

Abstract
Mitogen-activated protein (MAP) kinase family members, including extracellular signal–regulated kinase (ERK), c-Jun NH2-terminal kinase ( JNK), and p38 MAP kinase, have been implicated in coupling the B cell antigen receptor (BCR) to transcriptional responses. However, the mechanisms that lead to the activation of these MAP kinase family members have been poorly elucidated. Here we demonstrate that the BCR-induced ERK activation is reduced by loss of Grb2 or expression of a dominant-negative form of Ras, RasN17, whereas this response is not affected by loss of Shc. The inhibition of the ERK response was also observed in phospholipase C (PLC)-γ2–deficient DT40 B cells, and expression of RasN17 in the PLC-γ2–deficient cells completely abrogated the ERK activation. The PLC-γ2 dependency of ERK activation was most likely due to protein kinase C (PKC) activation rather than calcium mobilization, since loss of inositol 1,4,5-trisphosphate receptors did not affect ERK activation. Similar to cooperation of Ras with PKC activation in ERK response, both PLC-γ2–dependent signal and GTPase are required for BCR-induced JNK and p38 responses. JNK response is dependent on Rac1 and calcium mobilization, whereas p38 response requires Rac1 and PKC activation.
Keywords: mitogen-activated protein kinase family, Ras, Rac1, protein kinase C, calcium
The B cell antigen receptor (BCR)1 complex has important functions in the binding and internalization of antigen as well as in transducing signals through the plasma membrane that lead to cell activation, proliferation, and apoptosis. One immediate consequence of triggering the BCR is the activation of protein tyrosine kinases and the resulting induction of phospholipase C (PLC)-γ2–mediated hydrolysis of inositol phospholipids to generate diacylglycerol and inositol 1,4,5-trisphosphate (IP3), which induce protein kinase C (PKC) activation and elevate intracellular calcium ([Ca2+]i), respectively (1–4). Supporting these sequential events, PLC-γ2–deficient DT40 B cells exhibit neither PKC activation nor calcium mobilization after BCR ligation (5), whereas the PKC activation still occurs in IP3 receptor (IP3R)-deficient DT40 cells (6). Thus, combined analyses using PLC-γ2– and IP3R-deficient DT40 cells have allowed us to dissect the requirement of PKC and calcium mobilization in BCR-mediated signaling pathways.
BCR stimulation also leads to extracellular signal–regulated kinase (ERK) activation, which in turn activates transcription factors (7–11). Analogous to receptor tyrosine kinases, the interaction of Grb2, Shc, and Sos, which have been implicated in the activation of Ras, is thought to be a potential pathway for the activation of ERK after BCR ligation (12–14). In addition to this potential pathway, there is evidence that the ERK cascade is activated by treatment with PMA and ionomycin in cultured 3T3 fibroblasts (15). However, it is not known whether BCR-induced ERK activation requires PKC activation and/or [Ca2+]i increase. Moreover, there is no direct evidence for the involvement of Grb2, Shc, and Ras in BCR-induced ERK activation.
In addition to ERK, other mitogen-activated protein (MAP) kinase family members c-Jun NH2-terminal kinase (JNK) and p38 are known to be stimulated by BCR cross-linking, although less effectively than is ERK (11, 16, 17). In a fibroblast cotransfection system, Rac1 has been shown to stimulate JNK and p38 (18–20). Furthermore, recent experiments have demonstrated that Vav, upon tyrosine phosphorylation, can activate members of the Rho family of GTPases (21–23). Although these observations provide the intriguing possibility that the activated Rac1 by Vav leads to activation of JNK and p38, any link between BCR-regulated Rac1 and the activation of JNK and p38 has not yet been explored.
Different activation patterns of these MAP kinase family members (ERK, JNK, p38) may lead to differential expression of genes and contrasting cellular phenotypes, such as growth or apoptosis in B cells. This concept is supported by the evidence obtained from the cultured B cell system and the anti–hen egg lysozyme transgenic mice system. Using the human B lymphoma cell line, B104, it has been suggested that JNK and p38, not ERK, are involved in membrane IgM-induced apoptosis (16). Moreover, in transgenic mice, both ERK and JNK are activated by foreign antigen in naive B cells, whereas ERK, but not JNK, is activated by self-antigen in tolerant B cells (24).
In this study, we focus upon how BCR activates ERK, JNK, and p38. RasN17 inhibited the BCR-induced ERK activation without affecting JNK and p38 responses. Conversely, the JNK and p38 responses were abrogated by expression of a dominant-negative form of Rac1 and Rac1N17, but not of RasN17. In addition to demonstrating requirement of these GTPases, our results reveal the importance of the PLC-γ2 pathway in activation of these MAP kinase family members.
Materials and Methods
Cells, Expression Constructs, and Antibodies.
Wild-type and various mutant DT40 cells were cultured in RPMI 1640 supplemented with 10% FCS, 1% chicken serum, 50 μM 2-ME, 2 mM l-glutamine, and antibiotics. The cDNA of RasN17 (provided by T. Urano, Department of Biochemistry, Nagoya University School of Medicine, Nagoya, Japan) and Myc-tagged Rac1N17 (reference 25; provided by G. Bokoch, Department of Immunology and Cell Biology, Scripps Research Institute, La Jolla, CA) were cloned into expression vector pApuro (26). These cDNAs were transfected by electroporation at 550 V, 25 μF, and selected in the presence of 0.5 μg/ml puromycin (Sigma Chemical Co., St. Louis, MO). Expression of transfected cDNA was confirmed by Western blot analysis. The mAb used for the stimulation of BCR was M4, which recognizes chicken IgM (27). Anti-Grap Ab were obtained by immunizing rabbits with bacterially expressed glutathione S-transferase (GST) fusion protein containing chicken Grap SH2 domain. The following Abs were purchased: anti-ERK2 and anti-p38 from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); anti-JNK1 and anti-Myc from PharMingen (San Diego, CA); anti-Grb2 Ab from Transduction Labs., Inc. (Lexington, KY); anti-Shc from Upstate Biotechnology, Inc. (Lake Placid, NY); and anti-Ras from Oncogene (Cambridge, MA).
Generation of Various Deficient DT40 Cells.
Based on published sequence of Grap (28) and Shc (29), each chicken cDNA was cloned by reverse transcriptase-PCR method using RNA from DT40 B cells. Chicken Grb2 cDNA sequence has already been published (30). Genomic clones of Grb2, Grap, and Shc were obtained by PCR using oligonucleotides designed from each cDNA sequence and genomic DNA as a template. The targeting vectors, pGrb2-neo and pGrb2-hisD, were constructed by replacing the genomic fragment containing exons that correspond to chicken Grb2 SH2 domain, amino acid residues 59–166 (30), with neo and hisD cassettes. These cassettes were flanked by 2.3 and 1.8 kb of Grb2 sequence on the 5′ and 3′ sides, respectively. Selection was done in the presence of G418 (2 mg/ml) and clones were screened by Southern blot analysis. The pGrb2–hisD was again transfected into the neo-targeted clone and selected with both G418 (2 mg/ml) and histidiol (1 mg/ml). Introduction of a single copy of each targeting vector was verified by reprobing the blots with internal neo or hisD probe. The bsr and bleo targeting constructs of Grap were made by replacing the genomic fragment containing exons corresponding to human Grap SH2 domain, amino acid residues 59–100 (28), with bsr and bleo cassettes. These constructs spanned 0.8 (5′ side) and 1.2 kb (3′ side) of Grap sequence. They were sequentially transfected into DT40 cells by electroporation to obtain a null mutant. Selection for drug-resistant clones was carried out by using blastcidin S (50 μg/ml) and phleomycin (0.3 mg/ml). The constructs (pShc–neo and pShc–hisD) were designed for neo and hisD cassettes to replace exons that correspond to human Shc amino acid residues 101– 140 (29). The neo and hisD cassettes were flanked by 0.9 and 1.3 kb of Shc sequence on the 5′ and 3′ sides, respectively. Selection was performed as described above for Grb2. In this study, we also used DT40 cells deficient in PLC-γ2 and all three isoforms of IP3R, all of which have been described previously (5, 6).
Northern Blot Analysis.
RNA was prepared from wild-type and mutant DT40 cells using the guanidium thiocyanate method. Total RNA (20 μg) was separated in a 1.2% formaldehyde gel, transferred to Hybond-N+ nylon membrane (Nycomed Amersham, Arlington Heights, IL), and probed with 32P-labeled cDNAs.
Western Blot Analysis.
Wild-type, Grb2-, Grap-, and Shc-deficient cell lysates were separated by SDS-PAGE gel, transferred to nitrocellulose membrane, and detected by appropriate Abs and the ECL system (Nycomed Amersham).
In Vitro Kinase Assay.
After incubation with the indicated stimuli, 2–5 × 106 cells were lysed in 500 μl of lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 2% Triton X-100, 100 μM sodium vanadate, 10 mM sodium pyrophosphate, 2 mM PMSF, 10 μg/μl leupeptin, and 2 μg/μl aprotinin) for 30 min on ice. Cell debris were removed by centrifugation at 16,000 g for 15 min at 4°C. Precleared lysates were immunoprecipitated by 1 μg anti-ERK2 Ab, 1 μg anti-JNK1 Ab, or 1 μg anti-p38 Ab with 40 μl protein G–Sepharose (Amersham Pharmacia Biotech, Piscataway, NJ). The beads were pelleted by centrifugation and then washed three times with lysis buffer, two times with washing buffer (50 mM Hepes, pH 7.4, 10 mM MgCl2, 100 μM sodium vanadate, and 2 mM dithiothreitol). Immunoprecipitates were divided and half of them were used for Western blot analysis. The remaining half were washed once with kinase assay buffer (20 mM Hepes, pH 7.4, 10 mM MgCl2, 10 mM MnCl2, 2 mM dithiothreitol, and 10 μM sodium vanadate). Immune complexes were suspended in 30 μl kinase assay buffer containing γ-[32P]ATP (>3,000 Ci/mmol; NEN™ Life Science Products, Boston, MA) and 5 μM cold ATP. 5 μg of GST–Elk, GST–c-Jun (provided by M. Hibi, Biomedical Research Center, Osaka University Medical School, Suita, Japan) and GST–ATF2 fusion protein were added as substrates for ERK2, JNK, and p38, respectively. After 20 min of incubation at 30°C, the reaction was terminated by the addition of SDS sample buffer followed by boiling for 5 min. The samples were separated by SDS-PAGE gel, dried, and subjected to autoradiography.
Results
Requirement for Grb2, not Shc, in BCR-mediated ERK Activation.
To determine the necessity or redundancy of Grb2 and Shc in the BCR-induced ERK activation, we generated DT40 B cells deficient in these molecules by the gene targeting method. Since Grap has recently been shown to possess similar structural characteristics to Grb2 and this protein is prominently expressed in lymphoid cells (28, 31), we also established Grap-deficient DT40 cells. Lack of Grb2, Grap, and Shc was verified by Northern and Western blot analyses (Fig. 1, A and B). The level of cell surface expression of BCR on these targeted DT40 cells was the same as that of parental DT40 cells (data not shown).
Figure 1.

Disruption of Grb2, Grap, and Shc genes in DT40 cells. (A) RNA expression of Grb2, Grap, and Shc was analyzed by Northern blot analysis using each chicken cDNA probe (top) or β-actin (bottom) (reference 64). Positions of 28S and 18S RNA are shown. (B) Grb2, Grap, and Shc protein expression in wild-type and targeted DT40 cells. Each protein was detected by Western blotting analysis using anti-Grb2, anti-Grap, and anti-Shc Abs. (C) Expression of dominant-negative Ras and dominant-negative Rac1.
RNA analysis by reverse transcriptase-PCR method and Western blot analysis (32) indicated that ERK2 is the predominant isoform expressed in DT40 B cells. After BCR stimulation, DT40 cells were lysed and immunoprecipitated with anti-ERK2 Ab. The immunoprecipitates were assayed for its in vitro kinase activity by the ability to phosphorylate the substrate Elk-1. This anti-ERK2 Ab does not recognize JNK or p38 (Fig. 2). Consistent with previous reports (9, 10, 33, 34), activation of ERK2 in DT40 cells was maximal at 1 and 3 min, after which activity declined to a lower level (Figs. 3 and 4). Compared with wild-type cells, the BCR-induced ERK2 response was reduced approximately twofold by loss of Grb2. However, the ERK2 response was not reduced in Shc-deficient DT40 cells (Fig. 3), demonstrating that Grb2, but not Shc, is required for the BCR-induced ERK2 activation. Moreover, despite the structural similarity between Grb2 and Grap, Grap-deficient DT40 B cells exhibited no decrease in BCR-mediated ERK2 activation (Fig. 3), implicating that Grb2 and Grap have distinct function(s) in BCR signaling.
Figure 2.

Cross-reactivity of various Abs. DT40 cell extracts were immunoprecipitated by anti-ERK2 Ab, anti-p38 Ab, and anti-JNK Ab. These immunoprecipitates were separated by 9% SDS-PAGE gels, transferred to nitrocellulose membranes, and incubated with indicated Abs (Blot Ab).
Figure 3.

BCR-induced ERK2 activation in various DT40 cells. Various DT40 cells were stimulated with M4 (4 μg/ ml) for indicated time. ERK2 was immunoprecipitated and the precipitates were assayed for kinase activity using GST–Elk1 fusion protein as a substrate. The kinase reaction products were resolved by 12.5% SDS-PAGE and their phosphorylation was quantified by autoradiography.
Figure 4.
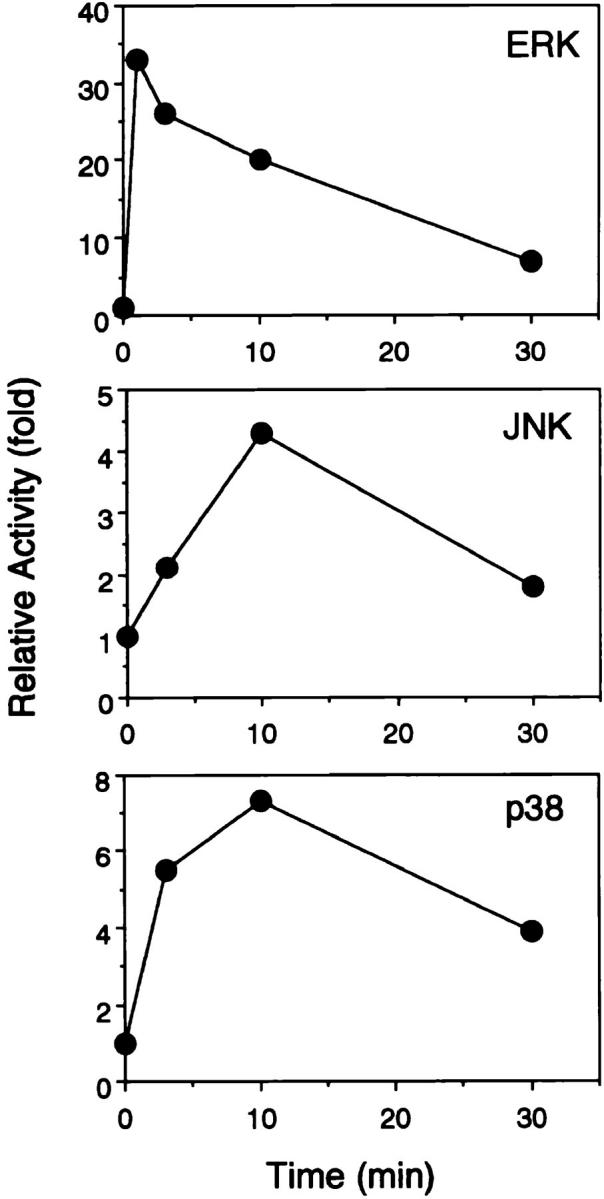
Time kinetics of ERK2, JNK, and p38 responses upon BCR engagement. After BCR stimulation, DT40 cell lysates were immunoprecipitated with anti-ERK2 Ab, anti-JNK1 Ab, or anti-p38 Ab. The kinase activities were assayed using GST–Elk1, GST–c-Jun, and GST–ATF2 as substrates for ERK2, JNK, and p38, respectively.
Since Grb2 is essential for Ras activation in receptor-type tyrosine kinase signaling, we decided to test whether Grb2 is involved in the ERK2 response along a Ras-dependent pathway in BCR signaling. As shown in Fig. 5, expression of RasN17 (Fig. 1 C) inhibited ERK2 activation by threefold compared with wild-type cells, suggesting that Ras activation, perhaps via Grb2, is required for BCR- induced ERK2 activation. The residual ERK2 response by loss of Grb2 or expression of RasN17 implicates that an additional signal(s) is required for the maximum ERK2 activation and that this additional signal is able to stimulate ERK2 to some extent even in the absence of Ras activation.
Figure 5.

Involvement of PLC-γ2 and Ras on BCR-induced ERK2 activation. BCR-stimulated DT40 cells were lysed and immunoprecipitated with anti-ERK2 Ab. Stimulation conditions and in vitro kinase assay were described as in Fig. 3.
Both PKC and Ras Pathways Are Required for ERK Activation.
Recent work has implicated PKC and/or [Ca2+]i increase as a cofactor for ERK activation (7, 15). To determine whether PKC and [Ca2+]i increase are required for BCR-induced ERK activation, we used PLC-γ2– and IP3R-deficient DT40 cells. We have previously shown that the BCR-induced [Ca2+]i increase is completely abrogated in both PLC-γ2– and IP3R-deficient DT40 cells, whereas PKC activation is abolished only in PLC-γ2–deficient cells (6). BCR ligation stimulated ERK2 activation even in the IP3R-deficient cells, whereas this activation was markedly inhibited by loss of PLC-γ2 (Fig. 3). These data suggest that the PKC pathway rather than the calcium pathway is required for the BCR-induced ERK2 activation. Furthermore, the extent of the inhibition by loss of PLC-γ2 was more than that in DT40 cells expressing RasN17 (Fig. 5), suggesting that the PKC pathway may play a more dominant role than Ras in BCR-mediated ERK2 activation.
The residual ERK2 activation in PLC-γ2–deficient DT40 cells was completely abolished by expression of RasN17 in this mutant DT40 cell (Fig. 5). Together, these data suggest that Ras activation and the PKC-dependent pathway may work synergistically for the maximum BCR-induced ERK2 activation.
Requirement for Rac1 in BCR-induced JNK and p38 Activation.
After BCR stimulation, the activities of JNK and p38 were determined by their ability to phosphorylate the substrates c-Jun and ATF-2, respectively. The kinetics of JNK activation were distinct from that of ERK2, being marked at 10 min and being declined by 30 min. The activation of p38 was maximal at 3 and 10 min, and more sustained than that of JNK (Figs. 4 and 6). Contrary to ERK2 activation, expression of RasN17 did not significantly affect the JNK and p38 responses upon BCR engagement (Fig. 6). Consistent with these observations, Grb2-deficient DT40 cells also showed normal JNK and p38 responses (data not shown). These findings prompted us to examine the involvement of Rac1 in BCR-induced JNK and p38 activation, since recent experiments in fibroblasts have shown that Rac1, rather than Ras, is an efficient activator of a cascade leading to JNK and p38 activation (18–20). Expression of Rac1N17 in DT40 cells (Fig. 1 C) markedly inhibited both JNK and p38 activation after BCR cross-linking (Fig. 6), whereas ERK2 activation was not affected (data not shown). These results indicate that Rac1 but not Ras is required for JNK and p38 activation after BCR ligation.
Figure 6.
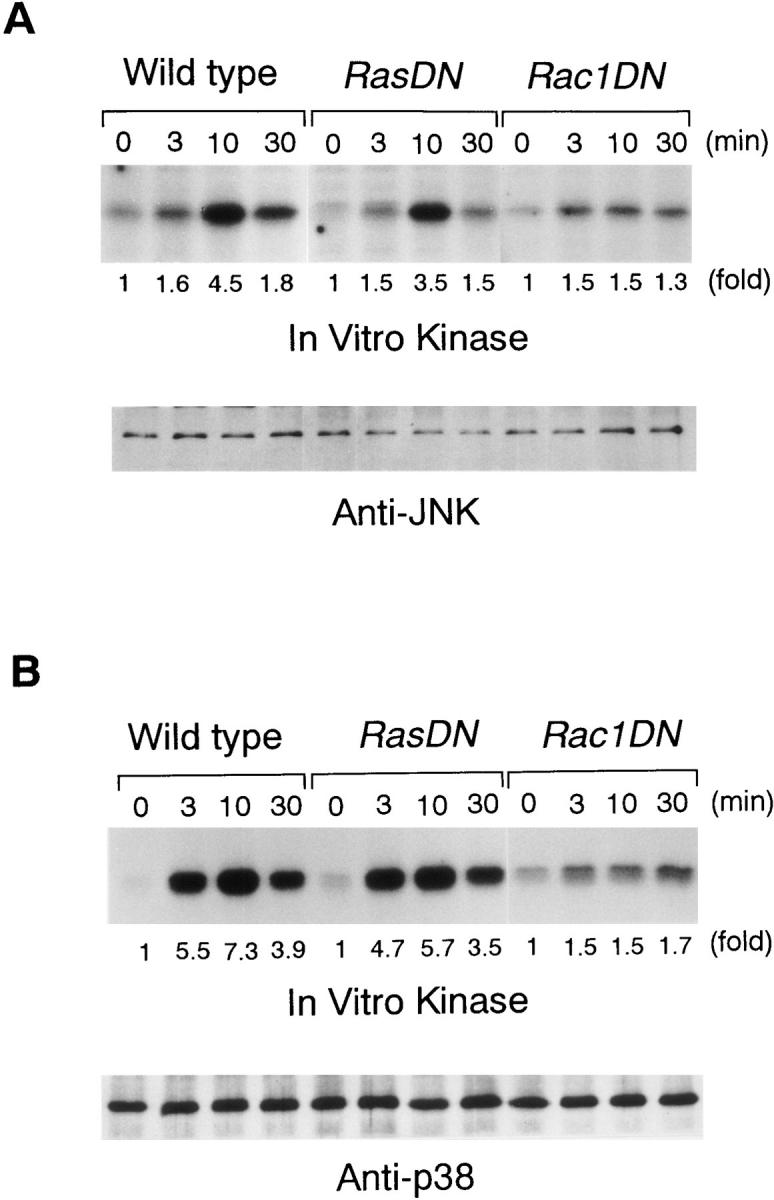
Effect of RasN17 and Rac1N17 on JNK and p38 responses. DT40 cells were stimulated with M4 (4 μg/ml) for indicated times. Cell lysates were immunoprecipitated with anti-JNK1 Ab and anti-p38 Ab, and kinase activities were assayed using GST–c-Jun and GST–ATF2 as substrates, respectively. After electrophoresis, the labeled GST–c-Jun and GST–ATF2 bands were visualized by autoradiography.
PLC-γ2–dependent Signaling Is Required for JNK and p38 Activation.
A calcium ionophore has been shown to activate JNK in B cells (16, 35). This suggests that other activators of JNK, such as ligated BCR, may use [Ca2+]i increase for maximum JNK activation. Likewise, p38 response also might be dependent on the [Ca2+]i increase in B cells. To test these possibilities, we examined the BCR-induced activation of JNK and p38 in PLC-γ2– and IP3R-deficient DT40 cells. As shown in Fig. 7, both PLC-γ2– and IP3R-deficient DT40 cells exhibited impaired JNK responses, suggesting the dependency of the JNK response on the calcium pathway rather than the PKC pathway. However, a residual JNK response was still observed even in the IP3R-deficient DT40 cells.
Figure 7.
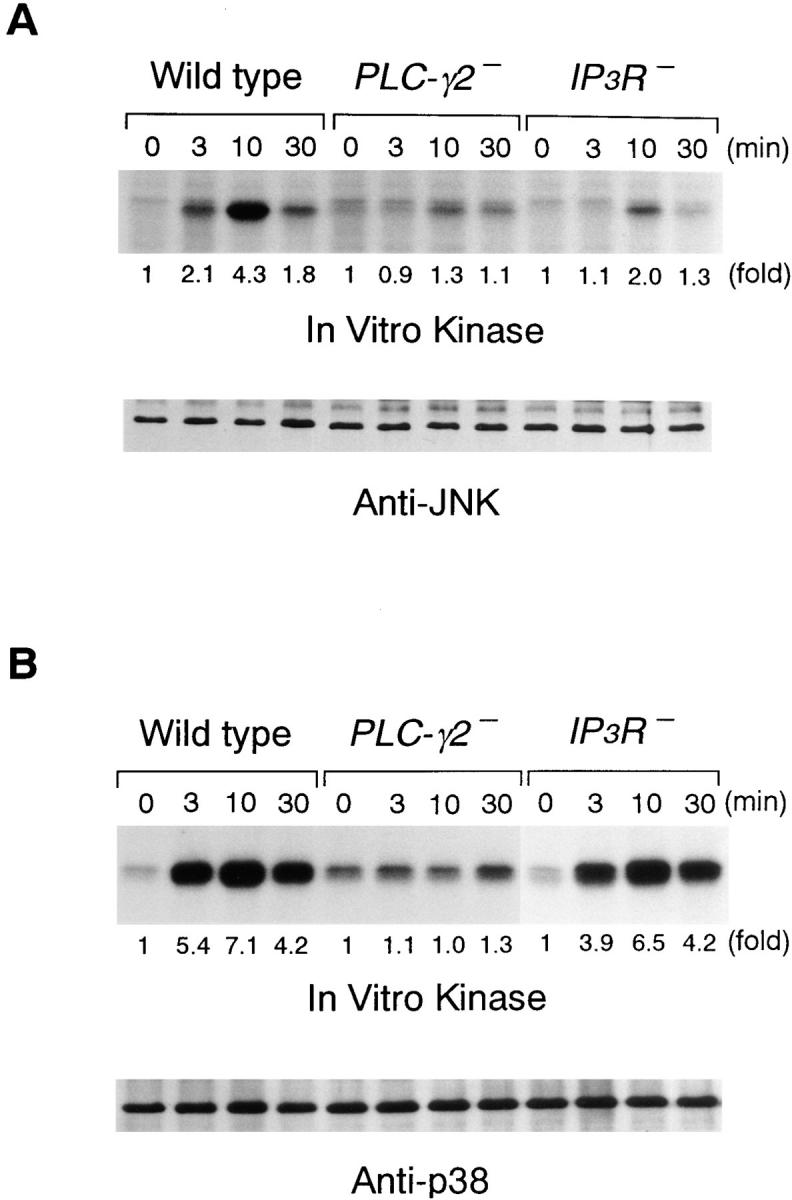
BCR-induced JNK and p38 responses in PLC-γ2– and IP3R-deficient DT40 cells. Cells were stimulated with M4 (4 μg/ml) for indicated times and cell lysates were immunoprecipitated with anti-JNK1 and anti-p38 Ab. The kinase assays were done as in Fig. 6.
Activation of p38 upon BCR engagement was completely abolished by loss of PLC-γ2, whereas this response was still observed in IP3R-deficient DT40 cells (Fig. 7). These results suggest that the BCR-induced p38 response requires a dominantly PKC pathway.
Discussion
In this study, we have addressed three questions regarding the regulation of ERK, JNK, and p38 by BCR, potentially leading to altered transcription of genes. First, are distinct GTPases required for the BCR-induced ERK, JNK, and p38 responses? Second, particularly in the ERK response, is either Grb2 or Shc, or both of them, essential for this response? Third, are GTPases the sole regulators of BCR-induced activation of these MAP kinase family members?
Since the two SH3 domains of Grb2 bind to proline-rich residues near the COOH terminus of Sos, Grb2 is thought to mediate the translocation of Sos to the plasma membrane, allowing Sos to activate membrane-bound Ras. In the case of the epidermal growth factor (EGF) receptor, translocation of Grb2 to membrane can occur upon binding of its SH2 domain to the autophosphorylated tail of the EGF receptor (36–40). However, because the BCR complex does not possess Grb2 binding sites despite activating Ras, an alternate model for Ras activation has been proposed. According to this model, BCR ligation leads to the recruitment of Shc to the plasma membrane, whereupon it undergoes tyrosine phosphorylation of the Grb2 binding site, thereby recruiting Grb2 to the plasma membrane (38). Indeed, BCR stimulation leads to tyrosine phosphorylation of Shc and to the assembly of an Shc–Grb2–Sos complex (8, 41–43). However, the data presented here indicate that Grb2 but not Shc is required for BCR-induced ERK response. These findings indicate the dispensability of Shc in Ras activation in BCR signaling, and raise the previous question of how Grb2 is recruited to the plasma membrane after BCR ligation. One candidate adaptor molecule might be LAT, an integral membrane protein that is phosphorylated and bound to Grb2 (44). Although LAT is expressed predominantly in T cells, B cells might express a similar molecule that is required for Grb2 recruitment to the membrane. This candidate molecule is tyrosine phosphorylated presumably by Syk, since the accompanying study has shown that Syk, but not Lyn, is a responsible kinase for BCR-induced ERK activation (32).
The extent of inhibition by RasN17 of BCR-mediated ERK2 activation was almost the same level as that observed in Grb2-deficient DT40 cells, suggesting that the Grb2-deficient phenotype in BCR signaling can be accounted for by loss of Ras activation. In contrast to the almost complete inhibition of EGF receptor–induced ERK response by expression of RasN17 (45), we observed residual BCR-induced ERK2 activation despite loss of Grb2 or expression of Ras N17. These observations provide important implications. First, the sensitivity to requirement of Ras for ERK activation might vary depending on receptor systems. In this regard, TCR-induced ERK response appears to be more strictly dependent on Ras activation than is BCR (46). Second, the BCR uses an additional signaling pathway to stimulate ERK even in the absence of Ras activation, and probably this additional signaling pathway, in addition to Ras, is required for maximum BCR-induced ERK activation. This additional signal is provided by PKC activation, since PLC-γ2–deficient DT40 cells, but not IP3R-deficient, exhibited profound loss in ERK2 activation by BCR ligation. Furthermore, expression of RasN17 in PLC-γ2– deficient cells led to the complete abrogation of BCR- induced ERK2 activation, suggesting the synergistic action of Ras and the PKC pathway for BCR-induced ERK activation. As we have recently shown that PKC activation is negatively regulated by Lyn in B cells (47), the exaggerated activation of ERK in lyn −/− mice (48) might be explained by the enhanced activation of the PKC-dependent pathway.
In contrast to the ERK response, the JNK and p38 responses are not affected by expression of RasN17. Conversely expression of Rac1N17 abolished the JNK and p38 activation without affecting the ERK response in B cells. This difference presumably reflects that distinct exchange factors, Sos and Vav, participate in ERK and JNK/p38 responses through activating Ras and Rac1, respectively, in BCR signaling. Supporting this notion, recent experiments have shown that Vav, upon tyrosine phosphorylation, stimulates Rac1 (21–23). Our results reveal that not only Rac1- but also PLC-γ2–dependent signal is required for BCR- induced JNK/p38 response; JNK is dependent on calcium mobilization rather than PKC activation, whereas p38 requires the PKC pathway. Requirement of calcium for JNK activation agrees with the data that JNK response, but not p38 response, is inhibited by treatment of BAPTA-AM, a chelator of both intracellular and extracellular calcium (32). The requirement of both Rac1 and PLC-γ for JNK activation is well conserved between B and T cells. Indeed, the data presented in this study and the accompanying paper (32) are consistent with the findings that JNK activation in T cells requires calcium-dependent calcineurin and Syk, in addition to Rac1 (49, 50).
Although this study has not addressed the mechanism by which PKC and calcium regulate p38 and JNK activation by BCR, modulation of the signaling pathway upstream of Rac1 by PKC and calcium seems to be unlikely, as Vav behaves as a tyrosine phosphorylation–dependent exchange factor for Rac1 (21–23). Rather, distinct requirement for PKC and calcium for p38 and JNK, respectively, might suggest that downstream molecules of Rac1 are potential targets of PKC and calcium. Analogous to Raf, which is recruited by Ras (51), and which is a substrate of PKC (52), another kinase participating in the cascade leading to p38 and JNK response might be a substrate of PKC and a calcium-dependent kinase, respectively.
Regulation of ERK, JNK, and p38 by PKC and calcium might provide insights into the mechanism by which CD19 costimulates the activation of these MAP kinase family members by BCR. Although coclustering CD19 and BCR leads to enhanced activation of ERK, the CD19-mediated enhancement of JNK and p38 responses is more dramatic (53). Since CD19 associates with Vav and phosphatidylinositol 3 kinase (54–57), coligation of CD19 with BCR brings these molecules into the close proximity of the BCR signaling complex. This may lead to further enhancement of tyrosine phosphorylation of Vav by Lyn and Syk, leading to its enhanced activation towards Rac1. Recently, phosphatidylinositol 3 kinase has been reported to participate in the enhanced calcium mobilization (58–60), which potentially could lead to augmented JNK activation. Conversely, inhibitory receptors such as PIR-B and FcγRIIB on B cells are predicted to inhibit these MAP kinase family members, as these receptors downregulate the BCR-induced PLC-γ2 activation and calcium mobilization (61–63).
Acknowledgments
We would like to thank Dr. G. Bokoch, Dr. T. Urano, Dr. M. Hibi, and Dr. V.-M. Wasenius (Department of Radiotherapy and Oncology, Helsinki University Central Hospital, Helsinki, Finland) for providing us with Rac1N17 cDNA, RasN17 cDNA, GST–c-Jun plasmid, and chicken Grb2 cDNA, respectively; and Dr. A. Craxton, Dr. M. Ishiai, and Dr. H. Sugawara (Omiya Medical Center, Omiya, Japan) for assistance in establishing the p38 MAPK in vitro kinase assay, purification of GST–Grap, and cloning of chicken Shc cDNA, respectively.
This work was supported by grants to T. Kurosaki from the Ministry of Education, Science, Sports, and Culture of Japan, the Naito Foundation, and the Toray Science Foundation; and to E.A. Clark from the National Institutes of Health (grants GM-42508 and GM-37905).
Abbreviations used in this paper
- BCR
B cell antigen receptor
- EGF
epidermal growth factor
- ERK
extracellular signal–regulated kinase
- GST
glutathione S-transferase
- IP3
inositol 1,4,5-triphosphate
- JNK
c-Jun NH2-terminal kinase
- MAP
mitogen-activated protein
- PKC
protein kinase C
- PLC
phospholipase C
References
- 1.DeFranco AL. The complexity of signaling pathways activated by the BCR. Curr Opin Immunol. 1997;9:296–308. doi: 10.1016/s0952-7915(97)80074-x. [DOI] [PubMed] [Google Scholar]
- 2.Reth M, Wienands J. Initiation and processing of signals from the B cell antigen receptor. Annu Rev Immunol. 1997;15:453–479. doi: 10.1146/annurev.immunol.15.1.453. [DOI] [PubMed] [Google Scholar]
- 3.Kurosaki T. Molecular mechanisms in B cell antigen receptor signaling. Curr Opin Immunol. 1997;9:309–318. doi: 10.1016/s0952-7915(97)80075-1. [DOI] [PubMed] [Google Scholar]
- 4.Pleiman CM, D'Ambrosio D, Cambier JC. The B–cell antigen receptor complex: structure and signal transduction. Immunol Today. 1994;15:393–399. doi: 10.1016/0167-5699(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 5.Takata M, Homma Y, Kurosaki T. Requirement of phospholipase C-γ2 activation in surface immunoglobulin M–induced B cell apoptosis. J Exp Med. 1995;182:907–914. doi: 10.1084/jem.182.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugawara H, Kurosaki M, Takata M, Kurosaki T. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO (Eur Mol Biol Organ) J. 1997;16:3078–3088. doi: 10.1093/emboj/16.11.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8:402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- 8.Saxton TM, van Oostveen I, Bowtell D, Aebersold R, Gold MR. B cell antigen receptor cross-linking induces phosphorylation of the p21rasoncoprotein activators SHC and mSOS1 as well as assembly of complexes containing SHC, GRB-2, mSOS1, and a 145-kDa tyrosine-phosphorylated protein. J Immunol. 1994;153:623–636. [PubMed] [Google Scholar]
- 9.Kim K-M, Alber G, Weiser P, Reth M. Differential signaling through the Ig-α and Ig-β components of the B cell antigen receptor. Eur J Immunol. 1993;23:911–916. doi: 10.1002/eji.1830230422. [DOI] [PubMed] [Google Scholar]
- 10.Tordai A, Franklin RA, Patel H, Gardner AM, Johnson GL, Gelfand EW. Cross-linking of surface IgM stimulates the Ras/Raf-1/MEK/MAPK cascade in human B lymphocytes. J Biol Chem. 1994;269:7538–7543. [PubMed] [Google Scholar]
- 11.Li Y-Y, Baccam M, Waters SB, Pessin JE, Bishop GA, Koretzky GA. CD40 ligation results in protein kinase C–independent activation of ERK and JNK in resting murine splenic B cells. J Immunol. 1996;157:1440–1447. [PubMed] [Google Scholar]
- 12.Nagai K, Takata M, Yamamura H, Kurosaki T. Tyrosine phosphorylation of Shc is mediated through Lyn and Syk in B cell receptor signaling. J Biol Chem. 1995;270:6824–6829. doi: 10.1074/jbc.270.12.6824. [DOI] [PubMed] [Google Scholar]
- 13.Harmer SL, DeFranco AL. Shc contains two Grb2 binding sites needed for efficient formation of complexes with SOS in B lymphocytes. Mol Cell Biol. 1997;17:4087–4095. doi: 10.1128/mcb.17.7.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crowley MT, Harmer SL, DeFranco AL. Activation-induced association of a 145-kDa tyrosine-phosphorylated protein with Shc and Syk in B lymphocytes and macrophages. J Biol Chem. 1996;271:1145–1152. doi: 10.1074/jbc.271.2.1145. [DOI] [PubMed] [Google Scholar]
- 15.Chao T-SO, Foster DA, Rapp UR, Rosner MR. Differential Raf requirement for activation of mitogen-activated protein kinase by growth factors, phorbol esters, and calcium. J Biol Chem. 1994;269:7337–7341. [PubMed] [Google Scholar]
- 16.Graves JD, Draves KE, Craxton A, Saklatvala J, Krebs EG, Clark EA. Involvement of stress-activated protein kinase and p38 mitogen-activated protein kinase in mIgM-induced apoptosis of human B lymphocytes. Proc Natl Acad Sci USA. 1996;93:13814–13818. doi: 10.1073/pnas.93.24.13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sutherland CL, Heath AW, Pelech SL, Young PR, Gold MR. Differential activation of the ERK, JNK, and p38 mitogen-activated protein kinases by CD40 and the B cell antigen receptor. J Immunol. 1996;157:3381–3390. [PubMed] [Google Scholar]
- 18.Minden A, Lin A, Claret F-X, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 19.Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- 20.Coso OA, Chiariello M, Yu J-C, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 21.Crespo P, Bustelo XR, Aaronson DS, Coso OA, Lopez-Barohona M, Barbacid M, Gutkind JS. Rac-1 dependent stimulation of the JNK/SAPK signaling pathway by Vav. Oncogene. 1996;13:455–460. [PubMed] [Google Scholar]
- 22.Crespo P, Schuebel KE, Ostrom AA, Gutkind JS, Bustelo XR. Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vavproto-oncogene product. Nature. 1997;385:169–172. doi: 10.1038/385169a0. [DOI] [PubMed] [Google Scholar]
- 23.Teramoto H, Salem P, Robbins KC, Bustelo XR, Gutkind JS. Tyrosine phosphorylation of the vavproto-oncogene products links FcεRI to the Rac1-JNK pathway. J Biol Chem. 1997;272:10751–10755. doi: 10.1074/jbc.272.16.10751. [DOI] [PubMed] [Google Scholar]
- 24.Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, Lewis RS, Goodnow CC. Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity. 1997;6:419–428. doi: 10.1016/s1074-7613(00)80285-x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, Bokoch GM. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- 26.Takata M, Sabe H, Hata A, Inazu T, Homma Y, Nukada T, Yamamura H, Kurosaki T. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+mobilization through distinct pathways. EMBO (Eur Mol Biol Organ) J. 1994;13:1341–1349. doi: 10.1002/j.1460-2075.1994.tb06387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C-LH, Lehmeyer JE, Cooper MD. Evidence for an IgD homologue on chicken lymphocytes. J Immunol. 1982;129:2580–2585. [PubMed] [Google Scholar]
- 28.Feng G-S, Ouyang Y-B, Hu D-P, Shi Z-Q, Gentz R, Ni J. Grap is a novel SH3-SH2-SH3 adaptor protein that couples tyrosine kinases to the Ras pathway. J Biol Chem. 1996;271:12129–12132. doi: 10.1074/jbc.271.21.12129. [DOI] [PubMed] [Google Scholar]
- 29.Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Grignani F, Pawson T, Pelicci PG. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- 30.Wasenius V-M, Meriläinen J, Lehto V-P. Sequence of a chicken cDNA encoding a GRB2 protein. Gene. 1993;134:299–300. doi: 10.1016/0378-1119(93)90111-f. [DOI] [PubMed] [Google Scholar]
- 31.Trüb T, Frantz JD, Miyazaki M, Band H, Shoelson SE. The role of a lymphoid-restricted, Grb2-like SH3-SH2-SH3 protein in T cell receptor signaling. J Biol Chem. 1997;272:894–902. doi: 10.1074/jbc.272.2.894. [DOI] [PubMed] [Google Scholar]
- 32.Jiang A, Craxton A, Kurosaki T, Clark EA. Different protein tyrosine kinases are required for B cell antigen receptor–mediated extracellular signal–regulated kinase, c-Jun NH2-terminal kinase 1, and p38 mitogen-activated protein kinase activation. J Exp Med. 1998;188:1297–1306. doi: 10.1084/jem.188.7.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casillas A, Hanekom C, Williams K, Katz R, Nel AE. Stimulation of B-cells via the membrane immunoglobulin receptor or with phorbol myristate 13 acetate induces tyrosine phosphorylation and activation of a 42-kDa microtubule-associated protein-2 kinase. J Biol Chem. 1991;266:19088–19094. [PubMed] [Google Scholar]
- 34.Gold MR, Sanghera JS, Stewart J, Pelech SL. Selective activation of p42 mitogen-activated protein (MAP) kinase in murine B lymphoma cell lines by membrane immunoglobulin cross-linking. Evidence for protein kinase C–independent and –dependent mechanisms of activation. Biochem J. 1992;287:269–276. doi: 10.1042/bj2870269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 36.Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullich A, Skolnik EY, Bar-Sagi D, Schlessinger J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70:431–442. doi: 10.1016/0092-8674(92)90167-b. [DOI] [PubMed] [Google Scholar]
- 37.Chardin P, Camonis JH, Gale NW, Aelst LV, Schlessinger J, Wigler MH, Bar-Sagi D. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science. 1993;260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- 38.Egan SE, Giddings BW, Brooks MW, Buday L, Sizeland AM, Weinberg RA. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 39.Gale NW, Kaplan S, Lowenstein EJ, Schlessinger J, Bar-Sagi D. Grb2 mediates the EGF-dependent activation of guanine nucleotide exchange on Ras. Nature. 1993;363:88–92. doi: 10.1038/363088a0. [DOI] [PubMed] [Google Scholar]
- 40.Rozakis-Adcock M, Fernley R, Wade J, Pawson T, Bowtell D. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature. 1993;363:83–85. doi: 10.1038/363083a0. [DOI] [PubMed] [Google Scholar]
- 41.Kumar G, Wang S, Gupta S, Nel A. The membrane immunoglobulin receptor utilizes a Shc/Grb2/hSOS complex for activation of the mitogen-activated protein kinase cascade in a B-cell line. Biochem J. 1995;307:215–223. doi: 10.1042/bj3070215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lankester AC, van Schijndel GMW, Rood PML, Verhoeven AJ, van Lier RAW. B cell antigen receptor cross-linking induces tyrosine phosphorylation and membrane translocation of a multimeric Shc complex that is augmented by CD19 co-ligation. Eur J Immunol. 1994;24:2818–2825. doi: 10.1002/eji.1830241136. [DOI] [PubMed] [Google Scholar]
- 43.Smit L, de Vries-Smits AMM, Bos JL, Borst J. B cell antigen receptor stimulation induces formation of a Shc-Grb2 complex containing multiple tyrosine-phosphorylated proteins. J Biol Chem. 1994;269:20209–20212. [PubMed] [Google Scholar]
- 44.Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE. LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell. 1998;92:83–92. doi: 10.1016/s0092-8674(00)80901-0. [DOI] [PubMed] [Google Scholar]
- 45.Howe LR, Leevers SJ, Gómez N, Nakielny S, Cohen P, Marshall CJ. Activation of the MAP kinase pathway by the protein kinase raf. Cell. 1992;71:335–342. doi: 10.1016/0092-8674(92)90361-f. [DOI] [PubMed] [Google Scholar]
- 46.Izquierdo M, Leevers SJ, Marshall CJ, Cantrell D. p21rascouples the T cell antigen receptor to extracellular signal–regulated kinase 2 in T lymphocytes. J Exp Med. 1993;178:1199–1208. doi: 10.1084/jem.178.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katsuta H, Tsuji S, Niho Y, Kurosaki T, Kitamura D. Lyn-mediated down-regulation of B cell antigen receptor signaling: Inhibition of protein kinase C activation by Lyn in a kinase-independent fashion. J Immunol. 1998;160:1547–1551. [PubMed] [Google Scholar]
- 48.Chan VWF, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity. 1997;7:69–81. doi: 10.1016/s1074-7613(00)80511-7. [DOI] [PubMed] [Google Scholar]
- 49.Jacinto E, Werlen G, Karin M. Cooperation between Syk and Rac1 leads to synergistic JNK activation in T lymphocytes. Immunity. 1998;8:31–41. doi: 10.1016/s1074-7613(00)80456-2. [DOI] [PubMed] [Google Scholar]
- 50.Werlen G, Jacinto E, Xia Y, Karin M. Calcineurin preferentially synergizes with PKC-θ to activate JNK and IL-2 promoter in T lymphocytes. EMBO (Eur Mol Biol Organ) J. 1998;17:3101–3111. doi: 10.1093/emboj/17.11.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- 52.Schönwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal–regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tooze RM, Doody GM, Fearon DT. Counterregulation by the coreceptors CD19 and CD22 of MAP kinase activation by membrane immunoglobulin. Immunity. 1997;7:59–67. doi: 10.1016/s1074-7613(00)80510-5. [DOI] [PubMed] [Google Scholar]
- 54.Weng W-K, Jarvis L, LeBien TW. Signaling through CD19 activates a Vav/mitogen-activated protein kinase pathway and induces formation of a CD19/Vav/phosphatidylinositol 3-kinase complex in human B cell precursors. J Biol Chem. 1994;269:32514–32521. [PubMed] [Google Scholar]
- 55.Chalupny NJ, Kanner SB, Schieven GL, Wee SF, Gilliland LK, Aruffo A, Ledbetter JA. Tyrosine phosphorylation of CD19 in pre-B and mature B cells. EMBO (Eur Mol Biol Organ) J. 1993;12:2691–2696. doi: 10.1002/j.1460-2075.1993.tb05930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tuveson DA, Carter RH, Soltoff SP, Fearon DT. CD19 of B cells as a surrogate kinase insert region to bind phophatidylinositol 3-kinase. Science. 1993;260:986–989. doi: 10.1126/science.7684160. [DOI] [PubMed] [Google Scholar]
- 57.O'Rourke LM, Tooze R, Turner M, Sandoval DM, Carter RH, Tybulewicz VLJ, Fearon DT. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity. 1998;8:635–645. doi: 10.1016/s1074-7613(00)80568-3. [DOI] [PubMed] [Google Scholar]
- 58.Bolland S, Pearse R, Kurosaki T, Ravetch JV. SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity. 1998;8:509–516. doi: 10.1016/s1074-7613(00)80555-5. [DOI] [PubMed] [Google Scholar]
- 59.Hippen KL, Buhl AM, D'Ambrosio D, Nakamura K, Persin C, Cambier JC. FcγRIIB1 inhibition of BCR-mediated phosphoinositide hydrolysis and Ca2+mobilization is integrated by CD19 dephosphorylation. Immunity. 1997;7:49–58. doi: 10.1016/s1074-7613(00)80509-9. [DOI] [PubMed] [Google Scholar]
- 60.Kiener PA, Lioubin MN, Rohrschneider LR, Ledbetter JA, Nadler SG, Diegel ML. Co-ligation of the antigen and Fc receptors gives rise to the selective modulation of intracellular signaling in B cells. Regulation of the association of phosphatidylinositol 3-kinase and inositol 5′-phosphatase with the antigen receptor complex. J Biol Chem. 1997;272:3838–3844. doi: 10.1074/jbc.272.6.3838. [DOI] [PubMed] [Google Scholar]
- 61.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 62.Bléry M, Kubagawa H, Chen C-C, Vély F, Cooper MD, Vivier E. The paired Ig-like receptor PIR-B is an inhibitory receptor that recruits the protein-tyrosine phosphatase SHP-1. Proc Natl Acad Sci USA. 1998;95:2446–2451. doi: 10.1073/pnas.95.5.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maeda A, Kurosaki M, Ono M, Takai T, Kurosaki T. Requirement of tyrosine phosphatase SHP-1 and SHP-2 for PIR-B–mediated inhibitory signal. J Exp Med. 1998;187:1355–1360. doi: 10.1084/jem.187.8.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kost TA, Theodorakis N, Hughes SH. The nucleotide sequence of the chick cytoplasmic β-actin gene. Nucleic Acids Res. 1983;11:8287–8301. doi: 10.1093/nar/11.23.8287. [DOI] [PMC free article] [PubMed] [Google Scholar]