory molecules such as B7, which are much higher on mature than on immature DCs (23–27).
Although signals regulating the complex traffic pattern of DCs are not fully understood, chemokines are probably involved. Chemokines are small molecular mass proteins (8–10 kD) that regulate leukocyte migration and activation (28–31). They are secreted by activated leukocytes themselves and by stromal cells, including endothelial cells and epithelial cells upon inflammatory stimuli (28–31). Responses to chemokines are mediated by seven transmembrane–spanning G protein–coupled receptors (30, 32, 33). Several chemokines such as monocyte chemotactic protein (MCP)-3, MCP-4, macrophage inflammatory protein (MIP)-1α, MIP-1β, RANTES (regulated on activation, normal T cell expressed and secreted), stromal cell–derived factor (SDF)-1, thymus-expressed chemokine ( Teck), and macrophage-derived chemokine have been reported to attract DCs in vitro (34–38). Moreover, signals provided by TNF-α and LPS are known to induce in vivo migration of resident DCs from the tissues to the draining lymphoid organs (20, 37–40). However, the relationship between signals inducing DC migration in vivo and their responses to chemokines is not known.
In this study, we have taken advantage of the well-established control of CD34+ hematopoietic progenitor cell (HPC; 41–43) and monocyte (22, 44) cultures to study the chemokine responsiveness of DCs during their differentiation and maturation. Focusing on CC chemokine receptor (CCR)6 (45–48) and CCR7 (49) and their known ligands (47, 50–52), we found that chemokine receptor expression and the subsequent chemokine responsiveness are indeed integral parts of the maturation process of DCs. In addition, in situ localization of chemokines active on immature versus mature DCs provides a likely explanation for at least part of the DC traffic pattern.
Materials and Methods
Hematopoietic Factors, Reagents, and Cell Lines.
Recombinant human (rh)GM-CSF (specific activity: 2 × 106 U/mg; Schering-Plough Research Institute, Kenilworth, NJ) was used at a saturating concentration of 100 ng/ml. rhTNF-α (specific activity: 2 × 107 U/mg; Genzyme, Boston, MA) was used at an optimal concentration of 2.5 ng/ml (42). rhSCF (specific activity: 4 × 105 U/mg, R&D Systems, Abington, UK) was used at an optimal concentration of 25 ng/ml. rhIL-4 (specific activity: 2 × 107 U/mg, Schering-Plough Research Institute) was used at a saturating concentration of 50 U/ml. Recombinant human chemokines MIP-1α (specific activity: 2 × 105 U/mg, 9 × 1012 U/M), RANTES (specific activity: 104 U/mg, 8 × 1010 U/M), MIP-3α (specific activity: 4 × 105 U/mg, 3 × 1012 U/M) and MIP-3β (specific activity: 104 U/mg, 9 × 1010 U/M) were obtained from R&D Systems. LPS was used at 10 ng/ml (Sigma Chemical Co., St. Louis, MO).
The murine CD40 ligand transfected cell line (CD40L L cells) was produced in the laboratory and used as stimulator of DC maturation (53).
Generation of DCs from Cord Blood CD34+ HPCs.
Umbilical cord blood samples were obtained after full-term delivery. Cells bearing CD34+ antigen were isolated from mononuclear fractions through positive selection as previously described (42, 54), using anti-CD34+ mAb (Immu-133.3, Immunotech, Marseille, France), goat anti–mouse IgG-coated microbeads (Miltenyi Biotec GmBH, Bergisch Gladbach, Germany), and Minimacs separation columns (Miltenyi Biotec GmBH). In all experiments the isolated cells were 80–99% CD34+. After purification, CD34+ cells were cryopreserved in 10% DMSO.
Cultures were established in the presence of stem cell factor, GM-CSF, and TNF-α as previously described (42) in endotoxin-free medium consisting of RPMI 1640 (GIBCO BRL, Gaithersburg, MD) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Life Technologies, Pontoise, France), 10 mM Hepes, 2 mM l-glutamine, 5 × 10−5 M β-mercaptoethanol, and 100 μg/ml gentamycin (Schering-Plough, Levalois, France) (referred to as complete medium). After thawing, CD34+ cells were seeded for expansion in 25–75 cm2 culture vessels (Linbro; ICN Biomedicals, Aurora, OH) at 2 × 104 cells/ml. Optimal conditions were maintained by splitting these cultures at days 5 and 10 with medium containing fresh GM-CSF and TNF-α (cell concentration: 1–3 × 105 cells/ml). At day 12, between 70 and 90% of the cells were CD1a+ DCs.
Isolation of Immature and Mature DCs According to CD86 Expression by FACS® Sorting.
After 12 d of culture in the presence of GM-CSF and TNF-α, cells were collected and labeled with FITC-conjugated OKT6 (CD1a; Ortho Diagnostic System, Raritan, NJ) and PE-conjugated IT2.2 (CD86; PharMingen, San Diego, CA). Cells were separated according to CD1a and CD86 expression into immature CD1a+CD86− and mature CD1a+CD86+ DC populations using a FACStarplus® (laser setting: power 250 mW, excitation wavelength 488 nm; Becton Dickinson, Sunnyvale, CA). All the procedures of staining and sorting were performed in presence of 0.5 mM EDTA to avoid cell aggregation. Reanalysis of the sorted populations showed a purity >98%.
Generation of DCs from Peripheral Blood Monocytes.
Monocyte s were purified by immunomagnetic depletion (Dynabeads; Dynal, Oslo, Norway) after preparation of PBMCs followed by a 52% Percoll gradient. The depletion was performed with anti-CD3 (OKT3), anti-CD19 (4G2), anti-CD8 (OKT8), anti-CD56 (NKH1, Coulter Corp., Hialeah, FL), and anti-CD16 (ION16, Immunotech) mAbs. Monocyte-derived dendritic cells were produced by culturing purified monocytes for 6–7 d in the presence of GM-CSF and IL-4 (44).
Induction of Maturation of In Vitro–generated DCs.
CD34+ HPCs were cultured until day 6 in presence of GM-CSF plus TNF-α and in presence of GM-CSF alone from days 6 to 12 in order to preserve their immaturity. Immature DCs from CD34+ HPCs or monocyte-derived DCs were activated for 3–72 h in the presence of TNF-α (2.5 ng/ml), LPS (10 ng/ml), or CD40L transfected L cells (one L cell for five DCs) as described elsewhere (55).
Purification of CD11c+ DCs from Peripheral Blood or Tonsils.
CD11c+ DCs were prepared as previously described from peripheral blood or tonsils (56). In brief, tonsils obtained from children undergoing tonsillectomy were finely minced and digested with collagenase IV and DNase I (Sigma Chemical Co.). The collected cells were centrifuged through Ficoll-Hypaque with SRBCs (BioMérieux, Lyon, France) for 15 min at 500 rpm, then for 30 min at 2,000 rpm. Mononuclear cells from peripheral blood were isolated by Ficoll-Hypaque. CD3+ T cells (OKT3), CD19+ B cells (4G7), and CD14+ monocytes (MOP9) were removed from the resulting low density cells by magnetic beads (anti–mouse Ig-coated Dynabeads [Dynal]). A second depletion was performed with anti-NKH1, anti–glycophorine A (Immunotech), and anti-CD20 (1F54). The remaining cells were stained with the following mAbs: anti-CD1a FITC (OKT6); anti-CD14 FITC, anti-CD57 FITC, anti-CD16 FITC, anti-CD7 FITC, anti-CD20 FITC, anti-CD3 FITC (Becton Dickinson); anti-CD4 PE-Cy5 (Immunotech), and anti-CD11c PE (Becton Dickinson). CD4+CD11c+lineage− DCs were isolated by cell sorting using a FACStarPlus® (laser setting: power 250 mW, excitation wavelength 488 nm). All the procedures of depletion, staining, and sorting were performed in presence of 0.5 mM EDTA. Reanalysis of the sorted population showed a purity >97%.
Chemotaxis Assay.
Cell migration was evaluated using a chemotaxis microchamber technique (48-well Boyden microchamber; Neuroprobe, Pleasanton, CA; reference 57). In brief, MIP-3α and MIP-3β, MIP-1α, and RANTES were diluted to concentrations ranging from 1 to 1,000 ng/ml in RPMI 1640 medium, and were added to the lower wells of the chemotaxis chamber. 105 cells/well (or 5 × 104 cells/well for CD11c+ DCs) in 50 μl of RPMI 1640 medium were applied to the upper wells of the chamber, with a standard 5-μm pore polyvinylpyrrolidone-free polycarbonate filter (Neuroprobe) separating the lower wells. The chamber was incubated at 37°C in humidified air with 5% CO2 for 1 h. Then, cells that had migrated to the underside of the filter were stained with Field's A and Field's B (BDH Chemicals, Ltd., Abingdon, Essex, UK) and counted using an image analyzer (software: Vision Explorer and ETC 3000; Graphtek, Mirmande, France) in two randomly selected low power fields (magnification: ×20). Each assay was performed in duplicate and the results were expressed as the mean ± SD of migrating cells per two fields.
Extraction of Total RNA and Synthesis of cDNA.
Cells were prepared as described above, and total RNA was extracted by the guanidinium thiocyanate method as mentioned by the manufacturer (RNAgents total RNA isolation system; Promega, Madison, WI). After DNAse I treatment (RQ1 RNAse free DNAse; Promega), RNA was quantified by spectrophotometry and quality was evaluated by electrophoresis in formaldehyde denaturing conditions. First strand cDNA was synthetized from total RNA extracted in RNAse-free conditions. The reaction was performed with 5 μg of total RNA, 25 ng/μl oligo dT12-18 primers (Pharmacia, Orsay, France) and the Superscript kit (SuperScript II RNase H− Reverse Transcriptase; GIBCO BRL), as described by the manufacturer. For all samples, synthesis of cDNA was controlled and calibrated by reverse transcriptase (RT)-PCR using β-actin primers for 21 cycles.
RT-PCR Analysis.
Semi-quantitative PCR was performed in a Perkin Elmer 9,600 thermal cycler, in a final volume of 100 μl reaction mixture containing 2.5 U AmpliTaq enzyme (5 U/μl, Perkin Elmer, Paris, France) with its 1× buffer, 0.2 mM of each dNTP (Perkin Elmer), 5% DMSO, and 1 μM of each forward and reverse primer. CCR6 and CCR7 primers (sequence data available from EMBL/GenBank/DDBJ under accession numbers Z79784 and L08176, respectively) were designed within regions of lowest homology between the chemokine receptors. +80/ CCR6 5′-ATTTCAGCGATGTTTTCGACTC-3′ forward primer, −1081/CCR6 5′-GGAGAAGCCTGAGGACTTGTA-3′ reverse primer, +154/CCR7 5′-GATTACATCGGAGACAACACC-3′ forward primer and −1202/CCR7 5′-TAGTCCAGGCAGAAGAGTCG-3′ reverse primer were used for RT-PCR and sequencing. For both chemokine receptors, the reaction mixture was subjected to 30 and 35 cycles of PCR with the following conditions: 94°C for 1 min, 61.5°C for 2 min, and 72°C for 3 min. PCR products were visualized on 1.2% agarose gels containing 0.5 μg/ml ethidium bromide. Reaction products migrating at the predicted size (1,021 bp for CCR6 and 1,067 bp for CCR7) were gel purified and subcloned into pCRII TA cloning vector (Invitrogen, Leek, The Netherlands) for sequencing verification on an ABI 373A Sequencer (Applied Biosystems, Inc., Foster City, CA) using dye terminator technology. Two other oligonucleotides, −622/CCR6 5′-GCTGCCTTGGGTGTTGTATTT-3′ and +662/CCR7 5′-AGAGGAGCAGCAGTGAGCAA-3′, were used as probes for hybridization with the PCR products separated on 1.2% agarose gel and blotted onto Hybond N+ membranes (Amersham, Les Ulis, France).
Calcium Fluorimetry.
Intracellular Ca2+ concentration was measured using the fluorescent probe Indo-1, according to the technique reported by Grynkiewicz et al. (58). In brief, cells were washed in PBS and resuspended at 107 cells/ml in complete RPMI 1640 medium (see above). Then, cells were incubated for 45 min at room temperature with 3 μg/ml Indo-1 AM (Molecular Probes, Inc., Eugene, OR) in the dark. After incubation, cells were washed and resuspended in HBSS/1% FCS at 107 cells/ml. Before measurement of intracellular Ca2+ concentration, cells were diluted 10-fold in HBSS/10 mM Hepes/1.6 mM CaCl2 preheated at 39°C. Samples were excited at 330 nm with continuous stirring and the Indo-1 fluorescence was measured as a function of time at 405 nm (dye is complexed with Ca2+) and 485 nm (Ca2+-free medium) in a 810 Photomultiplier Detection System (software: Felix; Photon Technology International, Monmouth Junction, NJ). Results are expressed as the ratio of values obtained at the two emission wavelengths.
In Situ Hybridization.
In situ hybridization was performed as previously described (59). Two-couple primers were used for amplifying by RT-PCR the majority of the open reading frame of MIP-3α and MIP-3β genes (sequence data available from EMBL/GenBank/DDBJ under accession numbers D86955 and U77180, respectively). +77/MIP-3α 5′-TTGCTCCTGGCTGCTTTG-3′ forward primer and −425/MIP-3α 5′-ACCCTCCATGATGTGCAAG-3′ reverse primer, +25/MIP-3β 5′-CTGCTGGTTCTCTGGACTTC-3′ forward primer and −439/ MIP-3β 5′-CACACTCACACTCACACACAC-3′ reverse primer were used as described above with an annealing temperature at 62°C. Then, PCR products were cloned into pCRII TA cloning vector (Invitrogen) for the generation of sense and antisense probes with the adapted promoters. Sense and antisense 35S-labeled probes of MIP-3α and MIP-3β were obtained by run off transcription of the 367-bp and 435-bp fragments, respectively. 6-μm human tonsil sections were fixed in acetone and 4% paraformaldehyde followed by 0.1 M triethanolamine/0.25% acetic anhydride. The sections were hybridized overnight, RNAse A treated and exposed for 24 d. After development sections were stained with hematoxylin.
Results
Differential Responsiveness to MIP-3α and MIP-3β during Development of CD34+-derived DCs.
As a way to understand the regulation of DC traffic, we have studied the response to various chemokines of DCs at different stages of maturation. DCs were generated from CD34+ HPCs cultured in the presence of GM-CSF plus TNF-α, and tested at different days of culture for their ability to migrate in response to chemokines in Boyden microchambers. MIP-3α and MIP-3β recruited two to three times more CD34+-derived DCs than did MIP-1α or RANTES (Fig. 1 A, mean of 20 experiments). However, MIP-3α and MIP-3β attracted DCs collected at different time points of the culture (Fig. 1 B). The response to MIP-3α was already detectable at day 4, maximal at days 5–6, and lasted until day 10. At days 13–14, the response to MIP-3α was usually lost. In contrast, the response to MIP-3β, which could not be detected before day 10, peaked at day 13, and persisted beyond day 15 (data not shown). Of note, at early time points (Fig. 2, Day 6), when most of the cells in culture were still DC precursors (CD1a−CD86−), the response to MIP-3α could be detected at concentrations of 1–10 ng/ml (depending on the experiment). In contrast, 4 d later, when almost all cells were immature DCs (CD1a+CD86−), ≥300 ng/ml were needed to attract the cells (Fig. 2, Day 10), suggesting a progressive desensitization of the cells during maturation. Relatively high concentrations of MIP-3β (300 ng/ml) were also needed to recruit mature DCs (CD1a+CD86+) (Fig. 2, Day 13). Checkerboard analysis established that MIP-3α and MIP-3β induced chemotaxis and not chemokinesis of DCs (data not shown).
Figure 1.
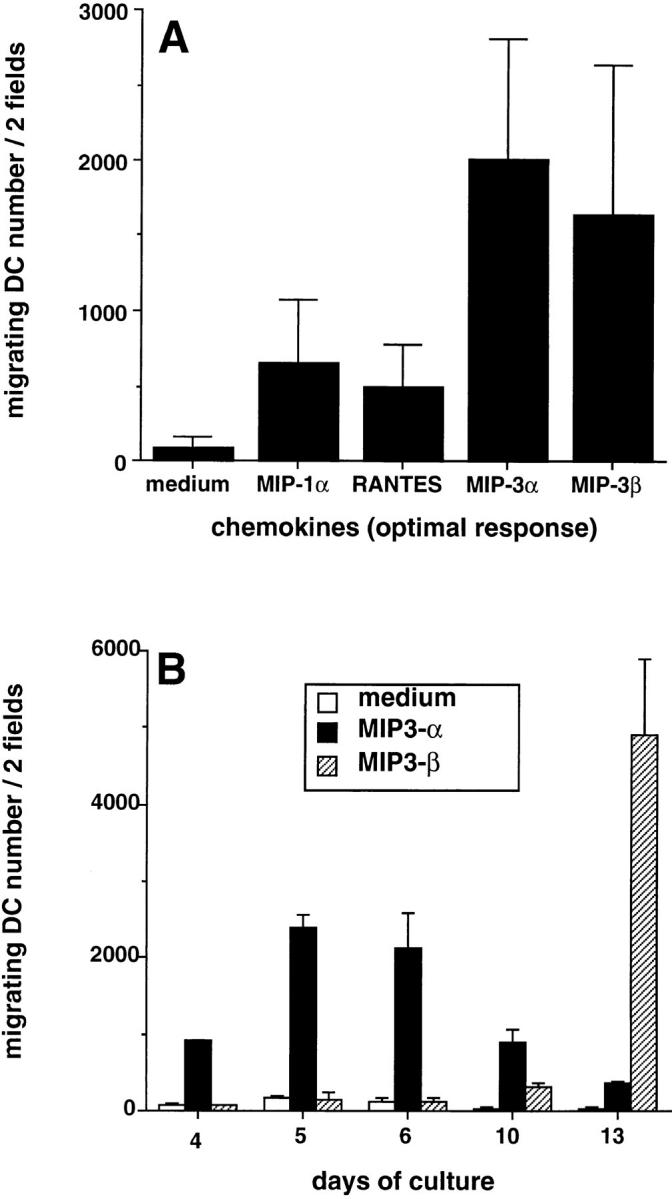
Responses to MIP-3α and MIP-3β during development/ maturation of DCs from CD34+ HPCs. CD34+ HPCs were cultured in presence of GM-CSF plus TNF-α for 13 d. (A) Migration assays were performed between days 10 and 13. Results represent the means of optimal responses for each chemokines obtained in 20 independent experiments. (B) At the indicated time points, aliquots of cells were recovered and tested for their response to MIP-3α (10–1,000 ng/ml) and to MIP-3β (10–1,000 ng/ml), the optimal concentrations being shown. Results are representative of more than three experiments. Migration assays were performed in Boyden microchambers. Results are expressed as number of migrating cells per two low power fields (original magnification: ×20).
Figure 2.

Dose responses to MIP-3α and to MIP-3β of CD34+-derived DCs at different stages of development/maturation. CD34+ HPCs were cultured in presence of GM-CSF plus TNF-α for 13 d. Expression of CD1a (FITC) and CD86 (PE) is shown at days 6, 10 and 13 of culture (top). Dose responses to MIP-3α and to MIP-3β are shown at the same time points (bottom). Migration assays were performed in Boyden microchambers. Results are expressed as number of migrating cells per two low power fields (original magnification: ×20). Results are representative of more than three experiments.
To confirm the relation between the stage of maturation and the response to MIP-3α and MIP-3β, CD34+-derived DCs were FACS® sorted at day 10 of culture according to CD86 expression into immature DCs (CD1a+CD86−) and mature DC (CD1a+CD86+). CD1a+CD86− responded exclusively to MIP-3α, whereas CD1a+CD86+ responded mainly to MIP-3β (Fig. 3). These observations also confirmed that the cells recruited by MIP-3α and MIP-3β were indeed DCs (CD1a+). The correlation between DC maturation and chemokine responsiveness was further illustrated when the immaturity of DCs was preserved by removing TNF-α from days 6 to 12 and when their maturation was synchronized by addition of TNF-α, LPS, or CD40L. As shown in Fig. 4 A, response to MIP-3α had strongly decreased after 48 h of maturation with TNF-α, LPS, and CD40L. Meanwhile, the response to MIP-3β was induced by all three signals, CD40L and LPS being more potent than TNF-α. In kinetics experiments, the response to MIP-3α decreased by 50–70% after only 24 h of CD40 activation and was completely lost at 72 h. The response to MIP-3β was already maximal after 24 h of CD40 activation (Fig. 4 B) and required relatively high concentration of chemokine (100–300 ng/ml) at 48 h (Fig. 4 C).
Figure 3.

CD1a+CD86− immature DCs and CD1a+CD86+ mature DCs respond to MIP-3α and MIP-3β, respectively. CD34+ HPCs were cultured in the presence of GM-CSF plus TNF-α. At day 10, DCs were FACS® sorted according to CD86 expression into CD1a+CD86− immature DCs and CD1a+CD86+ mature DCs, and their response to MIP-3α (10–1,000 ng/ml) and MIP-3β (10–1,000 ng/ml) were determined by migration assays in Boyden microchambers. Results are expressed as index of migrating cells (ratio chemokine/medium), the optimal concentrations being shown. Results are representative of three experiments.
Figure 4.
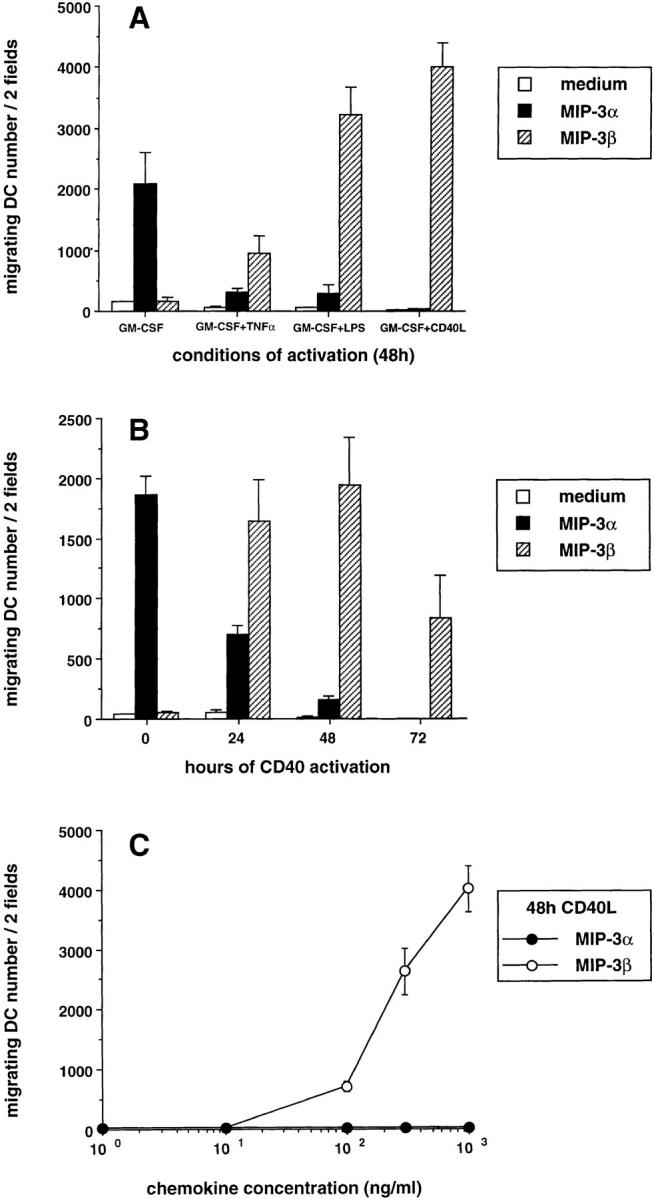
Different signals triggering DC maturation block the response to MIP-3α and induce that to MIP-3β. CD34+ HPCs were cultured in presence of GM-CSF plus TNF-α until day 6 and in presence of GM-CSF alone from day 6 to 12 to preserve their immaturity. (A) At day 12, DCs were cultured for 48 h in presence of GM-CSF with or without TNF-α (2.5 ng/ml), LPS (10 ng/ml), or CD40L and assessed for their response to MIP-3α (10–1,000 ng/ml) and to MIP-3β (10–1,000 ng/ml), the optimal concentrations being shown. (B) At day 12, DCs were cultured for 72 h in the presence of GM-CSF and CD40L and their response to MIP-3α (10–1,000 ng/ml) and to MIP-3β (10–1,000 ng/ml) were assessed at the indicated time points, the optimal concentrations being shown. (C) At day 12, DCs were cultured for 48 h in presence of GM-CSF and CD40L. Dose responses to MIP-3α and MIP-3β are shown. Migration assays were performed in Boyden microchambers. Results are expressed as number of migrating cells per two low power fields (original magnification: ×20). Results are representative of more than three experiments.
Taken together, these results establish that immature CD34+-derived DCs respond to MIP-3α, whereas mature DCs respond to MIP-3β.
Responses to MIP-3α and MIP-3β Parallel the Expression of Their Respective Receptors, CCR6 and CCR7, on CD34+-derived DCs.
To define the mechanisms of regulation of MIP-3α and MIP-3β responsiveness, the expression of their respective receptors, CCR6 (45–48) and CCR7 (49) mRNA, was studied by semi-quantitative RT-PCR. During DC development from CD34+ HPCs, CCR6 mRNA was first detected at day 6 and increased up to day 10, after which it decreased and became barely detectable at day 14. In contrast, CCR7 mRNA appeared at day 10 and steadily increased up to day 14 (Fig. 5 A). Moreover, CD40L-dependent maturation induced progressive downregulation of CCR6 mRNA that became almost undetectable after 72 h, and upregulation of CCR7 mRNA as early as 24 h after (Fig. 5 B). Similar results were obtained after either LPS- or TNF-α–induced DC maturation (data not shown). The upregulation of CCR7 mRNA after activation was confirmed by Southern blot analysis of cDNA libraries (data not shown).
Figure 5.


CCR6 mRNA downregulation and CCR7 mRNA upregulation during CD34+-derived DC activation/maturation induced by TNF-α or CD40L. (A) CD34+ HPCs were cultured in the presence of GM-CSF plus TNF-α for 14 d. At the indicated time points, aliquots of cells were recovered, mRNA was extracted, and expressions of CCR6 and CCR7 mRNA were determined. (B) CD34+ HPCs were cultured in the presence of GM-CSF plus TNF-α until day 6 and in the presence of GM-CSF alone from day 6 to 12 to preserve their immaturity. At day 12, DCs were cultured for 72 h in presence of GM-CSF and CD40L and the expression of CCR6 and CCR7 mRNA were assessed at the indicated time points. Expression of CCR6 and CCR7 mRNA were determined by semi-quantitative RT-PCR (30, 35 cycles). The results at 35 cycles are shown and PCR for β-actin are shown at 21 cycles as an internal mRNA control. Results are representative of three experiments.
In line with the migration assays and the regulation of CCR6 and CCR7 expression, MIP-3α induced a Ca2+ flux exclusively in resting/immature DC (Fig. 6 A), and MIP-3β induced the same in mature DCs only (independently of the maturation signal, CD40 activation is shown on Fig. 6 B). Maximal Ca2+ fluxes were observed with 30 ng/ml of MIP-3α and 30 ng/ml of MIP-3β, on immature and mature DCs, respectively (Fig. 6, C and D).
Figure 6.

MIP-3α and MIP-3β induce Ca2+ flux on only immature and mature DCs, respectively. CD34+ HPCs were cultured in the presence of GM-CSF plus TNF-α until day 6 and in the presence of GM-CSF alone from day 6 to 12 (immature/resting DCs). At day 12, DCs were cultured for 48 h in presence of GM-CSF and CD40L (mature/ activated DC). Immature/resting DC (A) and mature/CD40-activated DCs (B) were tested for their response to MIP-3α (1,000 ng/ml) and to MIP-3β (1,000 ng/ml) in Ca2+ flux. Dose responses in Ca2+ flux are shown for MIP-3α and MIP-3β on, respectively, immature/resting DCs (C) and mature/ CD40-activated DCs (D). Results are representative of three experiments.
These results show that changes in responsiveness to MIP-3α and MIP-3β are linked to the regulation of CCR6 and CCR7 mRNA expression, and suggest that CCR6 and CCR7 are the major functional receptors expressed on DCs for MIP-3α and MIP-3β, respectively.
The Response to MIP-3β Is Also Induced upon Maturation of Monocyte-derived DCs.
Monocyte-derived DCs, generated by culturing monocytes in presence of GM-CSF+IL-4 for 6 d, are typically immature DCs (CD1a+, CD14−, CD80low, CD86low, and CD83−) (22, 44). They migrated in response to MIP-1α and RANTES but not to MIP-3α (data not shown) or MIP-3β (Fig. 7). The lack of response of monocyte-derived DCs to MIP-3α is in accordance with the absence of CCR6 expression on those cells (45, 46). Upon maturation induced by TNF-α, LPS, or CD40L, responses to MIP-1α and RANTES were lost, whereas response to MIP-3β was induced (Fig. 7, A and B). As with CD34+-derived DCs, the response to MIP-3β correlated with the upregulation of CCR7 mRNA expression observed upon maturation induced by TNF-α (Fig. 8 A), LPS (data not shown), or CD40L (Fig. 8 B). Again, upregulation of CCR7 occurred at early time points (3 h) after TNFR or CD40 signaling. Moreover, migration and chemokine receptor expression data were in agreement with Ca2+ flux results (data not shown).
Figure 7.


The response to MIP-3β is induced upon maturation of monocyte-derived DCs. DCs were generated by culturing monocytes in presence of GM-CSF+IL-4 for 6 d. (A) DCs were cultured in the presence of GM-CSF (resting) or in the presence of GM-CSF and CD40L (activated). Dose responses to MIP-1α, RANTES, and MIP-3β were determined after 48 h. (B) DCs were cultured for 48 h in the presence of GM-CSF with or without TNF-α (2.5 ng/ml), LPS (10 ng/ml), or CD40L and assessed for their responses to MIP-1α (10–1,000 ng/ ml) and to MIP-3β (10–1000 ng/ml), the optimal concentrations being shown. Migration assays were performed in Boyden microchambers. Results are expressed as number of migrating cells per two low power fields (original magnification: ×20). Results are representative of more than three experiments.
Figure 8.


CCR7 is rapidly induced upon CD40 and TNF-α activation of monocyte-derived DCs. DCs were generated by culturing monocytes in presence of GM-CSF+IL-4 for 6 d. DCs were activated in presence of GM-CSF plus TNF-α or GM-CSF+CD40L. At the indicated time points, the expression of CCR7 mRNA was assessed by semi-quantitative RT-PCR (30, 35 cycles). Results at 35 cycles are shown and PCR for β-actin are shown at 21 cycles as an internal mRNA control. Results are representative of three experiments.
Those results extend to monocyte-derived DCs the concept that upon maturation, DCs lose their responsiveness to various chemokines while becoming sensitive to a single chemokine, MIP-3β.
Peripheral Blood CD11c+ DCs Migrate in Response to MIP-3β after Maturation.
We next studied the chemotactic activities of MIP-3α and MIP-3β on immature CD11c+ DCs isolated from peripheral blood (or tonsils). Freshly isolated DCs did not migrate in response to MIP-3α, or to MIP-3β (Fig. 9 A), an observation that correlates with the absence of CCR6 (data not shown) and CCR7 mRNA expression in these cells (Fig. 9 B). However, the maturation that is known to occur after overnight culture with GM-CSF (56) turned on the response of CD11c+ DC to MIP-3β but not to MIP-3α. Once more, the response to MIP-3β correlated with the induction of CCR7 mRNA expression (Fig. 9 B).
Figure 9.
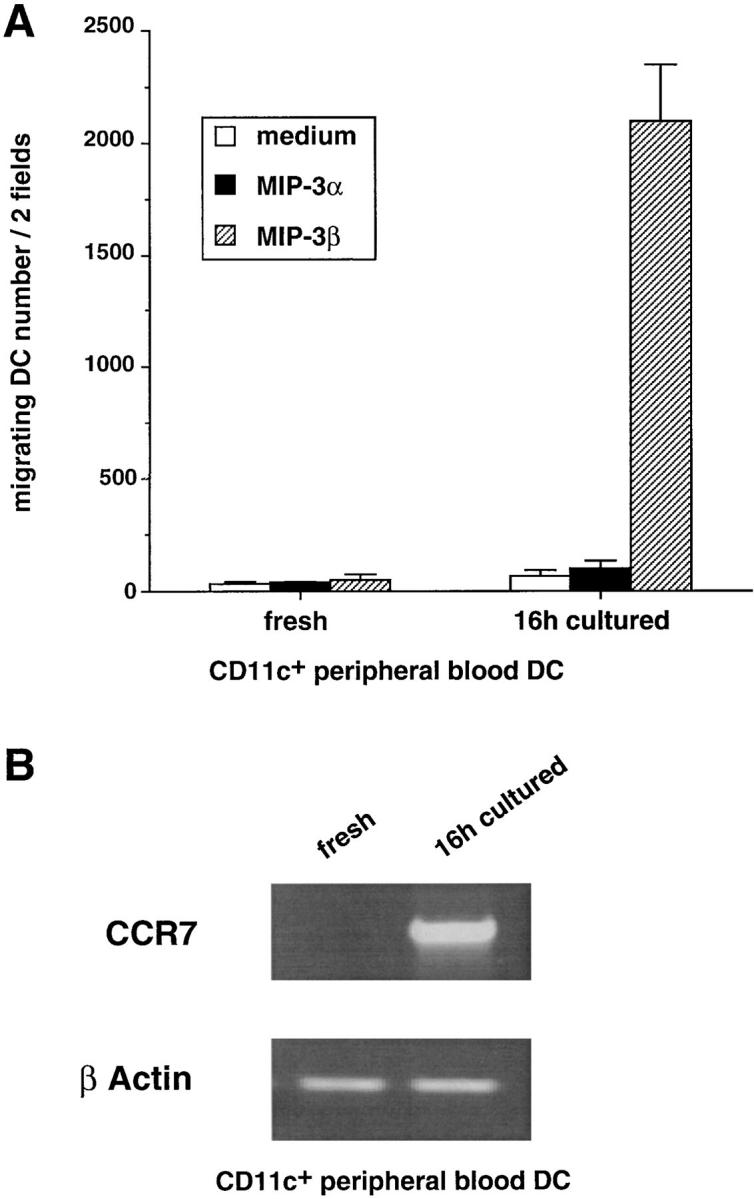
CD11c+ peripheral blood DCs respond to MIP-3β but not to MIP-3α upon spontaneous maturation. CD11c+ DCs were FACS® sorted from PBLs according to their lack of lineage markers and expression of CD4 and CD11c (see Materials and Methods). (A) The responses to MIP-3α (500 ng/ml) and to MIP-3β (500 ng/ml) were determined after FACS® sorting and after 16-h culture with GM-CSF. Migration assays were performed in Boyden microchambers. Results are expressed as number of migrating cells per two low power fields (original magnification: ×20). Results are representative of four experiments. (B) The expression of CCR7 mRNA was assessed by semiquantitative RT-PCR. Results at 35 cycles are shown, and PCR for β actin is shown at 21 cycles as an internal mRNA control. Results are representative of three experiments.
Therefore, although immature CD11c+ DCs freshly isolated from blood cannot respond to MIP-3α, these results show that maturation-dependent responsiveness to MIP-3β also applies to ex vivo–isolated DCs.
In Vivo MIP-3α Is Expressed in Inflamed Epithelium and MIP-3β within T Cell–rich Areas of Tonsils.
The physiological relevance of these findings was then addressed through the analysis of MIP-3α and MIP-3β mRNA expressions by in situ hybridization on sections of inflamed tonsils. mRNA for MIP-3α was detected at high levels in inflamed epithelial crypts but not in T cell–rich areas or in B cell follicles (Fig. 10, A–C). In fact, MIP-3α expression was restricted to cells lining the epithelial crypts (Fig. 10 D). In contrast, expression of MIP-3β mRNA was restricted to T cell–rich areas (Fig. 10, F–H). The strongest signal was present in scattered cells, with a distribution overlapping that of IDCs (Fig. 10, F–I). Outside the paracortical area, no signal could be detected in B cell follicles or in epithelial crypts (Fig. 10, F–H). Fig. 10, A and F (serial sections), shows clear absence of MIP-3β expression within epithelial crypts where MIP-3α was abundantly present. Sense probes for MIP-3α and MIP-3β did not generate background hybridization (Fig. 10, E and J).
Figure 10.
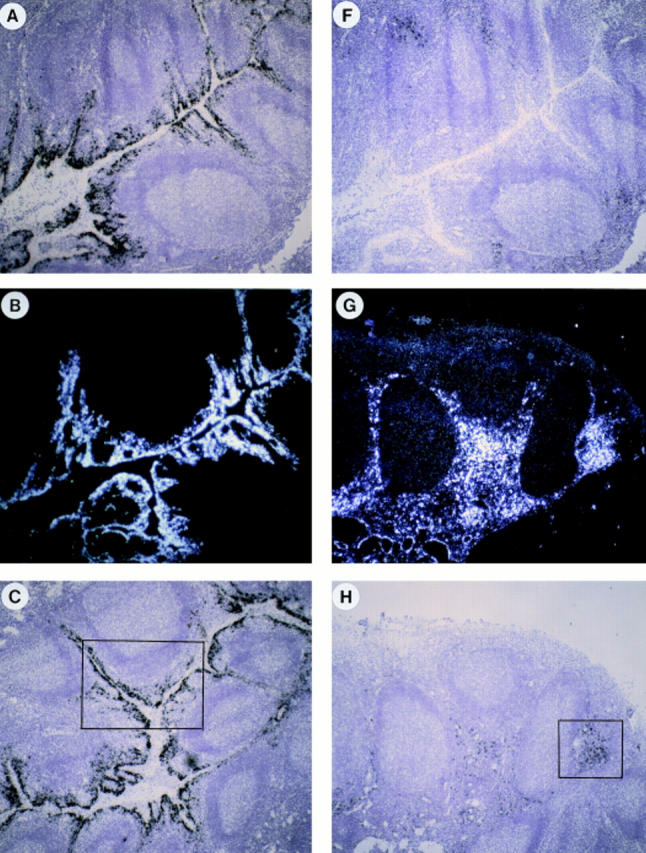
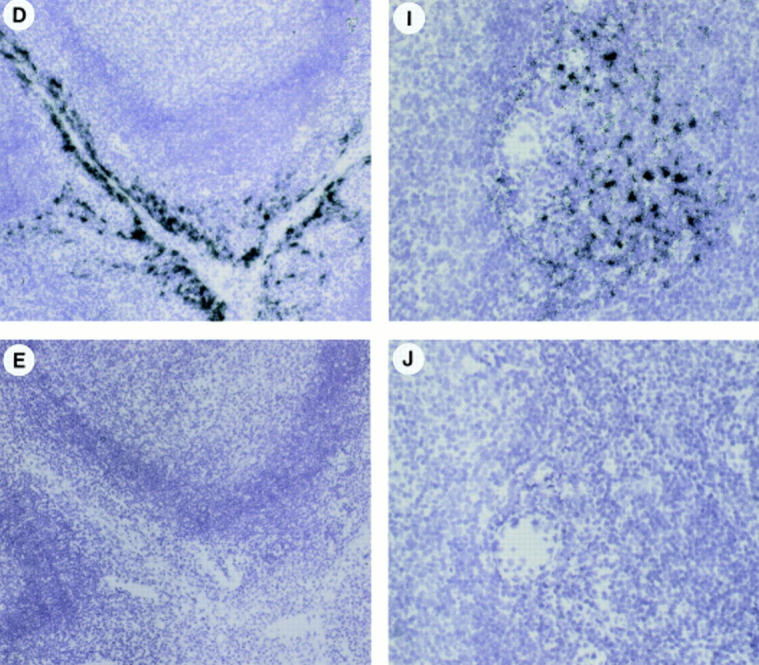
In vivo MIP-3α is expressed in inflamed epithelium and MIP-3β within T cell–rich areas of tonsils. Human tonsil sections were hybridized with antisense (A–D, F–I) and sense (E and J) 35S-labeled RNA probes for MIP-3α (A–E) and MIP-3β (F–J), and were exposed for 24 d. A and F, D and E, and I and J are serial cryostat sections. D and I are higher magnification of inset areas of C and H, respectively. (B and G) Darkfield illuminations are the same areas shown in A and H, respectively. MIP-3α mRNA is strongly expressed within epithelial crypts, no signal is seen within the T cell–rich areas nor the B cell–rich areas (A–C). MIP-3α mRNA is expressed in cells lining the epithelial crypts (D). MIP-3β mRNA is detected within the T cell– rich areas, no signal is seen within the B cell follicles nor in the epithelial crypts (F–H). MIP-3β mRNA is strongly expressed in scattered cells spread over the T cell areas around blood vessels (I). The sense probes do not generate background hybridization (E and J). Original magnification: A–C and F–H, ×40; D and E, ×100; (I and J), ×200.
Therefore, MIP-3α expression is restricted to inflamed epithelium at the site of antigen entry, where immature DCs should be recruited. In contrast, MIP-3β is only detected in paracortical areas, where mature IDCs home and generate primary T cell responses.
Discussion
Chemokine responsiveness appears to be one important functional feature that characterizes the stage of DC maturation. Immature DCs respond to MIP-3α, RANTES, and MIP-1α. Upon maturation, DCs lose their responsiveness to these chemokines to become sensitive to a single chemokine, MIP-3β.
Taking advantage of culture conditions that tightly control DC maturation, we have found that immature DCs, which very efficiently capture and process the antigens, migrate in response to MIP-1α, RANTES, and MIP-3α, in line with previous reports (34–36, 45). But in agreement with recent observations (60), mature DCs that have lost their aptitude to internalize antigens (21, 44, 61) cannot be recruited by these chemokines anymore. However, along with the ability to prime naive T cells, they have acquired responsiveness to MIP-3β. Furthermore, microenvironmental signals, like TNF-α and LPS, or T cell signals, such as CD40L, induce loss of response to MIP-3α, MIP-1α, and RANTES and acquisition of response to MIP-3β. These same signals induce DC maturation characterized by a rapid loss of antigen uptake capacities (21, 44, 62, 63). The link between DC maturation and migration pattern is further supported by the observation that the same signals, such as TNF-α and LPS that induce DC maturation in vitro, also trigger migration in vivo (20, 37–40). Thus, the modulation of chemokine responsiveness is part of a coordinated regulation of functions that defines the stage of DC maturation. Such a connection between maturation/activation and chemokine responsiveness is not unique to DCs. In particular, resting and activated (64) or naive and memory (65, 66) T cells are not recruited by the same chemokines. Also memory and naive B cells respond to stromal cell–derived factor (SDF)-1, whereas germinal center B cells do not (67). Altogether, these observations suggest that the recruitment of immune cells in vivo would be dependent on their stages of development and maturation/activation.
The mechanism underlying the modulation of chemokine responsiveness during activation/maturation of DC includes the regulation of the chemokine receptor expression. Therefore, the change in expression levels of CCR6 and CCR7 contributes to the functional shifts characterizing DC maturation. Other molecules linked to function are also regulated, particularly molecules involved in antigen uptake (Fc receptors, mannose receptors, and other lectins) or skin homing (E-cadherin), which are downregulated upon maturation. In contrast, molecules involved in migration (matrix metalloproteases [our observations]), in antigen presentation (MHC class II, protease/protease inhibitor, DC-LAMP [de Saint Vis, B., manuscript in preparation), in costimulation (CD80, CD86) (68) or in T cell commitment (IL-10, IL-12) (69) are upregulated. CCR6 gene expression is progressively downregulated on CD34+-derived DCs after activation/maturation with TNF-α, LPS, or CD40L. The downregulation of CCR6 mRNA expression upon DC activation is in line with a previous study showing downregulation of CCR1, CCR2, and CCR5 mRNA on monocytes after TNF-α or LPS activation (70). Also, on T cells, CD3 activation induces a rapid loss of CCR1 and CCR2 (64) and PHA activation induces downregulation of CCR6 (47). However, in addition to the downregulation of CCR6 mRNA expression, which is not completely extinct on mature DCs, other mechanisms might be involved in the loss of MIP-3α responsiveness (31). Signals inducing maturation might directly interfere with CCR6 signalization or indirectly cause CCR6 desensitization through engagement by endogenous MIP-3α, which is effectively produced by DCs upon CD40 activation (our observations).
In contrast to CCR6, CCR7 mRNA, absent on immature DCs, is induced upon maturation and the rapidity of this induction (detected within 3 h) probably reflects a direct effect of DC activation through CD40 or TNFR. Similar activation-dependent upregulation of CCR7 had been previously reported on T and B cells (71, 72). In this context, CCR7 expression is also induced on B and T cells after EBV infection (72) or herpesvirus infection (73), respectively. Furthermore, the expression of other receptors is activation-dependent. In particular, on T cells, expression of CCR1, CCR2A, and CCR2B is IL-2 dependent (64), and CXCR4 expression is induced by PHA activation (66). In addition, IL-2 increases the response of NK cells to chemokines (74, 75).
Most of the known chemokines bind several receptors. However, with regards to the correlation between the migration profiles, the expression of chemokine receptors, and Ca2+ flux responses, it is likely that CCR6 and CCR7 are the major functional receptors for MIP-3α and MIP-3β, respectively, expressed on DCs. MIP-3α response was observed on immature CD34+-derived DCs, but not on monocyte-derived DCs, in line with absence of CCR6 expression (45, 46). The reason for this lack of CCR6 expression is under investigation and might reflect either that monocyte-derived DCs represent a different DC subset, or that IL-4 blocks CCR6 expression.
Although MIP-3α appears as the most potent chemokine in inducing migration of immature CD34+-derived DCs, the DC population responding to MIP-3α in vivo has not yet been identified. Immature CD11c+ blood DCs are unresponsive, but Langerhans cells or their direct precursors (CD34+CLA+; reference 76) represent potential target populations. In this regard, precursor cells (days 4–6 of culture) are responsive to low concentrations of MIP-3α (1–10 ng/ml) compared with their more differentiated but still immature progenies (day 10 of culture). In contrast, the response to MIP-3β appears to be a general feature of mature DCs, including monocyte-derived and CD11c+ blood DCs.
The biological activities of MIP-3α and MIP-3β on other immune cells have been poorly characterized thus far. MIP-3α and MIP-3β have been shown to induce arrest of lymphocytes rolling under flow conditions, MIP-3α acting mainly on memory CD4 T cells and MIP-3β on most lymphocytes (77). Furthermore, MIP-3α induces migration of T cells but is inactive on monocytes and neutrophils (reference 45 and our observations) and MIP-3β attracts T and B lymphocytes but not granulocytes and monocytes (78).
Regarding in vivo expression, MIP-3α mRNA is mainly expressed in lung, appendix, liver, and some lymphoid organs (47, 50–52) and by many cell types both of hematopoietic and nonhematopoietic (fibroblasts, keratinocytes, endothelial cells) origins (47, 50–52, and our observations). Moreover, the production of MIP-3α is induced by inflammatory stimuli such as LPS or TNF-α by endothelial cells and monocytes (50, 52, and our observations). Now, we show by in situ hybridization that MIP-3α is expressed in tonsil by inflamed epithelium, a site known to be infiltrated by immature DCs. In contrast, the expression of MIP-3β mRNA is restricted to lymphoid organs and to cells of hematopoietic origin, in particular DCs as well as B lymphocytes and monocytes (49, 52, and our observations). In tonsil, MIP-3β mRNA as detected by in situ hybridization, is expressed by scattered cells within T cell–rich areas where mature IDCs home.
Thus, it is tempting to speculate that upon local injury, immature DCs or their precursors, expressing CCR6, would be attracted to the site of inflammation through the local production of chemokines like MIP-3α. Such accumulation of DCs has been observed in the airway epithelium of rat after intratracheal antigen instillation (3). After antigen uptake, TNF-α– or LPS-induced maturation would turn off MIP-3α responsiveness, thereby enabling emigrating DCs to escape the local gradient of MIP-3α. Simultaneous acquisition of CCR7 would drive maturing DCs into the paracortical area of lymphoid organs in response to the production of MIP-3β by cells spread over the T cell zone. As MIP-3β can attract mature DCs and lymphocytes (77, 78), it is likely to play a key role in helping antigen-loaded DCs to meet specific T cells.
Acknowledgments
We are grateful to I. Durand and E. Garcia for FACS® sorting; C. Massacrier for cell purification; T. Schall and J. Hedrick for scientific and technical expertise, S. Bourdarel and M. Vatan for editorial assistance; D. Lepot for the setting of the image analyzer and of the spectrofluorimeter; and the doctors and colleagues from clinics and hospitals in Lyon who provided us with umbilical cord blood samples and tonsils. We would like to address a special dedication to express all our gratitude to Dr. J. Chiller for his support in this specific project.
Abbreviations used in this paper
- CCR
CC chemokine receptor
- DC
dendritic cell
- HPC
hematopoietic progenitor cell
- IDC
interdigitating cell
- MCP
monocyte chemotactic protein
- MIP
macrophage inflammatory protein
- RANTES
regulated on activation, normal T cell expressed and secreted
- rh
recombinant human
- RT
reverse transcriptase
Footnotes
M.C. Dieu is recipient of a grant from Fondation Marcel Mérieux, Lyon, France.
M.-C. Dieu and B. Vanbervliet contributed equally to this paper.
References
- 1.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan G, Walsh G, Guido LS, Meyn P, Burkhardt RA, Abalos RM, Barker J, Frindt PA, Fajardo TT, Celona R, Cohn ZA. Novel responses of human skin to intradermal recombinant granulocyte/macrophage-colony–stimulating factor: Langerhans cell recruitment, keratinocyte growth, and enhanced wound healing. J Exp Med. 1992;175:1717–1728. doi: 10.1084/jem.175.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McWilliam AS, Nelson D, Thomas JA, Holt PG. Rapid dendritic cell recruitment is a hallmark of the acute inflammatory response at mucosal surfaces. J Exp Med. 1994;179:1331–1336. doi: 10.1084/jem.179.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inaba K, Schuler G, Witmer MD, Valinksy J, Atassi B, Steinman RM. Immunologic properties of purified epidermal Langerhans cells. Distinct requirements for stimulation of unprimed and sensitized T lymphocytes. J Exp Med. 1986;164:605–613. doi: 10.1084/jem.164.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Streilein JW, Grammer SF. In vitro evidence that Langerhans cells can adopt two functionally distinct forms capable of antigen presentation to T lymphocytes. J Immunol. 1989;143:3925–3933. [PubMed] [Google Scholar]
- 6.Romani N, Koide S, Crowley M, Witmer-Pack M, Livingstone AM, Fathman CG, Inaba K, Steinman RM. Presentation of exogenous protein antigens by dendritic cells to T cell clones. Intact Protein is presented best by immature, epidermal Langerhans cells. J Exp Med. 1989;169:1169–1178. doi: 10.1084/jem.169.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puré E, Inaba K, Crowley MT, Tardelli L, Witmer-Pack MD, Ruberti G, Fathman G, Steinman RM. Antigen processing by epidermal Langerhans cells correlates with the level of biosynthesis of MHC class II molecules and expression of invariant chain. J Exp Med. 1990;172:1459–1469. doi: 10.1084/jem.172.5.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuler G, Steinman RM. Murine epidermal Langerhans cells mature into potent immunostimulatory dendritic cells in vitro. J Exp Med. 1985;161:526–546. doi: 10.1084/jem.161.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Austyn JM, Kupiec-Weglinski JW, Hankins DF, Morris PJ. Migration patterns of dendritic cells in the mouse. Homing to T cell–dependent areas of spleen, and binding within marginal zone. J Exp Med. 1988;167:646–651. doi: 10.1084/jem.167.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kupiec-Weglinski JW, Austyn JM, Morris PJ. Migration patterns of dendritic cells in the mouse. Traffic from blood, and T cell–dependent and independent entry to lymploid tissues. J Exp Med. 1988;167:632–645. doi: 10.1084/jem.167.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen CP, Steinman RM, Witmer-Pack MD, Hankins DF, Morris PJ, Austyn JM. Migration and maturation of Langerhans cells in skin transplants and explants. J Exp Med. 1990;172:1483–1494. doi: 10.1084/jem.172.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fossum S. Lymph-borne dendritic leukocytes do not recirculate, but enter the lymph node paracortex to become interdigitating cells. Scand J Immunol. 1988;27:97–105. doi: 10.1111/j.1365-3083.1988.tb02326.x. [DOI] [PubMed] [Google Scholar]
- 13.Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P. Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. J Exp Med. 1987;166:1654–1667. doi: 10.1084/jem.166.6.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kripke ML, Munn CG, Jeevan A, Tang J-M, Bucana C. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J Immunol. 1990;145:2833–2838. [PubMed] [Google Scholar]
- 15.Inaba K, Metlay JP, Crowley MT, Steinman RM. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J Exp Med. 1990;172:631–640. doi: 10.1084/jem.172.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu LM, MacPherson GG. Antigen acquisition by dendritic cells: intestinal dendritic cells acquire antigen administered orally and can prime naive T cells in vivo. J Exp Med. 1993;177:1299–1307. doi: 10.1084/jem.177.5.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sornasse T, Flamand V, De Becker G, Bazin H, Tielemans F, Thielemans K, Urbain J, Leo O, Moser M. Antigen-pulsed dendritic cells can efficiently induce an antibody response in vivo. J Exp Med. 1992;175:15–21. doi: 10.1084/jem.175.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heufler C, Koch F, Schuler G. Granulocyte/ macrophage colony–stimulating factor and interleukin 1 mediate the maturation of murine epidermal Langerhans cells into potent immunostimulatory dendritic cells. J Exp Med. 1988;167:700–705. doi: 10.1084/jem.167.2.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Streilein JW, Grammer SF, Yoshikawa T, Demidem A, Vermeer M. Functional dichotomy between Langerhans cells that present antigen to naive and memory/ effector T lymphocytes. Immunol Rev. 1990;117:159–184. doi: 10.1111/j.1600-065x.1990.tb00572.x. [DOI] [PubMed] [Google Scholar]
- 20.De Smedt T, Pajak B, Muraille E, Lespagnard L, Heinen E, De Baetselier P, Urbain J, Leo O, Moser M. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J Exp Med. 1996;184:1413–1424. doi: 10.1084/jem.184.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol. 1997;9:10–16. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- 23.Romani N, Lenz A, Glassl H, Stossel H, Stanzl U, Majdic O, Fritsch P, Schuler G. Cultured human Langerhans cells resemble lymphoid dendritic cells in phenotype and function. J Invest Dermatol. 1989;93:600–609. doi: 10.1111/1523-1747.ep12319727. [DOI] [PubMed] [Google Scholar]
- 24.Larsen CP, Ritchie SC, Pearson TC, Linsley PS, Lowry RP. Functional expression of the costimulatory molecule, B7/BB1, on murine dendritic cell populations. J Exp Med. 1992;176:1215–1220. doi: 10.1084/jem.176.4.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Doherty U, Steinman RM, Peng M, Cameron PU, Gezelter S, Kopeloff I, Swiggard WJ, Pope M, Bhardwaj N. Dendritic cells freshly isolated from human blood express CD4 and mature into typical immunostimulatory dendritic cells after culture in monocyte-conditioned medium. J Exp Med. 1993;178:1067–1078. doi: 10.1084/jem.178.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teunissen MBM, Wormeester J, Krieg SR, Peters PJ, Vogels IMC, Kapsenberg ML, Bos JD. Human epidermal Langerhans cells undergo profound morphological and phenotypical changes during in vitro culture. J Invest Dermatol. 1990;94:166–173. doi: 10.1111/1523-1747.ep12874439. [DOI] [PubMed] [Google Scholar]
- 27.Inaba K, Witmer-Pack M, Inaba M, Hathcock KS, Sakuta H, Azuma M, Yagita H, Okumura K, Linsley PS, Ikehara S, Muramatsu S, et al. The tissue distribution of the B7-2 costimulator in mice: abundant expression on dendritic cells in situ and during maturation in vitro. J Exp Med. 1994;180:1849–1860. doi: 10.1084/jem.180.5.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oppenheim JJ. Overview of chemokines. Adv Exp Med Biol. 1993;351:183–186. doi: 10.1007/978-1-4615-2952-1_19. [DOI] [PubMed] [Google Scholar]
- 29.Schall TJ, Bacon KB. Chemokines, leucocyte trafficking, and inflammation. Curr Opin Immunol. 1994;6:865–873. doi: 10.1016/0952-7915(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 30.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 31.Baggiolini M, DeWold B, Moser B. Interleukin-8 and related chemotactic cytokines-CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- 32.Premack BP, Schall TJ. Chemokine receptors: gateways to inflammation and infection. Nat Med. 1996;2:1174–1178. doi: 10.1038/nm1196-1174. [DOI] [PubMed] [Google Scholar]
- 33.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 34.Sozzani S, Sallusto F, Luini W, Zhou D, Piemonti L, Allavena P, van Damme J, Valitutti S, Lanzavecchia A, Mantovani A. Migration of dendritic cells in response to formyl peptides, C5a, and a distinct set of chemokines. J Immunol. 1995;155:3292–3295. [PubMed] [Google Scholar]
- 35.Sozzani S, Luini W, Borsatti A, Polentarutti N, Zhou D, Piemonti L, D'Amico G, Power CA, Wells TN, Gobbi M, Allavena P, Mantovani A. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J Immunol. 1997;159:1993–2000. [PubMed] [Google Scholar]
- 36.Xu LL, Warren MK, Rose WL, Gong W, Wang JM. Human recombinant monocyte chemotactic protein and other CC chemokines bind and induce directional migration of dendritic cells in vitro. J Leukocyte Biol. 1996;60:365–371. doi: 10.1002/jlb.60.3.365. [DOI] [PubMed] [Google Scholar]
- 37.MacPherson GG, Jenkins CD, Stem MJ, Edwards C. Endotoxin-mediated dendritic cell release from the intestine. Characterization of released dendritic cells and TNF dependence. J Immunol. 1995;154:1317–1322. [PubMed] [Google Scholar]
- 38.Roake JA, Rao AS, Morris PJ, Larsen CP, Hankins DF, Austyn JM. Dendritic cell loss from nonlymphoid tissues after systemic administration of lipopolysaccharide, tumor necrosis factor, and interleukin 1. J Exp Med. 1995;181:2237–2247. doi: 10.1084/jem.181.6.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cumberbatch M, Kimber I. Dermal tumour necrosis factor-alpha induces dendritic cell migration to draining lymph nodes, and possibly provides one stimulus for Langerhans' cell migration. Immunology. 1992;75:257–263. [PMC free article] [PubMed] [Google Scholar]
- 40.Cumberbatch M, Kimber I. Tumour necrosis factor-alpha is required for accumulation of dendritic cells in draining lymph nodes and for optimal contact sensitization. Immunology. 1995;84:31–35. [PMC free article] [PubMed] [Google Scholar]
- 41.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-α cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360:258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 42.Caux C, Vanbervliet B, Massacrier C, Dezutter-Dambuyant C, de Saint-Vis B, Jacquet C, Yoneda K, Imamura S, Schmitt D, Banchereau J. CD34+hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF+TNF-α. J Exp Med. 1996;184:695–706. doi: 10.1084/jem.184.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caux, C., C. Dezutter-Dambuyant, Y.J. Liu, and J. Banchereau. 1998. Isolation and propagation of human dendritic cells. In Methods in Microbiology: Immunological Methods. Volume 25. D. Kabelitz and K. Ziegler, editors. Academic Press Ltd. 506–538.
- 44.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony–stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor α. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Power CA, Church DJ, Meyer A, Alouani S, Proudfoot AE, Clark-Lewis I, Sozzani S, Mantovani A, Wells TN. Cloning and characterization of a specific receptor for the novel CC chemokine MIP-3α from lung dendritic cells. J Exp Med. 1997;186:825–835. doi: 10.1084/jem.186.6.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greaves DR, Wang W, Dairaghi DJ, Dieu MC, de Saint-Vis B, Franz-Bacon K, Rossi D, Caux C, McClanahan T, Gordon S, et al. CCR6, a CC chemokine receptor that interacts with macrophage inflammatory protein 3α and is highly expressed in human dendritic cells. J Exp Med. 1997;186:837–844. doi: 10.1084/jem.186.6.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baba M, Imai T, Nishimura M, Kakizaki M, Takagi S, Hieshima K, Nomiyama H, Yoshie O. Identification of CCR6, the specific receptor for a novel lymphocyte-directed CC chemokine LARC. J Biol Chem. 1997;272:14893–14898. doi: 10.1074/jbc.272.23.14893. [DOI] [PubMed] [Google Scholar]
- 48.Liao F, Alderson R, Su J, Ullrich SJ, Kreider BL, Farber JM. STRL22 is a receptor for the CC chemokine MIP-3α. Biochem Biophys Res Commun. 1997;236:212–217. doi: 10.1006/bbrc.1997.6936. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida R, Imai T, Hieshima K, Kusuda J, Baba M, Kitaura M, Nishimura M, Kakizaki M, Nomiyama H, Yoshie O. Molecular cloning of a novel human CC chemokine EBI1-ligand chemokine that is a specific functional ligand for EBI1, CCR7. J Biol Chem. 1997;272:13803–13809. doi: 10.1074/jbc.272.21.13803. [DOI] [PubMed] [Google Scholar]
- 50.Hromas R, Gray PW, Chantry D, Godiska R, Krathwohl M, Fife K, Bell GI, Takeda J, Aronica S, Gordon M, et al. Cloning and characterization of exodus, a novel beta-chemokine. Blood. 1997;89:3315–3322. [PubMed] [Google Scholar]
- 51.Hieshima K, Imai T, Opdenakker G, Van Damme J, Kusuda J, Tei H, Sakaki Y, Takatsuki K, Miura R, Yoshie O, Nomiyama H. Molecular cloning of a novel human CC chemokine liver and activation-regulated chemokine (LARC) expressed in liver. Chemotactic activity for lymphocytes and gene localization on chromosome 2. J Biol Chem. 1997;272:5846–5853. doi: 10.1074/jbc.272.9.5846. [DOI] [PubMed] [Google Scholar]
- 52.Rossi DL, Vicari AP, Franz-Bacon K, McClanahan TK, Zlotnik A. Identification through bioinformatics of two new macrophage proinflammatory human chemokines: MIP-3alpha and MIP-3beta. J Immunol. 1997;158:1033–1036. [PubMed] [Google Scholar]
- 53.Garrone P, Neidhardt EM, Garcia E, Galibert L, van Kooten C, Banchereau J. Fas ligation induces apoptosis of CD40-activated human B lymphocytes. J Exp Med. 1995;182:1265–1273. doi: 10.1084/jem.182.5.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caux C, Saeland S, Favre C, Duvert V, Mannoni P, Banchereau J. Tumor necrosis factor-alpha strongly potentiates interleukin-3 and granulocyte-macrophage colony-stimulating factor–induced proliferation of human CD34+ hematopoietic progenitor cells. Blood. 1990;75:2292–2298. [PubMed] [Google Scholar]
- 55.Caux C, Massacrier C, Vanbervliet B, Dubois B, van Kooten C, Durand I, Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J Exp Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grouard G, Durand I, Filgueira L, Banchereau J, Liu YJ. Dendritic cells capable of stimulating T cells in germinal centers. Nature. 1996;384:364–367. doi: 10.1038/384364a0. [DOI] [PubMed] [Google Scholar]
- 57.Bacon KB, Camp RDR, Cunningham FM, Woollard PM. Contrasting in vitro lymphocyte chemotactic activity of the hydroxyl enantiomers of 12-hydroxy-5, 8, 10, 14-cicosatetraenoic acid. Br J Pharmacol. 1988;95:966–974. doi: 10.1111/j.1476-5381.1988.tb11727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 59.Peuchmaur M, Emilie D, Crevon MC, Solal-Celigny P, Maillot MC, Lemaigre G, Galanaud P. IL-2 mRNA expression in Tac-positive malignant lymphomas. Am J Pathol. 1990;136:383–390. [PMC free article] [PubMed] [Google Scholar]
- 60.Delgado E, Finkel V, Baggiolini M, Clark-Lewis I, Mackay CR, Steinman RM, Granelli-Piperno A. Mature dendritic cells respond to SDF-1 but not to several beta-chemokines. Immunobiology. 1998;198:2–12. doi: 10.1016/s0171-2985(98)80073-9. [DOI] [PubMed] [Google Scholar]
- 61.Caux C, Massacrier C, Vanbervliet B, Dubois B, Durand I, Cella M, Lanzavecchia A, Banchereau J. CD34+hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to granulocyte-macrophage colony–stimulating factor plus tumor necrosis factor α: II. Functional analysis. Blood. 1997;90:1458–1470. [PubMed] [Google Scholar]
- 62.Sallusto F, Nicolo C, de Maria R, Corinti S, Testi R. Ceramide inhibits antigen uptake and presentation by dendritic cells. J Exp Med. 1996;184:2411–2416. doi: 10.1084/jem.184.6.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782–787. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 64.Loetscher P, Seitz M, Baggiolini M, Moser B. Interleukin-2 regulates CC chemokine receptor expression and chemotactic responsiveness in T lymphocytes. J Exp Med. 1996;184:569–577. doi: 10.1084/jem.184.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adema GJ, Hartgers F, Verstraten R, de Vries E, Marland G, Menon S, Foster J, Xu Y, Nooyen P, McClanahan T, et al. A dendritic-cell–derived CC chemokine that preferentially attracts naive T cells. Nature. 1997;387:713–717. doi: 10.1038/42716. [DOI] [PubMed] [Google Scholar]
- 66.Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA. 1997;94:1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bleul CC, Schultze JL, Springer TA. B lymphocyte chemotaxis regulated in association with microanatomic localization, differentiation state, and B cell receptor engagement. J Exp Med. 1998;187:753–762. doi: 10.1084/jem.187.5.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caux C, Vanbervliet B, Massacrier C, Azuma M, Okumura K, Lanier LL, Banchereau B. B70/B7-2 is identical to CD86 and is the major functional ligand for CD28 expressed on human dendritic cells. J Exp Med. 1994;180:1841–1847. doi: 10.1084/jem.180.5.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Saint-Vis B, Fugier-Vivier I, Massacrier C, Vanbervliet B, Aït-Yahia S, Banchereau J, Liu YJ, Lebecque S, Caux C. The cytokine profile expressed by human dendritic cells is dependent on cell subtype and mode of activation. J Immunol. 1998;160:1666–1676. [PubMed] [Google Scholar]
- 70.Sica A, Saccani A, Borsatti A, Power CA, Wells TN, Luini W, Polentarutti N, Sozzani S, Mantovani A. Bacterial lipopolysaccharide rapidly inhibits expression of C-C chemokine receptors in human lymphocytes. J Exp Med. 1997;185:969–974. doi: 10.1084/jem.185.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schweickart VL, Raport CJ, Godiska R, Byers MG, Eddy RLJ, Shows TB, Gray PW. Cloning of human and mouse EBI1, a lymphoid-specific G-protein– coupled receptor encoded on human chromosome 17q12-q21.2. Genomics. 1994;23:643–650. doi: 10.1006/geno.1994.1553. [DOI] [PubMed] [Google Scholar]
- 72.Birkenbach M, Josefsen K, Yalamanchili R, Lenoir G, Kieff E. Epstein-Barr virus–induced genes: first lymphocyte-specific G protein–coupled peptide receptors. J Virol. 1993;67:2209–2220. doi: 10.1128/jvi.67.4.2209-2220.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hasegawa H, Utsunomiya Y, Yasukawa M, Yanagisawa K, Fujita S. Induction of G protein–coupled peptide receptor EBI 1 by human herpesvirus 6 and 7 infection in CD4+T cells. J Virol. 1994;68:5326–5329. doi: 10.1128/jvi.68.8.5326-5329.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Godiska R, Chantry D, Raport CJ, Sozzani S, Allavena P, Leviten D, Mantovani A, Gray PW. Human macrophage-derived chemokine (MDC), a novel chemoattractant for monocytes, monocyte-derived dendritic cells, and natural killer cells. J Exp Med. 1997;185:1595–1604. doi: 10.1084/jem.185.9.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Polentarutti N, Allavena P, Bianchi G, Giardina G, Basile A, Sozzani S, Mantovani A, Introna M. IL-2–regulated expression of the monocyte chemotactic protein-1 receptor (CCR2) in human NK cells: characterization of a predominant 3.4-kilobase transcript containing CCR2B and CCR2A sequences. J Immunol. 1997;158:2689–2694. [PubMed] [Google Scholar]
- 76.Strunk D, Rappersberger K, Egger C, Strobl H, Krömer E, Elbe A, Maurer D, Stingl G. Generation of human dendritic cells/Langerhans cells from circulating CD34+hematopoietic progenitor cells. Blood. 1996;87:1292–1302. [PubMed] [Google Scholar]
- 77.Campbell JJ, Hedrick J, Zlotnik A, Siani MA, Thompson DA, Butcher EC. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 1998;279:381–384. doi: 10.1126/science.279.5349.381. [DOI] [PubMed] [Google Scholar]
- 78.Kim CH, Pelus LM, White JR, Applebaum E, Johanson K, Broxmeyer HE. CK beta-11/macrophage inflammatory protein-3 beta/EB11-ligand chemokine is an efficacious chemoattractant for T and B cells. J Immunol. 1998;160:2418–2424. [PubMed] [Google Scholar]