

Abstract
The mechanism by which progesterone causes localized suppression of the immune response during pregnancy has remained elusive. Using human T lymphocytes and T cell lines, we show that progesterone, at concentrations found in the placenta, rapidly and reversibly blocks voltage-gated and calcium-activated K+ channels (KV and KCa, respectively), resulting in depolarization of the membrane potential. As a result, Ca2+ signaling and nuclear factor of activated T cells (NF-AT)-driven gene expression are inhibited. Progesterone acts distally to the initial steps of T cell receptor (TCR)-mediated signal transduction, since it blocks sustained Ca2+ signals after thapsigargin stimulation, as well as oscillatory Ca2+ signals, but not the Ca2+ transient after TCR stimulation. K+ channel blockade by progesterone is specific; other steroid hormones had little or no effect, although the progesterone antagonist RU 486 also blocked KV and KCa channels. Progesterone effectively blocked a broad spectrum of K+ channels, reducing both Kv1.3 and charybdotoxin–resistant components of KV current and KCa current in T cells, as well as blocking several cloned KV channels expressed in cell lines. Progesterone had little or no effect on a cloned voltage-gated Na+ channel, an inward rectifier K+ channel, or on lymphocyte Ca2+ and Cl− channels. We propose that direct inhibition of K+ channels in T cells by progesterone contributes to progesterone-induced immunosuppression.
Keywords: T lymphocyte, K+ channel, calcium signaling, gene expression, nuclear factor of activated T cells
Immunosuppression within the uterus is crucial for the survival of the fetus (1, 2). Although the maternal immune system becomes sensitized to paternal antigens during pregnancy, fetal cells and placental trophoblasts bearing those antigens do not elicit a cytolytic immune response (3–5). High concentrations of progesterone in the placenta inhibit the maternal immune response against the fetal allograft (6, 7). The immunosuppressive effects of progesterone were demonstrated in vivo by prolonged survival of xenografts near silastic implants containing progesterone at concentrations typically found in the placenta (3, 6). In vitro assays have established that progesterone inhibits lymphocyte activation and proliferation in response to allogeneic cells or mitogens (8–10). In contrast, progesterone does not inhibit the effector functions of previously activated cytolytic T cells (11). These data suggest that progesterone may interfere with the early phases of T cell activation.
Antigen presentation and TCR ligation stimulate tyrosine kinases, leading to the generation of inositol 1,4,5-trisphosphate (IP3)1 and a consequent rise in the cytoplasmic calcium concentration ([Ca2+]i). Elevated [Ca2+]i activates calcineurin, a phosphatase which then dephosphorylates a cytoplasmic transcription factor, the nuclear factor of activated T cells (NF-AT). Dephosphorylated NF-AT moves into the nucleus where it promotes the expression of the IL-2 gene (12). A sustained elevation in [Ca2+]i requiring Ca2+ influx across the plasma membrane is necessary for the retention of NF-AT in the nucleus and efficient transcription of IL-2 (13–16). In lymphocytes, the opening of Ca2+ release–activated Ca2+ (CRAC) channels initiates Ca2+ influx after the depletion of Ca2+ stores by IP3 (17–19). Once the CRAC channels are open, the transmembrane concentration gradient for Ca2+ and the membrane potential (E m) provide the driving force for Ca2+ entry. E m is set by the interplay between several ion channels in the T cell membrane. By itself, the Ca2+ current through CRAC channels would diminish the driving force for calcium entry by reducing E m. However, currents through voltage-gated K+ (KV) channels and Ca2+-activated K+ (KCa) channels enhance Ca2+ entry by driving E m to a negative voltage. Chloride channels may also play a role in maintaining a negative E m during T cell activation (20, 21). The four major types of ion channels found in T cells are possible targets for immunosuppressive agents. In particular, the KV channel encoded by Kv1.3 is required for normal lymphocyte activation both in vitro and in vivo (14, 22–25).
We have determined the effects of progesterone on lymphocyte ion channels, Ca2+ signaling, and gene expression. By combining functional assays of gene expression with patch-clamp and Ca2+-imaging measurements, we demonstrate that progesterone blocks lymphocyte K+ channels, interferes with TCR-induced [Ca2+]i signaling, and inhibits gene expression. We propose that progesterone acts as an endogenous immunosuppressant by directly and reversibly blocking K+ channels.
Materials and Methods
Chemicals and Solutions.
Salts and other reagents were obtained from Sigma Chemical Co. (St Louis, MO) unless otherwise noted. Thapsigargin (TG) was obtained from Alexis Corp. (San Diego, CA). Progesterone (10 mM) and PMA (3 mM) stocks were prepared in DMSO.
Cell Culture.
B3Z cells and K897 cells were provided along with the antigenic peptide fragment SIINFEKL by Dr. N. Shastri (University of California, Berkeley, CA). B3Z cells are a murine, CD8+, T cell hybridoma with a known antigen specificity for OVA/Kb-MHC and containing a β-galactosidase reporter gene construct (lacZ) under the control of the NF-AT promoter (26). The corresponding antigen-presenting K897 cells had been transfected with Kb class I MHC as described (27). The human leukemia T cell line Jurkat E6-1 was obtained from the American Type Culture Collection (Rockville, MD). Human peripheral T lymphocytes were collected from venous blood of healthy volunteers and isolated using a Ficoll-Hypaque density gradient as described previously (28). A population of activated T cell blasts was prepared in culture by treating the resting cells with 10 μg/ml PHA (PHA-P; Difco Laboratories, Inc., Detroit, MI). Cell lines expressing the cloned KV channels Kv1.1, Kv1.2, Kv1.3, Kv1.4, Kv1.5, and Kv3.1 and a voltage-gated Na+ channel hSKM1 were maintained in culture. All cells were grown in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 10 mM Hepes, 2 mM glutamine, 1 mM pyruvate, 50 μM 2-ME. Cells were cultured in 25-ml flasks (Costar Corp., Cambridge, MA) at 37°C, 5% CO2 in a humidified incubator. For experiments on inward rectifier K+ channels, rat basophilic leukemia (RBL) cells were maintained in Eagle's MEM supplemented with 10% FCS and 1% l-glutamine. RBL cells were plated onto glass coverslips 1–2 d before use.
LacZ Reporter Gene Assay.
The fraction of B3Z cells expressing β-galactosidase was measured using flow cytometry (FACScan®; Becton Dickinson, San Jose, CA) as described previously (13). In brief, 5 × 104 cells in RPMI without serum were placed in individual wells of 24-well plates activated either by 1 μM TG plus 50 nM PMA or by antibodies to the CD3-ε subunit of the TCR complex. In the latter, wells were coated with 10 μg/ml anti–CD3-ε antibodies (PharMingen, San Diego, CA) overnight and rinsed briefly with PBS before use. Cells were activated in the incubator for a total of 4 h before being resuspended and loaded by osmotic shock with the fluorogenic substrate, fluorescein-di-β-galactopyranoside (FDG; Molecular Probes, Inc., Eugene, OR). The fluorescence of lacZ + cells was at least fivefold greater than autofluorescence. The effect of progesterone on lacZ gene expression was quantified using a MUG (4-methylumbelliferyl β-d-galactopyranoside) assay (29). In brief, B3Z cells were plated at 105 cells per well in 96-well plates, and the fluorescence produced by cell lysates in a solution containing 3 mM MUG was measured in a multi-well plate reader (CytoFluor Series 4000; PerSeptive Biosystems, Framingham, MA).
Patch-clamp Recordings.
Membrane currents were measured using the whole-cell configuration of the patch-clamp technique (28, 30), and membrane potential was measured using the perforated patch method in current-clamp mode with nystatin to permeabilize the cells (31). An EPC-9 amplifier (HEKA, Lambrecht/ Pfalz, Germany) interfaced to a Macintosh Quadra 700 computer was used for pulse application and data recording. Membrane voltages were corrected for liquid junction potentials, and current recordings were corrected for leak and capacitative currents. Patch pipettes were pulled from Accu-fill 90 Micropets (Becton Dickinson, Parsippany, NJ) using a P87 micropipette puller (Sutter Instruments Co., Novato, CA). Pipettes were coated with Sylgard (Dow Corning Corp., Midland, MI) and heat polished to final resistances of 2–5 MΩ. Patch-clamp experiments were performed at room temperature (20–25°C). Unless otherwise indicated, the membrane currents were filtered at 1.5 kHz. Data analysis was performed using the program Pulse (HEKA). Mammalian Ringer contained (in mM): 160 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 10 Hepes (pH 7.4; osmolality 290–310 mosmol/kg). The ionic composition of the pipette solutions used in the individual experiments is reported in the figure legends.
[Ca2+]i Measurement.
[Ca2+]i was measured ratiometrically using fura-2, as described previously (13). In brief, cells were loaded with 3 μM fura-2/AM (Molecular Probes, Inc.) for 30–40 min at room temperature (20–25°C). The cells were then washed three times with RPMI/10% FCS and stored in the dark. Illumination was provided by a xenon arc-lamp (Carl Zeiss, Jena, Germany) and transmitted through a filter wheel unit (Lambda 10; Axon Instruments, Inc., Foster City, CA) containing 350- and 385-nm excitation filters. The filtered light was reflected by a 400-nm dichroic mirror through a 63× oil-immersion objective to illuminate cells. Emitted light >480 nm was received by a SIT camera (C2400; Hamamatsu Photonics, Bridgewater, NJ) and the video information relayed to an image processing system (Videoprobe; ETM Systems, Petaluma, CA). Full field-of-view 8-bit images, averaged over 16 frames, were collected at 350- and 385-nm wavelengths. Digitally stored 350/385 ratios were constructed from background-corrected 350- and 385-nm images. Single-cell measurements of [Ca2+]i were calculated from the 350/385 ratios using the equation of Grynkiewicz et al. (32) and a K d of 250 nM for fura-2. The minimum 350/385 ratio was measured in single cells after incubation for 10 min in Ca2+-free Ringer containing 2 mM EGTA. Maximum ratio values were obtained after perfusion with Ringer containing 10 mM Ca2+, 1 μM TG, and 10 μM ionomycin.
Data Analysis.
Numerical values for single-cell [Ca2+]i traces were analyzed with Origin (Microcal Software, Inc., Northampton, MA). Statistical analysis was performed on data sets using Excel version 5.0 (Microsoft Corp., Redmond, WA). Data are reported as mean ± SD. Analysis of variance (ANOVA) or Student's t test was used to compare mean values. Pairs of means were considered statistically different if P < 0.05.
Results
Progesterone Suppresses Gene Expression Driven by NF-AT.
The murine T cell hybridoma, B3Z, recognizes an octapeptide fragment from ovalbumin (SIINFEKL) and expresses the lacZ reporter construct under transcriptional control of the NF-AT response element of the IL-2 promoter (26, 27). Several treatments that increase [Ca2+]i lead to NF-AT–driven lacZ expression in B3Z cells (13), including TCR engagement or stimulation with TG, a specific inhibitor of the endoplasmic reticulum Ca2+- ATPase (33). By flow cytometry in the present series of experiments, cross-linking the TCR with anti–CD3-ε antibodies or stimulating with TG plus PMA produced lacZ expression in the majority of B3Z cells (60 ± 4%, n = 7; or 72 ± 10%, n = 3, respectively). Progesterone reduced NF-AT–mediated lacZ gene expression in a concentration- dependent manner, with an IC50 value of 22 ± 2.1 μM in cells stimulated by TG/PMA (Fig. 1 A, filled circles). The progesterone antagonist RU 486 also inhibited gene expression with slightly lower potency (Fig. 1 A, open squares). Progesterone reduced lacZ expression when B3Z cells were stimulated by TG alone or with PMA, by immobilized anti–CD3-ε antibody, or by antigen presentation (Fig. 1 B). Thus, at concentrations normally obtained in the placenta (34, 35), progesterone inhibits NF-AT–mediated gene expression when driven by four treatments that increase [Ca2+]i. Our results with NF-AT–driven lacZ reporter gene expression are consistent with levels of progesterone or RU 486 shown previously to inhibit activation of human T cells in vitro (8, 9).
Figure 1.


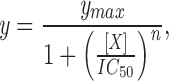 |
Progesterone Inhibits [Ca2+]i Signals in T Cells after TCR Engagement or TG Stimulation.
Upon contact with K897 cells preloaded with SIINFEKL, B3Z cells responded with an initial Ca2+ transient from a resting [Ca2+]i of 180 ± 86 nM to a peak of 2.5 ± 0.5 μM (n = 20), followed by sustained Ca2+ oscillations (Fig. 2 A). In the absence of preloaded antigen, K897 cells did not elicit Ca2+ signaling in B3Z cells (data not shown). Application of 50 μM progesterone reversibly suppressed antigen-induced Ca2+ oscillations (Fig. 2 A).
Figure 2.

Progesterone inhibits [Ca2+]i oscillations induced by TCR ligation. (A) [Ca2+]i responses from four representative B3Z cells activated by contact with SIINFEKL-presenting K897 cells illustrate that [Ca2+]i oscillations were reversibly inhibited by the application of 50 μM progesterone to the bath (bar). (B) [Ca2+]i oscillations in four B3Z cells activated by settling onto coverslips coated with anti–CD3-ε antibodies in the absence (a) or presence (b) of 50 μM progesterone.
To determine if progesterone directly inhibits TCR-initiated signaling or interferes with costimulatory pathways, we activated the TCR complex by cross-linking CD3. Settling B3Z cells onto chambers coated with anti–CD3-ε antibodies elicited an initial Ca2+ transient followed by vigorous Ca2+ oscillations that continued for at least 40 min (Fig. 2 B). In the presence of 50 μM progesterone, most cells produced only the initial Ca2+ transient lasting 271 ± 178 s, or a transient followed by severely attenuated oscillations (n = 14; Fig. 2 C). These data demonstrate that progesterone blocks Ca2+ signaling after TCR engagement. Since the initial Ca2+ transient results from IP3- mediated release of Ca2+ from intracellular stores, and the sustained Ca2+ signal requires Ca2+ influx, these data also suggest that progesterone inhibits Ca2+ influx but not the steps that lead to Ca2+ release from intracellular stores.
TG inhibits the Ca2+ reuptake pump, leading to depletion of the intracellular Ca2+ stores and Ca2+ influx while bypassing the initial steps of TCR signaling and IP3 generation (36, 37). In resting human T cells, the addition of TG to the bathing solution increased [Ca2+]i from 72 ± 8 nM to a stable plateau level of 1.2 ± 0.1 μM (n = 76; Fig. 3 A). Progesterone reversibly inhibited the sustained Ca2+ signal with an IC50 value of 28 ± 2.7 μM (Fig. 3, A and B). Thus, progesterone blocks Ca2+ influx subsequent to emptying of the Ca2+ stores.
Figure 3.


Progesterone reduces the [Ca2+]i plateau evoked by TG stimulation in human T cells. (A) Average [Ca2+]i is plotted against time (n = 76) before and during stimulation with 1 μM TG (arrow). TG stimulated Ca2+ influx, resulting in a stable rise in [Ca2+]i. Addition of 30 μM progesterone (bar) reversibly reduced the [Ca2+]i plateau obtained after TG stimulation. (B) The concentration dependence of the reduction of [Ca2+]i by progesterone is plotted for TG-stimulated cells. Calcium levels were normalized by subtracting the resting [Ca2+]i and dividing [Ca2+]i in the presence of progesterone and TG by [Ca2+]i in the presence of TG alone. Data are presented as mean ± SD and were fitted to a Hill equation illustrated by the smooth curve (IC50 = 28 ± 2.7 μM and n = 1.3 ± 0.2).
Progesterone Depolarizes the Membrane Potential, Reducing the Driving Force for Ca2+ Entry.
Ca2+ influx depends upon the opening of CRAC channels and upon an electrochemical gradient for Ca2+ entry across the cell membrane. The negative membrane potential supported by K+ channels provides the electrical component of the driving force favoring Ca2+ entry; membrane depolarization would reduce this driving force. By direct measurement during perforated-patch current-clamp recording, we found that progesterone depolarizes the membrane potential, E m. KV channels normally maintain the resting membrane potential of T lymphocytes near −60 mV (38). Fig. 4 A illustrates that membrane hyperpolarization accompanies TG stimulation (from −61 ± 1.2 mV, n = 3; to −78 ± 2.0 mV, n = 11). The hyperpolarization that follows TG stimulation is produced by activation of KCa channels during the rise in [Ca2+]i, driving E m towards the K+ equilibrium potential of ∼−80 mV. Progesterone (50 μM; application bar, Fig. 4 A) not only reversed the hyperpolarization, but also resulted in depolarization to an average of −41 ± 4.3 mV (n = 13, P < 0.0001). These results suggest that progesterone may block both KV and KCa channels. If the reduction of [Ca2+]i by progesterone is secondary to decreased K+ current, then restoration of a K+ flux across the cell membrane should reverse this effect of progesterone. We tested this hypothesis by using the K+ ionophore, valinomycin, to hyperpolarize E m. In the experiment shown in Fig. 4 B, 30 μM progesterone reduced the plateau in [Ca2+]i that followed TG stimulation. The subsequent addition of valinomycin in the continued presence of progesterone resulted in an increase of [Ca2+]i. Thus, Ca2+-imaging and current-clamp experiments demonstrate that progesterone reduces the driving force for Ca2+ entry, an effect that can be reversed by reestablishing K+ efflux. These data suggest that progesterone may affect [Ca2+]i and gene expression by blocking K+ channels.
Figure 4.
Progesterone reduces the driving force for Ca2+ influx. (A) Current-clamp recordings using the perforated-patch technique were performed in B3Z cells to determine the effects of progesterone on E m. This panel presents a representative single-cell recording. The addition of 1 μM TG (arrow) hyperpolarized the cell from the resting potential (−60 mV) to near the K+ equilibrium potential (∼−80 mV), enhancing the driving force for Ca2+ influx. Subsequent application of 50 μM progesterone depolarized the cell to −26 mV, resulting in a reduction of the driving force for Ca2+ influx. For perforated-patch recordings, the tips of the pipettes were filled with the following solution (in mM): 120 K2SO4, 16 KCl, 5 MgSO4, 10 Hepes (pH 7.2). A stock solution of nystatin in DMSO (25 μg/ml) was prepared daily and subsequently diluted in the pipette solution to a final concentration of 100 μg/ml. After sonication, this solution was used for backfilling the pipettes as described previously (reference 31). (B) Measurements of average [Ca2+]i from B3Z cells (n = 37). After the addition of 1 μM TG (arrow), [Ca2+]i rose from a resting level of 70 ± 30 nM to a plateau of 1.3 ± 0.4 μM. Progesterone (30 μM) reduced the [Ca2+]i to approximately half of plateau concentration, an effect that was completely reversed by the addition of 2 μM valinomycin. Application bars, The additions of progesterone and valinomycin to the bath.
Progesterone Blocks Voltage-gated K+ and Ca2+-activated K+ Channels in T Cells.
Whole-cell recording with the patch-clamp technique permits direct measurement of several types of channel activity in T cells. A voltage step from a holding potential of −80 to +30 mV elicits rapidly activating KV currents, which reach a peak and then slowly inactivate. KV currents consist of two components, a predominant fraction encoded by homotetramers of the Shaker-related Kv1.3 gene and a much smaller fraction of unknown molecular identity (for a review, see reference 24). The two components can be readily distinguished by their biophysical and pharmacological properties. Kv1.3 channels are blocked by nanomolar concentrations of a scorpion toxin, charybdotoxin (CTX), and undergo cumulative (use-dependent) C-type inactivation during repetitive depolarization. The smaller, CTX-resistant current does not exhibit use-dependent inactivation. Application of progesterone rapidly and reversibly reduces KV currents in human or B3Z T cells, with an IC50 of 29 ± 2 μM for the peak K+ current (Fig. 5). Progesterone accelerates the decline of K+ current during a depolarizing pulse, and thus the block is more potent when evaluated at the end of a 200-ms pulse (Fig. 5 C, filled triangles). The apparent increased rate of channel inactivation (Fig. 5, A and B) suggests that progesterone may preferentially bind to and block the open or inactivated Kv1.3 channel. To determine if steady-state inactivation enhances the block by progesterone, we inactivated a significant fraction of Kv1.3 channels by decreasing the holding potential from −80 to −50 mV. As shown in Fig. 6 A, progesterone (30 μM) reduced the peak Kv1.3 currents more potently when the holding potential was −50 mV (70% block; see open circle in Fig. 5 C) than when the holding potential was −80 mV (45% block), demonstrating that channel inactivation effectively enhances the block of Kv1.3 currents by progesterone. During antigen-induced oscillations of [Ca2+]i, Kv1.3 channels would undergo repetitive cycles of activation, inactivation, and recovery as the membrane potential fluctuates. Activation cycles can result in frequency or use-dependent inhibition if the interval between depolarizations is less than the time required for full recovery from inactivation; normally, pulse intervals of >20 s allow full recovery. In the presence of progesterone, Kv1.3 currents steadily declined during repetitive pulsing, because channel inactivation recovers 10-fold more slowly, resulting in accumulation of channels in the inactivated state (Fig. 6, B and C).
Figure 5.


Progesterone blocks KV channels. Whole-cell currents were measured in human T cells (A) and B3Z cells (B) during 200-ms voltage pulses from a holding potential of −80 mV to +30 mV applied every 30 s. The pipette solution contained (in mM): 160 K+ aspartate, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 Hepes (pH 7.2). Currents are shown before and during bath application of 10, 30, or 100 μM progesterone. (C) Concentration dependence for the reduction of KV currents by progesterone. Peak current amplitudes in human T cells (▵) and B3Z cells (□) were analyzed and plotted against progesterone concentration. Data points for B3Z and human T cells overlie each other at most concentrations. For human T cells, the current at the end of the 200-ms pulse (▴) was also plotted. To determine the effect of depolarization on progesterone block, E m was held at −50 mV (○). Data were normalized to control currents measured in the absence of progesterone and presented as mean ± SD. The line represents a fit using a Hill equation with an IC50 of 29 ± 2 μM and n = 2.1 ± 0.4.
Figure 6.
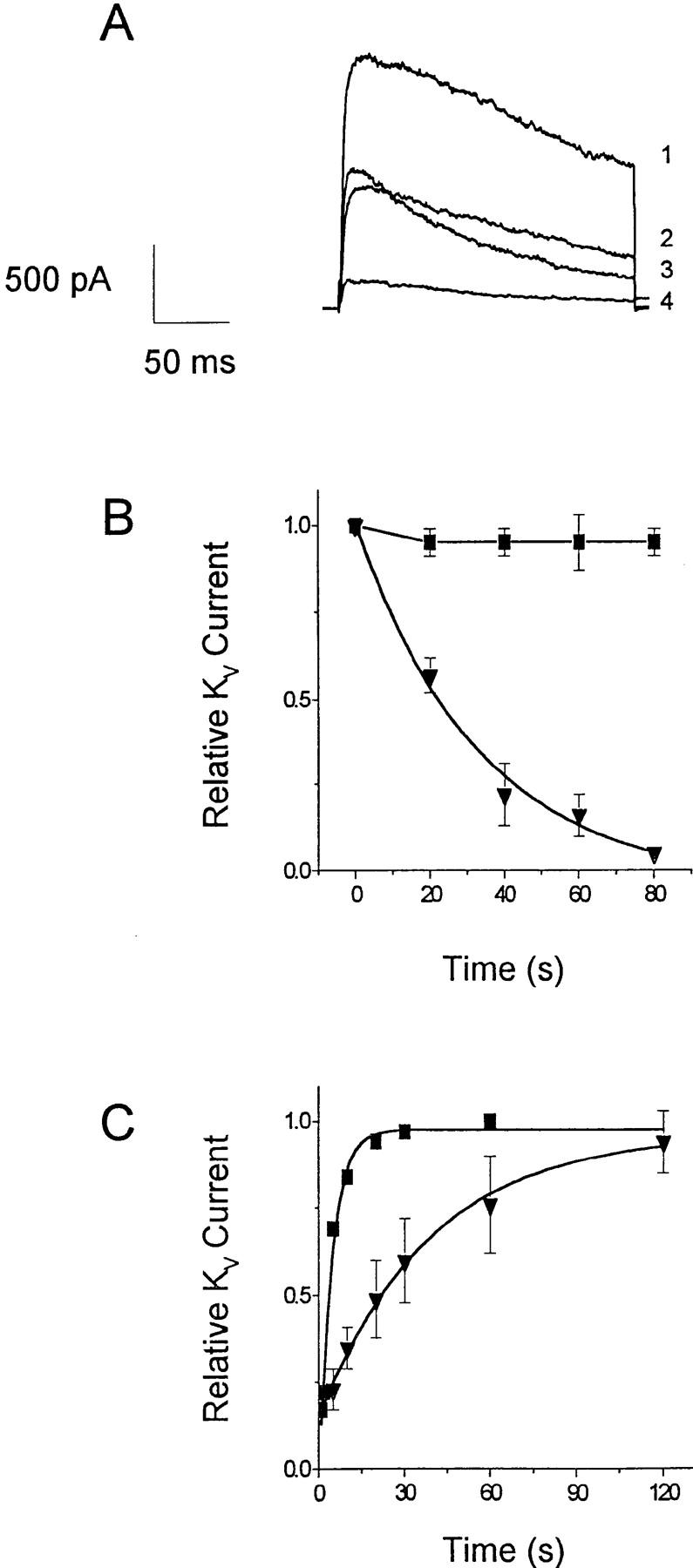
Inactivation enhances KV block by progesterone. (A) KV currents were elicited by steps to +30 mV from holding potential of −80 mV (1, 3) or −50 mV (2, 4) in the absence (1, 2) or presence (3, 4) of 30 μM progesterone. Solutions used are as in Fig. 5. (B) Use-dependent block of voltage-gated K+ currents. Current responses were elicited by repetitive voltage pulses from −80 to +30 mV separated by 20 s. Normalized peak current amplitudes in the absence (▪) or presence (▾) of 67 μM progesterone were plotted against time. (C) Time course of recovery from inactivation in a B3Z cell before and during application of 60 μM progesterone. Pairs of 200-ms pulses to +30 mV were applied from a holding potential of −80 mV. The graph shows the ratio of peak current during the second pulse to the peak current during the first pulse, plotted as a function of the time interval between the pulses. Data were fitted by single exponentials with time constants of 4 s for control (▪) and 39 s for progesterone (▾).
We also examined the effects of progesterone on the smaller component of KV current by selectively blocking the Kv1.3 component with 100 nM CTX. At this dose, we expect a residual Kv1.3 current of only 2% that in the absence of CTX. However, in 11 cells examined in this series of experiments and in previous work (21), 5–20% of the KV current remains in the presence of CTX. This residual CTX-resistant current does not undergo use-dependent inactivation during repetitive depolarizing pulses (Fig. 7 A). Fig. 7 B demonstrates that the larger, use-dependent, CTX-sensitive component of KV current is blocked either by progesterone or by RU 486. As shown in Fig. 7 C, most of the CTX-resistant current is also blocked by 50 μM progesterone, whereas RU 486 at the same concentration is less effective. We conclude that progesterone blocks both the predominant Kv1.3 component and the CTX- resistant component of KV current in T cells, and that RU 486 blocks primarily the Kv1.3 component.
Figure 7.

Progesterone blocks CTX-resistant KV channels. Pipette solution as in Fig. 5. (A) Current responses of the total KV current (•) and the CTX-resistant current (▪) to repetitive voltage pulses from −80 to +30 mV separated by 1 s. At this rate of pulsing, Kv1.3 channels undergo cumulative inactivation, as shown by normalized peak current amplitudes in the absence of CTX (▪). With 100 nM CTX present (•), the remaining current does not decline during repetitive pulsing. (B) Progesterone or RU 486 (50 μM) blocks the KV component. (C) Progesterone blocks the CTX-resistant component of KV current more than RU 486. 100 nM CTX was preapplied in Ringer in order to block Kv1.3 channels.
KCa channels hyperpolarize the membrane potential of T cells during the [Ca2+]i signal, effectively counteracting the depolarizing effects of Ca2+ influx. KCa channels are voltage-independent and highly sensitive to a rise in [Ca2+]i, with half-activation at ∼400 nM and a steep [Ca2+]i dependence, suggesting that at least four Ca2+ ions must bind to open the channel (for a review, see reference 24). Based upon biophysical characterization and its expression in T cells, it is likely that the KCa channel in T cells is encoded by the gene hKCa4 (39). Intracellular dialysis of B3Z or activated human T cells with solutions containing 1 μM Ca2+ activated a large K+ current that was evident in voltage ramps (Fig. 8 A). Progesterone blocked KCa channels with an IC50 value of 113 ± 9 μM (Fig. 8 B). Thus, progesterone blocks the KV currents more potently than the KCa currents in T cells. In contrast, the progesterone antagonist RU 486 was consistently more potent than progesterone in blocking KCa current (Fig. 8 C). These experiments demonstrate some degree of selectivity of the two components of KV current and the KCa current for progesterone and RU 486.
Figure 8.


Concentration-dependent inhibition of KCa current in B3Z cells by progesterone. (A) KCa currents were activated by dialyzing the cell with a solution containing (in mM) 140 K+ aspartate, 2 MgCl2, 7.8 CaCl2, 10 EGTA, 5 Hepes (pH 7.2). The nominal free Ca2+ concentration of this solution was 1 μM, assuming a dissociation constant for EGTA and Ca2+ of 10−7 at pH 7.2. Ca2+-activated K+ currents were evoked by voltage ramps of 200-ms duration from −120 to +50 mV every 30 s. Application of progesterone at different concentrations (indicated at the right of each trace) inhibited the KCa current. (B) Concentration–response curve for progesterone block of KCa currents (•). The slope conductance between −100 and −60 mV was used as a measure of the KCa conductance to avoid contamination by KV currents. Data were normalized to the conductance measured in the absence of progesterone and presented as mean ± SD. The line represents the fit to a Hill equation with IC50 = 113 μM and n = 1.2. Block of KCa channels by 60 μM RU 486 (□) is shown for comparison. (C) Comparison of progesterone and RU 486. Slope conductance values at −80 mV illustrate activation and block of the KCa current by RU 486 and progesterone, each applied at 60 μM.
Selectivity of Progesterone for KV Channels: Progesterone Does Not Block CRAC or Chloride Channels in T Cells.
The results from patch-clamp experiments suggested that progesterone, RU 486, and perhaps other steroid hormones might block several K+ channel types, albeit with rather low affinity. Therefore, we screened a panel of steroids on the KV and KCa channels in T lymphocytes. Most of the compounds tested either had no effect or were less effective than progesterone in blocking KV or KCa currents (Table 1). In addition, we screened several channel types, including both cloned and native channels expressed in a variety of cell lines, for block by progesterone, as summarized in Table 2. Progesterone blocks several Kv1 family members, including Kv1.3 expressed in lymphocytes as the predominant KV current, as well as Kv3.1 expressed in the brain and in certain subsets of mouse thymocytes. In contrast, progesterone had very little effect on a cloned voltage-gated Na+ channel found in skeletal muscle, or on a strongly inward rectifying K+ channel found in RBL cells. We conclude that progesterone is a broad spectrum, low-affinity K+ channel blocker.
Table 1.
Percent Inhibition of KV and KCa Channels by Steroids (60 μM)
| Progesterone | RU 486 | Estradiol | Testosterone | Cortisol | DHEA | OHP | Aldosterone | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KV | 77 ± 8 | 61 ± 15 | 24 ± 7 | 17 ± 12 | 13 ± 4 | 10 ± 6 | 12 ± 6 | 12 ± 13 | ||||||||
| KCa | 34 ± 11 | 55 ± 12 | 8 ± 10 | 4 ± 7 | 0.0 ± 0.1 | 0.4 ± 0.7 | 2 ± 4 | 6 ± 7 |
Currents through KV channels in human T cells were evoked by depolarizing pulses to +30 mV, as in Fig. 5. KCa channels in activated human T cells were opened by 1 μM [Ca2+]i, with currents measured during voltage-ramp stimuli as in Fig. 8. Data are presented as mean ± SD, with at least three separate experiments for each compound tested at 60 μM, and represent the percent inhibition of peak KV currents or slope conductances of KCa currents near −80 mV. DHEA, Dehydroepiandrosterone; OHP, 17α-hydroxy-4-pregnene-3,20-dione.
Table 2.
Percent Inhibition of Various Channel Types by Progesterone
| Cloned channels | ||||||||||||||
| Gene | Kv1.1 | Kv1.2 | Kv1.3 | Kv1.4 | Kv1.5 | Kv3.1 | hSKM1 | |||||||
| Cell type | L929 | NGK1 | L929 | LTK | MEL | L929 | HEK | |||||||
| 31 ± 6 | 45 ± 10 | 44 ± 9 | 23 ± 8 | 25 ± 2 | 53 ± 3 | 11 ± 1 | ||||||||
| Native channels | ||||||||||||||
| Channel | KV | KV | KV (CTX) | KCa | CRAC | Cl− | KIR | |||||||
| Cell type | Human T | B3Z | Human T | Human T, act. | Jurkat T | Jurkat T | RBL | |||||||
| 50 ± 6 | 40 ± 10 | 70 ± 20 | 34 ± 11 | 0 | 0 | 4 ± 12 |
The panel of cloned KV channels was provided by Dr. K. George Chandy (Department of Physiology and Biophysics, University of California, Irvine), as described (reference 55). The voltage-gated Na+ channel clone hSKM1, originally derived from skeletal muscle, was made by Dr. Frank Lehmann-Horn (Department of General Physiology, University of Ulm, Germany). Peak Na+ currents were evaluated at +10 mV. KV currents were evoked by depolarizing pulses, as in Fig. 5. CTX-resistant KV channels [KV (CTX)] were studied in the presence of 100 nM CTX, as in Fig. 7. KCa channels were activated by 1 μM [Ca2+]i, as in Fig. 8. CRAC and Cl− channels were activated by Ca2+-store depletion or by cell swelling, respectively, as in Fig. 9. Inwardly rectifying K+ (KIR) currents were evaluated during voltage-ramp stimuli. In each case, progesterone was tested at 30 μM, except for KV (CTX), KCa, and Cl− channels tested at 50 μM. Data are presented as mean ± SD, with at least three separate experiments for each channel type.
In further experiments on Jurkat T lymphocytes, we evaluated effects of progesterone on CRAC and Cl− channels to determine if modulation of these channel types might contribute to the inhibition of Ca2+ signaling. During whole-cell patch-clamp recordings, intracellular dialysis with heavily buffered low-Ca2+ solutions passively depleted the intracellular Ca2+ stores and activated CRAC channels (Fig. 9 A). After maximal activation, 30 μM progesterone (Fig. 9 B, application bar) had no effect on the amplitude or the current-voltage characteristics (n = 6 cells). This experiment rules out direct CRAC channel block as a possible contributor to the inhibition of Ca2+ signaling by progesterone; instead, it appears that progesterone blocks Ca2+ signaling by inhibiting K+ channels, indirectly reducing the driving force for Ca2+ entry. Cl− channels have also been implicated in lymphocyte signaling mechanisms by helping to maintain E m during T cell activation (20, 21). However, superfusion of B3Z or Jurkat cells with 50 μM progesterone did not affect the amplitude or the current-voltage characteristics of Cl− currents induced by cell swelling (Fig. 9, C and D).
Figure 9.

Progesterone does not affect CRAC or Cl− channels. (A) Whole-cell recordings of Ca2+ current through CRAC channels measured during voltage ramps from −100 to +40 mV for a duration of 200 ms. The pipette solution contained (in mM) 128 Cs+ aspartate, 12 BAPTA, 0.9 CaCl2, 3.16 MgCl2, Hepes (pH 7.2). The external solution had the following composition (in mM): 150 NaMeSO3, 20 CaCl2, 10 glucose, 10 Hepes (pH 7.4). (B) Time-dependent changes in the amplitude of the current measured at −80 mV. Arrows, Representative currents recorded before store depletion (1), and after maximal induction activation of CRAC channels, while superfusing cells with control solution (2) or with an external solution containing 30 μM progesterone (3). (C) Whole-cell recordings of swelling-activated Cl− current (ICl) measured during voltage ramps from −120 to +40 mV for a duration of 200 ms. The pipette solution contained (in mM) 140 Cs+ aspartate, 2 MgCl2, 4 MgATP, 1 CaCl2, 10 EGTA, 10 Hepes (pH 7.2). For the induction of ICl, the pipette solution was made hypertonic (390–400 mosmol) by the addition of 100 mM glucose. Currents are shown before the onset of cell swelling (1), and after maximal induction of ICl, while superfusing cells with control solution (2) or with an external solution containing 50 μM progesterone (3). (D) Time-dependent changes in the slope conductance measured at the reversal potential for Cl−.
Discussion
In this report, we demonstrate by patch-clamp measurement that progesterone directly blocks KV and KCa channels, but not Ca2+ or Cl− channels in T lymphocytes. Furthermore, we show that K+ channel blockade is associated with membrane depolarization, inhibition of Ca2+ signaling, and a reduction of NF-AT–driven gene expression. Since NF-AT links activation of the TCR to IL-2 production, interruption of these signals would inhibit production of the major proliferative cytokine for T cells. We propose that K+ channel blockade provides a mechanism contributing to the immunosuppressive effects of progesterone.
The rapid onset and reversibility of KV channel block by progesterone is incompatible with changes in mRNA or protein synthesis, suggesting that these effects are not mediated by the classical steroid receptor pathway (40). The progesterone antagonist RU 486 is nearly as potent as progesterone in blocking both KV channels and gene expression, also implicating a nongenomic action of progesterone. Furthermore, channel inactivation enhances block by progesterone, suggesting that the state of the channel modulates the affinity of progesterone. In T cells, the direct block of KV channels by dihydroquinolines and benzhydryl piperidines is also enhanced by inactivation (41, 42). In addition, progesterone blocks Kv1.3 channels exogenously expressed in a cell line (Table 2). Progesterone also blocks other KV and KCa channels with low affinity. These data suggest that progesterone blocks K+ channels directly, rather than acting via the classical nuclear progesterone receptor pathway.
Channel blockade by progesterone is not without precedent. Previous studies demonstrated that progesterone blocks voltage-gated Ca2+ channels in smooth muscle cells and a variety of K+ channels in MDCK cells and hepatocytes (43–45). Several transmitter-activated channels are also suppressed by progesterone in the micromolar concentration range (46–49). In contrast to its effects on somatic cells, progesterone activates Ca2+ influx in sperm (50, 51). We found no evidence for progesterone-induced Ca2+ influx in T cells.
Our data provide the first evidence that an endogenous hormone may act as an immunosuppressant by blocking K+ channels. Inhibition of K+ channels has been shown to reduce IL-2 production and T cell activation in vitro (22, 23, 52). Moreover, recent studies demonstrated that the peptide scorpion toxin margatoxin, a specific blocker of Kv1.3 channels, inhibits delayed-type hypersensitivity reaction and reduces response to allogeneic challenge in vivo (25). The depolarization and reduction of the driving force for Ca2+ entry resulting from K+ channel inhibition are sufficient to account for the reduction of Ca2+ signals and NF-AT–driven gene expression. CRAC channels are inwardly rectifying, and a modest depolarization can reduce Ca2+ entry significantly, reducing the rise in [Ca2+]i below the threshold for gene expression (for a review, see reference 24). At high concentrations, progesterone reduces Ca2+ signaling and gene expression almost to control levels, below a plateau level seen with 100 nM CTX (21, 53, and data not shown). Progesterone, although acting with low affinity, may reduce Ca2+ signaling and gene expression to a greater extent than CTX because progesterone also inhibits CTX-resistant KV channels. The block of K+ channels by progesterone or RU 486 can also account for previous results showing that progesterone or RU 486 inhibits activation of human T cells in vitro (8, 9), as well as the reduction of the number of CD3+ cells in the placenta compared with maternal blood (11, 54).
During pregnancy, immunoregulatory mechanisms must operate locally at the placental interface and be readily reversible to preserve the systemic immune competence of the mother. Several mechanisms involving progesterone may contribute to fetal–maternal protection, including altered expression of MHC class I proteins in fetal tissue, altered T cell subsets, or elaboration of immunosuppressive factors (2). Biochemical measurements have estimated progesterone concentrations to be 20 μM within the placenta (34, 35); concentrations in the vicinity of trophoblasts producing progesterone must be even higher. Average progesterone levels found in the placenta would be sufficient to block lymphocyte K+ channels and thereby mediate a highly localized and reversible immunosuppression without compromising the maternal immune system. The affinity of progesterone for K+ channels ensures that this mechanism would only be effective in the region of potential contact between allogeneic cells, where progesterone is present at high concentrations.
Acknowledgments
We thank Dr. Patricia Schmidt for helpful discussions at the onset of this project, Dr. K. George Chandy for providing clones and a panel of stable cell lines, and Dr. Lu Forrest for tissue culture support.
This work is supported by National Institutes of Health grants GM41514 and NS14609 (to M.D. Cahalan), a Schroedinger Stipendium (to H.H. Kerschbaum), and a fellowship of the Alexander von Humboldt Foundation (to C. Eder).
Abbreviations used in this paper
- [Ca2+]i
intracellular free calcium concentration
- CRAC
Ca2+ release–activated Ca2+
- CTX
charybdotoxin
- Em
membrane potential
- IP3
inositol 1,4,5-trisphosphate
- KCa
Ca2+-activated K+
- KV
voltage-gated K+
- NF-AT
nuclear factor of activated T cells
- TG
thapsigargin
Footnotes
H.H. Kerschbaum's present address is Department of Physiology, Institute for Zoology, University of Salzburg, 5020 Salzburg, Austria. Claudia Eder's current address is Department of Neurophysiology, Institute of Physiology Humboldt University, D-10019 Berlin, Germany. P.A. Negulescu's present address is Aurora Biosciences, 11010 Torreyana Rd., San Diego, CA 92121.
G.R. Ehring and H.H. Kerschbaum contributed equally to this work.
References
- 1.Stites DP, Siiteri PK. Steroids as immunosuppressants in pregnancy. Immunol Rev. 1983;75:117–138. doi: 10.1111/j.1600-065x.1983.tb01093.x. [DOI] [PubMed] [Google Scholar]
- 2.Pepe GJ, Albrecht ED. Actions of placental and fetal adrenal steroid hormones in primate pregnancy. Endocr Rev. 1995;16:608–648. doi: 10.1210/edrv-16-5-608. [DOI] [PubMed] [Google Scholar]
- 3.Beer AE, Sio JO. Placenta as an immunological barrier. Biol Reprod. 1982;26:15–27. doi: 10.1095/biolreprod26.1.15. [DOI] [PubMed] [Google Scholar]
- 4.Gurka G, Rocklin RE. Reproductive immunology. JAMA (J Am Med Assoc) 1987;258:2983–2987. [PubMed] [Google Scholar]
- 5.Hunt JS. Immunologically relevant cells in the uterus. Biol Reprod. 1994;50:461–466. doi: 10.1095/biolreprod50.3.461. [DOI] [PubMed] [Google Scholar]
- 6.Siiteri PK, Febres F, Clemens LE, Chang RJ, Gondos B, Stites D. Progesterone and maintenance of pregnancy: is progesterone nature's immunosuppressant? . Ann NY Acad Sci. 1977;286:384–397. doi: 10.1111/j.1749-6632.1977.tb29431.x. [DOI] [PubMed] [Google Scholar]
- 7.Siiteri PK, Stites DP. Immunologic and endocrine interrelationships in pregnancy. Biol Reprod. 1982;26:1–14. doi: 10.1095/biolreprod26.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Clemens LE, Siiteri PK, Stites DP. Mechanism of immunosuppression of progesterone on maternal lymphocyte activation during pregnancy. J Immunol. 1979;122:1978–1985. [PubMed] [Google Scholar]
- 9.Van Voorhis BJ, Anderson DJ, Hill JA. The effects of RU 486 on immune function and steroid-induced immunosuppression in vitro. . J Clin Endocrinol Metab. 1989;69:1195–1199. doi: 10.1210/jcem-69-6-1195. [DOI] [PubMed] [Google Scholar]
- 10.Monterroso VH, Hansen PJ. Regulation of bovine and ovine lymphocyte proliferation by progesterone: modulation by steroid receptor antagonists and physiological status. Acta Endocrinol. 1993;129:532–535. doi: 10.1530/acta.0.1290532. [DOI] [PubMed] [Google Scholar]
- 11.Pavia C, Siiteri PK, Perlman JD, Stites DP. Suppression of murine allogeneic cell interactions by sex hormones. J Reprod Immunol. 1979;1:33–38. doi: 10.1016/0165-0378(79)90027-5. [DOI] [PubMed] [Google Scholar]
- 12.Crabtree GR, Clipstone NA. Signal transmission between the plasma membrane and nucleus of T lymphocytes. Annu Rev Biochem. 1994;63:1045–1083. doi: 10.1146/annurev.bi.63.070194.005145. [DOI] [PubMed] [Google Scholar]
- 13.Negulescu PA, Shastri N, Cahalan MD. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc Natl Acad Sci USA. 1994;91:2873–2877. doi: 10.1073/pnas.91.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis RS, Cahalan MD. Potassium and calcium channels in lymphocytes. Annu Rev Immunol. 1995;13:623–653. doi: 10.1146/annurev.iy.13.040195.003203. [DOI] [PubMed] [Google Scholar]
- 15.Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+signals and immunosuppression. Nature. 1996;383:837–840. doi: 10.1038/383837a0. [DOI] [PubMed] [Google Scholar]
- 16.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 17.Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+current in human leukemic T cells. Cell Regul. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+stores. Proc Natl Acad Sci USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Premack BA, McDonald TV, Gardner P. Activation of Ca2+ current in Jurkat T cells following the depletion of Ca2+ stores by microsomal Ca2+-ATPase inhibitors. J Immunol. 1994;152:5226–5240. [PubMed] [Google Scholar]
- 20.Phipps DJ, Branch DR, Schlichter LC. Chloride-channel block inhibits T lymphocyte activation and signalling. Cell Signalling. 1996;8:141–149. doi: 10.1016/0898-6568(95)02039-x. [DOI] [PubMed] [Google Scholar]
- 21.Kerschbaum HH, Negulescu PA, Cahalan MD. Ion channels, Ca2+signaling, and reporter gene expression in antigen-specific mouse T cells. J Immunol. 1997;159:1628–1638. [PubMed] [Google Scholar]
- 22.Chandy KG, DeCoursey TE, Cahalan MD, McLaughlin C, Gupta S. Voltage-gated potassium channels are required for human T lymphocyte activation. J Exp Med. 1984;160:369–385. doi: 10.1084/jem.160.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin CS, Boltz RC, Blake JT, Nguyen M, Talento A, Fischer PA, Springer MS, Sigal NH, Slaughter RS, Garcia ML. Voltage-gated potassium channels regulate calcium-dependent pathways involved in human T lymphocyte activation. J Exp Med. 1993;177:637–645. doi: 10.1084/jem.177.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cahalan MD, Chandy KG. Ion channels in the immune system as targets for immunosuppression. Curr Opin Biotechnol. 1997;8:749–756. doi: 10.1016/s0958-1669(97)80130-9. [DOI] [PubMed] [Google Scholar]
- 25.Koo GC, Blake JT, Talento A, Nguyen M, Lin S, Sirotina A, Shah K, Mulvany K, Hora DJ, Cunningham P, et al. Blockade of the voltage-gated potassium channel Kv1.3 inhibits immune responses in vivo. . J Immunol. 1997;158:5120–5128. [PubMed] [Google Scholar]
- 26.Karttunen J, Shastri N. Measurement of ligand-induced activation in single viable T cells using the lacZ reporter gene element of the human interleukin 2 enhancer. Proc Natl Acad Sci USA. 1991;88:3972–3976. doi: 10.1073/pnas.88.9.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shastri N, Gonzalez F. Endogenous generation and presentation of the ovalbumin peptide/Kbcomplex to T cells. J Immunol. 1993;150:2724–2736. [PubMed] [Google Scholar]
- 28.Cahalan MD, Chandy KG, DeCoursey TE, Gupta S. A voltage-gated potassium channel in human T lymphocytes. J Physiol (Lond) 1985;358:197–237. doi: 10.1113/jphysiol.1985.sp015548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fiering SN, Roederer M, Nolan GP, Micklem DR, Parks DR, Herzenberg LA. Improved FACS-Gal: flow cytometric analysis and sorting of viable eukaryotic cells expressing reporter gene constructs. Cytometry. 1991;12:291–301. doi: 10.1002/cyto.990120402. [DOI] [PubMed] [Google Scholar]
- 30.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch Eur J Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 31.Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 33.Thastrup O, Dawson AP, Scharff O, Foder B, Cullen PJ, Drobak BK, Bjerrum PJ, Christensen SB, Hanley MR. Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions. 1989;27:17–23. doi: 10.1007/BF02222186. [DOI] [PubMed] [Google Scholar]
- 34.Runnebaum B, Runnebaum H, Stober I, Zander J. Progesterone 20 alpha-dihydroprogesterone and 20 beta-dihydroprogesterone levels in different compartments from the human foeto-placental unit. Acta Endocrinol. 1975;80:558–568. doi: 10.1530/acta.0.0800558. [DOI] [PubMed] [Google Scholar]
- 35.Runnebaum B, Stober I, Zander J. Progesterone, 20 alpha-dihydroprogesterone and 20 beta-dihydroprogesterone in mother and child at birth. Acta Endocrinol. 1975;80:569–576. [PubMed] [Google Scholar]
- 36.Gouy H, Cefai D, Christensen SB, Debre P, Bismuth G. Ca2+ influx in human T lymphocytes is induced independently of inositol phosphate production by mobilization of intracellular Ca2+ stores. A study with the Ca2+endoplasmic reticulum-ATPase inhibitor thapsigargin. Eur J Immunol. 1990;20:2269–2275. doi: 10.1002/eji.1830201016. [DOI] [PubMed] [Google Scholar]
- 37.Mason MJ, Mahaut-Smith MP, Grinstein S. The role of intracellular Ca2+ in the regulation of the plasma membrane Ca2+permeability of unstimulated rat lymphocytes. J Biol Chem. 1991;266:10872–10879. [PubMed] [Google Scholar]
- 38.Verheugen JA, Vijverberg HP, Oortgiesen M, Cahalan MD. Voltage-gated and Ca2+-activated K+channels in intact human T lymphocytes. Noninvasive measurements of membrane currents, membrane potential, and intracellular calcium. J Gen Physiol. 1995;105:765–794. doi: 10.1085/jgp.105.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Logsdon NJ, Kang J, Togo JA, Christian EP, Aiyar J. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J Biol Chem. 1997;272:32723–32726. doi: 10.1074/jbc.272.52.32723. [DOI] [PubMed] [Google Scholar]
- 40.Revelli A, Massobrio M, Tesarik J. Nongenomic actions of steroid hormones in reproductive tissues. Endocr Rev. 1998;19:3–17. doi: 10.1210/edrv.19.1.0322. [DOI] [PubMed] [Google Scholar]
- 41.Michne WF, Guiles JW, Treasurywala AM, Castonguay LA, Weigelt CA, Oconnor B, Volberg WA, Grant AM, Chadwick CC, Krafte DS. Novel inhibitors of potassium ion channels on human T lymphocytes. J Med Chem. 1995;38:1877–1883. doi: 10.1021/jm00011a007. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen A, Kath JC, Hanson DC, Biggers MS, Canniff PC, Donovan CB, Mather RJ, Bruns MJ, Rauer H, Aiyar J, et al. Novel nonpeptide agents potently block the C-type inactivated conformation of Kv1.3 and suppress T cell activation. Mol Pharmacol. 1996;50:1672–1679. [PubMed] [Google Scholar]
- 43.Bielefeldt K, Waite L, Abboud FM, Conklin JL. Nongenomic effects of progesterone on human intestinal smooth muscle cells. Am J Physiol. 1996;271:G370–G376. doi: 10.1152/ajpgi.1996.271.2.G370. [DOI] [PubMed] [Google Scholar]
- 44.Waldegger S, Beisse F, Apfel H, Breit S, Kolb HA, Haussinger D, Lang F. Electrophysiological effects of progesterone on hepatocytes. Biochim Biophys Acta. 1995;1266:186–190. doi: 10.1016/0167-4889(94)00236-8. [DOI] [PubMed] [Google Scholar]
- 45.Breit S, Kolb H, Apfel H, Haberland C, Schmitt M, Heaussinger D, Graf J, Lang F. Regulation of ion channels in rat hepatocytes. Pflügers Arch Eur J Physiol. 1998;435:203–210. doi: 10.1007/s004240050502. [DOI] [PubMed] [Google Scholar]
- 46.Wu FS, Gibbs TT, Farb DH. Inverse modulation of gamma-aminobutyric acid- and glycine-induced currents by progesterone. Mol Pharmacol. 1990;37:597–602. [PubMed] [Google Scholar]
- 47.Bertrand D, Valera S, Bertrand S, Ballivet M, Rungger D. Steroids inhibit nicotinic acetylcholine receptors. Neuroreport. 1991;2:277–280. doi: 10.1097/00001756-199105000-00016. [DOI] [PubMed] [Google Scholar]
- 48.Valera S, Ballivet M, Bertrand D. Progesterone modulates a neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 1992;89:9949–9953. doi: 10.1073/pnas.89.20.9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergeron R, de Montigny C, Debonnel G. Potentiation of neuronal NMDA response induced by dehydroepiandrosterone and its suppression by progesterone: effects mediated via sigma receptors. J Neurosci. 1996;16:1193–1202. doi: 10.1523/JNEUROSCI.16-03-01193.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foresta C, Rossato M, Di Virgilio F. Ion fluxes through the progesterone-activated channel of the sperm plasma membrane. Biochem J. 1993;294:279–283. doi: 10.1042/bj2940279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blackmore PF. Rapid non-genomic actions of progesterone stimulate Ca2+influx and the acrosome reaction in human sperm. Cell Signalling. 1993;5:531–538. doi: 10.1016/0898-6568(93)90048-q. [DOI] [PubMed] [Google Scholar]
- 52.Price M, Lee SC, Deutsch C. Charybdotoxin inhibits proliferation and interleukin 2 production in human peripheral blood lymphocytes. Proc Natl Acad Sci USA. 1989;86:10171–10175. doi: 10.1073/pnas.86.24.10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hess S, Oortgiesen M, Cahalan MD. Calcium oscillations in human T and natural killer cells depend upon membrane potential and calcium influx. J Immunol. 1993;150:2620–2633. [PubMed] [Google Scholar]
- 54.Roussev RG, Higgins NG, McIntyre JA. Phenotypic characterization of normal human placental mononuclear cells. J Reprod Immunol. 1993;25:15–29. doi: 10.1016/0165-0378(93)90039-k. [DOI] [PubMed] [Google Scholar]
- 55.Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]