

Abstract
In normal hemopoietic cells that are dependent on specific growth factors for cell survival, the expression of the basic helix-loop-helix transcription factor SCL/Tal1 correlates with that of c-Kit, the receptor for Steel factor (SF) or stem cell factor. To address the possibility that SCL may function upstream of c-kit, we sought to modulate endogenous SCL function in the CD34+ hemopoietic cell line TF-1, which requires SF, granulocyte/macrophage colony–stimulating factor, or interleukin 3 for survival. Ectopic expression of an antisense SCL cDNA (as-SCL) or a dominant negative SCL (dn-SCL) in these cells impaired SCL DNA binding activity, and prevented the suppression of apoptosis by SF only, indicating that SCL is required for c-Kit–dependent cell survival. Consistent with the lack of response to SF, the level of c-kit mRNA and c-Kit protein was significantly and specifically reduced in as-SCL– or dn-SCL– expressing cells. c-kit mRNA, c-kit promoter activity, and the response to SF were rescued by SCL overexpression in the antisense or dn-SCL transfectants. Furthermore, ectopic c-kit expression in as-SCL transfectants is sufficient to restore cell survival in response to SF. Finally, enforced SCL in the pro–B cell line Ba/F3, which is both SCL and c-kit negative is sufficient to induce c-Kit and SF responsiveness. Together, these results indicate that c-kit, a gene that is essential for the survival of primitive hemopoietic cells, is a downstream target of the transcription factor SCL.
Keywords: SCL, TAL1, c-kit, apoptosis, Steel factor
CL, also known as TAL1 or Tcl-5, is associated with 25% of chromosomal rearrangements in childhood T cell acute lymphoid leukemia (1–6). SCL codes for a hemopoietic-specific transcription factor of the basic helix-loop-helix family, and can enhance or suppress transcription (7). SCL expression is detected in primitive pluripotent hemopoietic precursors (8, 9), in a subset of CD34+ cells, in mast cells and megakaryocytes (8, 10, 11) and in maturing erythroid cells (9, 12–16). Consistent with a role for SCL in establishing the hemopoietic lineage, a targeted disruption of the SCL gene specifically abrogates blood cell formation, resulting in embryonic lethality at day E8.5 (17, 18). Moreover, scl−/− embryonic stem cells fail to contribute to hemopoietic stem cells and blood cells in chimeric mice, further suggesting a critical role of SCL for the development of all hemopoietic lineages (19, 20). Together, these results suggest that SCL is required for mesodermal cell commitment into hemopoietic stem cells and/or is essential for the survival of hemopoietic stem cells.
SCL binds DNA only upon interaction with the ubiquitous basic helix-loop-helix transcription factors E2A or HEB (7, 21). The SCL-E2A heterodimer preferentially binds the TAL1 consensus AACAGATGGT defined by in vitro binding site selection (CASTing). SCL represses the E2A-dependent activity directed from the TAL1 consensus in front of a minimal promoter, but relieves the inhibition conferred by Id1 on E2A (7). However, the TAL1 consensus has not been found in any known hemopoietic promoter. SCL also associates with Lmo-2 or RBTN-2, a RING finger protein (22–25) in a multiprotein complex that includes E2A, GATA-1, an erythroid zinc finger transcription factor (26), and a novel LIM-binding protein, Ldb1 (24), and selectively binds an Ebox-GATA motif also defined by cycle amplification and selection of targets (CASTing). Although similar motives are found on two erythroid promoters, those of the glycophorin A and B genes and the porphobilinogen deaminase gene, their contribution to promoter activity in response to SCL has not been established (24). Finally, in T-ALL, SCL upregulates the expression of a cell surface marker, TALLA-1 (27). However, it is not clear whether TALLA-1 is a direct target of SCL. Thus, despite observations that indicate a crucial role for SCL in normal hemopoiesis and T cell leukemogenesis, no natural binding site or SCL target gene has been as yet reported.
The survival of hemopoietic cells both in culture and in vivo is critically dependent on the presence of hemopoietic growth factors that act to suppress apoptosis (28–30). The importance of such growth factors for blood cell development was first revealed by the genetics of hereditary anemias in mice. Perhaps the best studied examples are white spotting and Steel mice that are severely anemic due to mutations in the genes encoding the tyrosine kinase receptor c-kit (31) or its ligand Steel factor (SF),1 respectively (32, 33). Severe mutations result in lethality in homozygotes (for review see references 34, 35), indicating a crucial role for these two genes in hemopoiesis. We and others have previously shown that SF suppresses apoptosis in early hemopoietic cells that express the surface antigen CD34 (32, 36), and in the CD34+ cell line TF-1 (28). SF has also been shown to synergize with IL-7 in delaying apoptosis in primitive thymocytes (37–39).
The expression pattern of SCL in primary hemopoietic cells parallels that of c-kit (8), suggesting that SCL functions in concert with c-Kit. Furthermore, a two- to fourfold augmentation of SCL protein was observed in maturing erythroid progenitors stimulated with SF (16). However, it is not known whether upregulation of SCL is a cause or consequence of c-Kit activation, cell survival, or cell proliferation. Because of the correlation between c-Kit and SCL expression, we directly investigated the relationship of SCL function relative to c-Kit through the attenuation of SCL protein levels in TF-1 cells using an antisense SCL (as-SCL) expression vector, or through expression of a dominant negative SCL (dn-SCL). We reasoned that SCL function is likely to be determined by the nature of its binding partners, which have yet to be defined in CD34+ cells. We therefore chose to investigate this through the disruption of endogenous SCL function in the CD34+ hemopoietic cell line TF-1, which should express appropriate SCL binding partners. Moreover, TF-1 has retained one of the most important characteristics of primary hemopoietic cells (9, 40), i.e., their requirement in hemopoietic growth factors for cell survival (28, 41). Our approach to defining SCL function and SCL target(s) was to disrupt SCL in TF-1 cells and to screen stable transfectants with a functional assay, i.e., cell survival in response to different growth factors.
Materials and Methods
Cell Lines, Growth Factors, and Antibodies.
The TF-1 cell line (40) was a gift from Dr. T. Kitamura (DNAX, Palo Alto, CA). The cells were maintained in IMDM (GIBCO BRL, Gaithersburg, MD) supplemented with 10% FCS (GIBCO BRL) and 5 ng/ml human GM-CSF. The cells were passaged every second day at 1.5 × 105/ml. Jurkat T cells were also maintained in IMDM supplemented with FCS (10%), and were passaged three times weekly at a concentration of 1.5 × 105/ml.
Purified recombinant human GM-CSF was a gift from Dr. Steve Clark (Genetics Institute, Cambridge, MA) and purified recombinant SF was from Dr. K. Langley (Amgen, Thousand Oaks, CA). The monoclonal antibody 2TL136 specific for human SCL was provided by Dr. Danièle Mathieu-Mahul (INSERM, Marseille, France; references 10, 42). The monoclonal antibody ACK2 is specific for murine c-Kit (GIBCO BRL). The monoclonal mouse anti–human c-Kit was from Cedarlane Labs. (Hornby, Ontario, Canada). The monoclonal mouse anti–rabbit eIF-4E cross-reacts with the human protein (Transduction Labs., Lexington, KY). The monoclonal anti-CD45 (clone 4B2; ATCC number HB 196, American Type Culture Collection, Rockville, Maryland) was used at 1:10 dilution of hybridoma supernatant. Monoclonal anti–hGM-CSF receptor β chain was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY) and used at a 1:100 dilution. Annexin V was purchased from Biodesign (Kennebunk, ME), and used at a concentration of 400 mg/ml.
Retrovirus Production and Infection.
The human SCL cDNA, the dn-SCL devoid of DNA binding domain (12) and the as-SCL were all cloned in the EcoRI site of the murine stem cell virus (MSCV)-neo vector (43). High titer amphotropic viruses (106 PFU/ml) were produced by transient transfection into BING cells (44). The cells were irradiated and used for coculture with TF-1 cells for 48 h. Murine c-kit cloned in the LXSN retroviral vector was a gift from Dr. A. Bernstein (Samuel Lunenfeld Research Institute, Toronto, Ontario, Canada). Ecotropic viruses were produced by transfection into the BOSC23 packaging cell line (44).
For retroviral infection, 106 exponentially growing cells (TF-1 or A31) were presensitized with polybrene at 2 μg/ml for 24 h and cocultured with virus-producing clones for another 24 h. Nonadherent TF-1 cells were separated from the infected fibroblasts. A polyclonal population was analyzed 7 d after selection in G418 at 1 mg/ml. For long-term experiments, cloning was performed through limiting dilution (12).
Plasmids and Transfection.
The human SCL cDNA was cloned in the antisense orientation under the control of the metallothionein promoter by digesting the plasmid pMSCL (12) with EcoRI and religation. The EcoRI sites come from the plasmid polylinker.
Stable TF-1 transfectants harboring the as-SCL were obtained through Lipofectin-mediated DNA transfer (GIBCL BRL) as previously described (9, 12). Cells were cloned immediately after gene transfer by limiting dilution, and the selective pressure was applied the following day, at a concentration of 1 mg/ml G418 (GIBCO BRL). GM-CSF was present throughout the gene transfer procedure. After selection, G418 concentrations were lowered to 400 μg/ml in order to expand the cells and prepare a large stock of frozen cells immediately after characterization. Each cell line was kept for not more than 2 mo in culture.
Nuclear Staining for SCL.
3 × 105 TF-1 and as-SCL transfectants were fixed in 500 μl of Bouin's fixative for 15 min at room temperature. The cells were then pelleted at 200 g for 5 min and resuspended in 200 μl of 0.2% Triton X-100, followed immediately with 800 μl of PBS. The cells were pelleted again, washed with 1 ml of PBS, and resuspended in 100 μl of 1:10 dilution of anti-SCL monoclonal antibody (clone BTL73, provided by Dr. Danièle Mathieu-Mahul). After 30 min of incubation on ice, cells were washed twice with 1 ml of PBS followed by a 10-min wash in 1 ml of PBS, resuspended in 100 μl of FITC-coupled goat anti–mouse antibody at the recommended dilution (Caltech, San Francisco, CA), and incubated for 30 min at 4°C. The cells were washed as above before flow cytometry analysis.
Surface Marker Staining.
For surface marker staining, 3–5 × 105 cells were washed once with PBS supplemented with 2% FBS and 0.05% sodium azide (staining buffer), labeled with the primary antibody for 30 min on ice in a total volume of 100 ml, washed three times with 1 ml of staining buffer, and labeled with a biotinylated goat anti–mouse antibody (Cedarlane, Labs.) in staining buffer supplemented with 1% normal goat serum (Sigma-Aldrich, Oakville, Ontario, Canada) for 30 min on ice. After washing, the cells were incubated with PE-conjugated streptavidin (Sigma) for an additional 30 min on ice, washed, and analyzed on the FACScan® flow cytometer (Becton Dickinson, San Jose, CA).
Northern Blotting Analysis.
Cells were homogenized in the presence of guanidium and RNA isolated by acid-phenol extraction as previously described (9, 45). Northern blotting and hybridization were performed as previously described (9). Probes used were the 1.2-kb Hind–XbaI human SCL cDNA fragment (nucleotides 109–1293; reference 12), the 1.4-kb SalI–XbaI fragment of human GATA-1 cDNA (26), a 1.5- kb DraI–KpnI human c-kit cDNA fragment from human c-kit–BS plasmid (gift from Dr. J. Simms, Immunex, Seattle, WA), and the 1.2-kb PstI– XbaI fragment of the rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA (46). Blots were exposed to a PhosphorImager screen for quantitation.
Western Blotting Analysis.
Cell lysates were prepared as described previously (42). Protein concentrations were determined with the Bio-Rad Protein Assay reagent (Bio-Rad, Hercules, CA). Equal amounts of proteins (20 μg) were loaded on a 12% SDS-polyacrylamide gel and transferred to nitrocellulose using the mini “Transblot” (Bio-Rad) for 1 h at 100 V.
Membranes were blocked in 5% nonfat dry milk and 1% BSA, incubated with 2TL13G hybridoma tissue culture medium (monoclonal anti-SCL) diluted 1:300 for 2.5 h at room temperature, washed, incubated with a goat anti–mouse alkaline phosphatase– linked antibody (Bio-Rad), diluted 1:1,000 for 1.5 h, and washed extensively. Western blots were then developed by incubating membranes in the dark with 10 ml of BCIP (165 μg/ml) and NBT (330 μg/ml) (GIBCO BRL) for 2 min, and the reaction was terminated by several washes with water as described previously (42). In parallel, Western blotting was performed with a monoclonal antibody against the eukaryotic translation initiation factor (eIF-4E) (Transduction Labs.) as a control for protein loading.
Electrophoretic Mobility Shift Assays
Nuclear extracts were prepared as previously described (47). Protein concentrations were assayed with the Bio-Rad Protein Assay reagent. Binding reactions for electrophoretic mobility shift assays were performed as previously described (42). In brief, the binding reactions were allowed to proceed at room temperature for 15 min in the presence of 0.5 μg of poly(dI-dC) as nonspecific competitor DNA in 20 mM Hepes (pH 7.5), 50 mM KCl, 1 mM dithiothreitol, 1 mM EDTA, 5% glycerol, 10 μg BSA, 100,000 cpm double-stranded synthetic oligonucleotide, and 25 μg nuclear extract in a total volume of 10 μl. 50-fold molar excess of unlabeled oligonucleotides were used for self-competition and added before nuclear extracts. For antibody supershift assays, 200 ng of affinity-purified anti-E2A (Santa Cruz Biotechnology, Santa Cruz, CA), 1 μl of the monoclonal anti-SCL antibody (BTL73; reference 48), or 1 μl of an unrelated antibody were added to the binding reaction. Protein complexes were resolved by electrophoresis on 4% polyacrylamide gel buffered in 0.25× Tris-borate– EDTA, 195 mM glycine (pH 8.5) at 4°C. The sequences of the probes (coding strand) are: ACCTGAA CAGATG GTCGGCT TAL1 consensus (21); CTAGGGAG CACCTG CCAGGTG GCTGGCCC murine c-kit probe (49); and an E box probe derived from the rat POMC E box also referred to as DE2c (50).
c-kit Promoter Constructs, Transfection Protocols, and Luciferase Assay.
The human c-kit promoter was cloned by PCR from genomic DNA to generate a fragment that extends from 1146 bp upstream of the transcription initiation site to 43 bp downstream. In brief, genomic DNA was amplified in two steps, the first PCR providing a fragment that extends from position −634 to +60 (PCR1), and the second PCR a fragment that covers −1146 to −379 (PCR2). PCR1 was cut with SalI and BamHI and PCR2 with HindIII (site added) and SalI, and the fragments were cloned into the HindIII and BglII sites of the promoterless luciferase expression vector pXPII to generate kit 1146. The chimeric construct was verified through sequencing. TF-1 and A31 cell lines were transfected by electroporation. Cells were passaged 24 h before gene transfer at a concentration of 3 × 105 cells/ml. Exponentially growing cells were then concentrated at 2.5 × 107 cells/ml and electroporated at 900 μF and 350 mV using a Bio-Rad electroporator with 12 μg of reporter DNA and 1.5 μg of CMV β-galactosidase used as an internal control for the experiment, with or without different molar ratios of MSCV-SCL as shown; the total amount of transfected DNA was then filled to 25 μg with pGEM4 as a carrier. Cell lysates were prepared 15 h after transfection, normalized for β-galactosidase content, and assayed for luciferase activity. Rous Sarcoma virus–driven luciferase (RSV-luc) was used as an external control for all the transfections and pXP2 as a negative control as described previously (47).
Apoptosis Assays.
Analysis of DNA fragmentation by agarose gel electrophoresis was performed as previously described (28). In brief, equal cell numbers were lysed in Sarkosyl buffer and treated with Proteinase K and RNase A before loading on a 1.2% agarose gel (20 cm), followed by a 16-h electrophoresis at 40 V. Cell viability was independently confirmed through trypan blue exclusion and counting (28, 29), or the double fluorochrome staining assay, as described below. Apoptosis was also assessed by flow cytometry analysis of cells labeled with Annexin V-FITC (1 μg/ml), shown previously to be an early marker of apoptosis (51, 52). Immediately before acquisition, 1 ml of propidium iodide solution (1 mg/ml) was added in order to detect dead cells.
Double Fluorochrome Staining Assay.
A stock solution of dye mix containing 100 μg/ml acridine orange and 100 μg/ml ethidium bromide was prepared in PBS. Cell were resuspended at 5 × 105 to 5 × 106 in IMDM. 1 μl of dye mix was added to 25 μl of cell suspension, and 10 μl of the mixture was placed on a microscope slide for examination by fluorescent microscopy (Leica, Wetzlar, Germany). Cells with bright green chromatin were considered viable cells while those with bright orange chromatin or collapsed chromatin were considered dead cells. A minimum of 200 cells were counted.
Results
Decreased SCL Protein Levels and c-Kit Levels in TF-1 Cells Expressing an as-SCL.
To address SCL function and define SCL target genes in primitive hemopoietic cells, we chose to disrupt SCL function in the CD34+/c-kit+ cell line TF-1. To this end, we took two complementary approaches. The first one consists of expressing an as-SCL that interferes with SCL protein levels, and the second one involves a dn-SCL devoid of its DNA binding domain (12), which prevents SCL function without affecting protein levels. We reasoned that loss of SCL function may be detrimental to the cells and therefore decided to analyze the cells as early as 1 wk after gene transfer, at a time when the selective pressure (G418) is already optimal. To minimize cell loss during the selective pressure and, consequently, the generation time required for in vitro expansion, we chose retroviral infection as a method for high efficiency gene delivery for which we typically obtain 30– 50% transfer efficiency.
We first determined the efficiency of as-SCL in disrupting SCL protein levels, which were assessed through immunostaining of permeabilized cells with the monoclonal anti-TAL1 antibody BTL73 (Fig. 1). As expected, TF-1 cells express high levels of endogenous SCL. In contrast, 1 wk after transduction of as-SCL the endogenous SCL protein level was significantly decreased, with little if any overlap between the two populations. The cells were further analyzed for expression of several cell surface markers. Data shown in Fig. 1 indicate that the fluorescence intensity for c-Kit (CD117) was at least three- to fourfold lower in dn-SCL and as-SCL transfectants when compared with parental TF-1 cells (data not shown) or control cells expressing the vector alone (MSCV ). In contrast, the level of CD45 was not affected by as-SCL or dn-SCL expression and that of CD116 (GM-CSF receptor α chain) was either similar or slightly higher. Together, the results indicate that decreased SCL protein levels (as-SCL) or decreased SCL function (dn-SCL) specifically lower c-Kit levels without affecting other surface markers.
Figure 1.
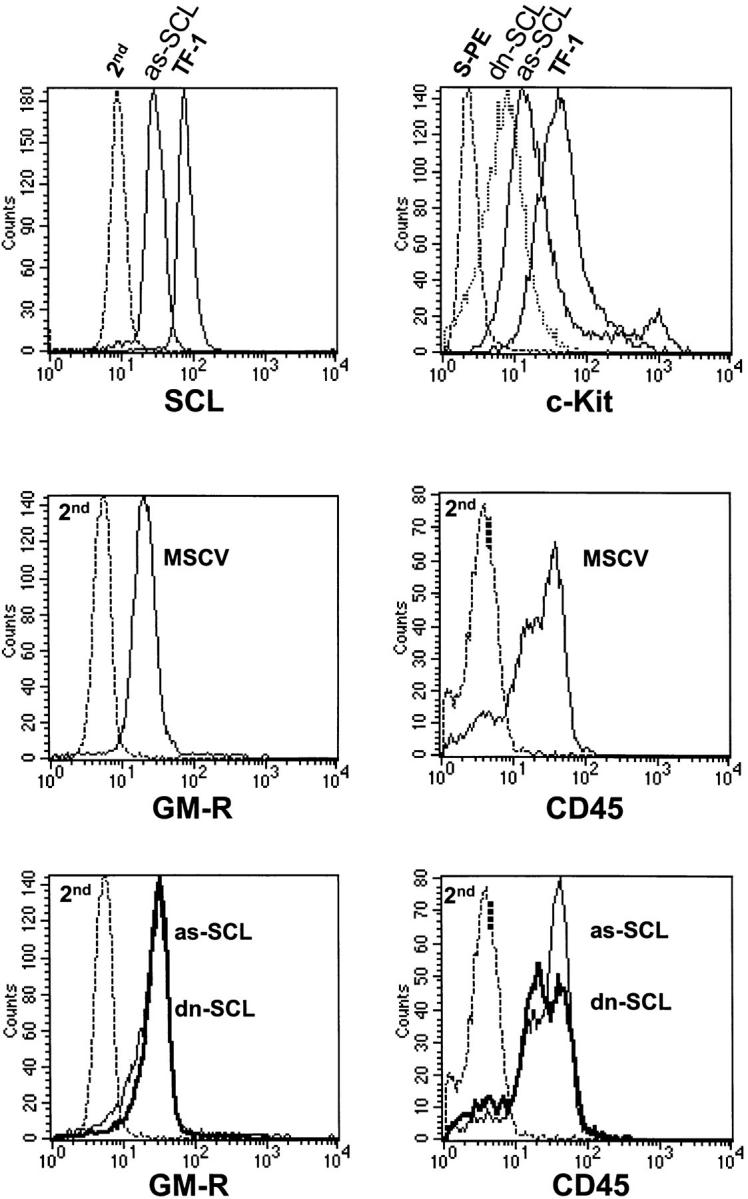
Flow cytometry analysis of TF-1 transfectants expressing as-SCL or dn-SCL. (top left) Decreased SCL protein in TF-1 cells expressing as-SCL. TF-1 cells were infected with a retrovirus harboring the human SCL cDNA cloned in the antisense orientation and maintained as a polyclonal population in GM-CSF containing medium in the presence of G418 (1 mg/ml). 1 wk after infection, as-SCL transfectants were fixed and permeabilized with Triton X-100 for nuclear staining with the monoclonal anti-TAL1 at a final dilution of 1:10, followed by labeling with a goat anti–mouse antibody coupled to FITC (Sigma). Cells were washed extensively for 10 min after each labeling step and analyzed by flow cytometry. Parental TF-1 cells were stained in parallel as positive controls. Both cell types were also stained with the second antibody alone (2nd) as negative controls. In contrast to the shift in SCL nuclear staining between as-SCL transfectants and parental TF-1 cells, those of MSCV (vector alone) transfectants and TF-1 cells were comparable (data not shown). (top right) Decreased c-Kit expression in TF-1 cells expressing as-SCL or dn-SCL. The same cells as in the top left panel were analyzed for surface labeling with a biotinylated monoclonal antibody against human c-Kit and streptavidin-coupled PE (S-PE). Cells labeled with S-PE alone serve as negative controls. (middle and bottom) Surface expression of CD45 and CD116 (GM-CSF receptor [GM-R] α chain) is unaltered in TF-1 transfectants. Cells were labeled with a mouse anti–human CD45 and anti–human CD116 (GM-R), followed by FITC-labeled goat anti–mouse IgG. Labeling with the second antibody alone (2nd) is shown.
Since the GM-CSF receptor is not affected by SCL levels, we proceeded to derive stable TF-1 cell lines expressing dn-SCL (TF1-dn) following long term selection in G418 in the presence of GM-CSF as a survival factor, in order to directly address SCL function at the molecular and cellular levels. We also established clones that express as-SCL driven by a weaker promoter, the mouse metallothionein promoter, instead of the strong promoter contained within the retroviral LTR. Despite this, most of the 50 clones screened displayed weak levels of transgene expression, consistent with the reported difficulty in obtaining stable as-SCL clones in another hemopoietic cell line, K562 (9, 53; and Kirsch, I.R., unpublished data). The two highest antisense expressor clones, A30 and A31, were selected.
We next confirmed that SCL DNA binding activity was indeed reduced in the antisense clone A31 and the dn-SCL–expressing transfectants (TF1-dn) through electrophoretic mobility shift assays using the TAL1 consensus probe (reference 21; Fig. 2). TF-1 nuclear extracts produced four specific complexes (C1–C4), as shown by self-competition (Fig. 2, lanes 11 and 12). In contrast, a probe with divergent sequences (mDE2C) did not compete for binding (data not shown), indicating that these complexes were specific. All four complexes were also found using nuclear extracts from Jurkat cells (Fig. 2, lanes 9 and 10) and TF-1 transfectants harboring the vector alone (TF1-neo, lanes 1 and 6). The identity of each complex (C1–C4) was verified by antibody supershifting. Preincubation of TF1-neo extracts with either anti-SCL or anti-E2A antibody supershifted the two most slowly migrating complexes, C1 and C2 (Fig. 2, lanes 1, 2, and 6–8), indicating that these contain SCL/E2A heterodimers as previously described (21, 24). These complexes were absent in HL-60 cells, which were included as a negative control (lane 16). In the antisense-expressing clone A31, the SCL-containing complex C1 decreased significantly, whereas the C2 complex was only marginally affected (Fig. 2, compare lanes 3 and 5), suggesting that under conditions where SCL is limiting, the smaller complex, C2, is preferentially formed over C1. Rescuing clone A31 wild-type SCL (A31-SCL) restored C1 and C2 binding to the TAL1 probe (Fig. 2, lanes 3 and 4). In all cell lines, the two fastest migrating complexes, C3 and C4, were not supershifted with anti-SCL or anti-E2A, suggesting that these did not contain the corresponding proteins. These complexes were not affected in TF1-neo but were decreased in A31, possibly as a consequence of clonal variation, since they were also decreased in A31-SCL, a subclone of A31.
Figure 2.

Decreased SCL DNA binding activity in TF-1 cells expressing as-SCL (A31) or a dn-SCL (TF1-dn). TF-1 cells and the different transfectants were maintained in GM-CSF containing medium. Exponentially growing cells were treated with ZnCl2 for 24 h before preparation of nuclear extracts. A double-stranded 32P-labeled TAL1 consensus probe was incubated with 25 μg nuclear extracts from TF-1, TF1-neo, A31, A31-SCL, TF1-dn, Jurkat, and HL-60 as described in Materials and Methods. Unlabeled double-stranded oligonucleotide probes used as competitors were added at 50-fold molar excess. SCL-containing complexes (black arrows) formed on the TAL1 consensus probe were supershifted ( gray arrows) by the monoclonal antibody anti-SCL/TAL1 or anti-E2A, which were added to the binding reaction. These complexes were displaced by a 50-fold molar excess of self (lanes 4, 13, and 17 ) and of the c-kit probe but not by a control probe with divergent sequences, mDE2C (data not shown).
In the transfectant expressing a dn-SCL, which is a polyclonal population, both SCL-containing complexes, C1 and C2, were significantly decreased relative to TF1-neo whereas the non-SCL-containing complexes C3 and C4 were not affected (Fig. 2, compare lanes 5 and 6). Together, our results indicate that ectopic expression of as-SCL or dn-SCL significantly interferes with the formation of the two SCL-containing complexes, C1 and C2, on the TAL1 probe.
SCL Protein Levels Specify c-kit Expression.
As observed with transient transfectants (Fig. 1), flow cytometry analysis of the A31 and TF-1 cells (Fig. 3 A) or TF1-dn and TF1-neo (data not shown) using a monoclonal antibody to human c-Kit indicated a quantitative decrease in c-Kit levels in A31 and TF1-dn as compared with control cells. There was a direct correlation with c-kit mRNA levels as determined through Northern blotting (Fig. 3 B). Thus, parental TF-1 cells or TF-1 cells harboring the vector alone (TF1-pac and TF1-neo) exhibited comparable levels of c-kit mRNA (Fig. 3 B). In contrast, c-kit expression was significantly lower in A31 and T-dn (Fig. 3, B and C). To exclude the possibility that c-kit mRNA expression was affected by the site(s) of integration in these antisense clones, we attempted to rescue c-kit expression by wild-type SCL through retrovirus-mediated gene transfer (MSCV; reference 43), using puromycin resistance as a second selective marker. Expression of the puromycin resistance gene alone did not affect c-kit expression in TF-1 cells (Fig. 3 B, TF1-pac) nor in A31 cells (data not shown). In contrast, elevating SCL expression in clone A31 restored c-kit expression (Fig. 3 B, A31-SCL) and further increased c-kit mRNA levels in parental TF-1 cells (data not shown). Together, the results indicate that the targeted attenuation of SCL protein levels and DNA binding activity directly leads to decreased c-kit expression.
Figure 3.

Reduced c-kit expression in clones expressing an as-SCL: rescue with wild-type SCL. Cells were maintained in culture with 200 pM GM-CSF in the presence of ZnCl2 for 24 h. (A) Indirect immunofluorescence detection of c-Kit by flow cytometry with the monoclonal antibody against human c-Kit indicates decreased surface expression in A31 and in TF1-dn as compared with parental TF-1 cells or TF1-neo (data shown for A31 and TF-1). Cells labeled with the second antibody alone (goat anti–mouse FITC) served as controls, which were similar for A31 and TF-1 (data shown for TF-1). (B and C ) RNA extraction and Northern blotting were performed as described in Materials and Methods. The blots were sequentially hybridized with full-length cDNA for human c-kit, GATA-1, Lyl-1 (data not shown), and GAPDH. The blots were exposed for 24 h to a PhosphorImager screen. (B and C) After normalization with GAPDH and taking TF-1 as 1, c-kit mRNA levels were: 1.05 (TF1-pac), 0.39 (A31), 1.82 (A31-SCL), 1 (TF1-neo), and 0.5 (TF1-dn). (B) Viable cells were determined through double fluorochrome staining as described in Materials and Methods. (D) The c-kit promoter construct kit1146 driving the luciferase reporter gene are codelivered with SCL into exponentially growing TF1-dn cells by electroporation as described in Materials and Methods at the indicated activator to reporter ratio. Data are normalized to that of RSV-GH, cotransfected as an internal control, and corrected for the effect of SCL on RSV-luciferase, used as an external standard. Luciferase activity in the absence of activator was taken as 1. Data are the mean of two independent experiments that were performed in triplicates.
Sequence analysis of the c-kit promoter revealed two adjacent E boxes at −374 and −381 that are conserved between human and mouse (49), and fit the core binding motif of MyoD/E2A. Electrophoretic mobility shift assays indicate that SCL–E2A complexes bind these two E boxes (our unpublished results), suggesting that these E boxes may contribute to c-kit promoter activity in response to SCL. We directly tested SCL transactivation properties on a chimeric construct in which the luciferase reporter gene was placed under the control of the human c-kit promoter (kit1146). Since the endogenous levels of SCL are elevated in TF-1 cells, we used TF1-dn for transient transfection. The reporter construct kit1146 was codelivered with varying amounts of the SCL expression vector MSCV-SCL. Results shown in Fig. 3 D indicate that SCL induced a dose-dependent increase in c-kit promoter activity, which was twofold at an optimum ratio of activator to reporter. Conversely, cotransfection of dn-SCL and kit1146 in parental TF-1 cells caused a twofold decrease in luciferase activity (data not shown). Together, these results are consistent with the hypothesis that SCL positively regulates c-kit transcription through binding to two adjacent E boxes in the c-kit promoter.
Disrupted SCL Function Specifically Prevents SF-dependent but not GM-CSF or IL-3–dependent Cell Survival.
We have previously shown that both SF and GM-CSF suppress apoptosis in TF-1 cells. Parental cells and the different transfectants were, therefore, compared for their survival in response to optimal concentrations of SF, GM-CSF, or IL-3. Apoptotic death was revealed by the presence of a DNA ladder after agarose gel electrophoresis (data not shown). In the absence of growth factor, there was an intense DNA ladder, suggesting that the cells underwent apoptosis. At a concentration of 100 pM of SF, chosen to be near maximal suppression of apoptosis, parental TF-1 cells and the control TF1-neo line behaved similarly, with no detectable DNA degradation. Under identical conditions, apoptosis was evident in both antisense clones, A30 and A31 (data not shown), in which SCL protein levels (data not shown) and DNA binding activity (Fig. 2) were decreased. Cell viability was restored in clone A31-SCL, consistent with increased SCL DNA binding activity (Fig. 2, lane 4) and SCL protein levels (data not shown) in this clone, whereas control cells expressing the vector alone (A31-pac) behaved like the parental A31 cell line.
Cell viability was therefore quantitated using the double fluorochrome staining technique (Fig. 3 B). Full dose– response curves for SF and GM-CSF were performed in the two antisense clones, A30 and A31, as well as in control cells. Data are shown for growth factor concentrations that provide 80% survival in TF-1 cells (Fig. 3 B). As observed with agarose electrophoresis, TF-1 and TF1-neo lines remained viable with SF, whereas A30 and A31 underwent apoptosis. As above, ectopic SCL expression in clone A31 (A31-SCL) restored cell viability in SF-containing cultures, indicating that apoptotic death was due to as-SCL. In contrast, cell survival was the same in control and antisense clones in GM-CSF or IL-3 stimulated cultures (data not shown).
Apoptotic cells were also detected through Annexin V staining of membrane phosphatidyl serine, which is exteriorized during apoptotic death, both in stable clones (data not shown) and in transient transfectants 1 wk after G418 selection. Results shown in Fig. 4 indicate that parental cells and the different transfectants survive well in GM-CSF–containing cultures, consistent with their staining pattern for GM-CSF receptor (GM-R) observed in Fig. 1. In contrast, when the cells were maintained with SF, 80% of the population expressing dn-SCL or as-SCL was apoptotic, whereas both controls (TF-1 and MSCV transfectants) survived readily. Impaired survival in response to SF stimulation in these transfectants is directly correlated with decreased surface c-Kit expression as shown in Fig. 1. Together, the results suggest that SCL specifically regulates c-Kit expression and cell survival in response to SF.
Figure 4.
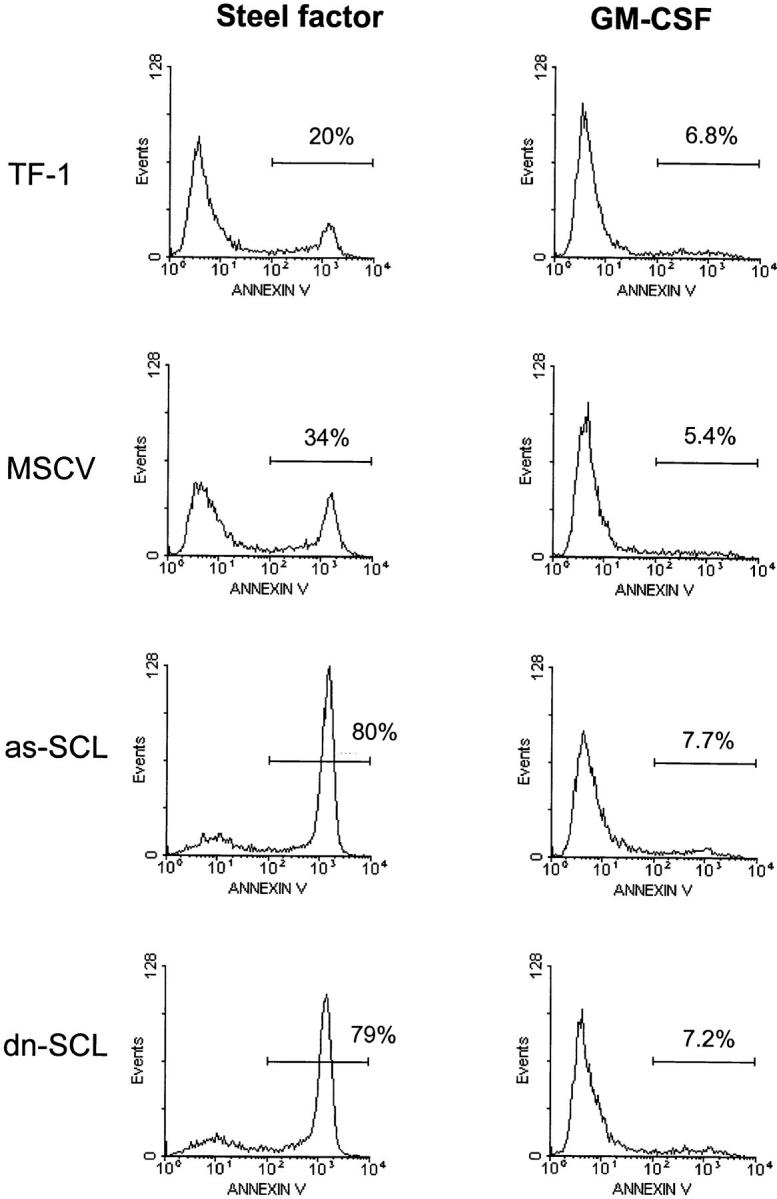
as-SCL or dn-SCL prevents cell survival in response to SF. Apoptotic cell death is analyzed quantitatively by staining with Annexin V 6 d after retroviral infection. The indicated transfectants were incubated for 44 h with a concentration of SF chosen to provide 70–80% survival in parental TF-1 cells (25% Annexin+ cells), or kept in GM-CSF containing medium. Cells were stained with propidium iodide and Annexin V–FITC as described in Materials and Methods. The proportions of Annexin+ cells are shown in each histogram. Results shown are typical of two independent infections.
Cell Survival in Response to SF Is Rescued by Ectopic SCL or Ectopic c-kit Expression.
As discussed above, the A31 subclone in which endogenous c-kit levels were restored after ectopic SCL expression, A31-SCL, survived well in cultures stimulated with SF. We, therefore, addressed the possibility whether c-kit expression by itself was sufficient to rescue SF-dependent cell viability. Murine c-kit was delivered to A31 cells through retrovirus-mediated gene transfer, using the LXSN retroviral vector. Despite a high homology between murine and human c-kit, ligand binding is species specific such that cells expressing human c-Kit require five- to eightfold higher concentrations of murine SF as compared with human SF (Fig. 5 C). After a 2-d selection in low concentrations of murine SF, c-kit–infected A31 and TF-1 cells (A31-kit and TF1-kit, respectively) were expanded with human GM-CSF. Flow cytometry analysis indicated that >75% of TF1-kit or A31-kit cells express murine Kit (Fig. 5 A). In GM-CSF–containing cultures, all clones behaved similarly (data not shown). As expected, TF1-kit responded better than parental TF-1 cells to murine SF (Fig. 5 C). As with human SF, the response of A31 cells to murine SF was significantly lower than that of TF-1 cells, even at saturating concentrations of murine SF. In contrast, the response of A31-kit to murine SF was similar to that of TF1-kit, suggesting that c-kit overexpression was sufficient to overcome the effect of reduced SCL expression. These observations are compatible with the view that the major survival function of SCL in TF-1 cells is mediated through c-kit expression.
Figure 5.

Survival response to SF rescued by ectopic murine c-kit expression in as-SCL transfectants. TF-1 cells and the as-SCL–expressing cells (A31 cells) were infected with MSCV harboring the murine c-kit gene, or with the pac resistance gene (control) as described in Materials and Methods. A31-Kit and TF1-Kit (A) as well as A31 and TF-1 (B) were labeled with ACK2, a monoclonal antibody specific for murine Kit, and a goat anti–rat Ig coupled to FITC (solid lines). Cells labeled with the second antibody alone served as negative controls (broken lines). For apoptosis assay, cells were maintained with the indicated concentrations of murine SF for 2 d. Viable cells were determined by trypan blue exclusion. (C ) The half efficient concentration of murine SF for the different clones were: TF-1, 17.8 ± 2.1; A31, 13 ± 4; TF1-kit, 3.5 ± 0.5; A31-kit, 4.3 ± 0.6.
SCL Induces c-Kit Expression in a Negative Cell Line.
Ba/F3 is a pro–B cell line that expresses the IL-3 receptor but not c-Kit (54, 55) or SCL (data not shown). Therefore, we transduced the human SCL cDNA into Ba/F3 cells through retroviral infection in order to address the possibility that SCL can induce c-kit expression in a negative cell line. Control cells were transduced with the MSCV vector alone. 2 wk after selection in G418, polyclonal populations were analyzed for surface expression of murine c-Kit by flow cytometry (Fig. 6 A). As expected, Ba/F3-MSCV cells were c-Kit negative. In contrast, Ba/F3-SCL cells were clearly c-Kit positive.
Figure 6.

SCL induces c-Kit expression in pro–B cell line Ba/F3. (top) After retroviral-mediated gene transfer, polyclonal populations with enforced SCL expression (Ba/F3-SCL) or control cells harboring the vector alone (Ba/F3-MSCV) were labeled with the monoclonal rat anti–murine c-Kit as in Fig. 5. (bottom) Viable cell counts from cultures stimulated with optimal concentrations of murine SF were determined through trypan blue exclusion. Cell counts for growth factor–deprived Ba/F3 and Ba/F3-MSCV (data not shown) were comparable to those observed with murine SF.
We next determined whether the cells have acquired responsiveness to murine SF (Fig. 6 B). In the absence of growth factor, Ba/F3-SCL transfectants continuously die over a 3-d period, with the same kinetics as parental Ba/F3 or MSCV transfectants maintained with or without SF. In contrast, when Ba/F3-SCL transfectants were maintained with SF, cell numbers remained at input levels for 2 d and declined slightly on day 3, consistent with the presence of surface c-Kit on these cells. Together, our observations indicate that enforced SCL expression induces c-Kit and confers SF responsiveness in Ba/F3 cells.
Discussion
Role of SCL as a Survival Gene: Upregulation of c-kit Expression.
This study provides evidence for a mechanism whereby SCL secures the survival of primitive hemopoietic cells through upregulation of the tyrosine kinase receptor c-Kit, which is itself essential for the generation of hemopoietic cells both in vivo and in vitro (17–20). Several lines of evidence indicate a direct link between SCL and cell survival in response to SF. First, the apoptotic phenotype is induced by both as-SCL and dn-SCL. Second, apoptosis in the antisense clone A31 is rescued by ectopic SCL expression in the sense orientation. Third, apoptosis is specific to the c-Kit pathway triggered by SF, and is not observed with GM-CSF or IL-3, which indicates exquisite biological specificity. Indeed, SF is essential for the normal development of hemopoietic cells, whereas IL-3 and GM-CSF are produced by activated T cells as part of an inflammatory/stress response, suggesting that SCL function is more important in steady-state hemopoiesis. Finally, enforced SCL expression in the pro–B cell line Ba/F3, which is c-Kit−, clearly induces c-Kit expression in these cells. In all, our study provides a direct link between SCL and the survival response of hemopoietic cells to SF, mostly through upregulation of c-kit transcription.
SCL and Transcription Regulation.
The conservation of two everted E boxes at positions −381 and −374 between the human and mouse promoter sequences (49) suggests a functional role in transcription regulation, which was confirmed through binding studies (data not shown) and promoter deletion analysis. Our gel shift assays indicate that SCL-containing complexes that are formed on the c-kit probe also include E2A, consistent with the view that SCL does not bind DNA on its own and requires interaction with E12, E47, or HEB, another ubiquitous basic helix-loop-helix (21, 24). Furthermore, SCL is also found in association with the LIM-only protein Rbtn2/Lmo2 (22, 23) in a multiprotein complex with GATA-1 and a LIM-binding protein, Ldb1 (24). An E box–GATA motif was identified through in vitro binding site selection with nuclear extracts instead of isolated proteins (24), which corresponds strikingly to the c-kit everted E box sequences at −381. Sequence analysis of the human c-kit promoter also reveals the presence of several potential GATA sites at −900. Although these are found upstream instead of downstream of the E box as in the CASTing experiment, it is possible that a looping mechanism favored by protein–protein association allows for optimal spatial arrangements such as those defined by CASTing.
Transactivation assays indicate that SCL represses the E2A-dependent activity of a promoter construct containing the TAL1 motif. It is only in the presence of Id that SCL was shown to relieve the inhibition conferred by Id1 on E2A (7). In addition, despite its high affinity binding to the Ebox-GATA motif, the SCL–E2A heterodimer has no activity on a reporter construct with a minimal promoter linked to two such motives, either in the absence or presence of Lmo2. This multiprotein complex, however, provides a twofold enhancement of luciferase activity driven by GATA-1 on the same construct (24). Thus, SCL appears to be a weak transactivator, consistent with our results indicating a twofold contribution to the activity of the proximal c-kit promoter in TF-1 cells. This twofold contribution was confirmed by three distinct approaches, Northern blotting for steady state c-kit mRNA levels, flow cytometry analysis of surface c-Kit protein, and analysis of the c-kit promoter driving the luciferase reporter gene. A recent report using semiquantitative reverse transcriptase PCR did not detect significant differences in c-kit mRNA levels in embryoid bodies grown from scl −/− embryonic stem and wild-type embryonic stem cells (56). However, it should be noted that flow cytometry analysis and luciferase activity can quantitatively reveal a two- to fourfold difference, whereas reverse transcriptase PCR remains semiquantitative and requires much larger differences for detection.
Implication for Leukemogenesis.
It was previously shown that loss of SCL function is associated with premature apoptosis upon nutrient deprivation in the lymphoid cell line Jurkat (42) and decreased colony formation in the erythroid cell line K562 (53), suggesting a role for SCL in mediating cell survival. As observed with the antiapoptotic gene bcl-2, which infrequently induces lymphomas in transgenic mice after a long latency period of 18 mo (57), elevating SCL expression in thymocytes is not sufficient to cause tumors in transgenic mice (58). Directing SCL expression in the thymus can nonetheless shorten the time of appearance of T cell tumors in LMO-2 (59), LMO-1 (58), or casein kinase II transgenic mice (48). Although these observations suggest a role for SCL in leukemogenesis, its target genes remain unknown.
The cells that repopulate the thymus and a subpopulation of triple negative thymocytes are c-Kit+ (37). Interestingly, SF is expressed by fetal thymic stromal cells and epithelial cells (38, 39, 60). More importantly, T cell differentiation in thymic lobe reconstitution with fetal liver precursors is inhibited by anti-Kit (37) and is impaired in white spotting mice, indicating the functional importance of c-Kit (61). In parallel, the presence of SCL mRNA in a subset of thymocytes (11) and the absolute requirement in SCL for the generation of T cells in vivo (19, 20) are indicative of the importance of SCL for thymocyte development. Our observations suggest that SCL may also specify c-Kit expression in primitive thymocytes to sustain cell survival in response to SF. Consistent with this, elevating SCL levels in thymocytes in double CD2-SCL and CD2-Lmo2 transgenics results in a twofold expansion of the double negative CD4−/CD8− thymocyte population relative to CD2-Lmo2 transgenics (48), which, in light of our results, may be due to an increase in c-Kit+ subpopulations. We propose that constitutive SCL expression in the T cell lineage caused by chromosomal rearrangements results in constitutive c-kit expression in primitive thymocytes and prolongs their survival, which may represent an initiating event in T cell ALL. Secondary events are probably involved in the full development of T cell leukemia (58, 62), a mechanism commonly observed in tumor biology (63).
Tissue-specific Transcription Factors and Cell Survival.
The regulation of cell survival is central to normal developmental processes and stress response. It is widely accepted that survival cues are crucial for the differentiation process (64), and emerging evidence suggests a novel role for tissue-specific transcription factors, otherwise important for driving a defined pattern of gene expression, in regulating cell survival. Thus, the zinc finger transcription factor GATA-1 is required for terminal maturation in the erythroid lineage (65). GATA-1 gene ablation in embryonic stem cells blocks the differentiation at the proerythroblast stage (65, 66) and causes apoptotic death (66). Similarly, GATA-4 is required for cardiomyocyte differentiation from embryonal carcinoma cells (67) and cell survival in the cardiomyocyte pathway but not the neuronal lineage. Hence, apoptosis may be linked to insufficient survival signals, but also to inefficient or abortive cell differentiation. Similarly, SCL is required early in hemopoiesis, possibly in specifying ventral mesoderm to the hemopoietic fate (19), and is shown here to be crucial for SF-mediated cell survival. Therefore, it is proposed that lineage-specific transcription factors orchestrate both lineage-specific gene expression and the survival of the specified cell types.
Acknowledgments
The authors wish to thank Dr. Robert Hawley (Oncology Division, Toronto General Hospital, Toronto, Ontario, Canada) for providing the MSCV retroviral vectors; Drs. Rafick P. Sékaly and Guy Sauvageau (IRCM, Montreal, Quebec, Canada) for critical reading of the manuscript; Dr. André Haman for help with Northern blotting; Dr. Richard Martin for the MSCV-dn-SCL construct; Dr. Don Lombardi for the pMTH as-SCL construct; and Magali Domin and Vivianne Jodoin for expert secretarial assistance.
This work was supported by grants from the National Cancer Institute of Canada with funds from the Canadian Cancer Society and a research traineeship from the Heart and Stroke Foundation of Canada to F. Charron. M. Nemer is a Senior Scientist of the Medical Research Council of Canada and T. Hoang is Senior Scientist from the Fonds de la Recherche en Santé du Québec and a Visiting Scientist of the National Cancer Institute (USA).
Abbreviations used in this paper
- as-SCL
antisense SCL
- dn-SCL
dominant negative SCL
- GAPDH
rat glyceraldehyde-3-phosphate dehydrogenase
- MSCV
murine stem cell virus
- SF
Steel factor
References
- 1.Aplan PD, Lombardi DP, Ginsberg AM, Cossman J, Bertness VL, Kirsch IR. Disruption of the human SCL locus by “illegitimate” V-(D)-J recombinase activity. Science. 1990;250:1426–1429. doi: 10.1126/science.2255914. [DOI] [PubMed] [Google Scholar]
- 2.Aplan PD, Raimondi SC, Kirsch IR. Disruption of the SCL gene by a t(1;3) translocation in a patient with T cell acute lymphoblastic leukemia. J Exp Med. 1992;176:1303–1310. doi: 10.1084/jem.176.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Begley CG, Aplan PD, Davey MP, Nakahara K, Tchorz K, Kurtzberg J, Hershfield MS, Haynes BF, Cohen DI, Waldmann TA, Kirsch IR. Chromosomal translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor delta-chain diversity region and results in a previously unreported fusion transcript. Proc Natl Acad Sci USA. 1989;86:2031–2035. doi: 10.1073/pnas.86.6.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernard O, Guglielmi P, Jonveaux P, Cherif D, Gisselbrecht S, Mauchauffe M, Berger R, Larsen CJ, Mathieu-Mahul D. Two distinct mechanisms for the SCL gene activation in the t(1; 14) translocation of T-cell leukemias. Genes Chromosomes Cancer. 1990;1:194–208. doi: 10.1002/gcc.2870010303. [DOI] [PubMed] [Google Scholar]
- 5.Chen Q, Cheng JT, Tasi LH, Schneider N, Buchanan G, Carroll A, Crist W, Ozanne B, Siciliano MJ, Baer R. The tal gene undergoes chromosome translocation in T cell leukemia and potentially encodes a helix-loop-helix protein. EMBO (Eur Mol Biol Organ) J. 1990;9:415–424. doi: 10.1002/j.1460-2075.1990.tb08126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finger LR, Kagan J, Christopher G, Kurtzberg J, Hershfield MS, Nowell PC, Croce CM. Involvement of the TCL5 gene on human chromosome 1 in T-cell leukemia and melanoma. Proc Natl Acad Sci USA. 1989;86:5039–5043. doi: 10.1073/pnas.86.13.5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu HL, Wadman I, Tsan JT, Baer R. Positive and negative transcriptional control by the TAL1 helix-loop-helix protein. Proc Natl Acad Sci USA. 1994;91:5947–5951. doi: 10.1073/pnas.91.13.5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brady G, Billia F, Knox J, Hoang T, Kirsch IR, Voura EB, Hawley RG, Cumming R, Buchwald M, Siminovitch K, et al. Analysis of gene expression in a complex differentiation hierarchy by global amplification of cDNA from single cells. Curr Biol. 1995;5:909–922. doi: 10.1016/S0960-9822(95)00181-3. [DOI] [PubMed] [Google Scholar]
- 9.Hoang T, Paradis E, Brady G, Billia F, Nakahara K, Iscove NN, Kirsch IR. Opposing effects of the basic helix-loop-helix transcription factor SCL on erythroid and monocytic differentiation. Blood. 1996;87:102–111. [PubMed] [Google Scholar]
- 10.Pulford K, Lecointe N, Leroy-Viard K, Jones M, Mathieu-Mahul D, Mason DY. Expression of TAL-1 proteins in human tissues. Blood. 1995;85:675–684. [PubMed] [Google Scholar]
- 11.Mouthon MA, Bernard O, Mitjavila MT, Romeo PH, Vainchenker W, Mathieu-Mahul D. Expression of tal-1 and GATA-binding proteins during human hematopoiesis. Blood. 1993;81:647–655. [PubMed] [Google Scholar]
- 12.Aplan PD, Nakahara K, Orkin SH, Kirsch IR. The SCL gene product: a positive regulator of erythroid differentiation. EMBO (Eur Mol Biol Organ) J. 1992;11:4073–4081. doi: 10.1002/j.1460-2075.1992.tb05500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Begley CG, Aplan PD, Denning SM, Haynes BF, Waldmann TA, Kirsch IR. The gene SCL is expressed during early hematopoiesis and encodes a differentiation-related DNA-binding motif. Proc Natl Acad Sci USA. 1989;86:10128–10132. doi: 10.1073/pnas.86.24.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green AR, Lints T, Visvader J, Harvey R, Begley CG. SCL is coexpressed with GATA-1 in hemopoietic cells but is also expressed in developing brain. Oncogene. 1992;7:653–660. [PubMed] [Google Scholar]
- 15.Green AR, Salvaris E, Begley CG. Erythroid expression of the ‘helix-loop-helix' gene, SCL. Oncogene. 1991;6:475–479. [PubMed] [Google Scholar]
- 16.Miller BA, Floros J, Cheung JY, Wojchowski DM, Bell L, Begley CG, Elwood NJ, Kreider J, Christian C. Steel factor affects SCL expression during normal erythroid differentiation. Blood. 1994;84:2971–2976. [PubMed] [Google Scholar]
- 17.Robb L, Lyons I, Li R, Hartley L, Kontgen F, Harvey RP, Metcalf D, Begley CG. Absence of yolk sac hematopoiesis from mice with a targeted disruption of the scl gene. Proc Natl Acad Sci USA. 1995;92:7075–7079. doi: 10.1073/pnas.92.15.7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shivdasani RA, Mayer EL, Orkin SH. Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature. 1995;373:432–434. doi: 10.1038/373432a0. [DOI] [PubMed] [Google Scholar]
- 19.Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt FW, Orkin SH. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86:47–57. doi: 10.1016/s0092-8674(00)80076-8. [DOI] [PubMed] [Google Scholar]
- 20.Robb L, Elwood NJ, Elefanty AG, Kontgen F, Li RL, Barnett LD, Begley CG. The SCL gene product is required for the generation of all hematopoietic lineages in the adult mouse. EMBO (Eur Mol Biol Organ) J. 1996;15:4123–4129. [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu HL, Huang L, Tsan JT, Funk W, Wright WE, Hu JS, Kingston RE, Baer R. Preferred sequences for DNA recognition by the TAL1 helix-loop-helix proteins. Mol Cell Biol. 1994;14:1256–1265. doi: 10.1128/mcb.14.2.1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valge-Archer VE, Osada H, Warren AJ, Forster A, Li J, Baer R, Rabbitts TH. The LIM protein RBTN2 and the basic helix-loop-helix protein TAL1 are present in a complex in erythroid cells. Proc Natl Acad Sci USA. 1994;91:8617–8621. doi: 10.1073/pnas.91.18.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wadman I, Li J, Bash RO, Forster A, Osada H, Rabbitts TH, Baer R. Specific in vivo association between the bHLH and LIM proteins implicated in human T cell leukemia. EMBO (Eur Mol Biol Organ) J. 1994;13:4831–4839. doi: 10.1002/j.1460-2075.1994.tb06809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wadman IA, Osada H, Grütz GG, Agulnick AD, Westphal H, Forster A, Rabbitts TH. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the Tal1, E47, GATA-1 and LDB1/NLI proteins. EMBO (Eur Mol Biol Organ) J. 1997;16:3145–3157. doi: 10.1093/emboj/16.11.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warren AJ, Colledge WH, Carlton MB, Evans MJ, Smith AJ, Rabbitts TH. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell. 1994;78:45–57. doi: 10.1016/0092-8674(94)90571-1. [DOI] [PubMed] [Google Scholar]
- 26.Tsai SF, Martin DI, Zon LI, D'Andrea AD, Wong GG, Orkin SH. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature. 1989;339:446–451. doi: 10.1038/339446a0. [DOI] [PubMed] [Google Scholar]
- 27.Ono Y, Fukuhara N, Yoshie O. Transcriptional activity of TAL1 in T cell acute lymphoblastic leukemia (T-ALL) requires RBTN1 or -2 and induces TALLA1, a highly specific tumor marker of T-ALL. J Biol Chem. 1997;272:4576–4581. doi: 10.1074/jbc.272.7.4576. [DOI] [PubMed] [Google Scholar]
- 28.Caceres-Cortes J, Rajotte D, Dumouchel J, Haddad P, Hoang T. Product of the steel locus suppresses apoptosis in hemopoietic cells. Comparison with pathways activated by granulocyte macrophage colony-stimulating factor. J Biol Chem. 1994;269:12084–12091. [PubMed] [Google Scholar]
- 29.Rajotte D, Haddad P, Haman A, Cragoe EJ, Jr, Hoang T. Role of protein kinase C and the Na+/H+antiporter in suppression of apoptosis by granulocyte macrophage colony-stimulating factor and interleukin-3. J Biol Chem. 1992;267:9980–9987. [PubMed] [Google Scholar]
- 30.Williams GT, Smith CA, Spooncer E, Dexter TM, Taylor DR. Haemopoietic colony stimulating factors promote cell survival by suppressing apoptosis. Nature. 1990;343:76–79. doi: 10.1038/343076a0. [DOI] [PubMed] [Google Scholar]
- 31.Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:87–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- 32.Abrahamson JL, Lee JM, Bernstein A. Regulation of p53-mediated apoptosis and cell cycle arrest by Steel factor. Mol Cell Biol. 1995;15:6953–6960. doi: 10.1128/mcb.15.12.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Copeland NG, Gilbert DJ, Cho BC, Donovan PJ, Jenkins NA, Cosman D, Anderson D, Lyman SD, Williams DE. Mast cell growth factor maps near the steel locus on mouse chromosome 10 and is deleted in a number of steel alleles. Cell. 1990;63:175–183. doi: 10.1016/0092-8674(90)90298-s. [DOI] [PubMed] [Google Scholar]
- 34.Fleischman RA. From white spots to stem cells: the role of the Kit receptor in mammalian development. Trends Genet. 1993;9:285–290. doi: 10.1016/0168-9525(93)90015-a. [DOI] [PubMed] [Google Scholar]
- 35.Russel ES. Hereditary anemias of the mouse: a review for geneticists. Adv Genet. 1979;20:357–459. [PubMed] [Google Scholar]
- 36.Li CL, Johnson GR. Stem cell factor enhances the survival but not the self-renewal of murine hematopoietic long-term repopulating cells. Blood. 1994;84:408–414. [PubMed] [Google Scholar]
- 37.Godfrey DI, Zlotnik A, Suda T. Phenotypic and functional characterization of c-kit expression during intrathymic T cell development. J Immunol. 1992;149:2281–2285. [PubMed] [Google Scholar]
- 38.Moll J, Eibel H, Schmid P, Sansig G, Botteri F, Palacios R, Van der Putten H. Thymic hyperplasia in transgenic mice caused by immortal epithelial cells expressing c-kit ligand. Eur J Immunol. 1992;22:1587–1594. doi: 10.1002/eji.1830220636. [DOI] [PubMed] [Google Scholar]
- 39.Palacios R, Nishikawa S. Developmentally regulated cell surface expression and function of c-kit receptor during lymphocyte ontogeny in the embryo and adult mice. Development (Camb) 1992;115:1133–1147. doi: 10.1242/dev.115.4.1133. [DOI] [PubMed] [Google Scholar]
- 40.Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T, Miyagawa K, Piao YF, Miyazono K, Urabe A, Takaku F. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J Cell Physiol. 1989;140:323–334. doi: 10.1002/jcp.1041400219. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez C, Lacasse C, Hoang T. Interleukin-1 beta suppresses apoptosis in CD34 positive bone marrow cells through activation of the type I IL-1 receptor. J Cell Physiol. 1996;166:387–396. doi: 10.1002/(SICI)1097-4652(199602)166:2<387::AID-JCP17>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 42.Leroy-Viard K, Vinit MA, Lecointe N, Jouault H, Hibner U, Roméo PH, Mathieu-Mahul D. Loss of TAL-1 protein activity induces premature apoptosis of Jurkat leukemic T cells upon medium depletion. EMBO (Eur Mol Biol Organ) J. 1995;14:2341–2349. doi: 10.1002/j.1460-2075.1995.tb07229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hawley RG, Fong AZ, Lu M, Hawley TS. The Hox-11 homeobox-containing gene of human leukemia immortalizes murine hematopoietic precursors. Oncogene. 1994;9:1–12. [PubMed] [Google Scholar]
- 44.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 46.Tso JY, Sun XH, Kao TH, Reece KS, Wu R. Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res. 1985;13:2485–2502. doi: 10.1093/nar/13.7.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Durocher D, Chen CY, Ardati A, Schwartz RJ, Nemer M. The atrial natriuretic factor promoter is a downstream target for Nkx-2.5 in the myocardium. Mol Cell Biol. 1996;16:4648–4655. doi: 10.1128/mcb.16.9.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelliher MA, Seldin DC, Leder P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIa. EMBO (Eur Mol Biol Organ) J. 1996;15:5160–5166. [PMC free article] [PubMed] [Google Scholar]
- 49.Tsujimura T, Morii E, Nozaki M, Hashimoto K, Moriyama Y, Takebayashi T, Kondo T, Kanakura Y, Kitamura Y. Involvement of transcriptional factor encoded by the mi locus in the expression of c-kit receptor tyrosine kinase in cultured mast cells of mice. Blood. 1996;88:1225–1233. [PubMed] [Google Scholar]
- 50.Therrien M, Drouin J. Cell-specific helix-loop-helix factor required for pituitary expression of the pro-opiomelanocortin gene. Mol Cell Biol. 1993;13:2342–2353. doi: 10.1128/mcb.13.4.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- 52.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Green AR, DeLuca E, Begley CG. Antisense SCL suppresses self-renewal and enhances spontaneous erythroid differentiation of the human leukaemic cell line K562. EMBO (Eur Mol Biol Organ) J. 1991;10:4153–4158. doi: 10.1002/j.1460-2075.1991.tb04993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohashi H, Kameda R, Nishikawa M, Kawagishi M, Liu YC. The c-kit receptor transduces the stem cell factor–triggered growth signal in murine interleukin-3– dependent cell line. Cytotechnology. 1994;16:27–35. doi: 10.1007/BF00761776. [DOI] [PubMed] [Google Scholar]
- 55.Nelson BH, Lord JD, Greenberg PD. Cytoplasmic domains of the interleukin-2 receptor beta and gamma chains mediate the signal for T-cell proliferation. Nature. 1994;369:333–336. doi: 10.1038/369333a0. [DOI] [PubMed] [Google Scholar]
- 56.Elefanty AG, Robb L, Birner R, Begley CG. Hematopoietic-specific genes are not induced during in vitro differentiation of scl-null embryonic stem cells. Blood. 1997;90:1435–1447. [PubMed] [Google Scholar]
- 57.Linette GP, Hess JL, Sentman CL, Korsmeyer SJ. Peripheral T-cell lymphoma in lckpr-bcl-2 transgenic mice. Blood. 1995;86:1255–1260. [PubMed] [Google Scholar]
- 58.Robb L, Rasko JE, Bath ML, Strasser A, Begley CG. scl, a gene frequently activated in human T cell leukaemia, does not induce lymphomas in transgenic mice. Oncogene. 1995;10:205–209. [PubMed] [Google Scholar]
- 59.Larson RC, Lavenir I, Larson TA, Baer R, Warren AJ, Wadman I, Nottage K, Rabbitts TH. Protein dimerization between Lmo2 (Rbtn2) and Tal1 alters thymocyte development and potentiates T cell tumorigenesis in transgenic mice. EMBO (Eur Mol Biol Organ) J. 1996;15:1021–1027. [PMC free article] [PubMed] [Google Scholar]
- 60.Wolf SS, Cohen A. Expression of cytokines and their receptors by human thymocytes and thymic stromal cells. Immunology. 1992;77:362–368. [PMC free article] [PubMed] [Google Scholar]
- 61.Rodewald HR, Kretzschmar K, Swat W, Takeda S. Intrathymically expressed c-kit ligand (stem cell factor) is a major factor driving expansion of very immature thymocytes in vivo. Immunity. 1995;3:313–319. doi: 10.1016/1074-7613(95)90116-7. [DOI] [PubMed] [Google Scholar]
- 62.Elwood NJ, Begley CG. Reconstitution of mice with bone marrow cells expressing the SCL gene is insufficient to cause leukemia. Cell Growth Differ. 1995;6:19–25. [PubMed] [Google Scholar]
- 63.Bishop JM. Molecular themes in oncogenesis. Cell. 1991;64:235–248. doi: 10.1016/0092-8674(91)90636-d. [DOI] [PubMed] [Google Scholar]
- 64.Linette GP, Korsmeyer SJ. Differentiation and cell death: lessons from the immune system. Curr Opin Cell Biol. 1994;6:809–815. doi: 10.1016/0955-0674(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 65.Pevny L, Lin CS, D'Agati V, Simon MC, Orkin SH, Costantini F. Development of hematopoietic cells lacking transcription factor GATA-1. Development (Camb) 1995;121:163–172. doi: 10.1242/dev.121.1.163. [DOI] [PubMed] [Google Scholar]
- 66.Weiss MJ, Orkin SH. Transcription factor GATA-1 permits survival and maturation of erythroid precursors by preventing apoptosis. Proc Natl Acad Sci USA. 1995;92:9623–9627. doi: 10.1073/pnas.92.21.9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grépin C, Nemer G, Nemer M. Enhanced cardiogenesis in embryonic stem cells overexpressing the GATA-4 transcriptional factor. Development (Camb) 1997;124:2387–2395. doi: 10.1242/dev.124.12.2387. [DOI] [PubMed] [Google Scholar]