

Abstract
The ability of influenza virus to evade immune surveillance by neutralizing antibodies (Abs) directed against its variable surface antigens provides a challenge to the development of effective vaccines. CD8+ cytotoxic T lymphocytes (CTLs) restricted by class I major histocompatibility complex molecules are important in establishing immunity to influenza virus because they recognize internal viral proteins which are conserved between multiple viral strains. In contrast, protective Abs are strain-specific. However, the precise role of effector CD8+ CTLs in protection from influenza virus infection, critical for understanding disease pathogenesis, has not been well defined. In transgenic mice with a very high frequency of antiinfluenza CTL precursors, but without protective Abs, CD8+ CTLs conferred protection against low dose viral challenge, but exacerbated viral pathology and caused mortality at high viral dose. The data suggest a dual role for CD8+ CTLs against influenza, which may present a challenge to the development of effective CTL vaccines. Effector mechanisms used by CD8+ CTLs in orchestrating clearance of virus and recovery from experimental influenza infection, or potentiation of lethal pathology, are discussed.
Keywords: CD8+ cytotoxic T lymphocytes, influenza A virus, T cell receptor–transgenic mice, interferon γ, influenza viral pneumonia
Influenza A virus possesses the ability to modify its surface antigens, hemagglutinin and neuraminidase (1–4), thereby permitting sequential reinfections of the same host. Such antigenic variation leads to worldwide epidemics and has prevented control of disease by vaccines designed to induce neutralizing Abs. The majority of CD8+ CTLs are directed against conserved internal viral proteins such as nucleoprotein (NP)1 of influenza A virus. These CD8+ CTLs are broadly cross-reactive, and recognize all major virus subtypes (for a review, see references 5–8). Thus, much effort has been directed towards development of a vaccine capable of inducing CD8+ CTL memory that recognizes peptide epitopes of conserved viral proteins. Since replication of mammalian influenza viruses is restricted to epithelial cells of the respiratory tract, and systemic exposure of the immune system to influenza consequently is limited (9, 10), the contribution of CD8+ CTLs in primary antiviral responses is not inherently obvious. The recurrent nature of influenza viral infections in humans (11) suggests that immunity mediated by CD8+ CTLs directed at conserved internal viral proteins is transient or only partially effective. Thus, CTL memory cells, which occur in relatively high frequency after influenza infection (6, 12–14), have marginal impact on morbidity and mortality caused by reinfection with heterosubtypic virus strains in humans (15–17). Observations on the role of CD8+ CTLs in heterosubtypic immunity in animals give varying conclusions. Thus, virus-specific CD8+ CTLs protect against challenge with influenza A infection in mice devoid of mature B cells and Abs (18– 20). Similarly, cloned CTLs specific for NP of influenza A virus can passively transfer protection (21). On the other hand, active immunization with recombinant NP or with NP expressing vectors is only weakly protective (22–27).
In our studies, we have taken a new approach towards evaluation of the physiological features of CD8+ CTLs in influenza infection. Tracking in situ CTL effector functions has been technically challenging, due mainly to the very low frequency and high TCR diversity of antigen-specific CTLs in normal animals. To overcome this problem, we used transgenic (Tg) mice expressing a uniform type of αβ TCR heterodimer (αβ4/β11; termed F5-Tg) derived from an NP-specific T cell clone obtained from C57BL/10 mice (28, 29). The F5-Tg TCR recognizes the NP peptide (amino acids 366–374) of influenza A virus A/NT/60/68 (H3N2) presented by MHC-Db class I molecules, and is expressed in ∼90% of peripheral T cells. Therefore, the mice possess a high frequency of antiviral CTL precursor cells. By staining cells with Abs specific for Vβ11, CD8, and markers for T cell activation, responsive Tg-CTLs can be identified and characterized directly in situ. We demonstrate here that CD8+ CTLs directed against the conserved NP of influenza A virus in the absence of protective Abs can potently block viral replication in situ and either promote survival or exacerbate a lethal influenza pneumonia. These results provide a clear demonstration that protection and pathology induced by antiviral CD8+ CTLs represent different balance situations between a pathogen and the host's immune system. This consideration is especially important in the lung, where disruption of lung structure and pulmonary function can have devastating consequences.
Materials and Methods
Mice.
Mice transgenic or deficient for recombination activating gene 1 (RAG-1−/−) were maintained by breeding with C57BL/10 (H-2b) mice under specific pathogen–free conditions. F5-Tg mice were described previously (28, 29). RAG-1−/− mice were maintained by breeding with each other, or with F5-Tg in order to obtain F5-Tg mice deficient for RAG-1 (30, 31). Animals were kept and experiments were performed in accordance with the institutional animal welfare guidelines of the United Kingdom and the United States.
Viruses.
Stock virus of influenza A/NT/60/68 (H3N2) virus or the X31 (H3N2) reassortant influenza virus was grown in the allantoic cavity of 10-d-old embryonated hen eggs, and were free of bacterial, mycoplasma, and endotoxin contamination. X31 is a reassortant virus with external virion proteins of A/Aichi/2/68 (H3N2) and the internal NP of A/PR/8/34 (H1N1). This reassortant cannot be recognized by the Tg-CTLs due to alterations (372D→ E, 373A→ T) within the relevant peptide epitope (32). Virus was quantified in a plaque assay on Madin-Darby canine kidney (MDCK) cells obtained from the American Type Culture Collection (Rockville, MD; references 33 and 34).
Virus Titers in Lung Tissue.
Viral lung titers were determined by 10-fold serial dilution of tissue extracts, and tested for infectivity of MDCK cells in 96-well plates as detected by hemagglutinating activity in the supernatants after a 48-h incubation at 37°C and 5% CO2. Virus titers were estimated according the method of Reed and Muench (35); the threshold of virus detection in the MDCK assay is 102 TCID50 (50% tissue culture infectious dose)/g lung tissue. Lung extracts that were negative in the MDCK assay were further tested by inoculation of 50 μl of undiluted extract in the allantoic cavity of 10-d-old embryonated hen eggs; the threshold of detection in this system was ≤20 egg infectious doses per lung.
Detection of Antiviral Abs in Sera of Infected Mice.
The titer of virus-specific Abs in serum was assayed by ELISA as described previously (36). Sera of infected mice were tested on plates coated with 1 μg of purified A/NT/60/68 or X31 influenza virus (37).
Flow Cytometry.
Cells isolated by bronchoalveolar lavage (BAL) were stained directly with FITC- or PE-coupled reagents or indirectly with biotinylated Abs followed by streptavidin-Tricolor (Caltag Laboratories, Inc., South San Francisco, CA), and analyzed with a FACScan® (Becton Dickinson, San Jose, CA). mAbs were against mouse CD8 (clone 53-6.7), CD4 (clone GK1.5), TCRVβ11 (clone KT11), CD44 (clone IM-7), IL-2R (clone 7D4), L-selectin (clone MEL-14), and macrophages/monocytes (clone F4/80 or clone M1/70). The Abs were prepared from hybridoma cell lines or purchased from PharMingen (San Diego, CA).
Cytotoxicity Assay.
Ex vivo cytolytic activities in BAL were tested directly in a standard cytotoxicity assay as described (31, 38). BAL cells obtained from two mice were pooled before being assayed directly on EL-4 (H-2b) target cells infected with virus or loaded with a synthetic peptide (amino acids 366–374 NP of A/ NT/60/68), in a 5-h cytotoxicity test. Tg-CTLs from the same sample were detected directly by flow cytometric analysis, and percentages of specific lysis were calculated at the highest Tg-CTL to target cell ratio.
Treatment with Anti–IFN-γ mAb.
F5–RAG-1−/− mice infected with influenza A virus intranasally were injected intraperitoneally with XMG1.2 anti–IFN-γ mAb (2 mg/mouse; donated by Dr. P.C. Doherty, St. Jude Children's Research Hospital, Memphis, TN [39]), starting 1 d before infection and continuing every other day throughout the experiment. Protocols using the same mAb concentration and conditions have been shown to sufficiently neutralize the antiviral effects of IFN-γ in vivo (40, 41).
Histology.
Lung tissues fixed in 10% buffered formalin were paraffin embedded and sectioned. Each lung specimen was stained with hematoxylin and eosin, and subjected to gross and microscopic pathologic analysis.
Results
Protection against Influenza Virus Is Associated with Activities of Antiviral Abs, Whereas CTL Responses Play a Peripheral Role in Local Immunity
Intranasal administration of ≤106 PFU of A/NT/60/68 of influenza A virus to F5-Tg mice resulted in a pulmonary infection and associated pathology that was regularly resolved within 2 wk (Fig. 1, A, E, and I). Viral replication peaked between days 2 and 4, followed by a rapid decline in virus lung titers by day 8. This correlated with increased levels of serum IgM and IgG antiviral Abs. Note that F5-Tg mice contain considerable numbers of mature CD4+ Th cells selected in the thymus via endogenous TCR α chains (due to less stringent allelic exclusion) and that production of IgG requires interaction of B cells with virus-specific CD4+ T cells (31, 42, 43). Inoculation of a high dose (107 PFU) of A/NT/60/68 caused rapid spread of virus in the lung; ∼40% of the animals died (Fig. 1 A). Tg-CTLs isolated by BAL and from tissues of the pulmonary-associated lymphatic system were capable of recognizing and lysing virally infected target cells, or cells pulsed with the Tg-CTL peptide epitope in standard cytotoxicity assays (data not shown). Maximal cytolytic activities correlated with reduction of virus in lungs by days 6–8. No evidence of Tg-CTL activation was observed in cells obtained from spleen or non–pulmonary-associated lymphatic system tissues tested directly in CTL assay or by staining with activation markers (data not shown). A similar course of infection was observed in control C57BL/10 (H-2b) inbred mice (Fig. 1, B, F, and J). To directly assess the contribution of antiviral CTLs in protection against influenza, F5-Tg mice were infected with the X31 reassortant virus, which cannot be recognized by the Tg-CTLs. When X31-infected F5-Tg mice were compared with control C57BL/10 mice (Fig. 1, C, G, and K, versus D, H, and L), no significant differences in survival rate, viral replication in lungs, or virus-specific Ab titers were observed. The overall kinetics of virus decline differed slightly, in that virus was detectable in F5-Tg mice until day 12. These observations indicate that in the presence of antiviral Abs, host CTLs specific for NP of influenza virus play only a peripheral role in local immunity.
Figure 1.

Protection of mice against influenza A virus infection was observed by reduction of viral titers in lungs and survival rate, correlating with levels of antiviral Abs. F5-Tg or C57BL/10 control mice were infected with A/NT/60/68 (A and B, E and F, and I and J) or X31 (C and D, G and H, and K and L) influenza A viruses. (A–D) Mice were infected with 107 PFU (filled circles) or ≤106 PFU (filled triangles) of influenza A virus, and percent survival is shown for groups of 10–15 mice. (E–H) Titer of antiviral IgG (open circles) or IgM (filled circles) in serum of mice infected with 106 PFU of influenza A virus was determined. Values shown for Ab activity are mean log (ELISA titer) ± SEM of three mice. (I–L) Virus in lungs was measured in separate groups of mice infected with 106 PFU of influenza A virus. The virus titer is shown as mean log10 TCID50 per gram of lung ± SEM of three to five mice.
Do CTLs Protect a Host against Influenza Virus in the Absence of Protective Abs?
We next evaluated the role of CTLs in influenza viral infection more stringently by assessing their immunoreactivity in the absence of antiviral Abs using RAG-1–deficient F5 mice (F5–RAG-1−/−). The repertoire of peripheral lymphocytes of these mice consists only of Tg-CTLs (31). F5–RAG-1−/− and RAG-1−/− (the latter lacking both B and T cells) control mice were inoculated intranasally with varying doses of A/NT/60/68 or X31 influenza virus, and CTL functions were examined in the following ways.
CTLs Confer Protection or Contribute to Influenza Pathology Depending on the Magnitude of Pulmonary Viral Load.
The ability of CTLs to limit the severity of acute lung infection was evaluated by measuring survival of F5–RAG-1−/− mice after infection with A/NT/60/68 (Fig. 2, left). At the highest dose given (107 PFU), all mice died between days 2 and 6 (Fig. 2 A, left). A progressive delay in the time of death and increased survival rate were observed when viral inoculum was decreased (Fig. 2, B–E), with complete protection observed at ≤104 PFU. All control RAG-1−/− mice infected with A/NT/60/68 succumbed to viral disease (0% survival; Fig. 2, left). Similarly, all F5–RAG-1−/− and RAG-1−/− mice infected with X31 died during the first 20 d, with comparable kinetics (Fig. 2, right). The fact that F5–RAG-1−/− mice infected with 107 PFU of A/NT/ 60/68 died significantly faster (days 2–6; P <0.0001 by Wilcoxon test) than RAG-1−/− mice (days 6–24) but survived a relatively low dose of infection (≤104 PFU) suggests that, depending on the magnitude of pulmonary viral load, CTLs can either confer protection or contribute to pathology in influenza virus infection.
Figure 2.

In the absence of protective antiviral Abs, CTLs in the lung tissue conferred protection against low viral challenge, but exacerbated disease at high viral doses. F5–RAG-1−/− (filled circles) or RAG-1−/− (open circles) mice were infected with A/NT/60/68 (left) or X31 (right) influenza A viruses. Percent survival is shown for groups of 10–15 mice. The virus was administrated intranasally at a dose of (A) 107, (B) 106, (C) 105, (D) 104, or (E) 102 PFU. Percent survival was significantly greater in F5–RAG-1−/− mice infected with 107 (P <0.0001 by Wilcoxon test) or 105 PFU (P <0.0001) of A/NT/60/68 than in control infected RAG-1−/− mice (A and B, left). No significant differences (P >0.09) were observed between the same strains of mice infected with X31 (A and B, right).
Protection against Influenza Virus or Lethal Viral Pathology Are Related to the Success or Failure of CTLs to Control the Viral Infection.
Susceptibility to influenza virus, often lethal for mice, is closely associated with progressive pulmonary viral infection. Therefore, comparative studies with F5– RAG-1−/− and RAG-1−/− infected mice were performed, correlating survival rate (Fig. 2) with virus titers in lungs (Fig. 3). Kinetic profiles of viral replication in the lungs were determined by measuring maximal viral titers and virus clearance rates (Fig. 3, A–D). Protection (increased survival; see Fig. 2) against influenza virus correlated with lower maximal levels and rapid decline in viral titers. Thus, F5–RAG-1−/− mice infected with A/NT/60/68 were protected only if they controlled viral replication (Fig. 3, C and D, left). In contrast, high viral lung titer was seen in mice that succumbed to infection (compare Figs. 2 A and 3 A). 104 PFU intranasal (i.n.) of A/NT/60/68 was a critical dose; about one fourth of the infected mice failed to clear the virus and died, whereas the rest of the mice eliminated the virus and were protected (compare Figs. 2 C and 3 B). The experiments in this section suggest that CTLs confer protection against influenza by blocking in situ viral replication. Their failure to control viral infection is closely associated with fatal disease.
Figure 3.

CTLs are protective against lethal influenza if they are able to control viral replication in lung tissue at the onset of viral infection. Virus titers in lung tissue of F5–RAG-1−/− (filled circles) or RAG-1−/− (open circles) mice infected with A/NT/60/68 (left) or X31 (right) influenza A viruses are expressed as mean log10 TCID50 per gram of lung of three to five mice. Mice are infected intranasally at doses of (A) 107, (B) 105, (C) 103, or (D) 10 PFU.
Characterization of Transgenic CTLs in Lung Tissue under Conditions of a Low versus High Viral Challenge.
The phenotype and functional status of the inflammatory cells recovered by BAL from F5–RAG-1−/−or RAG-1−/− mice were examined (Fig. 4). Under all conditions tested, primary inflammatory reactions were similar, consisting mainly of macrophages/monocytes (positive for F4/80 and Mac-1a antigen; data not shown). The total number of cells recovered by BAL (Fig. 4, open symbols) increased rapidly (days 2–5) and peaked between days 4 and 8 with maximal values that correlated with the dose of infection. However, transgenic CTLs (Vβ11+CD8+; Fig. 4, filled symbols) were found only in F5–RAG-1−/− mice infected with A/NT/ 60/68 (Fig. 4, A–C). The kinetics of appearance of transgenic CTLs in the lungs revealed that transgenic CTLs were detected earlier in mice infected with 107 PFU (day 2) than with 104 or 102 PFU (days 3–5). This difference was confirmed in two further experiments (our unpublished observations). Transgenic CTLs isolated on days 2–16 from lungs of mice infected with different doses of A/NT/60/68 and analyzed by flow cytometry were blast-sized and displayed activation status profiles (upregulation of IL-2R and CD44 antigen and downregulation of L-selectin) compared with naive transgenic CTLs (data not shown). As expected, CTLs were efficient in lysing target cells loaded with relevant viral peptide (A/NT/60/68 NP-366-374). Thus, cells obtained by BAL from two mice were pooled and assayed directly on EL-4 (H-2b) target cells loaded with peptide in a cytotoxicity assay, and percentages of specific lysis were calculated at highest transgenic CTL to target cell ratio. The lytic activity in F5–RAG-1−/− mice infected with 107 PFU of A/NT/60/68 was 15% (day 4, ratio 2:1), 30% (day 5, ratio 12:1), and 50% (day 8, ratio 25:1) compared with animals infected with 102 PFU, which exhibited 10% (day 5, ratio 4:1), 25% (day 8, ratio 12:1), and 60% (day 12, ratio 25:1). The same effector cells tested on unloaded target cells displayed cytotoxicity <2% at the highest E/T ratio. In addition, lytic activity in BAL from control F5–RAG-1−/− mice infected with X31, or from RAG-1−/− mice infected with A/NT/60/68, was undetectable (<2%). Thus, transgenic CTLs in lungs of mice that succumbed to lethal influenza were functionally active. Control F5–RAG-1−/− mice infected with X31, or RAG-1−/− mice infected with A/NT/60/68 or X31, developed a progressive pulmonary inflammation, but transgenic CTLs were undetectable (Fig. 4 D, and data not shown). Finally, because of the short time span between appearance of transgenic CTLs in lungs and lethal outcome of viral disease (2–4 d), it is unlikely that transgenic CTL escape variants in infected mice are responsible for these results (44, 45). Likewise, our findings are not compatible with anergy (46–49) or clonal deletion of transgenic CTLs (50, 51) as possible mechanisms for the inability of F5–RAG-1−/− mice to control infection with a relatively high dose of A/NT/60/68.
Figure 4.
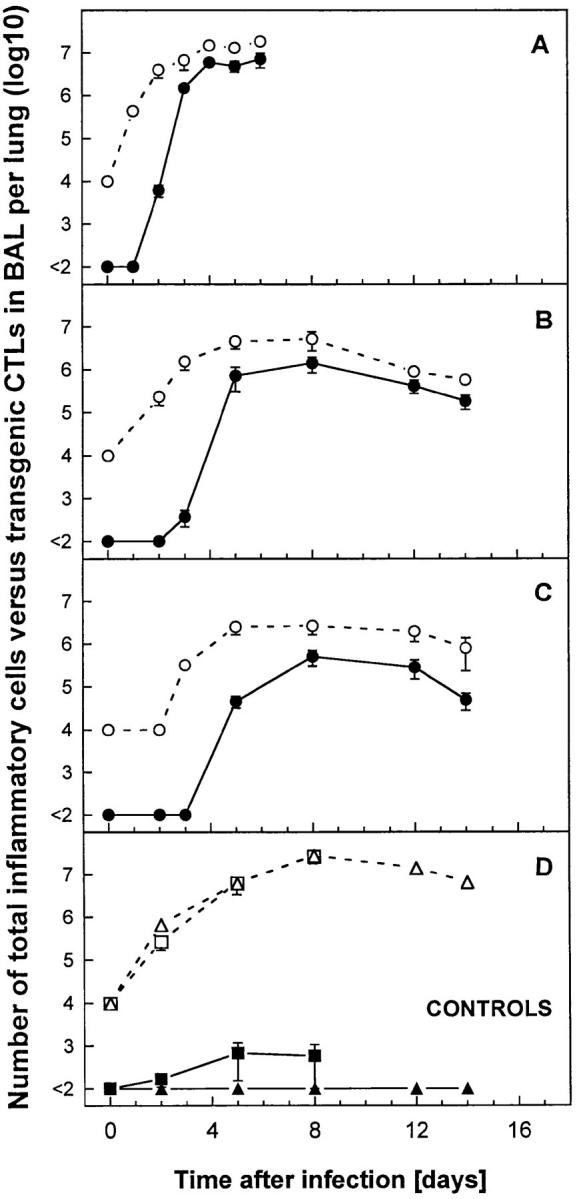
Kinetics of total inflammatory cells versus transgenic CTLs in BAL of mice infected with influenza A virus. F5–RAG-1−/− mice were infected intranasally with (A) 107, (B) 104, or (C) 102 PFU of A/ NT/60/68, or with (D) 107 PFU of control X31 virus (squares). In addition, (D) RAG-1−/− mice infected with 107 PFU of A/ NT/60/68 were included as controls (triangles). The numbers of inflammatory cells in BAL (open symbols) are indicated as mean ± SEM log10 per lung of three to five mice. BAL samples (total volume 1 ml per lung) containing <104 cells per ml (the limit of detection of our hemocytometer counting assay) were estimated as 104 cells per lung. Tg-CTLs (filled symbols) were detected in the same samples by staining cells with Abs specific for Vβ11, CD8 and analysis by flow cytometry. Absolute numbers of Tg-CTLs were calculated as percent transgenic positive cells by flow cytometry multiplied by total cell number. Populations <1% were considered undetectable.
Morphological Representations of CTL Activities In Vivo.
It is likely that the characteristics of cells obtained by BAL do not fully reflect the overall pulmonary inflammatory process. Therefore, lung tissue from control (uninfected) or virus-infected mice were analyzed histologically (Table 1). Hematoxylin and eosin–stained paraffin sections of lungs showed that the general course of lung pathology of F5– RAG-1−/− mice infected with A/NT/60/68 was partially influenced by the rate of pulmonary viral spread, but to a greater extent was determined by antiviral CTLs (Fig. 5). Indeed, Tg-CTLs in lungs of mice with a restricted viral infection (i.e., 102 PFU i.n.) tempered the severity of the disease (Fig. 5 A). Lung pathology was confined to a few foci of perivascular and peribronchial inflammation of mononuclear cells (macrophages/monocytes), containing numerous leukocytes/lymphoblasts. Although inflammation persisted beyond 2 wk, with gradual decline in magnitude, there was less evidence of epithelial necrosis and desquamation of affected tracheobronchiolar mucosa. In contrast, the activity of Tg-CTLs in lungs of mice with progressive viral infection (i.e., 107 PFU i.n.) had deleterious consequences for the host (Fig. 5 B). The entire architecture of lung tissue became profoundly altered within a few days as a result of extensive inflammation and edema, with thickening of intraalveolar septa and loss of alveoli, but with less evidence of hemorrhages. The pathologic process in control RAG-1−/− mice (107 PFU of A/NT/ 60/68) developed more slowly; there was less evidence of pathologic alterations in lung tissues during the first week of infection, and primary inflammatory reactions were confined to a few foci of infiltrating cells. However, in the course of infection the animals developed the characteristic features of fatal viral pneumonia (edematous lung tissues, congestion, and collapse of alveoli; data not shown). Lung tissues of F5–RAG-1−/− mice infected with X31 (107 PFU; Fig. 5 C) show the characteristic features of a progressive fatal pneumonia as described for F5–RAG-1−/− mice infected with 107 PFU of A/NT/60/68. Lung tissues of uninfected F5–RAG-1−/− mice were well aerated, without evidence of infiltrates or pathologic alterations (Fig. 5 D). Together, these results confirmed our initial observations suggesting a contribution of antiviral CTLs to pulmonary pathology as a result of overwhelming influenza viral infection.
Table 1.
Extent of the Pulmonary Inflammatory Process of TCR-transgenic Mice Infected with Influenza A Virus
| Mice | Virus | Time after infection | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 2 | Day 5 | Day 8 | Day 15 | Day 20 | ||||||||
| F5–RAG-1−/− | A/NT/60/68, 107 PFU | + | ++ | ++ | * | |||||||
| A/NT/60/68, 102 PFU | − | + | + | ± | ± | |||||||
| X31, 107 PFU | + | ++ | ++ | * | ||||||||
| RAG-1−/− | A/NT/60/68, 107 PFU | ± | + | ++ | ++ | ++ | ||||||
| X31, 107 PFU | + | ++ | ++ | * | ||||||||
++, Intensive pathology with cell inflammation encompassing several segments of lung tissues. Development of extensive lung edema and congestion. Thickened intraalveolar septa and loss of alveoli. +, Inflammatory reaction consisting of a few foci of peribronchiolar and perivascular infiltrates on medium and small airways. There was modest evidence of epithelial necrosis of affected tracheobronchiolar mucosa. ±, Lung tissue with a few infiltrates. No epithelial necrosis or desquamation. −, Lung tissue well aerated. No infiltrates or pathologic alteration of lung tissue.
, At the indicated time, all mice had died.
Figure 5.
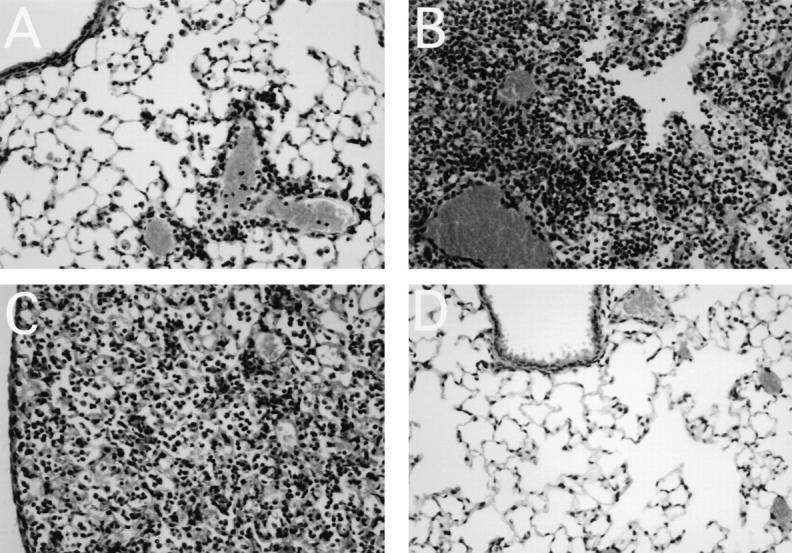
Histological examination of lung tissue. Lungs were taken on day 6 or 8 from F5–RAG-1−/− transgenic mice infected with (A) 102 PFU of A/NT/60/68, (B) 107 PFU of A/NT/60/68, or (C) 107 PFU of X31 influenza A virus. (D) Lung tissue of a control uninfected F5–RAG-1−/− mouse.
Effects of In Vivo Administration of Anti–IFN-γ mAb on the Mediation of Lethal and Sublethal Influenza by Antiviral CD8+ CTLs
To further define the mechanism(s) of virus-specific CD8+ T cell–mediated clearance or enhancement of inflammation, the effects of parenterally administrated anti– IFN-γ were examined. Although all F5-RAG mice infected with a high dose of A/NT/60/68 (107 PFU) died between days 2 and 6, a delay in the time of death and increased survival rate (∼50%) were observed when infected animals were treated with anti–IFN-γ mAb throughout the experiment (Fig. 6 A). Surprisingly, treated animals completely cleared virus from lung parenchyma by day 8 (Fig. 6 C). In contrast, control untreated infected mice were unable to eliminate the virus; however, significantly reduced viral lung titers were measured by day 6 and were maintained until the mice succumbed to infection (Fig. 6 C), indicating that antiviral CD8+ T cells were only partially efficient in controlling the infection. Treatment of mice with anti–IFN-γ mAb had no effect on the kinetics or magnitude of the effector Tg-CTL response in the lung parenchyma (Fig. 6 E). Identical total numbers of inflammatory cells were found in the BAL of anti–IFN-γ–treated mice and control mice (Fig. 6 G). However, histological analysis of the lung tissues revealed a restricted pattern of inflammation and significantly reduced pathologic features of pneumonia in the early stages of infection, with gradual decline in magnitude after anti–IFN-γ mAb treatment in comparison with control virus infected mice (data not shown). In mice given a sublethal dose of 102 PFU of influenza virus, blockade of IFN-γ had little effect on lethality, elimination of pulmonary virus, or kinetics and magnitude of Tg-CTL response in the lung parenchyma (Fig. 6, B, D, F, and H).
Figure 6.
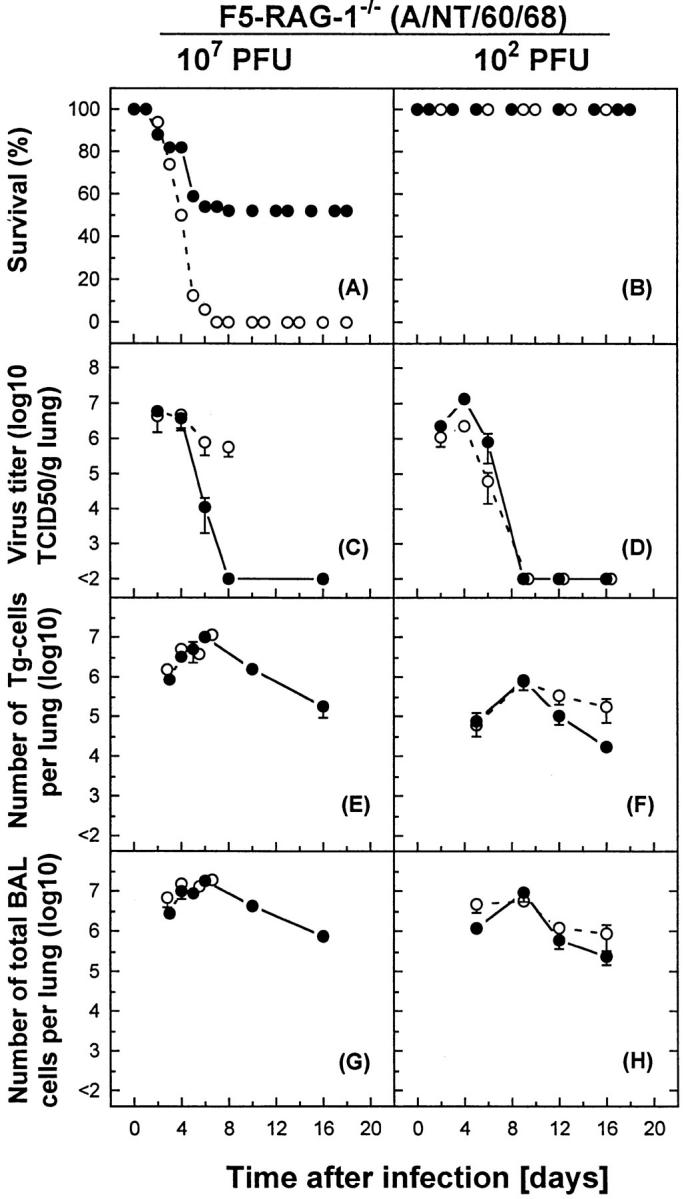
The effect of administration of anti–IFN-γ on mortality, elimination of pulmonary virus, and numbers of Tg-CTLs versus total cell counts in the BAL of influenza virus–infected F5–RAG-1−/− mice. F5– RAG-1−/− mice were infected intranasally with a lethal dose of 107 PFU (left) or a sublethal dose of 102 PFU (right) of A/NT/60/68. Mice were treated with anti–IFN-γ mAb (filled circles) as described in Materials and Methods, or with PBS as control (open circles). (A and B) Percent survival is shown for groups of 5–10 mice. (C and D) Virus titers in lung tissue were expressed as mean log10 TCID50 per gram of lung of three to five mice. (E and F) Tg-CTLs were detected in the same samples by staining cells with Abs specific for Vβ11, CD8 and analysis by flow cytometry, and were expressed as mean ± SEM log10 per lung of three mice. (G and H) The numbers of inflammatory cells in BAL were indicated as mean ± SEM log10 per lung of three mice.
Discussion
Many studies have demonstrated a major role for CD8+ CTLs in control of viral infection. However, it has been difficult to follow development of the specific CTL responses in situ in infected tissues. Here we report an innovative approach, using transgenic TCR mice specific for influenza virus (F5) and mice deficient in CD4+ and B cells (F5–RAG-1−/−), which allows the monitoring of specific CD8+ CTL responses directly in situ. This powerful tool provides a unique opportunity to study the in vivo fate, effector functions, and interactions of virus-specific CTLs in the lung tissue.
The results reported here elucidate some basic principles by which host CTLs amplify defenses against influenza virus. First, effector CTLs localized to sites of virus infection can have either beneficial or harmful effects on the infected host. In the absence of protective Abs, CTLs can potently block viral replication conferring protection against influenza virus, or they can contribute significantly to the genesis and progression of fatal disease. CTL-mediated effects are related to the magnitude of ongoing pulmonary viral infection, whereby the timing of CTL appearance in lung tissues seems to be the most critical factor. This dramatic example of CTL-mediated opposing effects (protection versus lethal pathology) during influenza virus infection adds to reports that CTLs may aggravate disease in viral infections (52–56). Second, the primary driving force underlining influenza pathology is the virus. Thus, unrestricted viral dissemination in lungs results in fatal pulmonary disease. The results of this study do not support the theory that pulmonary pathology is due to the intrinsic cytopathic effects of the virus. However, neither do the data suggest an “innocent bystander” role for the virus. In contrast, viral replication in the lungs is accompanied by an inflammatory process that is probably initiated by chemokines released from infected cells. These chemokines then attract inflammatory cells to the site of infection. Third, our studies are indicative of the dynamic process underlying the development of influenza viral CTL responses (57). Several lines of evidence suggest that the disease process is terminated rapidly if effector CTLs appear in the lungs before or very early after the onset of infection (8, 58–62). Our results suggest that this situation will be difficult to achieve by vaccination strategies aimed at increasing the frequency of antiviral CTL precursors. In support of this view, it has been found that both virgin and primed CTLs need a span of 4–5 d to become potent CTL effectors (7, 58, 63). Thus, the protective ability of CTLs is restricted to a delicate equilibrium between their effector activities and viral load in the lungs. Protective Abs recognizing minor changes in surface proteins within an influenza subtype may shift this balance by slowing down virus replication (and thus reducing viral load) in the onset of infection, thereby allowing CTLs to rapidly terminate viral replication in lungs. This may offer a simple explanation for why CTLs are not capable of preventing influenza epidemics, but on the other hand seem to provide limited protection from clinical disease (8, 64).
Multiple mechanisms may contribute to the protective and pathogenic effects shown by antiinfluenza CTLs. It is important to distinguish the role of soluble factors and cytokines, as well as possible qualitative differences in the CTLs themselves. Such information will be essential for developing a better understanding of viral pathogenesis and a more rational approach to therapeutic intervention in influenza and other respiratory viral infections. CD8+ T cells have been shown to mediate an in vivo antiviral effect either via direct lysis of infected host cells, or by release of cytokines that induce an antiviral effect (65, 66). The ultimate impact of these CD8+ T cell–mediated effector mechanisms on elimination of and recovery from influenza A virus infection, and on the outcome of pulmonary disease, is not well defined.
Regarding effector mechanisms used by CD8+ T cells in clearance of influenza virus, a recent study by Topham et al. using radiation chimeras suggested that target cell destruction mediated via Fas or perforin pathways is probably the primary mechanism used by CD8+ CTLs in clearance of the virus (67). On the other hand, studies with immunocompetent mice deficient in production of IFN-γ either by targeted gene disruption or parenteral administration of a neutralizing anti–IFN-γ Ab into mice lacking β2-microglobulin (the latter lack CD8+ T cells) indicated a less important role for IFN-γ in the clearance of influenza virus infection (39, 68). However, the data do not exclude the possibility that there may be some biologic redundancy in the immune response to influenza, and that other effector mechanisms (e.g., Abs) may influence the degree to which IFN-γ is required for prompt resolution of infection. The use of F5–RAG-1−/− mice provides an opportunity to address this issue more directly. The results reported here are in agreement with and extend the above findings. Although IFN-γ appears be nonessential for CD8+ T cell–mediated recovery from a sublethal influenza virus infection, this study shows clearly that it exerts a marked effect on the outcome of lethal infection. Thus, treatment of F5–RAG-1−/− mice with anti– IFN-γ mAb during a sublethal or lethal influenza infection had no effect on the kinetics or magnitude of effector CTL responses in the lung. However, it seems that IFN-γ secreted in high levels by activated Tg-CTLs does contribute to mortality after infection of F5–RAG-1−/− mice with a lethal dose of the virus. The most likely explanation for this effect is that Tg-CTLs release IFN-γ upon contact with infected MHC class I–positive cells. This results in increased vascular permeability (69), and promotes the development of massive lung edema and leukocyte migration and/or retention into lung parenchyma. Support for this hypothesis is provided by earlier observations showing that IFN-γ may also act as a typical inflammatory cytokine, which influences the overall increase in the number of cells found in the lung parenchyma but has no effect on either the preferential accumulation of CD8+ T cells or their cytolytic effector function (70). Our findings of reduced viral pathology in anti–IFN-γ–treated mice, despite the fact that the numbers of inflammatory cells in BAL were not different in comparison with control untreated mice, strengthen this concept. Thus, neutralization of IFN-γ during onset of viral infection may ameliorate the course of disease, allowing Tg-CTLs to clear the virus from the lung. It is also conceivable that IFN-γ acting as an immunomodulator increases MHC class I expression on virally infected cells and therefore promotes pathology based on cytodestructive Tg-CTL effects. Experiments with mice deficient in the CTL cytolytic pathways (perforin or Fas antigen) could directly address this issue.
In conclusion, our data suggest that suppression of virus replication in the early phase of infection is the most important feature in prevention of influenza virus disease. The challenge in creating a CTL-based vaccine (71–77) directed against heterosubtypic influenza virus strains is to raise the abundance of CTL precursor cells early in the infection in order to increase the protective response without exacerbating a pathology that is also CTL dependent. Finally, evaluation of the dynamic equilibrium established between the CTL immune response and viral infection is obviously a prerequisite for a better understanding of influenza pathogenesis, since inappropriate CTL activation intensifies the pathologic process (55, 78, 79).
Acknowledgments
The authors wish to thank Drs. Nahid Mivechi, Graeme Price, and Peter Openshaw for helpful discussions and suggestions. We also thank Rose Gonsalves and Wendy Hatton for advice and technical help, and Trisha Nordon and Farlyn Hudson for expert animal husbandry.
This work was supported by a grant from the European Union and Medical Research Council, UK.
Abbreviations used in this paper
- BAL
bronchoalveolar lavage
- i.n.
intranasal
- MDCK
Madin-Darby canine kidney
- NP
nucleoprotein
- RAG-1−/−
recombination activating gene 1–deficient
- TCID50
50% tissue culture infectious dose
- Tg
transgenic
References
- 1.Palese P, Young JF. Variation of influenza A, B, and C viruses. Science. 1982;215:1468–1474. doi: 10.1126/science.7038875. [DOI] [PubMed] [Google Scholar]
- 2.Webster, R.G., W.G. Laver, and G.M. Air. 1983. Antigenic variation among type A influenza viruses. In Genetics of Influenza Viruses. P. Palese and D.W. Kingsbury, editors. Springer-Verlag, Vienna/New York. 127–168.
- 3.Gorman OT, Bean WJ, Webster RG. Evolutionary processes in influenza viruses: divergence, rapid evolution, and stasis. Curr Top Microbiol Immunol. 1992;176:75–97. doi: 10.1007/978-3-642-77011-1_6. [DOI] [PubMed] [Google Scholar]
- 4.Yewdell JW, Webster RG, Gerhard W. Antigen variation in three distinct determinants of an influenza A haemagglutinin molecule. Nature. 1979;279:246–248. doi: 10.1038/279246a0. [DOI] [PubMed] [Google Scholar]
- 5.Townsend ARM, Bodmer H. Antigen recognition by class I-restricted T lymphocytes. Annu Rev Immunol. 1989;7:601–624. doi: 10.1146/annurev.iy.07.040189.003125. [DOI] [PubMed] [Google Scholar]
- 6.Ada GL, Jones PD. The immune response to influenza infection. Curr Top Microbiol Immunol. 1986;128:1–54. doi: 10.1007/978-3-642-71272-2_1. [DOI] [PubMed] [Google Scholar]
- 7.Doherty PC, Allan W, Eichelberger M, Carding SR. Roles of αβ and γδ T cell subsets in viral immunity. Annu Rev Immunol. 1992;10:123–151. doi: 10.1146/annurev.iy.10.040192.001011. [DOI] [PubMed] [Google Scholar]
- 8.McMichael A. Cytotoxic T lymphocytes specific for influenza virus. Curr Top Microbiol Immunol. 1994;189:75–91. doi: 10.1007/978-3-642-78530-6_5. [DOI] [PubMed] [Google Scholar]
- 9.Eichelberger MC, Wang ML, Allan W, Webster RG, Doherty PC. Influenza virus RNA in the lung and lymphoid tissue of immunologically intact and CD4-depleted mice. J Gen Virol. 1991;72:1695–1698. doi: 10.1099/0022-1317-72-7-1695. [DOI] [PubMed] [Google Scholar]
- 10.Roth MG, Fitzpatrick JP, Compans RW. Polarity of influenza and vesicular stomatitis virus maturation in MDCK cells: lack of a requirement for glycosylation of viral glycoproteins. Proc Natl Acad Sci USA. 1979;76:6430–6434. doi: 10.1073/pnas.76.12.6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kilbourne, E.D. 1987. Influenza. Plenum Medical Book Co., New York. 359 pp.
- 12.Owen JA, Dudzik KI, Klein L, Dorer DR. The kinetics and generation of influenza-specific cytotoxic T-lymphocyte precursor cells. Cell Immunol. 1988;111:247–252. doi: 10.1016/0008-8749(88)90067-6. [DOI] [PubMed] [Google Scholar]
- 13.Allan W, Tabi Z, Cleary A, Doherty PC. Cellular events in the lymph node and lung of mice with influenza. Consequences of depleting CD4+ T cells. J Immunol. 1990;144:3980–3986. [PubMed] [Google Scholar]
- 14.Muellbacher A. The long-term maintenance of cytotoxic T cell memory does not require persistence of antigen. J Exp Med. 1994;179:317–321. doi: 10.1084/jem.179.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy, B.R., and R.G. Webster. 1996. Orthomyxoviruses. In Fields Virology. 3rd ed., Vol. I. B.N. Fields, D.M. Knipe, and P.M. Howley, editors. 2 vols. Lippincott-Raven Publishers, Philadelphia. 1397–1445.
- 16.Steinhoff MC, Fries LF, Karron RA, Clements ML, Murphy BR. Effect of heterosubtypic immunity on infection with attenuated influenza A virus vaccines in young children. J Clin Microbiol. 1993;31:836–838. doi: 10.1128/jcm.31.4.836-838.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frank AL, Taber LH, Glezen WP, Paredes A, Couch RB. Reinfection with influenza A (H3N2) virus in young children and their families. J Infect Dis. 1979;140:829–836. doi: 10.1093/infdis/140.6.829. [DOI] [PubMed] [Google Scholar]
- 18.Epstein SL, Lo C-Y, Bennink JR. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J Immunol. 1998;160:322–327. [PubMed] [Google Scholar]
- 19.Graham MB, Braciale TJ. Resistance to and recovery from lethal influenza virus infection in B lymphocyte– deficient mice. J Exp Med. 1997;186:2063–2068. doi: 10.1084/jem.186.12.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topham DJ, Doherty PC. Clearance of an influenza A virus by CD4+ T cells is inefficient in the absence of B cells. J Virol. 1998;72:882–885. doi: 10.1128/jvi.72.1.882-885.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor PMA, Askonas BA. Influenza nucleoprotein-specific cytotoxic T-cell clones are protective in vivo. Immunology. 1986;58:417–420. [PMC free article] [PubMed] [Google Scholar]
- 22.Wraith DC, Vessey AE, Askonas BA. Purified influenza virus nucleoprotein protects mice from lethal infection. J Gen Virol. 1987;68:433–440. doi: 10.1099/0022-1317-68-2-433. [DOI] [PubMed] [Google Scholar]
- 23.Andrew ME, Coupar BEH. Efficacy of influenza hemagglutinin and nucleoprotein as protective antigens against influenza virus infection in mice. Scand J Immunol. 1988;25:81–85. doi: 10.1111/j.1365-3083.1988.tb02418.x. [DOI] [PubMed] [Google Scholar]
- 24.Tite JP, Hughes-Jenkins C, O'Callagham D, Dougan G, Russell SM, Gao X-M, Liew FY. Anti-viral immunity induced by recombinant nucleoprotein of influenza A virus. II. Protection from influenza infection and mechanism of protection. Immunology. 1990;71:202–207. [PMC free article] [PubMed] [Google Scholar]
- 25.Webster RG, Kawaoka Y, Taylor J, Weinberg R, Paoletti E. Efficacy of nucleoprotein and haemagglutinin antigens expressed in fowlpox virus as vaccine for influenza in chickens. Vaccine. 1991;9:303–308. doi: 10.1016/0264-410x(91)90055-b. [DOI] [PubMed] [Google Scholar]
- 26.Epstein SL, Misplon JA, Lawson CM, Subbarao EK, Connors M, Murphy BR. β2-microglobulin-deficient mice can be protected against influenza A infection by vaccination with vaccinia-influenza recombinants expressing hemagglutinin and neuraminidase. J Immunol. 1993;150:5484–5493. [PubMed] [Google Scholar]
- 27.Lawson CM, Bennink JR, Restifo NP, Yewdell JW, Murphy BR. Primary pulmonary cytotoxic T lymphocytes induced by immunization with a vaccinia virus recombinant expressing influenza A virus nucleoprotein peptide do not protect mice against challenge. J Virol. 1994;68:3505–3511. doi: 10.1128/jvi.68.6.3505-3511.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mamalaki C, Norton T, Tanaka Y, Townsend AR, Chandler P, Simpson E, Kioussis D. Thymic depletion and peripheral activation of class I major histocompatibility complex-restricted T cells by soluble peptide in T-cell receptor transgenic mice. Proc Natl Acad Sci USA. 1992;89:11342–11346. doi: 10.1073/pnas.89.23.11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mamalaki C, Elliott J, Norton T, Yannoutsos N, Townsend AR, Chandler P, Simson E, Kioussis D. Positive and negative selection in transgenic mice expressing a T-cell receptor specific for influenza nucleoprotein and endogenous superantigen. Dev Immunol. 1993;3:159–174. doi: 10.1155/1993/98015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spanopoulou E, Roman CAJ, Corcoran LM, Schlissel MS, Silver DP, Nemazee D, Nussenzweig MC, Shinton SA, Hardy RR, Baltimore D. Functional immunoglobulin transgenes guide ordered B-cell differentiation in RAG-1-deficient mice. Genes Dev. 1994;8:1030–1042. doi: 10.1101/gad.8.9.1030. [DOI] [PubMed] [Google Scholar]
- 31.Corbela P, Moskophidis D, Spanopoulou E, Mamalaki C, Tolaini M, Itano A, Lans D, Baltimore D, Robey E, Kioussis D. Functional commitment to helper T cell lineage precedes positive selection and is independent of T cell receptor MHC specificity. Immunity. 1994;1:269–276. doi: 10.1016/1074-7613(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 32.Townsend AR, Gotch FM, Davey J. Cytotoxic T cells recognize fragments of the influenza nucleoprotein. Cell. 1985;42:457–467. doi: 10.1016/0092-8674(85)90103-5. [DOI] [PubMed] [Google Scholar]
- 33.Barrett, T., and S.C. Inglis. 1991. Growth, purification and titration of influenza viruses. In Virology, A Practical Approach. B.W. Mahy, editor. IRL Press, Washington, DC. 119–150.
- 34.Simpson RW, Hirst GK. Genetic recombination among influenza viruses. I. Cross reactivation of plaque-forming capacity as a method for selecting recombinants from the progeny of crosses between influenza A strains. Virology. 1961;15:436–451. doi: 10.1016/0042-6822(61)90111-8. [DOI] [PubMed] [Google Scholar]
- 35.Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Hyg. 1936;27:493–497. [Google Scholar]
- 36.Moskophidis D, Lehmann-Grube F. The immune response of the mouse to lymphocytic choriomeningitis virus. IV. Enumeration of antibody-producing cells in spleens during acute and persistent infection. J Immunol. 1984;133:3366–3370. [PubMed] [Google Scholar]
- 37.Skehel JJ, Schild GC. The polypeptide composition of influenza A viruses. Virology. 1971;44:396–408. doi: 10.1016/0042-6822(71)90270-4. [DOI] [PubMed] [Google Scholar]
- 38.Brunner KT, Mauel J, Cerotini J-C, Chapuis B. Quantitative assay of the lytic action of immune lymphoid cells on 51Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology. 1968;14:181–196. [PMC free article] [PubMed] [Google Scholar]
- 39.Sarawar SR, Sangster M, Coffman RL, Doherty PC. Administration of anti-IFN-γ antibody to β2-microglobulin-deficient mice delays influenza virus clearance but does not switch the response to a T helper cell 2 phenotype. J Immunol. 1994;153:1246–1253. [PubMed] [Google Scholar]
- 40.Finkelman FD, Katona IM, Mosmann TR, Coffman RL. IFN-γ regulates the isotypes of Ig secreted during in vivo humoral immune responses. J Immunol. 1988;140:1022–1027. [PubMed] [Google Scholar]
- 41.Scott P. Host and parasite factors regulating the development of CD4+ T-cell subsets in experimental cutaneous leishmaniasis. Res Immunol. 1991;142:32–36. doi: 10.1016/0923-2494(91)90008-7. [DOI] [PubMed] [Google Scholar]
- 42.Burns WH, Billups LC, Notkins AL. Thymus dependence of viral antigens. Nature. 1975;256:654–662. doi: 10.1038/256654a0. [DOI] [PubMed] [Google Scholar]
- 43.Virelizier JL, Allison AC, Schild GC. Immune response to influenza virus in the mouse and their role in control of infection. Brit Med Bull. 1979;35:65–68. doi: 10.1093/oxfordjournals.bmb.a071544. [DOI] [PubMed] [Google Scholar]
- 44.Pircher H-P, Moskophidis D, Rohrer U, Burki K, Hengartner H, Zinkernagel RM. Viral escape by selection of cytotoxic T cell-resistant virus variants in vivo. Nature. 1990;346:629–633. doi: 10.1038/346629a0. [DOI] [PubMed] [Google Scholar]
- 45.Moskophidis D, Zinkernagel RM. Immunobiology of cytotoxic T-cell resistant variants: studies on lymphocytic choriomeningitis virus (LCMV) Semin Virol. 1996;7:3–11. [Google Scholar]
- 46.Schwarz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 47.Herman A, Kappler JW, Marrack P, Pullen AM. Superantigens: mechanism of T-cell stimulation and role in immune responses. Annu Rev Immunol. 1991;9:745–772. doi: 10.1146/annurev.iy.09.040191.003525. [DOI] [PubMed] [Google Scholar]
- 48.Rocha B, von Boemer H. Peripheral selection of the T cell repertoire. Science. 1991;251:1225–1228. doi: 10.1126/science.1900951. [DOI] [PubMed] [Google Scholar]
- 49.Schonrich G, Kalinke U, Momburg F, Malisen M, Schmitt-Verhulst AM, Malissen B, Hammerling GJ, Arnold B. Down-regulation of T cell receptors on self-reactive T cells as a novel mechanism for extrathymic tolerance induction. Cell. 1991;65:293–304. doi: 10.1016/0092-8674(91)90163-s. [DOI] [PubMed] [Google Scholar]
- 50.Moskophidis D, Lechner F, Pircher H-P, Zinkernagel RM. Virus persistence in acutely infected mice by exhaustion of antiviral cytotoxic T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- 51.Webb S, Morris C, Sprent J. Extrathymic tolerance of mature T lymphocytes: clonal elimination as consequence of immunity. Cell. 1990;63:1249–1256. doi: 10.1016/0092-8674(90)90420-j. [DOI] [PubMed] [Google Scholar]
- 52.Oehen S, Hengartner H, Zinkernagel RM. Vaccination for disease. Science. 1991;251:195–198. doi: 10.1126/science.1824801. [DOI] [PubMed] [Google Scholar]
- 53.Cannon MJ, Openshaw PJ, Askonas BA. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med. 1988;168:1163–1168. doi: 10.1084/jem.168.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Battegay M, Oehen S, Schulz M, Hengartner H, Zinkernagel RM. Vaccination with a synthetic peptide modulates lymphocytic choriomeningitis virus-mediated immunopathology. J Virol. 1992;66:1199–1201. doi: 10.1128/jvi.66.2.1199-1201.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim HW, Canchola JG, Brandt CD, Pyles G, Channock RM, Jensen K, Parrott RH. Respiratory syncytial virus disease in infants despite prior administration of antigen inactivated vaccine. Am J Epidemiol. 1969;89:422–434. doi: 10.1093/oxfordjournals.aje.a120955. [DOI] [PubMed] [Google Scholar]
- 56.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 57.Doherty, P.C. 1996. Immune response to viruses. In Clinical Immunology, Principles and Practice. Vol. I. R.R. Rich, T.A. Fleisher, B.D. Schwarz, W.T. Shearer, and W. Strober, editors. 2 vols. Mosby-Year Book, Inc., St. Louis. 535–549.
- 58.Doherty PC, Allan W, Boyle DB, Coupar BEH, Andrew ME. Recombinant vaccinia viruses and the development of immunization strategies using influenza virus. J Infect Dis. 1989;115:1119–1122. doi: 10.1093/infdis/159.6.1119. [DOI] [PubMed] [Google Scholar]
- 59.Yap KL, Ada GL, McKenzie IF. Transfer of specific cytotoxic T lymphocytes protects mice inoculated with influenza virus. Nature. 1978;273:238–239. doi: 10.1038/273238a0. [DOI] [PubMed] [Google Scholar]
- 60.Lin YL, Askonas BA. Biological properties of an influenza A virus–specific killer T cell clone. Inhibition of virus replication in vivo and induction of delayed-type hypersensitivity reactions. J Exp Med. 1981;154:225–234. doi: 10.1084/jem.154.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lukacher AE, Braciale VL, Braciale TJ. In vivo effector function of influenza virus–specific cytotoxic T lymphocyte clones is highly specific. J Exp Med. 1984;160:814–826. doi: 10.1084/jem.160.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wells MA, Ennis FA, Albrecht P. Recovery from a viral respiratory infection. II. Passive transfer of immune spleen cells to mice with influenza pneumonia. J Immunol. 1981;126:1042–1046. [PubMed] [Google Scholar]
- 63.Bennink J, Effros RB, Doherty PC. Influenza pneumonia: early appearance of cross-reactive T cells in lungs of mice primed with heterologous type A viruses. Immunology. 1978;35:503–509. [PMC free article] [PubMed] [Google Scholar]
- 64.McMichael AJ, Gotch FM, Noble GR, Beare PAS. Cytotoxic T-cell immunity to influenza. N Engl J Med. 1983a;301:13–17. doi: 10.1056/NEJM198307073090103. [DOI] [PubMed] [Google Scholar]
- 65.Kagi D, Hengartner H. Different roles of cytotoxic T cells in the control of infections with cytopathic versus noncytopathic viruses. Curr Opin Immunol. 1996;8:472–477. doi: 10.1016/s0952-7915(96)80033-1. [DOI] [PubMed] [Google Scholar]
- 66.Guidotti LG, Ishikawa T, Hobbs MV, Watzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 67.Topham DJ, Tripp RA, Doherty PC. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol. 1997;159:5197–5200. [PubMed] [Google Scholar]
- 68.Graham MB, Dalton DK, Giltinan D, Braciale VL, Stewart TA, Braciale TJ. Response to influenza infection in mice with a targeted disruption in the interferon γ gene. J Exp Med. 1993;178:1725–1732. doi: 10.1084/jem.178.5.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin SK, Maruta V, Burkart S, Gillis S, Kolb H. IL-1 and IFNγ increase vascular permeability. Immunology. 1988;64:301–305. [PMC free article] [PubMed] [Google Scholar]
- 70.Baumgarth N, Kelso A. In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J Virol. 1996;70:4411–4418. doi: 10.1128/jvi.70.7.4411-4418.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McMichael AJ, Gotch FM, Cullen P, Askonas BA, Webster RG. The human cytotoxic T cell response to influenza A vaccination. Clin Exp Immunol. 1981;43:276–285. [PMC free article] [PubMed] [Google Scholar]
- 72.Webster RG, Askonas BA. Cross-protection and cross-reactive cytotoxic T cells induced by influenza virus vaccines in mice. Eur J Immunol. 1980;10:396–401. doi: 10.1002/eji.1830100515. [DOI] [PubMed] [Google Scholar]
- 73.Bennink JR, Yewdell JW, Smith GL, Moller C, Moss B. Recombinant vaccinia virus primes and stimulates influenza haemagglutinin-specific cytotoxic T cells. Nature. 1984;311:578–579. doi: 10.1038/311578a0. [DOI] [PubMed] [Google Scholar]
- 74.Gao XM, Zheng B, Liew FY, Brett S, Tite JP. Priming of influenza virus-specific cytotoxic T lymphocytes in vivo by synthetic peptides. J Immunol. 1991;147:3268–3273. [PubMed] [Google Scholar]
- 75.Oukka M, Manuguerra JC, Livaditis N, Tourdot S, Riche N, Vergnon I, Cordopatis P, Kosmatopulos K. Protection against lethal infection by vaccination with nonimmunodominant peptides. J Immunol. 1996;157:3039–3045. [PubMed] [Google Scholar]
- 76.Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felger PL, Dwarki VJ, Gromkowski SH, Deck RR, DeWitt CM, Friedman A, et al. Heterolous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259:1745–1749. doi: 10.1126/science.8456302. [DOI] [PubMed] [Google Scholar]
- 77.Pertmer TM, Roberts TR, Haynes JR. Influenza virus nucleoprotein-specific immunoglobulin G subclass and cytokine responses elicited by DNA vaccination are dependent on the route of vector DNA delivery. J Virol. 1996;70:6119–6125. doi: 10.1128/jvi.70.9.6119-6125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fulginiti VA, Eller JJ, Downie AW, Kempe CH. Atypical measles in children previously immunized with inactivated measles virus vaccine. JAMA (J Am Med Assoc) 1967;202:1075–1080. doi: 10.1001/jama.202.12.1075. [DOI] [PubMed] [Google Scholar]
- 79.Wells MA, Albrecht P, Ennis FA. Recovery from a viral respiratory infection. I. Influenza pneumonia in normal and T-deficient mice. J Immunol. 1980;126:1036–1041. [PubMed] [Google Scholar]