

Abstract
Immunoglobulin (Ig)A provides the initial immune barrier to viruses at mucosal surfaces. Specific IgA interrupts viral replication in polarized epithelium during receptor-mediated transport, probably by binding to newly synthesized viral proteins. Here, we demonstrate by immunoelectron microscopy that specific IgA monoclonal antibodies (mAbs) accumulate within Sendai virus–infected polarized cell monolayers and colocalize with the hemagglutinin– neuraminidase (HN) viral protein in a novel intracellular structure. Neither IgG specific for HN nor irrelevant IgA mAbs colocalize with viral protein. Treatment of cultures with viral-specific IgA but not with viral-specific IgG or irrelevant IgA decreases viral titers. These observations provide definitive ultrastructural evidence of a subcellular compartment in which specific IgA and viral envelope proteins interact, further strengthening our hypothesis of intracellular neutralization of virus by specific IgA antibodies. Our results have important implications for intracellular protein trafficking, viral replication, and viral vaccine development.
Keywords: immunoglobulin A, Sendai virus, mucosal immunity, colocalization, hemagglutinin–neuraminidase protein
Many pathogens commonly invade the body through and replicate within mucous surfaces where IgA, the predominant antibody isotype found in mucosal secretions, forms the first layer of immune defense (1, 2). The importance of mucosal antibody is suggested by the observations that the synthetic rate of IgA exceeds that of all the other classes of antibodies combined and that resistance to viral infections correlates best with the presence of specific IgA antibody in mucosal secretions (3, 4). Traditionally, mucosal antibodies are thought to function extracellularly by complexing with viral envelope proteins, thereby preventing attachment of virions to the epithelium (5–8). Yet the unique active transport of polymeric immunoglobulins, mediated by the polymeric immunoglobulin receptor (pIgR)1 on the basolateral surface of secretory epithelium (9–11), may afford IgA antibody the opportunity to interact with intracellular antigens, including the synthetic products of viral pathogens.
Sendai virus, a prototypical paramyxovirus, is a natural respiratory pathogen of rodents. Similar to human parainfluenza viruses, Sendai virus contains a nonsegmented RNA genome encoding six major structural proteins (12, 13). We have shown that during transport through infected polarized epithelial cells, specific polymeric IgA acts intracellularly to interfere with virus replication, assembly, or release, presumably by binding to newly synthesized viral proteins (14–17). Such intracellular neutralization of virus by IgA might eradicate infection while avoiding cytolysis of infected epithelial cells. If the integrity of the epithelium is thus preserved, viral antigens would be prevented from gaining access into the body, and systemic sensitization would be forestalled.
The immunoelectron microscopy experiments described in this report document novel intracellular structures that are formed when specific polymeric IgA is added basolaterally to Sendai virus–infected polarized cell monolayers. These structures are not seen when infected monolayers are treated with specific IgG or irrelevant IgA. Thus, this previously undescribed cellular structure appears to be the site of intracellular neutralization of virus by specific IgA. The site of colocalization implies that optimal vaccination should focus upon selected viral proteins that transit through apical recycling endosomes.
Materials and Methods
Cell and Virus Culture.
Madin Darby Canine Kidney (MDCK) cells, stably transfected with the cDNA encoding pIgR derived from rabbit (obtained from Keith Mostov, University of California, San Francisco), were maintained in MEM containing 10% fetal bovine serum (Hyclone, Logan, UT), 100 μM nonessential amino acids, 2 mM glutamine, 20 μg/ml gentamicin, and 15 mM Hepes (all from GIBCO BRL, Gaithersburg, MD), and were grown in 5% CO2 at 37°C. Cells were grown to confluence on nitrocellulose filter inserts (Millicell; Millipore, Bedford, MA); polarization of the cells was confirmed by the electric potential determined by a Millicell ERS resistance meter (Millipore). Sendai virus strain 52 was grown in 10-d-old chicken embryos and the virus was harvested from allantoic fluid after extensive centrifugation (18).
mAbs.
Viral-specific IgA and IgG antibodies were produced and purified as previously described (14, 16, 19, 20). In brief, mice were immunized, twice intraduodenally and once orally by feeding tube with 100 μg of whole virus plus 5 μg of cholera toxin (List Laboratories, Campbell, CA). Mice were boosted by intravenous injection of 30 μg of antigen, and splenocytes of immunized animals were fused with SP2/0 cells as per published protocols (14, 16, 19, 20). Clones secreting antibody to the specific antigen were selected, subcloned, expanded, and frozen. Selected hybridoma cells were injected into BALB/c mice primed with 2,6,10,14-tetramethylpentadecane (Pristane; Sigma Chemical Co., St. Louis, MO) for the production of ascites.
Gold Labeling of Antibodies.
Gold particles (BB International, Cardiff, UK) were dialyzed in PBS before use. Antibody to be labeled was dialyzed at a concentration of 1 mg/ml against a 2 mM borax (Sigma Chemical Co.) buffer (pH 7.2) and centrifuged at 100,000 g for 1 h. After addition of antibody to the gold, BSA was added to a final concentration of 1% and the gold-antibody slurry was centrifuged at 60,000 g for 1 h. The sediment was washed twice by resuspending in Tris-buffered saline (TBS) with 1% BSA, (pH 7.2), followed by recentrifugation. The final pellet was resuspended in TBS/1% BSA/0.1% sodium azide and stored at 4°C until used.
Incubations and Immunoelectron Microscopy.
When confluent, polarized cell monolayers were apically infected with 10–30 PFU/cell of Sendai virus. 4 h later, ascites containing equivalent ELISA titers of IgA specific for the viral hemagglutinin–neuraminidase (HN) protein, IgG specific for the viral HN protein, or an irrelevant IgA (mineral oil plasmacytoma line 315) were added to the basolateral surface. 24 h after inoculation, cells were fixed in 2% paraformaldehyde for 30 min at 4°C and permeabilized by subsequent incubation in 0.25% saponin for 1 h at room temperature. To detect viral HN, monolayers experimentally treated with IgA were stained with a primary biotin-conjugated murine mAb for 1 h at room temperature and then 20 h at 4°C, followed by 5 nm gold-labeled sheep anti-biotin for 1 h at room temperature before embedding (Amersham Pharmacia Biotech, Arlington, IL); the primary antibody (murine IgG) was directed against a different epitope on the viral HN protein than the IgA antibody added to the basolateral surface during the experiment. Experimentally added IgA was detected by staining, initially with the IgG fraction of rabbit anti–mouse IgA for 1 h at room temperature and 20 h at 4°C, followed by 15 nm gold-labeled goat anti–rabbit IgG for 1 h at room temperature (Amersham Pharmacia Biotech). Similarly, detection of viral protein in cells experimentally treated with IgG used staining with a murine monoclonal IgA directed against a different epitope on the HN protein than the original IgG applied basolaterally to the cells. This was followed by treatment with the IgG fraction of rabbit anti–mouse IgA for 1 h at room temperature and 20 h at 4°C and 15 nm gold-labeled goat anti–rabbit IgG for 1 h at room temperature. IgG was detected by staining with 5 nm gold-labeled goat anti–mouse IgG for 1 h at room temperature. The monolayers were then embedded in Epon 812, ultra-thin sectioned, and counterstained with uranyl acetate and lead citrate. Sections were then examined with an electron microscope (Zeiss CEM 902; Carl Zeiss Inc., Thornwood, NY). In other experiments, polarized MDCK monolayers were apically infected with 30 PFU/cell and 4 h later incubated with 15 nm gold-labeled IgA anti-HN or IgG anti-HN or irrelevant IgA applied basolaterally. Additional control polarized MDCK cultures were also infected with 30 PFU/cell apically, but 4 h later were treated basolaterally with unlabeled IgA anti-HN or apically with 15 nm gold-labeled IgA anti-HN.
Morphometric Analysis of Cytoplasmic Viral Protein and Antibody, and Assessment of Colocalization.
A total of 100 coded electron micrographs from four separate experiments were subjected to morphometric analysis with computer assistance (Image II program; National Institutes of Health, Bethesda, MD; running on a Centris 650; Apple Computer, Cupertino, CA). For each micrograph, the area of the field of view, excluding extracellular area and areas occupied by cell nuclei or grid bars, was determined by digital planimetry; the cytoplasmic area depicted on each print was then calculated from the calibrated magnification of the particular micrograph (ranging from ×7,000 to ×30,000) and the print magnification (calibrated at ×2.6). Next, the total number of small (5-nm) and large (15-nm) gold particles were enumerated separately in each micrograph; these values were then divided by the cytoplasmic area in that micrograph to determine total particle density (expressed as particles/square micrometer). Finally, the number of small particles within a 225-nm radius of at least one large particle, and the number of large particles within a 225-nm radius of at least one small particle, were enumerated; again, the density of “small particles near to large particles” and of “large particles near to small particles” was calculated by dividing the number of each category of colocalized particle on each print by the cytoplasmic area within that print. The morphometric density data were subjected to two-way analysis of variance, classified according to the incubation conditions of the cells and the particular experiment; since interexperimental variation was not significant, the data of the four individual experiments were pooled, and a one-way classification (according to incubation conditions) is reported. In addition to expressing the morphometric data normalized to cytoplasmic area (particle densities), colocalization was also assessed by the fraction of all antibody label that lies in proximity (within 225 nm) to virus label, and vice versa, expressed as percentages of the total particles of the appropriate size in that micrograph. Two-way analysis of variance (classified by experiment and by incubation) of the percentages indicated no significant difference among experiments; all four experiments were pooled, and the results of a one-way classification are reported.
Results and Discussion
Although our previous data support an intersection of IgA antibody with a viral envelope glycoprotein (14–17), we here document definitively an intracellular interaction between IgA and viral proteins by immunoelectron microscopy (Figs. 1–3). The HN glycoprotein, which is responsible for adherence of virions to host epithelial cells, is synthesized in infected cells on the rough endoplasmic reticulum, glycosylated, processed through the Golgi apparatus, and finally transported to the apical surface, where it is inserted into the host cell membrane in anticipation of virion assembly and budding (21–23). As shown in Fig. 1, basolateral addition of IgA anti-HN (A), but not IgG anti-HN (B) or irrelevant IgA (C), reduces the expression of immunodetectable HN protein on the apical host cell membrane, and virion budding after 24 h. Collaterally, there is a reduction in the density of total immunostainable viral HN protein (Table 1) and in viral titer in the apical supernatant (data not shown) only if IgA anti-HN is added to the cells.
Figure 1.
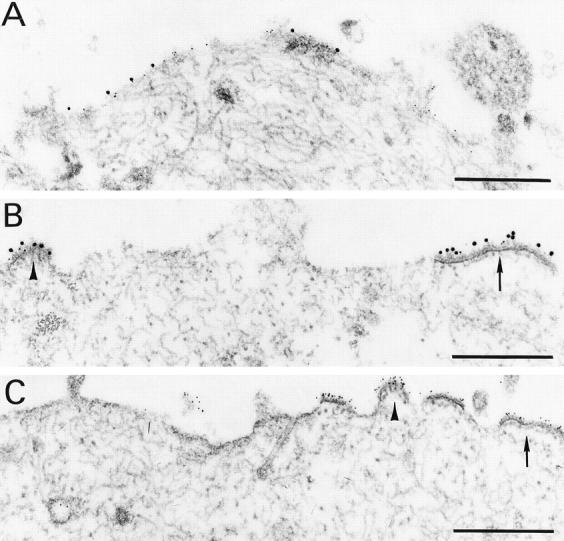
Treatment with specific IgA, but not specific IgG or irrelevant IgA, reduces the appearance of viral protein on the cell surface. Polarized monolayers of MDCK cells in culture well inserts, stably transfected with the pIgR derived from rabbit, were infected with Sendai virus at 10 PFU/cell. 4 h later, ascites containing equivalent ELISA titers of IgA specific for the viral HN protein (clone 37 HN; A), IgG specific for the viral HN protein (clone 20 HN; B), or an irrelevant IgA (mineral oil plasmacytoma line 315; C) were added to the basolateral surface as previously described (6). Productive viral infection is apparent in cells treated with specific IgG (B) or irrelevant IgA (C), with dense accretions of viral protein in patches on the apical portion of the cytoplasmic membrane (arrows), sometimes forming domed buds containing fibrillar chromatin-like material (arrowheads). Note that in A and C, small (5 nm) gold particles label the viral proteins, whereas in B, viral protein is detected by large (15 nm) gold particles. Essentially no intracellular Ig is identified in these latter specimens. By contrast, infected cells treated with specific IgA (A) show little viral protein at the cell surface and no accretions of viral protein or bud formations. Bar = 0.5 μm.
Figure 3.
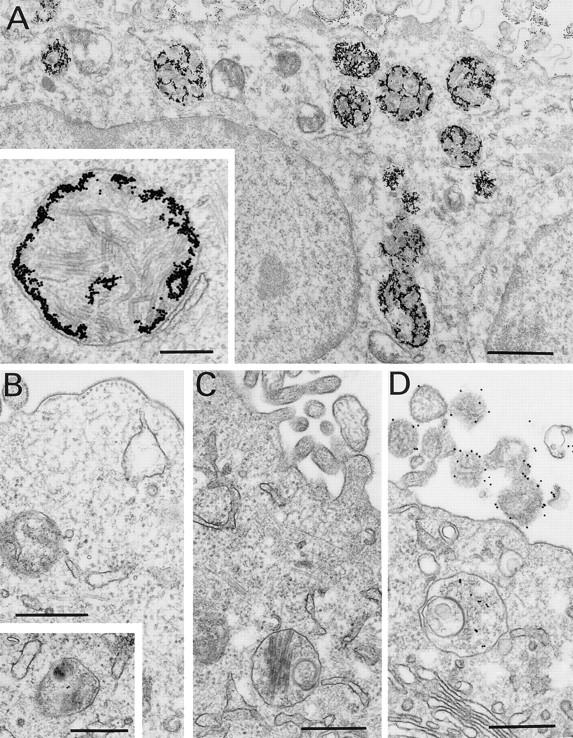
Specific IgA promotes the formation of membrane-delimited inclusions of multiple palisaded layers. Polarized MDCK cells were infected with 30 PFU/cell and 4 h later were incubated with 15 nm gold-labeled IgA anti-HN (A), IgG (data not shown), or irrelevant IgA (B), applied basolaterally. Other (control) polarized MDCK cells were also infected with 30 PFU/cell but 4 h later were treated basolaterally with unlabeled IgA anti-HN (C) or apically with 15 nm gold-labeled IgA anti-HN (D). Cells treated basolaterally with gold-labeled IgA anti-HN contain aggregates of numerous gold particles within numerous vesicular structures (A) that contain multi-lamellar structures (A, inset). In contrast, although a few gold particles are seen within vesicles in cells treated with IgG anti-HN (data not shown) or irrelevant IgA (B), the massive aggregates of gold associated with multilamellar structures, seen in A, are not visible. Infected cells treated with unlabeled IgA anti-HN (C) demonstrate vesicles similar in appearance to those in cells receiving gold-labeled specific IgA (A) but without gold particles, indicating that the formation of these structures is not due to the presence of gold. Although infected cells apically treated with gold-labeled IgA anti-HN exhibit a few gold particles in vesicles (D), large aggregates of gold and inclusions containing lamellae are not visualized, indicating that the initial reaction between IgA antibody and viral protein occurs within the cell during transcytosis and not near the cell surface after release into the apical supernatant upon subsequent re-uptake. Bars: A, 1 μm; inset to A, 0.25 μm; B–D, 0.5 μm.
Table 1.
Morphometric Assessment of Colocalization of Ig and Viral HN Protein
| Ig added | Virus added | Ig label | Viral HN protein label | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Colocalized density | Percentage colocalized | Total density | Percentage colocalized | |||||||
| specific IgA | yes | 113.7 ± 36* | 40.1 ± 1.9* | 0.50 ± 0.14 | 22.0 ± 1.6* | |||||
| irrevelant IgA | yes | 0.17 ± 0.2 | 0.28 ± 0.3 | 3.61 ± 0.78‡ | 1.8 ± 1.8 | |||||
| specific IgG | yes | 11.7 ± 8.1§ | 9.3 ± 1.9§ | 1.90 ± 1.2‡ | 6.2 ± 1.5§ | |||||
| specific IgA | no | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |||||
| none | no | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | |||||
Significantly (F > 4.8, P < 0.002) higher than all other groups.
Significantly (F = 6.4, P < 0.001) higher than infected cells treated with specific IgA and higher than both groups of uninfected cells.
Significantly (F > 4.8, P < 0.002) lower than infected cells treated with specific IgA and higher than all other groups.
As seen in Fig. 2, specific IgA (A), but not specific IgG (B) or irrelevant IgA (C), colocalizes with viral HN protein within multilamellar membrane-bound inclusions in the cell cytoplasm. Indeed, quantitative morphometry reveals a 10-fold higher density of specific IgA colocalized with viral protein relative to colocalized specific IgG (Table 1). Moreover, when expressed as a percentage of total Ig label, colocalized specific IgA was four times more abundant than colocalized specific IgG (Table 1). As these data imply, the total density of detectable intracellular specific IgA (430 ± 23 particles/100 μm2) was more than three times that of specific IgG (127 ± 83 particles/100 μm2). In contrast, irrelevant IgA did not accumulate within infected cells (total density = 27.7 ± 9.4 particles/100 μm2), and did not colocalize to viral label (Table 1).
Figure 2.
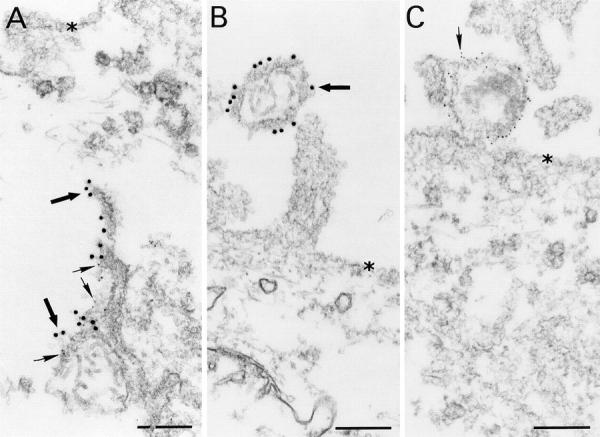
Colocalization of specific IgA and viral protein within the cytoplasm of Sendai virus–infected cells. In polarized MDCK cells infected with Sendai virus and treated by basolateral application of IgA anti-HN 4 h later (A, a replicate culture of Fig. 1 A), there is colocalization of numerous large (15 nm) gold particles labeling IgA (large arrows) and numerous small (5 nm) gold particles labeling viral protein (small arrows) to form multilamellar membrane structures, located deep within the cytosol. In infected cells treated with specific IgG (B) or irrelevant IgA (C), budding virions arising from the cell surface (indicated by asterisks) are identified by anti-viral HN protein staining (15-nm gold particles in B, large arrow; 5-nm gold particles in C, small arrow). The intracellular inclusions which develop in infected cells treated with specific IgA are only rarely identified in cells treated with specific IgG, and Ig does not frequently colocalize with viral protein. Neither infected cells treated with irrelevant IgA nor uninfected cells ever contain these structures. Bar = 0.25 μm.
In parallel experiments using antibodies labeled directly with gold particles before application to the basolateral surface of the cell (Fig. 3), infected cells treated with IgA anti-HN demonstrate massive accumulation of gold particles within innumerable membrane-delimited inclusions (Fig. 3 A) that contain multiple palisaded layers (Fig. 3 A, inset). This technique obviates the need for permeabilization of the cells. Smaller but similar organelles are rarely visible in infected cells treated with irrelevant IgA (Fig. 3 B), specific IgG (data not shown), or no antibody (data not shown). The relative abundance of these structures in cells treated with specific IgA indicates that IgA anti-HN promotes their formation. These inclusions are not the result of intracellular gold, since they are visible to a similar degree in infected cells treated with unlabeled IgA anti-HN, albeit without the gold particles (Fig. 3 C). To determine if accumulation of IgA results from endocytosis of free antibody or immune complexes formed on the apical cell surface by transported antibody, gold-labeled IgA anti-HN was added to the apical supernatant of infected cells. As shown (Fig. 3 D), only a few gold particles accumulate within the cells. The large quantities of gold and palisaded bodies seen in Fig. 3 A are not visualized, indicating that specific IgA is retarded in the infected cell during transcytosis, rather than being subject to significant re-uptake after release into the apical medium.
Scanning laser confocal microscopy with fluorescent antibodies discloses colocalization of antibody and viral HN protein in essentially all infected cells treated with polymeric IgA anti-HN applied basolaterally. The colocalization of specific IgA antibody and HN protein is seen only in the apical third of the polarized monolayer (data not shown), suggesting that the multilamellar inclusions arise from apical recycling endosomes (24–28). Colocalization is never observed in uninfected cells, nor in infected cells treated with irrelevant polymeric IgA or IgG anti-HN.
Finally, additional studies compared IgA mAbs directed against the HN viral envelope protein to those against the viral nucleoprotein (NP). In contrast to the HN protein, which is synthesized in the endoplasmic reticulum, the synthesis of NP occurs on free cytoplasmic ribosomes (29, 30). Upon addition to the basolateral surface, IgA anti-HN but not IgA anti-NP colocalizes with the respective viral protein by immunoelectron microscopy (data not shown) despite the fact that both IgA antibodies undergo effective transcytosis. Furthermore, the addition of IgA anti-HN reduces viral titers in the apical supernatants from infected monolayers, whereas IgA anti-NP does not (data not shown). These differences between IgA anti-HN and anti-NP antibodies are consistent with the different sites of synthesis and processing of the two viral proteins relative to the transcytotic pathway of the IgA antibody.
The current observations, in conjunction with our prior findings (14–17), strongly support the hypothesis that during transcytosis, specific IgA can complex with some viral proteins within polarized epithelial cells and thereby prevent virion assembly and release. The exact nature and site of this intracellular interaction remain to be defined. The current model of epithelial transcytosis does not postulate a unique receptor–ligand endosomal pathway for the transport of IgA from the basolateral to the apical cell surface. Rather, the pIgR–IgA complex travels through common endosomal compartments with other recycling proteins that undergo endocytosis (24–28). Initially, the pIgR–IgA complex is delivered to early basolateral endosomes, but is later routed to apical recycling endosomes, which are thought to be a key site of protein sorting. Thus, apical recycling endosomes are a potential location for IgA to intercept viral proteins, a view compatible with our confocal microscopic observations.
As demonstrated by these studies, the ability of specific IgA to interrupt viral replication depends on the mode of replication of the particular virus in question, and the viral protein that is recognized by the antibody. Viral glycoproteins that are synthesized on the rough endoplasmic reticulum and subsequently transported to the apical cell surface are probably most vulnerable to intracellular neutralization. Prevention of virion assembly and budding by IgA acting intracellularly may potentially forestall cytopathic effects and spare the cell, at least during some viral infections. Preservation of the integrity of the mucous membrane in this manner could maintain the epithelial barrier and retard systemic dissemination of viral antigens. Thus, the ability of IgA antibody to act within epithelial cells would synergistically reinforce its traditional extracellular function in affording humoral antiviral defense. These issues impact upon the strategy to develop mucosal vaccines and argue for continued investigation into humoral immune defense mechanisms in the mucosa.
Acknowledgments
We thank Alan M. Tartakoff and Michael E. Lamm for critical readings of the manuscript and Cecily Lewis for preparation of the manuscript.
This work was supported by National Institutes of Health grants AI-32588, AI-CA-36359, AI-37359, and AI-35827; a grant from the U.S. Agency for International Development (HRN-6001-A-00-2018-00); and a grant-in-aid for scientific research on priority areas from the Ministry of Education, Science, and Culture of Japan.
Abbreviations used in this paper
- HN
hemagglutinin-neuraminidase
- MDCK
Madin Darby canine kidney
- NP
nucleoprotein
- pIgR
polymeric immunoglobulin receptor
References
- 1.Lamm ME. Cellular aspects of immunoglobulin A. Adv Immunol. 1976;22:223–290. doi: 10.1016/s0065-2776(08)60550-7. [DOI] [PubMed] [Google Scholar]
- 2.Mestecky J, McGhee JR. Immunoglobulin A (IgA): molecular and cellular interactions involved in IgA biosynthesis and immune response. Adv Immunol. 1987;40:153–245. doi: 10.1016/s0065-2776(08)60240-0. [DOI] [PubMed] [Google Scholar]
- 3.Mills J, Van Kirk JE, Wright PF, Chanock RM. Experimental respiratory syncytial virus infection of adults. Possible mechanisms of resistance to infection and illness. J Immunol. 1971;107:123–130. [PubMed] [Google Scholar]
- 4.Liew FY SM, Russell, Appleyard G, Brand CM, Beale J. Cross-protection in mice infected with influenza A virus by the respiratory route is correlated with local IgA antibody rather than serum antibody or cytotoxic T-cell reactivity. Eur J Immunol. 1984;14:350–356. doi: 10.1002/eji.1830140414. [DOI] [PubMed] [Google Scholar]
- 5.Outlaw MC, Dimmock NJ. Mechanisms of neutralization of influenza virus on mouse tracheal epithelial cells by mouse monoclonal polymeric IgA and polyclonal IgM directed against the viral haemagglutinin. J Gen Virol. 1990;71:69–76. doi: 10.1099/0022-1317-71-1-69. [DOI] [PubMed] [Google Scholar]
- 6.Whitton, J.L., and M.B. Oldstone. 1996. Immune response to viruses. In Fundamental Virology. B.N. Fields., D.M. Knipe, and P.M. Howley, editors. Lippincott-Raven Publishers, New York. 311–340.
- 7.Murphy, B.R. 1994. Mucosal immunity to viruses. In Handbook of Mucosal Immunology. P.L. Ogra, M.E. Lamm, J.R. McGhee, J. Mestecky, W. Strober, and J. Bienenstock, editors. Academic Press, Inc., San Diego, CA. 333–343.
- 8.Crowe JE. The role of antibodies in respiratory viral immunity. Semin Virol. 1996;7:273–283. [Google Scholar]
- 9.Brandtzaeg P. Polymeric IgA is complexed with secretory component (SC) on the surface of human intestinal epithelial cells. Scand J Immunol. 1978;8:39–52. doi: 10.1111/j.1365-3083.1978.tb00494.x. [DOI] [PubMed] [Google Scholar]
- 10.Crago SS, Kulhavy R, Prince SJ, Mestecky J. Secretory component of epithelial cells is a surface receptor for polymeric immunoglobulins. J Exp Med. 1978;147:1832–1837. doi: 10.1084/jem.147.6.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuhn LC, Kraehenbuhl JP. Role of secretory component, a secreted glycoprotein, in the specific uptake of IgA dimer by epithelial cells. J Biol Chem. 1979;254:11072–11081. [PubMed] [Google Scholar]
- 12.Lamb, R., and A.D. Kolakofsky. 1996. Paramyxoviridae: the viruses and their replication. In Fundamental Virology. B.N. Fields, D.M. Knipe, and P.M. Howley, editors. Lippincott-Raven Publishers, New York. 577–604.
- 13.Lamb, R.A. 1996. Orthomyxoviridae: the viruses and their replication. In Fundamental Virology. B.N. Fields, D.M. Knipe, P.M. Howley, editors. Lippincott-Raven Publishers, New York. 605–648.
- 14.Mazanec MB, Kaetzel CS, Lamm ME, Nedrud JG. Intracellular neutralization of virus by immunoglobulin A antibodies. Proc Natl Acad Sci USA. 1992;89:6901–6905. doi: 10.1073/pnas.89.15.6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazanec MB, Nedrud JG, Kaetzel CS, Lamm ME. A three-tiered view of the role of IgA in mucosal defense. Immunol Today. 1993;14:430–435. doi: 10.1016/0167-5699(93)90245-G. [DOI] [PubMed] [Google Scholar]
- 16.Mazanec MB, Coudret CL, Fletcher DR. Intracellular neutralization of influenza virus by immunoglobulin A anti-hemagglutinin monoclonal antibodies. J Virology. 1995;69:1339–1343. doi: 10.1128/jvi.69.2.1339-1343.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazanec MB, Huang YT, Pimplikar SW, Lamm ME. Mechanisms of inactivation of respiratory viruses by IgA, including intraepithelial neutralization. Semin Virol. 1996;7:285–292. [Google Scholar]
- 18.Barrett, T., and S.C. Inglis. 1991. Growth, purification and titration of influenza viruses. In Virology: A Practical Approach. B.W.J. Mahy, editor. IRL Press, Washington, D.C. 119–150.
- 19.Mazanec MB, Nedrud JG, Lamm ME. Immunoglobulin A monoclonal antibodies protect against Sendai virus. J Virol. 1987;61:2624–2626. doi: 10.1128/jvi.61.8.2624-2626.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazanec MB, Nedrud JG, Lamm ME. Transport of serum IgA into pulmonary secretions in the mouse and its implications for vaccination strategies. J Immunol. 1989;142:4275–4281. [PubMed] [Google Scholar]
- 21.Scheid A, Choppin PW. Identification of biological activities of paramyxovirus glycoproteins. Activation of cell fusion, hemolysis, and infectivity of proteolytic cleavage of an inactive precursor protein of Sendai virus. Virology. 1974;57:475–490. doi: 10.1016/0042-6822(74)90187-1. [DOI] [PubMed] [Google Scholar]
- 22.Merz DC, Scheid A, Choppin PW. Immunological studies of the functions of paramyxovirus glycoproteins. Virology. 1981;109:94–105. doi: 10.1016/0042-6822(81)90474-8. [DOI] [PubMed] [Google Scholar]
- 23.Kristensson K, Orvell C. Cellular localization of five structural proteins of Sendai virus studied with peroxidase-labeled Fab fragments of monoclonal antibodies. J Gen Virol. 1983;64:1673–1678. doi: 10.1099/0022-1317-64-8-1673. [DOI] [PubMed] [Google Scholar]
- 24.Apodaca G, Bomsel M, Arden J, Breitfeld PP, Tang K, Mostov KE. The polymeric immunoglobulin receptor. J Clin Invest. 1991;87:1877–1882. doi: 10.1172/JCI115211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mostov K, Apodaca G, Aroeti B, Okamoto C. Plasma membrane protein sorting in polarized epithelial cells. J Cell Biol. 1992;116:577–583. doi: 10.1083/jcb.116.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mostov KE. Transepithelial transport of immunoglobulins. Annu Rev Immunol. 1994;12:63–84. doi: 10.1146/annurev.iy.12.040194.000431. [DOI] [PubMed] [Google Scholar]
- 27.Mostov KE, Cardone MH. Regulation of protein traffic in polarized epithelial cells. BioEssays. 1995;17:120–138. doi: 10.1002/bies.950170208. [DOI] [PubMed] [Google Scholar]
- 28.Apodaca G, Katz LA, Mostov KE. Receptor-mediated transcytosis of IgA in MDCK cells is via apical recycling endosomes. J Cell Biol. 1994;125:67–86. doi: 10.1083/jcb.125.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Portner A, Murti KG. Localization of P, NP, and M proteins on Sendai virus nucleocapsid using immunogold labeling. Virology. 1986;150:469–478. doi: 10.1016/0042-6822(86)90311-9. [DOI] [PubMed] [Google Scholar]
- 30.Deshpande KL, Portner A. Structural and functional analysis of Sendai virus nucleocapsid protein NP with monoclonal antibodies. Virology. 1984;139:32–42. doi: 10.1016/0042-6822(84)90327-1. [DOI] [PubMed] [Google Scholar]