

Abstract
Crry (complement receptor 1–related protein/gene y) is a key cellular complement regulator in rodents. It is also present in Fx1A, the renal tubular preparation used to immunize rats to induce active Heymann nephritis (HN), a model of membranous nephropathy. We hypothesized that rats immunized with anti-Fx1A develop autoantibodies (auto-Abs) to Crry as well as to the megalin-containing HN antigenic complex, and that anti-Crry Abs promote the development of injury in HN by neutralizing the complement regulatory activity of Crry. Rats immunized with Fx1A lacking Crry remained free of proteinuria and glomerular deposits of C3 during a 10-wk follow-up despite typical granular immunoglobulin (Ig)G deposits in glomeruli. Anti-Fx1A auto-Abs were present in their sera at levels that were not different from sera pooled from proteinuric rats with HN induced with nephritogenic Fx1A. Passive administration of sheep anti-Crry Abs to rats immunized with Crry-deficient Fx1A led to proteinuria and glomerular C3 deposition, which were not seen in such rats injected with preimmune IgG, nor in rats with collagen-induced arthritis injected with anti-Crry IgG. To directly examine the role of Crry in HN, rats were immunized with Crry-deficient Fx1A reconstituted with rCrry. This led to typical HN, with 8 out of 15 rats developing proteinuria within 14 wk. Moreover, the extent of glomerular C3 deposition correlated with proteinuria, and anti-Crry Abs were present in glomerular eluates. Thus, Crry is a key nephritogenic immunogen in Fx1A. Formation of neutralizing auto-Abs to Crry impairs its function, leading to unrestricted complement activation by Abs reactive with the HN antigenic complex on the epithelial cell surface.
Keywords: complement regulation, Heymann nephritis, Crry, autoantibodies, glomerulus
Heymann nephritis (HN)1 is a widely studied model of membranous nephropathy with many similarities to the human disease (1, 2). Active immunization of susceptible rat strains with fraction 1A (Fx1A), a crude renal tubular preparation, induces the production of IgG auto-Abs that accumulate in glomeruli. Within 8 wk of immunization with Fx1A, ∼60% of animals develop proteinuria, which is associated with glomerular deposition of C3 and C5b-9 (3, 4). As shown in passive HN, heterologous anti-Fx1A, when injected into rats, reacts with antigens on the glomerular epithelial cell surface and activates complement. In this model, the assembly of C5b-9 on glomerular epithelial cell membranes is directly pathogenic (5–7). Similar mechanisms are presumed to be operative in active HN.
Much effort has gone into identifying the antigen(s) contained within Fx1A that confer pathogenic potential to this immunogen. One well-described antigen was originally termed gp330 and is now known as megalin (8–10). Active immunization with megalin, or passive administration of anti-megalin Ab, leads to glomerular deposition of immune complexes. Yet despite the accumulation of IgG Ab in immune deposits, there is no glomerular C3 or C5b-9 deposition and animals do not develop abnormal proteinuria (2, 11).
Crry (complement receptor 1-related protein/gene y) is a rodent complement inhibitor that combines the functions of human decay-accelerating factor (CD55) and membrane cofactor protein (CD46). Although not a true genetic homologue, Crry is considered a functional analogue of these two proteins, and is widely expressed (12–17). Not surprisingly, given that Fx1A is a crude extract of renal tubules which contain Crry (18, 19), heterologous anti-Fx1A Abs contain reactivity towards Crry. These Abs inhibit the function of Crry in cultured glomerular epithelial cells leading to unrestricted complement activation (20, 21).
Since Crry is contained in Fx1A, we were interested in determining if autoAbs were generated to Crry and whether these were of pathogenic importance in HN. In this study, animals immunized with Fx1A lacking Crry did not develop abnormal proteinuria or glomerular C3 deposition despite the formation of anti-Fx1A Abs, which formed typical glomerular subepithelial deposits. Nephritogenicity of this preparation could be reconstituted by passive transfer of anti-Crry Abs or by inclusion of rCrry in the immunogen.
Materials and Methods
Fx1A and rCrry.
Fx1A was prepared from normal Sprague-Dawley rat kidneys by standard techniques (1). One batch was made from freshly isolated Sprague-Dawley rat kidneys, which led to typical HN upon active immunization of Lewis rats. The second Fx1A preparation was isolated from commercially obtained Sprague-Dawley rat kidneys (Pel-Freez Biologicals, Rogers, AR). This failed to induce classic disease manifestations of HN, the reasons for which are studied here.
The five NH2-terminal short consensus repeats of Crry (22, 23) containing the active sites of Crry were produced as a recombinant soluble protein in Pichia pastoris. rCrry was purified by sequential Mono Q and Mono P chromatography (Amersham Pharmacia Biotech, Piscataway, NJ; reference 24).
Anti-Crry Abs.
Serum was obtained from a single sheep hyperimmunized with rCrry. IgG was isolated by protein G affinity chromatography (Amersham Pharmacia Biotech). Preimmune IgG from the same sheep was used as a control in these studies. Anti-Crry F(ab′)2 was generated by pepsin treatment followed by size exclusion chromatography on a Sephacryl S-100HR column. No intact heavy chains were present by SDS-PAGE under reducing conditions.
Animal Studies.
HN was induced by immunizing 150-g female Lewis rats with Fx1A. 10 mg of Fx1A was mixed with 4 mg of Mycobacterium butyricum in 0.1 ml of PBS and emulsified with an equivalent volume of complete Freund's adjuvant (Difco, Detroit, MI), which was injected in equally divided doses into both hind footpads. Starting 6 wk after immunization, animals were housed biweekly in metabolic cages for urine collection. Sera were obtained at death by cardiac puncture. Renal tissue was obtained by survival renal biopsy and at death.
To have controls with an actively produced autoimmune disease, collagen-induced arthritis (CIA) was induced contemporaneously in Lewis rats. These animals were immunized with 0.2 mg of bovine type II collagen (Sigma Chemical Co., St. Louis, MO) in incomplete Freund's adjuvant twice over 7 d (25). This resulted in a polyarticular arthritis, which peaked in severity by 4 wk after immunization and then resolved.
Passive administration of anti-Crry Abs was performed by injecting animals with 10 mg of anti-Crry IgG or F(ab′)2 intravenously given in divided doses over 2 d. Control animals received 10 mg of preimmune IgG intravenously over 2 d.
Measures of Disease.
Urinary protein excretion was measured by a sulfosalicyclic acid precipitation method (26). Proteinuria was considered abnormal when excretion was ≥6 mg/d (1). Direct immunofluorescence (IF) was performed on cryostat sections for rat IgG and C3 (26). Semiquantitative scores for IF staining intensity were assigned to coded sections (27).
To measure glomerular anti-Fx1A and anti-Crry Abs, glomeruli were isolated from one kidney by sieving (6), after which Abs were eluted by sonication in 20 mM of citric acid, pH 3.2, in the presence of protease inhibitors (11). Glomerular protein content was determined by a bicinchoninic acid assay (Pierce Chemical Co., Rockford, IL).
ELISA.
ELISAs for anti-Fx1A and anti-Crry Abs were performed by coating polystyrene plates with 10 μg/ml detergent-solubilized Fx1A or rCrry. Diluted sera or glomerular eluates were added, which were detected with peroxidase-conjugated anti-rat IgG (Sigma Chemical Co.), followed by development with o-phenylene-diamine. Measurements were made at OD450, and data are presented as OD450 U/ml. When comparisons were made, samples were run in duplicates on a single ELISA plate. For measurements of Crry, polystyrene plates were coated with 20 μg/ml of anti-Crry IgG, after which serial dilutions of rCrry or detergent-solubilized Fx1A were added. Bound Crry was detected with biotinylated anti-Crry, followed by streptavidin– horseradish peroxidase. The ELISA was sensitive to 10 ng/ml Crry.
Statistics.
Statistical analyses were performed with Minitab software (College Park, MD). Data are expressed as mean ± SEM and were analyzed by Student's t test when two groups were compared or by analysis of variance for multiple groups.
Results
Two preparations of Fx1A were compared in our studies. These preparations differed in their capacity to induce classic HN, as defined by the development of abnormal proteinuria in the majority of animals starting 6–8 wk after immunization. The nephritogenic preparation of Fx1A isolated from freshly obtained kidneys contained 3.1% Crry (wt/wt), whereas the Fx1A that was prepared from freeze-thawed kidneys failed to induce disease manifestations of HN and lacked Crry by a sensitive ELISA. These results suggested that Crry was an important component in nephritogenic Fx1A.
The Fx1A preparation lacking Crry was further studied through the active immunization of 12 female Lewis rats. Animals were followed for 10 wk, during which time no significant proteinuria occurred (0.98 ± 0.12 and 2.14 ± 0.53 mg/d at 6 and 10 wk, respectively, after immunization). At this time, all animals underwent renal biopsy. By IF microscopy there was strongly positive granular staining for IgG outlining glomerular capillary walls in 10 out of 12 animals (Fig. 1 A), whereas the remaining 2 animals had only faintly apparent IgG deposits. In all animals, characteristic deposits of C3 were absent (Fig. 1 B).
Figure 1.
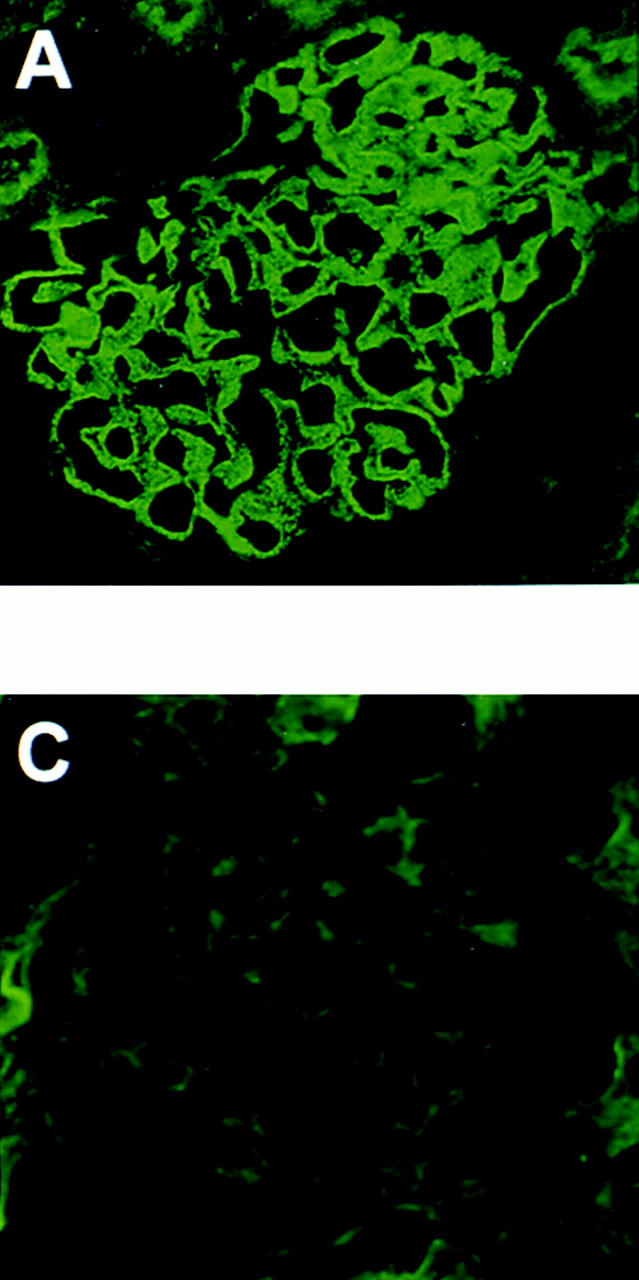
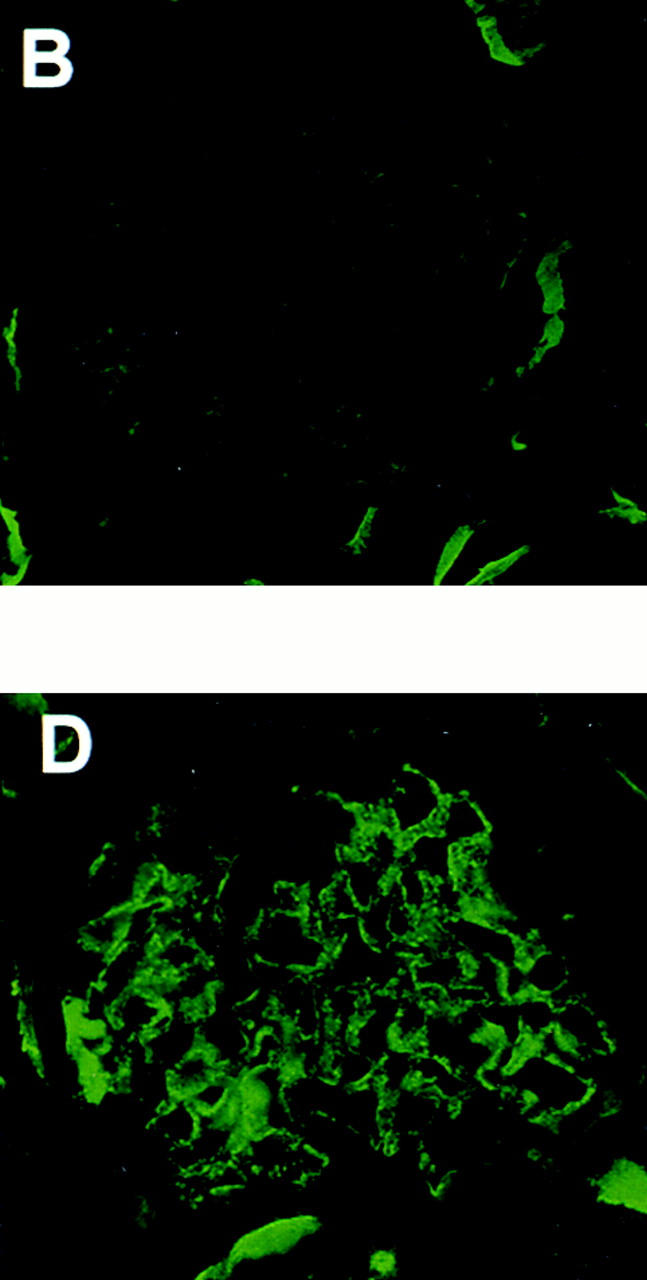
Representative IF micrographs in animals immunized with Fx1A lacking Crry. 10 wk after immunization, renal biopsies were performed and tissue was stained for IgG (A) and C3 (B). Animals were then injected with preimmune IgG (C) or anti-Crry IgG (D). 6 d later, animals were killed and renal tissue was stained for C3 (C and D). The IF micrographs shown in A, B, and D are from the same animal, and show typical granular staining for IgG in HN (A) and the appearance of a similar staining pattern for C3 only after injection of anti-Crry IgG (D).
To test if the absence of C3 deposits and proteinuria in HN rats immunized with Crry-deficient Fx1A was due to preservation of glomerular Crry complement regulatory activity, function-neutralizing anti-Crry Abs were administered passively. HN animals received either anti-Crry IgG or preimmune IgG as a control 10 wk after immunization with Crry-deficient Fx1A. For these studies, the 10 animals with strongly positive glomerular IgG staining were used. To exclude an effect of anti-Crry IgG to activate the classical pathway, anti-Crry F(ab′)2 was also used in these studies. As further controls, Lewis rats in which CIA had been induced 10 wk earlier were injected with anti-Crry IgG. HN animals injected with anti-Crry had a progressive rise in urinary protein which peaked 5 d after Ab injection, with five out of seven animals developing abnormal proteinuria (Fig. 2). In contrast, there was no significant proteinuria in either HN rats injected with preimmune IgG or CIA rats injected with anti-Crry IgG. In the three animals with heavy proteinuria (>60 mg/d), there was strong staining for C3 by IF microscopy in glomeruli (Fig. 1 D), whereas the remaining animals with <60 mg/d proteinuria continued to have absent C3 staining (Fig. 1 C).
Figure 2.

Effect of anti-Crry on urinary protein excretion in active HN. Animals were immunized with Fx1A lacking Crry (Active Heymann Nephritis) or with bovine type II collagen (CIA). 10 wk later, 5 mg of anti-Crry IgG or F(ab′)2, or preimmune IgG, were injected intravenously twice over a period of 24 h. 5 d after intravenous injection, urine was collected and urinary protein was measured by the sulfosalicyclic acid precipitation method (1). Proteinuria was considered abnormal when ≥6 mg/d (horizontal dashed line). Data for individual animals are shown. P = 0.022 by one-way analysis of variance.
To determine whether absence of Crry affected Ab production to Fx1A, anti-Fx1A Abs were measured in sera of rats immunized with Fx1A lacking Crry. These were present before anti-Crry injection (4.12 ± 0.55 OD450 U/ml) and were not different from anti-Fx1A levels in pooled sera from animals with active HN induced with nephritogenic Fx1A (4.72 OD450 U/ml). Anti-Crry Abs were also detectable (2.22 OD450 U/ml) in these latter sera, whereas in animals immunized with anti-Fx1A lacking Crry, anti-Crry levels were not significantly different from background (0.18 ± 0.12 OD450 U/ml). Thus, immunization of animals with an anti-Fx1A preparation lacking Crry leads to the production of anti-Fx1A Abs that deposit in glomeruli. Yet complement activation and proteinuria do not occur. Neutralization of Crry function via the passive administration of anti-Crry Abs can lead to complement activation and proteinuria in the presence of these glomerular-bound IgG Abs.
These results suggested that generation of an immune response to Crry is necessary for the development of proteinuria in HN. The next set of studies directly tested whether reconstituting Fx1A lacking Crry with rCrry could restore nephritogenicity. Therefore, female Lewis rats were immunized with Fx1A lacking Crry (n = 20) or with this same Fx1A preparation to which 0.5 mg of rCrry was added (n = 15). Animals were observed for 14 wk to ensure that the potential development of proteinuria was not missed. As in the previous studies, animals immunized with Fx1A lacking Crry did not develop proteinuria (Fig. 3). In contrast, in animals immunized with Fx1A reconstituted with rCrry there was progressive development of proteinuria, such that by 14 wk after immunization, 8 out of 15 animals had significant proteinuria. This proportion of proteinuric rats is very similar to that typically seen in active HN (1). At death, anti-Fx1A Ab levels were comparable in both groups (2.17 ± 0.23 and 2.17 ± 0.28 OD450 U/ml).
Figure 3.

Urinary protein excretion over time in animals immunized with Fx1A lacking Crry (Fx1A - rCrry, •) or immunized with this identical preparation reconstituted with rCrry (Fx1A + rCrry, ○). Data for individual animals are shown. Proteinuria was considered abnormal when ≥6 mg/d (horizontal dashed line). At 14 wk after immunization, urinary protein excretion was 42.4 ± 13.5 and 1.4 ± 0.3 mg/d in animals immunized with Fx1A with and without rCrry, respectively (mean ± SEM; P < 0.001 by Student's t testing).
In both groups of animals there was granular glomerular IgG staining by IF microscopy, yet in the animals immunized with Fx1A lacking Crry there was no glomerular C3 deposition. In contrast, animals immunized with Fx1A plus rCrry had glomerular C3 deposition. Furthermore, the semiquantitative score for C3 immunostaining correlated with proteinuria (r = 0.80, P < 0.001), suggesting that complement activation within glomeruli was pathogenic. Anti-Crry Abs were present in glomerular eluates of rats immunized with Fx1A plus rCrry (0.71 ± 0.19 OD450 U/μg glomerular protein). As expected, anti-Crry Abs were not present in glomeruli of rats immunized with the Fx1A preparation lacking Crry (0.06 ± 0.05 OD450 U/μg glomerular protein, which is not significantly different from background). Thus, in HN, active immunization with Fx1A containing Crry leads to formation of anti-Crry Abs that deposit in glomeruli.
Discussion
In this study we show that rats immunized with Fx1A lacking Crry do not develop proteinuria or C3 deposition in glomeruli, despite the generation of anti-Fx1A Abs that deposit in glomeruli. If Abs to Crry are reconstituted either via the passive administration of anti-Crry Abs or by the inclusion of rCrry in the immunogen, animals develop proteinuria to the extent and level seen in active HN induced with Crry-containing Fx1A. These findings show that Crry is a critical immunogen within Fx1A and is essential for its nephritogenic effect. Formation of Abs to Crry impairs the function of this intrinsic complement regulator. Since Crry is the key regulator of complement at the C3/C5 convertase stage, inhibition of its function leads to unrestricted complement activation. As shown in the passive HN model of membranous nephropathy, complement activation on the glomerular epithelial cell results in injury of this cell and impairment of the glomerular permselectivity barrier to the passage of protein.
In our past studies, we found that heterologous anti-Fx1A Abs reacted with glomerular epithelial cell antigens and could activate complement on the surface of these cells, leading to cellular injury (6). Subsequently, we observed that nephritogenic anti-Fx1A impaired intrinsic complement regulation in glomerular epithelial cells. Such an effect occurred at the level of complement C3/C5 convertases formed from either the classical or alternative pathways (20). At that time, the antigen responsible for these effects of anti-Fx1A was unknown. With the isolation of rat Crry as the antigen reactive with mAb 5I2 (28), it was possible to prove that anti-Fx1A reacted with rat Crry, consistent with its inhibiting intrinsic regulation of C3/C5 convertases (21). That heterologous anti-Fx1A Abs contains reactivity towards Crry is not surprising, given the widespread distribution of Crry, including in renal tubules (18, 19). Interestingly, we have also shown that heterologous anti-Fx1A has reactivity towards CD59, a regulator of C5b-9 formation, which can similarly be attributed to the presence of CD59 in renal tubular cells (18). The role for reactivity of heterologous anti-Fx1A with Crry or CD59 in passive HN is a subject of ongoing study.
Despite the identification of megalin as an unquestionably important immunogen within Fx1A, it is very clear that megalin is not the only nephritogenic component contained within Fx1A. Thus, either passive administration of antimegalin Ab or active immunization with megalin is insufficient to recapitulate the full disease of HN. This has stimulated a continuing search for additional antigenic component(s). A number of candidate antigens have been identified, including β1 integrins, dipeptidyl peptidase IV, aminopeptidase A, and an uncharacterized lipid antigen (29–31).
Although HN has many similarities to human membranous nephropathy, identifying target human antigens in this disease has proved elusive. For instance, as yet there is no identified analogue to megalin or the HN antigenic complex expressed in human glomeruli (8–10, 32). Human glomerular epithelial cells contain decay-accelerating factor and membrane cofactor protein (33, 34), which together contain the function of Crry. There are subepithelial IgG deposits in human membranous nephropathy, exactly as in HN. The evidence for the complement mediation of this disease in humans is strong. There is glomerular deposition of C3 and C5b-9, and the appearance of C5b-9 in the urine has prognostic importance (35–37). An intriguing possibility raised by these studies is that autoAbs to decay-accelerating factor and/or to membrane cofactor protein may form and accumulate in human membranous nephropathy. If so, it may be possible, with the use of recombinant complement regulators (38–42) to overcome the effects of these autoAbs and thereby prevent glomerular epithelial cell injury and proteinuria.
Acknowledgments
We thank Dr. Andrew Minto for preparing Fx1A.
This work was supported by National Institutes of Health grants DK30932 (to D.J. Salant) and DK41873 (to R.J. Quigg), and by a grant-in-aid from the National Kidney Foundation, Illinois Chapter (to R.J. Quigg). J.J. Alexander was supported by training grant DK07510 from the National Institutes of Health.
Abbreviations used in this paper
- CIA
collagen-induced arthritis
- Crry
complement receptor 1-related protein/gene y
- HN
Heymann nephritis
- IF
immunofluorescence
References
- 1.Salant DJ, Cybulsky AV. Experimental glomerulonephritis. Methods Enzymol. 1988;162:421–461. doi: 10.1016/0076-6879(88)62096-9. [DOI] [PubMed] [Google Scholar]
- 2.Kerjaschki D, Neale TJ. Molecular mechanisms of glomerular injury in rat experimental membranous nephropathy (Heymann nephritis) J Am Soc Nephrol. 1996;7:2518–2526. doi: 10.1681/ASN.V7122518. [DOI] [PubMed] [Google Scholar]
- 3.Salant DJ, Belok S, Madaio MP, Couser WG. A new role for complement in experimental membranous nephropathy in rats. J Clin Invest. 1980;66:1339–1350. doi: 10.1172/JCI109987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makker SP. Analysis of glomeruli-eluted Gp330 autoantibodies and of Gp330 antigen of Heymann nephritis. J Immunol. 1993;151:6500–6508. [PubMed] [Google Scholar]
- 5.Cybulsky AV, Rennke HG, Feintzeig ID, Salant DJ. Complement-induced glomerular epithelial cell injury: role of the membrane attack complex in rat membranous nephropathy. J Clin Invest. 1986;77:1096–1107. doi: 10.1172/JCI112408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quigg RJ, Cybulsky AV, Jacobs JB, Salant DJ. Anti-Fx1A produces complement-dependent cytotoxicity of glomerular epithelial cells. Kidney Int. 1988;34:43–52. doi: 10.1038/ki.1988.143. [DOI] [PubMed] [Google Scholar]
- 7.Baker PJ, Ochi RF, Schulze M, Johnson RJ, Campbell C, Couser WG. Depletion of C6 prevents development of proteinuria in experimental membranous nephropathy in rats. Am J Pathol. 1989;135:185–194. [PMC free article] [PubMed] [Google Scholar]
- 8.Kerjaschki D, Farquhar MG. The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci USA. 1982;79:5557–5581. doi: 10.1073/pnas.79.18.5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pietromonaco S, Kerjaschki D, Binder S, Ullrich R, Farquhar MG. Molecular cloning of a cDNA encoding a major pathogenic domain of the Heymann nephritis antigen gp330. Proc Natl Acad Sci USA. 1990;87:1811–1815. doi: 10.1073/pnas.87.5.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farquhar MG, Saito A, Kerjaschki D, Orlando RA. The Heymann nephritis antigenic complex: megalin (gp330) and RAP. J Am Soc Nephrol. 1995;6:35–47. doi: 10.1681/ASN.V6135. [DOI] [PubMed] [Google Scholar]
- 11.Raychowdhury R, Zheng G, Brown D, McCluskey RT. Induction of Heymann nephritis with a gp330/megalin fusion protein. Am J Pathol. 1996;148:1613–1623. [PMC free article] [PubMed] [Google Scholar]
- 12.Wong W, Fearon DT. p65: a C3b-binding protein on murine cells that shares antigenic determinants with the human C3b receptor (CR1) and is distinct from murine C3b receptor. J Immunol. 1985;134:4048–4056. [PubMed] [Google Scholar]
- 13.Paul MS, Aegerter M, O'Brien SE, Kurtz CB, Weis JH. The murine complement receptor gene family. I. Analysis of mCRY gene products and their homology to human CR1. J Immunol. 1989;142:582–589. [PubMed] [Google Scholar]
- 14.Foley S, Li B, Dehoff M, Molina H, Holers VM. Mouse Crry/p65 is a regulator of the alternative pathway of complement activation. Eur J Immunol. 1993;23:1381–1384. doi: 10.1002/eji.1830230630. [DOI] [PubMed] [Google Scholar]
- 15.Li B, Sallee C, Dehoff M, Foley S, Molina H, Holers VM. Mouse Crry/p65: Characterization of monoclonal antibodies and the tissue distribution of a functional homologue of human MCP and DAF. J Immunol. 1993;151:4295–4305. [PubMed] [Google Scholar]
- 16.Kim Y-U, Kinoshita T, Molina H, Hourcade D, Seya T, Wagner LM, Holers VM. Mouse complement regulatory protein Crry/p65 uses the specific mechanisms of both human decay-accelerating factor and membrane cofactor protein. J Exp Med. 1995;181:151–159. doi: 10.1084/jem.181.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quigg RJ, Morgan BP, Holers VM, Adler S, Sneed AE, Lo CF. Complement regulation in the rat glomerulus: Crry and CD59 regulate complement in glomerular mesangial and endothelial cells. Kidney Int. 1995;48:412–421. doi: 10.1038/ki.1995.309. [DOI] [PubMed] [Google Scholar]
- 18.Funabashi K, Okada N, Matsuo S, Yamamoto T, Morgan BP, Okada H. Tissue distribution of complement regulatory membrane proteins in rats. Immunology. 1994;81:444–451. [PMC free article] [PubMed] [Google Scholar]
- 19.Nomura A, Nishikawa K, Yuzawa Y, Okada H, Okada N, Morgan BP, Piddlesden SJ, Nadai M, Hasegawa T, Matsuo S. Tubulointerstitial injury induced in rats by a monoclonal antibody which inhibits function of a membrane inhibitor of complement. J Clin Invest. 1995;96:2348–2356. doi: 10.1172/JCI118291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quigg RJ, Cybulsky AV, Salant DJ. Effect of nephritogenic antibody on complement regulation in cultured rat glomerular epithelial cells. J Immunol. 1991;147:838–845. [PubMed] [Google Scholar]
- 21.Quigg RJ, Holers VM, Morgan BP, Sneed AE. Crry and CD59 regulate complement in rat glomerular epithelial cells and are inhibited by the nephritogenic antibody of passive Heymann nephritis. J Immunol. 1995;154:3437–3443. [PubMed] [Google Scholar]
- 22.Sakurada C, Seno H, Dohi N, Takizawa H, Nonaka M, Okada N, Okada H. Molecular cloning of the rat complement regulatory protein, 5I2 antigen. Biochem Biophys Res Commun. 1994;198:819–826. doi: 10.1006/bbrc.1994.1117. [DOI] [PubMed] [Google Scholar]
- 23.Quigg RJ, Lo CF, Alexander JJ, Sneed AE, Moxley G. Molecular characterization of rat Crry: widespread distribution of two alternative forms of Crry mRNA. Immunogenetics. 1995;42:362–367. doi: 10.1007/BF00179397. [DOI] [PubMed] [Google Scholar]
- 24.He C, Alexander JJ, Lim A, Quigg RJ. Production of the rat complement regulator, Crry, as an active soluble protein in Pichia pastoris. . Arch Biochem Biophys. 1997;341:347–352. doi: 10.1006/abbi.1997.9989. [DOI] [PubMed] [Google Scholar]
- 25.Ridge SC, Oronsky AL, Kerwar SS. Type II collagen-induced arthritis in rats. Methods Enzymol. 1988;162:355–360. doi: 10.1016/0076-6879(88)62090-8. [DOI] [PubMed] [Google Scholar]
- 26.Quigg RJ, Abrahamson DR, Cybulsky AV, Badalamenti J, Minto AWM, Salant DJ. Studies with antibodies to cultured rat glomerular epithelial cells: subepithelial immune deposit formation after in vivo injection. Am J Pathol. 1989;134:1125–1133. [PMC free article] [PubMed] [Google Scholar]
- 27.Quigg RJ, Lim A, Haas M, Alexander JJ, He C, Carroll MC. Immune complex glomerulonephritis in C4- and C3-deficient mice. Kidney Int. 1998;53:320–330. doi: 10.1046/j.1523-1755.1998.00723.x. [DOI] [PubMed] [Google Scholar]
- 28.Takizawa H, Okada N, Okada H. Complement inhibitor of rat cell membrane resembling mouse Crry/p65. J Immunol. 1994;152:3032–3038. [PubMed] [Google Scholar]
- 29.Adler S, Chen X. Anti-Fx1A antibody recognizes a β1-integrin on glomerular epithelial cells and inhibits adhesion and growth. Am J Physiol. 1992;262:F770–F776. doi: 10.1152/ajprenal.1992.262.5.F770. [DOI] [PubMed] [Google Scholar]
- 30.Susani M, Schulze M, Exner M, Kerjaschki D. Antibodies to glycolipids activate complement and promote proteinuria in passive Heymann nephritis. Am J Pathol. 1994;144:807–819. [PMC free article] [PubMed] [Google Scholar]
- 31.Salant, D.J., Y. Natori, and F. Shimizu. 1997. Glomerular injury due to antibody alone. In Immunologic Renal Diseases. E.G. Neilson and W.G. Couser, editors. Lippincott-Raven, Philadelphia. 359–375.
- 32.Raychowdhury R, Niles JL, McCluskey RT, Smith JA. Autoimmune target in Heymann nephritis is a glycoprotein with homology to the LDL receptor. Science. 1989;244:1163–1165. doi: 10.1126/science.2786251. [DOI] [PubMed] [Google Scholar]
- 33.Quigg RJ, Nicholson-Weller A, Cybulsky AV, Badalamenti J, Salant DJ. Decay accelerating factor regulates complement activation on glomerular epithelial cells. J Immunol. 1989;142:877–882. [PubMed] [Google Scholar]
- 34.Nakanishi I, Moutabarrik A, Hara T, Hatanaka M, Hayashi T, Syouji T, Okada N, Kitamura E, Tsubakihara Y, Matsumoto M, Seya T. Identification and characterization of membrane cofactor protein (CD46) in the human kidneys. Eur J Immunol. 1994;24:1529–1535. doi: 10.1002/eji.1830240711. [DOI] [PubMed] [Google Scholar]
- 35.Hinglais N, Kazatchkine MD, Bhakdi S, Appay MD, Mandet C, Grossetete J, Bariety J. Immunohistochemical study of the C5b-9 complex of complement in human kidneys. Kidney Int. 1986;30:399–410. doi: 10.1038/ki.1986.198. [DOI] [PubMed] [Google Scholar]
- 36.Schulze M, Donadio JV, Jr, Pruchno CJ, Baker PJ, Johnson RJ, Stahl RAK, Watkins S, Martin DC, Wurzner R, Gotze O, Couser WG. Elevated urinary excretion of the C5b-9 complex in membranous nephropathy. Kidney Int. 1991;40:533–538. doi: 10.1038/ki.1991.242. [DOI] [PubMed] [Google Scholar]
- 37.Lehto T, Honkanen E, Teppo AM, Meri S. Urinary excretion of protectin (CD59), complement SC5b-9 and cytokines in membranous glomerulonephritis. Kidney Int. 1995;47:1403–1411. doi: 10.1038/ki.1995.197. [DOI] [PubMed] [Google Scholar]
- 38.Weisman HF, Bartow T, Leppo MK, Marsh HC, Jr, Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML, Fearon DT. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 39.Kalli KR, Hsu P, Fearon DT. Therapeutic uses of recombinant complement protein inhibitors. Springer Semin Immunopathol. 1994;15:417–431. doi: 10.1007/BF01837368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore FD., Jr Therapeutic regulation of the complement system in acute injury states. Adv Immunol. 1994;56:267–299. doi: 10.1016/s0065-2776(08)60454-x. [DOI] [PubMed] [Google Scholar]
- 41.Couser WG, Johnson RJ, Young BA, Yeh CG, Toth CA, Rudolph AR. The effects of soluble recombinant complement receptor 1 on complement-mediated experimental glomerulonephritis. J Am Soc Nephrol. 1995;5:1888–1894. doi: 10.1681/ASN.V5111888. [DOI] [PubMed] [Google Scholar]
- 42.Higgins PJ, Jone-Long K, Lobell R, Sardonini C, Alessi MK, Yeh CG. A soluble chimeric complement inhibitory protein that possesses both decay-accelerating and factor I cofactor activities. J Immunol. 1997;158:2872–2881. [PubMed] [Google Scholar]