

Abstract
The molecular mechanisms that contribute to an eosinophil-rich airway inflammation in asthma are unclear. A predominantly T helper 2 (Th2)-type cell response has been documented in allergic asthma. Here we show that mice deficient in the p50 subunit of nuclear factor (NF)- κB are incapable of mounting eosinophilic airway inflammation compared with wild-type mice. This deficiency was not due to a block in T cell priming or proliferation in the p50−/− mice, nor was it due to a defect in the expression of the cell adhesion molecules VCAM-1 and ICAM-1 that are required for the extravasation of eosinophils into the airways. The major defects in the p50−/− mice were the lack of production of the Th2 cytokine interleukin 5 and the chemokine eotaxin, which are crucial for proliferation and for differentiation and recruitment, respectively, of eosinophils into the asthmatic airway. Additionally, the p50−/− mice were deficient in the production of the chemokines macrophage inflammatory protein (MIP)-1α and MIP-1β that have been implicated in T cell recruitment to sites of inflammation. These results demonstrate a crucial role for NF-κB in vivo in the expression of important molecules that have been implicated in the pathogenesis of asthma.
Keywords: allergic inflammation, eosinophils, nuclear factor κB, interleukin 5, eotaxin
Asthma is a chronic inflammatory disease of the lower airways that causes airway hyperresponsiveness to a wide variety of specific and non-specific stimuli. Both the prevalence and severity of asthma is increasing at an alarming rate in developed countries. Bronchoalveolar lavage (BAL)1 and biopsy of patients with mild to moderate asthma has provided impressive evidence for complex airway inflammation in asthma (1–3). The most striking and consistent pathophysiology, at least in atopic asthma, is damage to the bronchial epithelium and infiltration of the bronchial wall and lumen by eosinophils (2, 4–10). Eosinophils are conspicuous in the airways of the majority of asthmatics, from the mildest to the most severe, and the number of eosinophils in the peripheral blood and airways correlates with hyperresponsiveness and clinical severity (2, 4–11). Eosinophils can generate an array of potent proinflammatory mediators, including the cytotoxic proteins preformed and stored in the eosinophil granules such as major basic protein and eosinophil cationic protein, eicosanoid compounds including leukotriene (LT)C4, LTD4, platelet activating factor, and prostaglandins (PG)E1 and E2, and finally proinflammatory cytokines.
It is now generally accepted that the eosinophil-rich inflammatory response that mediates airway tissue damage in atopic individuals arises from aberrant T cell–mediated immune responses to a range of inhaled allergens that are seemingly ignored by nonresponsive “normal” individuals (3, 8, 12–14). Cytokine expression profiles of allergen-specific T cell clones isolated from atopics are skewed toward a Th2-like profile compared with a Th1-like profile in nonatopics (for review see references 15, 16). The cytokine IL-5, produced by Th2 cells, is now known to be the central mediator in the regulation of eosinophilic inflammation in asthma (1, 17–19). IL-5 is intimately associated with several features of eosinophil biology; it not only regulates the proliferation, differentiation, and activation of eosinophils (20–22), it also provides an essential signal for the rapid mobilization of eosinophils from the bone marrow and cooperates with the chemokine eotaxin in the homing of the eosinophils to sites of allergic inflammation (23). Due to the proposed central role of eosinophils in asthma and allergic disease, there is considerable interest in the molecular mechanisms that mediate the expression of genes that promote eosinophilia such as IL-5 and eotaxin.
The transcription factor nuclear factor (NF)-κB plays an important role in regulating many inflammatory processes (24–29). NF-κB proteins control the expression of multiple genes involved in immune responses such as those encoding proinflammatory cytokines and chemokines (24– 26, 28, 29). The NF-κB proteins belong to the Rel family of proteins. So far, five members of the NF-κB family have been described: c-Rel; NF-κB1 (p50); NF-κB2 (p52); RelA (p65); and RelB (24–26). The classic NF-κB complex is a heterodimer of two polypeptide subunits, p50 and RelA (p65). Most members of the NF-κB family can form homo- or heterodimers (except for RelB, which only forms heterodimers with p50 or p52) that bind slightly different κB motifs (24–26). (p50)2 has been shown to act as a dominant repressor of transcription from specific κB sites (30–32). We have recently shown that (p50)2 behaves as a selective repressor of RANTES (regulated on activation, normal T cell expressed and secreted) gene expression (33). It appears that in vivo (p50)2 functions as an activator in certain tissues such as the thymus but acts as a repressor in others present in peripheral lymphoid organs and in macrophages, such as T cells (34).
Studies of gene knockout animals indicate that the different Rel family proteins are not functionally redundant. For example, knockout of the RelA (p65) gene in mice causes embryonic lethality apparently due to extensive apoptosis in the liver (35). On the other hand, p50−/− mice develop normally and express normal mature B and T cell subsets (36). However, the B cells in these mice are severely limited in their functions. They do not proliferate in response to bacterial LPS (37), and furthermore, p50−/− B cells are defective in IgG3, IgE, and IgA class switching and display markedly reduced levels of germline CHγ3 and CHε RNA but normal levels of germline CHγ1 and CHα RNA (37).
Although gene knockout studies have eliminated notions of simple redundancy of the different NF-κB proteins, little is known about the specific role of these proteins in the pathogenesis of different inflammation-associated disease states in vivo. For example, whether the characteristic eosinophilic inflammation observed in asthma is an NF-κB– dependent phenomenon was not known before this study. We used p50−/− mice in a murine model of airway inflammation to investigate the role of NF-κB in the pathogenesis of eosinophilic airway inflammation, which, as we and others have shown, is predominantly a CD4+ T cell–dominated process (18, 38–41). We show that the p50−/− mice are devoid of eosinophilic airway inflammation upon antigenic stimulation. The lack of inflammation was not due to insufficient CD4+ T cell priming. Also, IL-2 production, which is believed to be an NF-κB–regulated process, was unimpaired in the p50−/− mice and the cells proliferated as efficiently as the wild-type cells in response to antigen. The absence of inflammation also was not due to a lack of expression of vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1, which have been shown to be important for eosinophil homing. A major deficiency in the p50−/− mice was the absence of IL-5 production by the CD4+ T cells upon antigenic stimulation. Another difference between the two types of mice was the lack of expression of the eosinophilic chemoattractant eotaxin at both the RNA and the protein level in the lungs of p50−/− mice. The expression of the chemokine genes macrophage inflammatory protein (MIP)-1α and MIP-1β, which have been implicated in T cell chemotaxis, was also deficient in the p50−/− mice. Taken together, this is the first report that demonstrates an essential role for NF-κB in the regulation of important molecules that regulate eosinophilic inflammation in vivo. Furthermore, our studies also demonstrate a dissociation between the expression of the IL-4 and IL-5 genes, both of which are expressed by Th2-type cells.
Materials and Methods
Mice.
The p50−/− and wild-type mice were on a C57BL6/ 129 background and the generation of the p50−/− mice has been described by Sha et al. (36). Mice between 6 and 8 wk of age were purchased from The Jackson Laboratory (Bar Harbor, ME). The mice were housed in a pathogen-free facility at Yale University School of Medicine.
Sensitization and Challenge of Mice.
Mice were sensitized and challenged essentially as described by Kung et al. (42). Mice were sensitized with 10 μg of OVA (Sigma Chemical Co., St. Louis, MO) and 1 mg of alum (Resorptar; Intergen Co., New York, NY) intraperitoneally on days 0 and 5. Sham-immunized mice received alum alone. On day 12, mice were challenged by exposure to an aerosol of 1% OVA in PBS twice for 1 h each at an interval of 4 h. Inhalation was carried out in a plastic chamber (27 × 20 × 10 cm). The inhalation chamber that we use has an attachment to allow entry of the aerosol from an ultrasonic nebulizer (1–5 μM particles by manufacturer's specifications, Model no. NE-U07; Omron, Vernon Hills, IL). The other end of the box has two small holes for the maintenance of continuous airflow.
BAL.
BAL was performed at 24 h after aerosol challenge. Mice were anesthetized and the lungs and heart were surgically exposed. The trachea were cannulated and the lungs were lavaged twice with 1-ml aliquots of PBS. The live cells (excluded trypan blue) recovered were counted in a hemocytometer. Cytospin preparations of BAL cells were stained with Diff-Quik (Baxter HealthCare Corp., Miami, FL) and cell differentials were enumerated based on morphology and staining profile.
Lung Histology.
Lungs were prepared for histology by perfusing the animal through the right ventricle with PBS to remove all blood. Lungs were then inflated to constant pressure with 1.0 ml of fixative instilled through a tracheostomy tube as described previously (43). For staining with hematoxylin and eosin (H&E), lungs were fixed in 10% formalin and embedded in paraffin. 5-μM sections were mounted on slides and stained with H&E according to established procedures (41, 43). For immunohistochemistry, lungs were perfused and fixed in 0.001 M periodate/ 0.075 M lysine/1% paraformaldehyde (PLP) and processed essentially as described previously (44). In brief, lungs were fixed in PLP overnight at 4°C with the tracheostomy tube left in place while in PLP. The tissue was washed three times for 5 min each in 0.1 M cold phosphate buffer. The tissue was then treated successively with 10 and 20% sucrose (in phosphate buffer) for 20 min each. Lungs were inflated via the tracheostomy tube with 1 ml of 40% OCT diluted in PBS (tissue freezing medium; Electron Microscopy Sciences, Fort Washington, PA). Lungs were embedded in 100% OCT in cryomold and plunged into a 2-methylbutane bath precooled on dry ice. Tissues were stored at −70°C until sectioning. Sections were mounted onto slides. Slides were soaked in wash buffer (0.1 M phosphate buffer plus 0.01% Triton X-100) for 10 min and blocked with 3% BSA in wash buffer. Biotinylated primary antibodies (anti–ICAM-1 antibody; catalog number 01542D; anti–VCAM-1 antibody; catalog number 01812D) and the isotype-matched control antibodies were all purchased from PharMingen (San Diego, CA). The anti–VCAM-1 antibody was used at a 1:50 dilution, whereas the anti–ICAM-1 antibody was used at a 1:150 dilution in 1% BSA. The antibodies were left on the slides for 1.5 h at 25°C. The slides were successively washed in wash buffer and 0.1 M Tris, pH 7.5. The tissues were incubated with streptavidin-alkaline phosphatase (Zymed, San Francisco, CA) for 30 min. The tissues were washed three times for 2 min each time in 0.1 M Tris, pH 7.5, and developed with Fast Red until staining was adequate. The slides were washed three times in double-distilled water and counterstained with hematoxylin.
Preparation of Nuclear Extracts from Whole Lung and Electrophoretic Mobility Shift Assays.
To prepare nuclear extracts from lung tissue, the lungs were snap frozen in liquid N2 and pulverized. The following steps all were carried out at 4°C. The lung powder was homogenized in 5 ml of solution A (10 mM Hepes, pH 7.9, 150 mM NaCl, 1 mM EDTA, 0.5 mM PMSF, and 0.6% NP-40) in a Dounce tissue homogenizer (Wheaton, Millville, NJ) and centrifuged at 800 g for 30 s to remove cellular debris. The supernatant was left on ice for 5 min and then centrifuged at 2000 g for 5 min. The pelleted nuclei were resuspended in 150–200 μl of solution B (20 mM Hepes, pH 7.9, 420 mM NaCl, 1.2 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM PMSF, 2 mM benzamidine, 25% glycerol, and a mixture of protease inhibitors (Boehringer Mannheim, Indianapolis, IN) for 20 min. The lysed nuclei were briefly centrifuged at 16,000 g. The supernatant was aliquoted, snap frozen in a dry ice/methanol bath and stored at −70°C until further use. A 32P-labeled oligonucleotide containing the NF-κB sequence present in the IgκB gene was used as the probe in electrophoretic mobility shift assays (EMSAs). The binding reactions were analyzed by electrophoresis on 6% native polyacrylamide gels (acrylamide/bisacrylamide = 30:1). Electrophoresis was carried out at 150 V in 0.5× TBE (1× = 0.05 M Tris base, 0.05 M boric acid, and 1.0 mM EDTA) at 4°C. Gels were dried and subjected to autoradiography.
RNA Isolation and Ribonuclease Protection Assays.
Total RNA was isolated from lung tissue using TRIzol Reagent (GIBCO BRL, Gaithersburg, MD) according to the manufacturer's instructions. Each whole fresh lung was homogenized in 4 ml of TRIzol reagent using Tissumizer (Tekmar Co., Cincinnati, OH) and centrifuged to remove cellular debris. The RNA pellet was resuspended in nuclease-free H2O. 10 μg of total lung RNA from each mouse was used in ribonuclease protection assay (RPA) using the RPA II Kit (Ambion, Austin, TX). The murine cytokine template set mCK-5 (PharMingen) was used to obtain radiolabeled antisense RNA probes for RANTES, eotaxin, MIP-1α, MIP-1β, MIP-2, IP-10, monocyte chemotactic protein (MCP)-1, L32, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). In vitro transcription was carried out by incubation in a buffer containing 10 mM ATP, 10 mM CTP, 10 mM GTP, 250 μCi α-[32P]UTP (800 Ci/mmol, 10 mCi/mmol; Amersham Pharmacia Biotech, Arlington Heights, IL) and T7 RNA polymerase in transcription buffer (MAXIscript Kit; Ambion). The mixture was incubated at 37°C for 60 min and then treated with DNase I at 37°C for 30 min. The mixture was extracted with a mixture of phenol and chloroform, and the RNA was precipitated with ethanol and collected by centrifugation at 4°C. The RNA was resuspended in 50 μl hybridization buffer and diluted to 3 × 105 cpm/μl, and 2 μl was used per reaction. The lung RNA samples (10 μg from each mouse) were dried in a vacuum evaporator and resuspended in 8 μl hybridization buffer. The RNA was annealed to the probe by incubating successively at 95°C for 3 min and at 56°C overnight in a total volume of 10 μl. RNase was added to each sample for removal of single-stranded RNA and RNase digestion was carried out at 30°C for 45 min. The protected RNA duplexes were purified by phenol/chloroform extraction and ethanol precipitation. The pelleted RNA was resuspended in 5–6 μl of gel loading buffer, incubated at 95°C for 3 min, quickly quenched on ice and analyzed by electrophoresis on 5% polyacrylamide/8 M urea gels. The gels were dried and subjected to autoradiography.
Cell Proliferation Assays.
Mice were sensitized as above by intraperitoneal injections of OVA plus alum on days 0 and 5. Spleens were harvested on day 12 and CD4+ T cells were prepared by negative selection using mAbs to CD8, class II MHC I-A, and anti-Ig–coated magnetic beads (Collaborative Research, Bedford, MA). Syngeneic T cell–depleted splenocytes were used as APCs that were prepared by negative selection using antibodies to CD4 (GK1.5), CD8, Thy-1, and treatment with rabbit complement and mitomycin C as described previously (41). 5 × 105 CD4+ T cells (pooled from two mice in each group) together with 5 × 105 APCs were cultured with OVA (0.1–100 μg/ml) in Bruff's medium supplemented with 5% FCS. Cultures were incubated for 72 h, supernatants were collected for cytokine analyses and cultures were pulsed with 1 μCi of methyl-[3H]thymidine/well. After incubation at 37°C for 24 h, triplicate wells were harvested onto glass filters and incorporated radioactivity was measured in a beta counter. The background was subtracted from the results.
Cytokine Assays.
IL-2, IL-4, IL-5, and IFN-γ protein levels in culture supernatants and in BAL and eotaxin protein levels in BAL were determined by ELISA. The lower limits of detection for the cytokines were: IL-2, 3 pg/ml; IL-4, 5 pg/ml; IL-5, 5 pg/ ml; IFN-γ, 15 pg/ml (Endogen, Cambridge, MA); and for the chemokine eotaxin, 3 pg/ml (R & D, Minneapolis, MN).
Reverse Transcription-PCR.
RNA isolated from the OVA-stimulated CD4+ T cells were used in reverse transcription (RT)- PCR. To synthesize cDNA, random primers (100 pmol) were annealed to 1 μg of total RNA by incubation at 68°C for 10 min. RT was carried out by adding dNTP, 8 U of an RNase inhibitor (RNasin), 0.2 mM dithiothreitol, and 200 U SuperScript II (GIBCO BRL) and buffer and by incubating at 37°C for 10 min, at 42°C for 50 min and finally at 94°C for 2 min. The cDNAs synthesized were used in PCR. PCR was performed using primer sets corresponding to the murine IL-4 and IL-5 genes. The sequences of the primer were: IL-4; 5′-CAT CGG CAT TTT GAA CGA GGT CA-3′ (forward) and 5′-CTT ATC GAT GAA TCC AGG CAT CG-3′ (reverse); IL-5; 5′-CTC ACC GAG CTC TGT TGA CAA G-3′ (forward) and 5′-GAA CTC TTG CAG GTA ATC CAG G-3′ (reverse). The reactions were assembled in HotStart tubes (Molecular Bio-Product, Inc., San Diego, CA) as follows: first the lower layer mix was added that included 10mM dNTP, 1× Pfu (Stratagene, La Jolla, CA) reaction buffer and primers (50 pmol). The mixture was heated to 90°C for 30 s and cooled down to room temperature, and then the upper layer that included cDNA (1–2 μl), 1.25 U of Pfu DNA polymerase (Stratagene) and 1× Pfu buffer was added. For PCR, the samples were first heated to 94°C for 2 min (for denaturation) and the PCR cycle conditions were: 94°C for 45 s, 64°C for 45 s, and 72°C for 1 min. The reactions were terminated by incubation at 72°C for 7 min to complete chain extensions. The products were analyzed by electrophoresis on 1% agarose gels.
Results
Activation of NF-κB Complexes By Antigen (OVA) in the Lungs of Wild-type but Not p50−/− Mice.
To investigate the effects of antigenic (OVA) stimulation on NF-κB activation in the whole lung, wild-type and p50−/− mice were sensitized and challenged with OVA (hereafter referred to as OVA/OVA). 2 h after challenge with OVA, mice were killed and nuclear extracts were prepared from the whole lung. The nuclear extracts were used in EMSAs using an oligonucleotide containing a consensus binding sequence for NF-κB. As shown in Fig. 1, lane 1, nuclear extracts prepared from sham-sensitized and OVA-challenged (Alum/ OVA) mice did not display appreciable DNA-binding activity. On the other hand, nuclear extracts prepared from the lungs of mice sensitized and challenged with OVA (OVA/OVA) formed three distinct complexes. In the case of p50−/− mice, no DNA–protein complex was observed with nuclear extracts from control mice (Alum/OVA). With extracts prepared from OVA/OVA mice, only the fastest migrating complex (shown by an arrow) was detected. We used specific antibodies to determine the composition of the different polypeptide complexes. Formation of complex I was affected by both anti-p50 and anti-p65 antibodies, suggesting that it contained the classic p50–p65 heterodimer (Fig. 1, lanes 6 and 10). Complex II formation was affected only by the anti-p50 antibody (Fig. 1, lane 6). Also, complex II comigrated with the complex formed with recombinant p50, confirming that it contained (p50)2. The intensity of the complex shown by an arrow was variable in different experiments. This complex was not supershifted by the anti-p50 or the anti-p65 antibody, nor by antibodies against other Rel proteins (data not shown), and most likely represents nonspecific binding. Oct-1–binding activity, used as a loading control, was comparable in all the lanes (data not shown). Thus, the EMSAs demonstrated the presence of active NF-κB dimers containing p50 in the lungs of antigen-challenged wild-type mice and their absence in the lungs of similarly challenged p50−/− mice.
Figure 1.

Induction of NF-κB activity in the lungs of p50+/+ but not p50−/− mice. Mice were either sensitized with OVA+ alum followed by an aerosol challenge with OVA (OVA/ OVA) or sham-sensitized with alum only and OVA-challenged (Alum/OVA). Nuclear extracts prepared from the lungs of animals subjected to Alum/OVA or OVA/OVA were used for EMSAs. A 32P-labeled oligonucleotide containing a consensus NF-κB– binding sequence was used to detect the κB-binding activities. Binding reactions were carried out in the presence or absence of specific antibodies (Ab) as indicated. In parallel reactions, binding reactions were also carried out with recombinant p50 (rp50; Promega Corp., Madison, WI). The probe was incubated with rp50 in the presence or absence of anti-p50 Ab (lane 9) or anti-p65 Ab (lane 10; used as a control). The p50 homodimer and the p50–p65 heterodimer are indicated as complexes I and II, respectively. Nonspecific binding to the probe is shown by an arrow and the intensity of this complex was variable in different experiments. Binding reactions were analyzed by electrophoresis on 6% native polyacrylamide gels as described previously (33, 90).
Absence of Antigen-Induced Eosinophilic Airway Inflammation in the Lungs of the p50−/− Mice.
To assess the effect of inhaled antigen on airway inflammation in the wild-type (+/+) and p50−/− (−/−) animals, mice were killed 24 h after aerosol challenge with OVA and the lungs were examined histologically. As shown in Fig. 2 a, wild-type mice displayed a prominent inflammatory response. Eosinophilic inflammation was widespread with both perivascular and peribronchiolar infiltration. On the other hand, the p50−/− mice had no eosinophilic inflammation in their airways upon antigen challenge (Fig. 2 b). In some p50−/− mice, a few patchy areas of a low degree of inflammation were noted in which eosinophils or neutrophils were barely detectable. The lungs of animals sham-sensitized with alum only and challenged with OVA (Fig. 2, c and d, respectively) were totally devoid of any inflammation.
Figure 2.

Absence of airway inflammation in p50−/− mice. Lung sections of p50+/+ or p50−/− mice either OVA/OVA-treated (a and b, respectively) or ALUM/OVA-treated (c and d, respectively) were subjected to H&E staining (50×). p50+/+ OVA/OVA-treated mice had impressive peribronchiolar and perivascular eosinophilic inflammation. No eosinophilic infiltrates were detected in lung tissue from p50−/− mice.
The impressive difference in lung inflammation between the wild-type and p50−/− mice was also observed in the BAL fluid (BALF) recovered from the animals. The total cell count in the BALF recovered from the p50−/− mice upon antigen challenge was 10-fold lower than that in the BALF derived from the wild-type mice (Fig. 3 A). After antigen provocation, although there was an ∼2,000-fold increase in the number of eosinophils in the airways of the wild-type mice (compared with sham-sensitized animals), the net increase in eosinophils was only 10-fold in the airways of the p50−/− mice (Fig. 3 A). In addition to a blunted eosinophil recruitment, neutrophil recruitment was also markedly attenuated in the airways of the p50−/− mice. Also, the number of lymphocytes recovered from the airways of the p50−/− mice was ∼5–6-fold less than that obtained from the wild-type mice (Fig. 3 A). The cells in the BALF recovered from the p50−/− mice contained predominantly monocytes/macrophages similar to those observed in the BALF derived from control mice that were sham-sensitized and challenged with OVA (Fig. 3 A).
Figure 3.
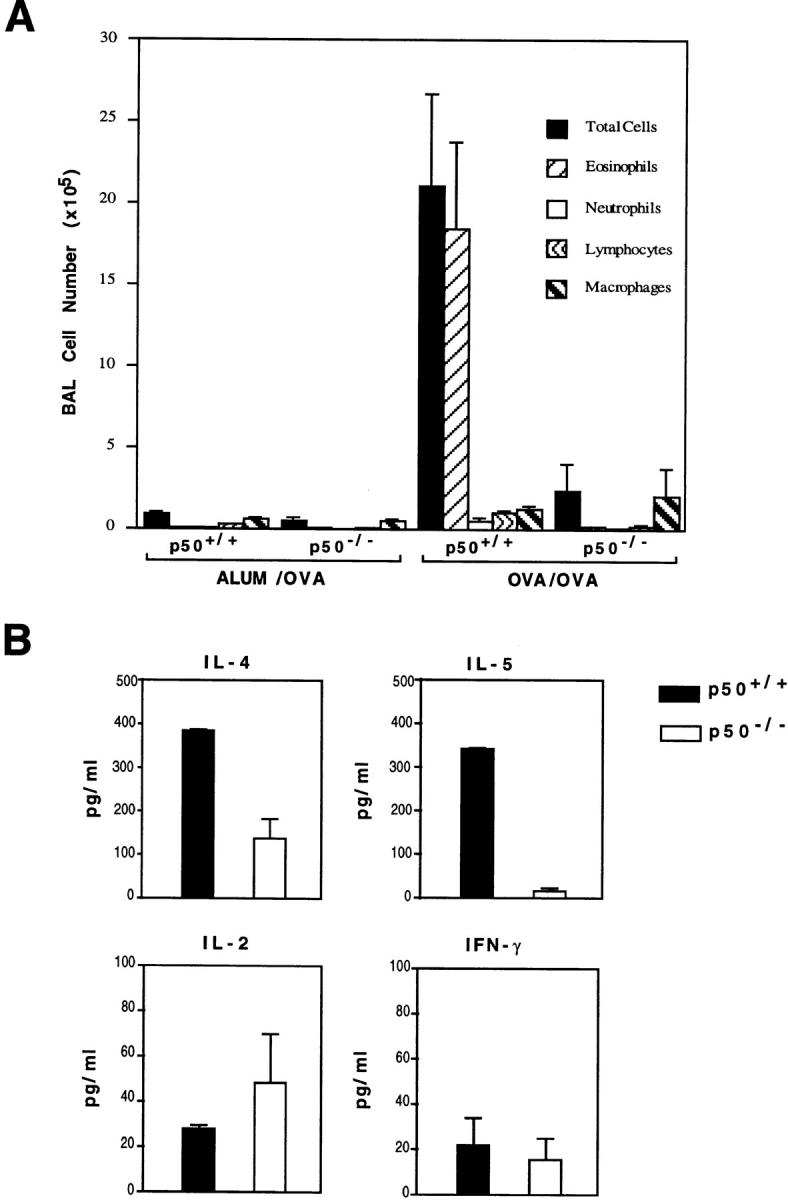
Decreased leukocyte infiltration and cytokine production in the airways of p50−/− mice. (A) Total and differential cell counts in the BAL of wild-type and p50−/− mice. Mean cell counts ± SEM are shown (n = 3 per group). Results shown are representative of three experiments. (B) Estimation of cytokines in BALF by ELISA. Results shown are mean ± SEM for three mice in each group.
We also examined cytokine levels in the airways of these mice. As shown in Fig. 3 B, the predominant cytokines present in the BALF obtained from the wild-type mice were of the Th2 type as was expected in this model. First, with the exception of IL-2, all other cytokines were present at lower levels in the airways of the p50−/− mice as was expected from the deficient airway inflammation in these mice after antigen challenge. Second, IL-4 was detectable in the BALF of p50−/− mice, albeit at lower levels compared with those in the wild-type mice. The lower level of IL-4 in the BALF of the p50−/− mice is probably due to fewer lymphocytes in the airways of these mice, as Th2 lymphocytes are the major source of this cytokine in asthma and allergic inflammation. The other Th2-type cytokine, IL-5, was barely detectable in the BALF of the p50−/− mice (Fig. 3 B). Thus, the production of IL-5 appeared to be more severely affected by the absence of the p50 subunit of NF-κB than the production of IL-4.
Basal Eosinophil Counts in the p50−/− Mice.
We investigated the basal level of eosinophils in the wild-type and the p50−/− mice. As shown in Table 1, the level of eosinophils in the bone marrow and peripheral blood of p50−/− mice was comparable to that in the controls.
Table 1.
Basal Eosinophil Counts in the p50+/+ and p50−/− Mice
| Bone marrow | Peripheral blood | |||
|---|---|---|---|---|
| % | % | |||
| p50+/+ mice | 7.2 ± 0 | 2.25 ± 0.75 | ||
| p50−/− mice | 5.0 ± 0.8 | 3.35 ± 0.35 |
Bone marrow cells were obtained by flushing the cavity of a single femur from each mouse with serum-free RPMI 1640 medium containing heparin (10 U/ml). Cells derived from the bone marrow and peripheral blood were suspended in lysis buffer (Pharm Lyse; PharMingen) for 2 min for lysis of the red blood cells. The cells were washed twice in PBS and cytospin preparations were stained with Diff-Quik. Shown are the number of eosinophils determined as a percentage of the total number of leukocytes ± SEM.
Expression of Cell Adhesion Molecules VCAM-1 and ICAM-1 in the Airways of Wild-type and p50−/− Mice.
We investigated the expression of the cell adhesion molecules VCAM-1 and ICAM-1 in the airways of wild-type and p50−/− mice by immunohistochemistry since both of these molecules have been implicated in eosinophil extravasation from the blood vessel into the airways (45). After OVA challenge, the animals were killed and the lungs were frozen for immunohistochemistry. As shown in Fig. 4 A, upon OVA challenge, strong expression of VCAM-1 was evident on the vascular endothelium in both the wild-type and p50-deficient mice (Fig. 4 A, a and b, respectively). No staining was observed in the lungs of the control animals sensitized with alum and challenged with OVA (Fig. 4. A, c and d). Also, no reaction was observed with the isotype-matched control antibody (Fig. 4 A, e and f, respectively). The expression of ICAM-1 was also similar in the wild-type and p50−/− mice (Fig. 4 B). Strong constitutive ICAM-1 expression on the alveolar epithelium and somewhat weaker expression on the vascular endothelium was noted in both wild-type and p50-deficient mice (Fig. 4 B, c and d, respectively) and there was no significant change in the pattern of staining after OVA challenge (Fig. 4 B, a and b). Again, no staining was observed with the control antibody (Fig. 4 B, e and f ).
Figure 4.

Expression of VCAM-1 and ICAM-1 in the lungs of both p50+/+ and p50−/− mice. Lung sections of ALUM/OVA or OVA/OVA-treated animals were stained with (A) anti–VCAM-1 or (B) anti–ICAM-1 mAb and counterstained with hematoxylin. In both cases, lung sections from OVA/ OVA- or ALUM/OVA-treated p50+/+ and p50−/− animals were incubated either with specific antibody (OVA/OVA, a and b respectively; ALUM/ OVA, c and d, respectively) or with isotype-matched control antibody (e and f, respectively). VCAM-1 staining (red) was observed on the vascular endothelium of both wild-type and p50−/− mice. In both groups of mice, strong constitutive ICAM-1 staining was observed on the alveolar epithelium and weaker staining on the vascular endothelium, and the staining pattern remained unchanged upon challenge with OVA. No staining was observed with the control antibodies in either case (lung sections used were from OVA/OVA mice). Filled arrowheads indicate endothelial staining and open arrowheads indicate epithelial staining.
Differential Chemokine Gene Expression in the Lungs of p50−/− and Wild-type Mice.
Since chemokines have been shown to be important for the recruitment of leukocytes to sites of inflammation, we investigated the expression of key chemokines that have been implicated in eosinophil chemotaxis. Mice were killed at different time points after OVA challenge, and RNA isolated from the lungs of these mice was subjected to RNase protection assays for the expression of different chemokine genes. The expression of RANTES mRNA was found to be constitutive in the lungs of both wild-type and p50−/− mice and the expression remained unchanged after challenge with OVA (Fig. 5 A). In the lungs of p50+/+ mice, the expression of mRNAs for eotaxin, MIP-1α, MIP-1β, IP-10, and MCP-1 was detectable early, at 2 h after OVA challenge, and the steady-state levels of these messages were higher at 4 h after challenge (data not shown). At 24–36 h after challenge, eotaxin and MIP-1α mRNAs continued to be expressed at a high level in the wild-type mice. In the p50−/− mice a different pattern of chemokine mRNA expression was detected. The expression of eotaxin or MIP-1α or MIP-1β mRNA was barely detectable in these mice at all time points. However, the expression of IP-10 mRNA was stronger in the p50−/− mice compared with that in the wild-type mice at all time points tested. The expression of MCP-1 mRNA was comparable in the wild-type and p50−/− mice at all time points. Given the importance of eotaxin as an eosinophil chemoattractant, we have also tested the level of eotaxin protein in the BALF in the OVA-challenged wild-type and p50−/− mice. As shown in Fig. 5 B, eotaxin protein was readily detected at 24 h in the BALF recovered from the wild-type mice but was barely detectable in that obtained from the p50−/− mice.
Figure 5.

Absence of induction of eotaxin, MIP-1α, and MIP-1β mRNA in the lungs of p50−/− mice upon challenge with OVA. (A) Chemokine mRNA expression was determined by RNase protection assays using the multiprobe template set mCK-5 (PharMingen) as described in Materials and Methods. Each lane is representative of four mice and the experiment was repeated three times with consistent results. (B) Estimation of eotaxin protein in BALF by ELISA. Results shown are mean ± SEM for four mice in each group. The significance of difference between the two groups was analyzed using Student's unpaired t test. Differences in means were considered significant if P < 0.05. *P < 0.0001 compared with protein levels in p50+/+ mice.
Proliferative Responses and Cytokine Production by CD4+ T Cells in Wild-type and p50−/− Mice.
We next examined whether the absence of airway inflammation in the p50-deficient mice in response to antigen challenge was due to a defect in the ability of CD4+ T cells to respond to antigen. Wild-type and p50−/− mice were immunized with OVA intraperitoneally and recall proliferative responses of spleen CD4+ T cells were analyzed by [3H]thymidine incorporation. CD4+ T cells from the p50−/− mice displayed a lower basal incorporation (∼50%) compared with those isolated from the wild-type mice. However, they proliferated as efficiently as the wild-type cells in response to antigen (Fig. 6 A).
Figure 6.

Similar proliferative responses of CD4+ T cells isolated from p50+/+ and p50−/− mice. (A) p50+/+ (open symbols) and p50−/− (filled symbols) mice were sensitized with OVA+alum on days 0 and 5, and on day 12 splenic T cells isolated from these mice were stimulated in vitro with different concentrations of OVA (0.1–100 μg/ml). Cells were pulsed with [3H]thymidine and incorporated counts were determined. Results shown are mean ± SEM. Shown is a representative experiment of two. (B) Culture supernatants of cells stimulated with 100 μg/ml of OVA for 72 h were used for cytokine estimations by ELISA and the cells were harvested for RNA isolation for RT-PCR analysis. (C) PCR was carried out for 35 cycles. The IL-4 and IL-5 PCR products were 240 and 251 bp, respectively.
Cytokine production was measured in culture supernatants and RNA was isolated from the OVA-restimulated cells for analysis by RT-PCR. IL-2, IL-4, and IFN-γ were detected in the supernatants of cells derived from both wild-type and p50−/− mice (Fig. 6 B). Interestingly, CD4+ T cells from the p50−/− mice produced ∼5–7-fold more IL-2 than did cells from wild-type mice. The most striking difference was the total absence of IL-5 production by p50-deficient T cells (Fig. 6 B). RT-PCR analysis of RNA isolated from the OVA-stimulated CD4+ T cells revealed IL-4 RNA expression in both wild-type and p50-deficient T cells (Fig. 6 C). The same RNA samples when analyzed for IL-5 mRNA expression revealed expression in the wild-type CD4+ T cells with barely detectable expression in the p50-deficient T cells (Fig. 6 C).
Discussion
The molecular mechanisms that regulate eosinophilic inflammation in asthma is an area of intense investigation in many laboratories. We have recently shown that the transcription factor GATA-3 is expressed in Th2 but not Th1 cells and plays a critical role in IL-5 gene expression (46– 48). In this study, we have investigated the role of NF-κB in the elicitation of eosinophilic inflammation in a murine model of allergic airway inflammation. We show that the absence of NF-κB abolishes airway inflammation. This results from the absence of IL-5 and eotaxin gene expression in the p50−/− mice, both of which are key mediators of eosinophilic inflammation in the airways. We also show markedly attenuated lymphocyte recruitment in the lungs of p50−/− mice that, in part, may be due to deficient expression of the chemokine genes MIP-1α and MIP-1β in the p50−/− mice. These results provide the first in vivo evidence that NF-κB is required for the development of eosinophilic inflammation in response to inhaled allergens.
Airway inflammation in asthma and allergic disease is a complex phenomenon driven predominantly by Th2-type cells. The inflammation is characterized by the recruitment of leukocytes, predominantly eosinophils, and their subsequent migration from the vasculature into the tissue where they cause severe damage to the bronchiolar epithelium (2, 4–10). Th2, epithelial, and endothelial cells play major roles in this cascade by secreting cytokines and chemokines and by expressing cell surface adhesion molecules. Since multiple in vitro studies have implicated NF-κB in the expression of many of these molecules (24–26, 28, 29), we examined the role of this transcription factor in eosinophilic inflammation after antigen provocation. We show that NF-κB plays a critical regulatory role in promoting eosinophilia in vivo.
The phenotype of the p50−/− mice with respect to inflammation was in sharp contrast to the recently reported phenotype of animals lacking the precursor p105 but expressing p50 (34). p50 is generated by proteolytic removal of the COOH terminus of a precursor protein p105. The murine nfkb1 gene produces two transcripts, a longer 4.0-kb transcript that encodes the full-length precursor p105 and a shorter 2.6-kb mRNA that encodes the IκBγ protein that is identical to the COOH terminus of p105 that is cleaved off to generate mature p50 (49, 50). Mice targeted to lack p105 but containing p50 showed multiple abnormalities, including inflammation in the lungs and liver composed largely of lymphocytes, increased susceptibility to opportunistic infections, splenomegaly, lymph node enlargement, and lymphoid hyperplasia (34). Since the COOH terminus of p105 has an inhibitory effect on p50 homodimer activity, mice lacking p105 had an enhanced p50 homodimer activity (34). Although excess (p50)2 present in p105−/− mice resulted in decreased cytokine production (IL-2 and IL-4) by stimulated T cells isolated from these mice, T cell–dependent immune responses overall were not seriously impaired in the p105−/− mice (34). In contrast, we demonstrate a severe impairment of T cell–dependent eosinophilic inflammation in the p50−/− mice. It should be noted that although the p105−/− mice have excess (p50)2 together with, potentially, a small increase in p50–p65 heterodimers, the p50−/− mice are devoid of both (p50)2 and p50–p65 dimers. Thus, the net effect in the p50−/− mice is a combination of deficient p50 homo- and heterodimeric activity.
The absence of eosinophilic inflammation in the p50−/− mice did not result from a deficiency in the expression of the cell adhesion molecules VCAM-1 and ICAM-1, which have been shown to be important for the extravasation of eosinophils into the lungs in models of airway inflammation. Many in vitro studies have implicated NF-κB in the transcriptional regulation of VCAM-1 (51–53) and ICAM-1 (53–55) genes in endothelial cells. Therefore, it was surprising that p50 deficiency did not abolish the expression of these molecules since these mice lack not only (p50)2 but also the p50–p65 heterodimer, which is the predominant NF-κB transactivator. In addition to the p50–p65 heterodimer, (p65)2 has been shown also to be a potent transactivator, although it is found in much lower abundance than the p50–p65 heterodimer (30). Indeed, a few studies propose a more important role for the (p65)2 rather than the p50–p65 heterodimer in IL-1 or TNF-induced VCAM-1 (56, 57) and ICAM-1 (58) gene expression. Interestingly, in one study, IL-4–induced VCAM-1 gene expression was shown to involve an NF-κB–independent mechanism (59). Our studies suggest that the expression of VCAM-1 and ICAM-1 in this allergen model of airway inflammation is not critically dependent on the p50–p65 heterodimer.
The lungs of the p50−/− mice were indistinguishable from the lungs of either sham-immunized or unchallenged animals and did not display an easily discernible increase in eosinophil, neutrophil, or lymphocyte infiltration after antigen challenge. Leukocyte infiltration into sites of inflammation is now known to involve cytokine-inducible chemoattractants called chemokines. Chemokines that have been implicated in the recruitment of activated CD4+ T cells to inflammatory sites include RANTES, MIP-1α, MIP-1β (60), and IP-10 (the murine homologue is Crg-2; reference 61). In this model we and others have detected a low level constitutive expression of RANTES mRNA that did not increase after antigen challenge (62). The induction of MIP-1α and MIP-1β mRNA was abolished in the p50−/− mice, suggesting that the induction of these chemokines in vivo is an NF-κB–dependent process, at least in this model. The lack of expression of the MIP-1α and MIP-1β genes may be partly responsible for the reduced number of lymphocytes in the lungs of p50-deficient mice. In contrast to lack of induction of eotaxin, MIP-1α and MIP-1β mRNA in the p50-deficient animals, the steady-state level of IP-10 mRNA was greater in the lungs of antigen-challenged p50−/− mice compared with that in the lungs of similarly challenged wild-type mice. We have not determined the cellular source of IP-10 in the lungs of these mice, but the likely candidates are activated macrophages and/or endothelial cells. CXCR3, the receptor for IP-10, has been shown to be predominantly expressed by human Th1 cells (63). If the same is true in mice, then despite increased expression of IP-10 in p50−/− mice, this chemokine was ineffective in the recruitment of Th2-type cells to the lung due to the absence of the cognate receptor on the Th2 cells. Recently, a subset of human Th2 cells were shown to express the chemokine receptor CCR3 (63–66), the best described ligand for which is eotaxin. CCR3 is also highly expressed on the cell surface of both murine and human eosinophils (67–69) and accounts for the specific chemotactic effects of eotaxin on eosinophils. Although a role for eotaxin in Th2 cell chemotaxis has not been demonstrated, it is suggested that eotaxin may control the trafficking of a subset of Th2s that stimulate growth and activation of colocalized eosinophils and basophils (65, 66, 70). Thus, the absence of key chemokines that have been shown to be important for the recruitment of activated CD4+ T cells probably explains the reduced T cell infiltration into the lungs of antigen-challenged p50−/− mice.
An important difference between the wild-type mice and the p50−/− mice was the lack of eotaxin and IL-5 gene expression in the p50-deficient animals. Eotaxin has been shown to be a specific chemoattractant for eosinophils in different species including the guinea pig, mouse, and human (71–73). The in vivo specificity and potency of eotaxin for eosinophil chemotaxis were demonstrated in studies showing eosinophil recruitment into tissue either after instillation of eotaxin into the airways or after injection into the skin (73–77). Eotaxin is produced in large quantities by the type I cells in the alveolar epithelium, although other cells such as endothelial cells and the infiltrating eosinophils are also known to produce eotaxin (73–75). In vivo, the production of eotaxin has been shown to be dependent on T cells (62). Using the OVA model of airway inflammation, MacLean et al. have shown a profound inhibition of eotaxin but not RANTES or MIP-1α production in mice treated with anti-CD3 antibody (62). The molecular basis for this effect is unclear. However, it is possible that eotaxin production upon allergen challenge is induced by a T cell– derived cytokine, most likely a Th2 cytokine. Since, in addition to eotaxin, the p50−/− mice also do not produce IL-5, it is possible that IL-5 is a necessary stimulus for the induction of eotaxin gene expression in this model of airway inflammation. A nonmutually exclusive explanation for the lack of eotaxin gene transcription in p50−/− mice is that eotaxin gene expression is directly regulated by NF-κB. Indeed, the 5′ flanking region of the eotaxin gene contains NF-κB sites, although their functional significance has yet to be determined (78). It is now believed that eosinophil chemotaxis involves cooperation between IL-5 and eotaxin as shown in the guinea pig and mouse (23, 76, 79, 80). IL-5 and eotaxin are both produced in the lung, the major source of IL-5 being Th2 cells. IL-5 secreted by Th2 cells is transported by the blood to the bone marrow. A major role of IL-5 at this step is to trigger a rapid mobilization of eosinophils and progenitors from the bone marrow into the blood since this process is inhibited upon treatment with the anti–IL-5 antibody TRFK5 (79). The role of eotaxin is to recruit the eosinophils from the vasculature. Thus, given the important role of IL-5 and eotaxin in eosinophil recruitment to the lungs in allergic inflammation, the combined deficiency of both of these factors in the p50-deficient animals could explain the total abrogation of airway eosinophilia in these mice.
In this study, we also show that the absence of NF-κB influences T cell cytokine gene expression. Different in vitro studies have suggested a role for NF-κB in the expression of the IL-2 gene. Although the p50–p65 heterodimer has been shown to interact with an NF-κB site in the IL-2 promoter, (p50)2 has been shown to function as a repressor of IL-2 gene expression (31). In our studies, not only was IL-2 produced by the p50-deficient cells, the amount of IL-2 secreted by these cells was ∼5–7-fold greater than that produced by the wild-type T cells. This suggests, first, that (p65)2 or other Rel dimers can substitute for the classic NF-κB dimer for transactivation of the IL-2 gene in this model. Second, our studies support the data of Kang et al. (31) that an essential turn-off signal for IL-2 gene transcription is (p50)2 since the p50−/− mice produced considerably more IL-2 than did the wild-type mice. Conversely, in p105−/− mice, which have excess (p50)2, IL-2 gene expression was found to be three- to sixfold lower than that in wild-type mice (34). Although no difference was noted between the wild-type and p50-deficient T cells with respect to the net fold-stimulation of the cells upon antigen challenge, the basal [3H]thymidine incorporation by the p50-deficient cells was ∼50% of that of the wild-type cells. This may be due to a lower basal expression of the IL-2 or the IL-2Rα gene (whose expression is also believed to be under NF-κB control) in the absence of the classic NF-κB dimer. We show that the absence of p50 profoundly inhibits IL-5 gene expression in CD4+ T cells. Presently, it is unclear whether this is due to a primary or secondary effect of NF-κB on IL-5 gene expression.
An important outcome of this study is the differential effect of NF-κB on IL-4 and IL-5 gene expression. There is increasing interest in the dissociation of IL-4 and IL-5 gene expression in T cells in various disease situations and in T cell lineages (for review see references 81, 82). For example, in intrinsic asthmatics, IL-5 but not IL-4 production is elevated (83). In contrast, in leukemic Sezary cells, IL-4 has been shown to be upregulated with concomitant decrease in IL-5 production (84). Again, in a study of human T cells using intracellular staining techniques, IL-4 and IL-5 were found to be predominantly made by different cells (85). There is no evidence to date that supports different lineage of cells for IL-4 and IL-5 production. However, it is reasonable to speculate that once cells have been committed to the Th2 lineage, different microenvironmental factors activate different sets of transcription factors such as NF-κB, GATA-3 (46, 47, 86, 87), c-maf (88), and NF-AT (89), which have differential effects on IL-4 and IL-5 gene expression. We have recently shown that ectopically expressed GATA-3 is sufficient for IL-5 but not IL-4 gene expression (48). Thus, our studies provide clear evidence that although IL-4 and IL-5 are often coordinately expressed, the absence of transcription factors such as NF-κB can cause selective impairment of IL-5 but not IL-4 gene expression. The attenuation of both IL-5 and eotaxin gene expression in p50−/− mice establishes a central role for NF-κB in airway eosinophilia in allergic inflammation.
Acknowledgments
The authors would like to thank Irena Schvayetsky for excellent technical assistance with immunohistochemistry.
This work was supported by the National Institutes of Health grants HL52014 and HL60207 (to P. Ray), AI31137 and HL56843 (to A. Ray), P50-HL56389 (to A. Ray and R. Homer), and KO8-HL03308 (to L. Cohn).
Abbreviations used in this paper
- BAL
bronchoalveolar lavage
- BALF
BAL fluid
- EMSA
electrophoretic mobility shift assay
- H&E
hematoxylin and eosin
- ICAM
intercellular adhesion molecule
- MCP
monocyte chemotactic protein
- MIP
macrophage inflammatory protein
- NF
nuclear factor
- PLP
paraformaldehyde
- RANTES
regulated on activation, normal T cell expressed and secreted
- RT
reverse transcription
- VCAM
vascular cell adhesion molecule
Footnotes
This paper is dedicated to the memory of our beloved mentor and friend Dr. Jyotirmoy Das of Indian Institute of Chemical Biology, India.
References
- 1.Hamid Q, Azzawi M, Ying S, Moqbel R, Wardlaw AJ, Corrigan CJ, Bradley B, Durham SR, Collins JV, Jeffery PK, et al. Expression of mRNA for interleukin 5 in mucosal bronchial biopsies from asthma. J Clin Invest. 1991;87:1541–1546. doi: 10.1172/JCI115166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker C, Kaegi MK, Braun P, Blaser K. Activated T cells and eosinophilia in bronchoalveolar lavages from subjects with asthma correlated with disease severity. J Allergy Clin Immunol. 1991;88:935–942. doi: 10.1016/0091-6749(91)90251-i. [DOI] [PubMed] [Google Scholar]
- 3.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 4.de Monchy JGR, Kaufmann HF, Venge P, Koeter GH, Jansen HM, Sluiter HJ, de Vries K. Bronchoalveolar eosinophilia during allergen-induced late asthmatic reactions. Am Rev Respir Dis. 1985;131:373–376. doi: 10.1164/arrd.1985.131.3.373. [DOI] [PubMed] [Google Scholar]
- 5.Gleich GJ, Flavahan NA, Fujisawa T, Vanhoutte PM. The eosinophil as a mediator of damage to respiratory epithelium: a model for bronchial hyperreactivity. J Allergy Clin Immunol. 1988;81:776–781. doi: 10.1016/0091-6749(88)90931-1. [DOI] [PubMed] [Google Scholar]
- 6.Frick WE, Sedgwick JB, Busse WW. The appearance of hypodense eosinophils in antigen-dependent late phase asthma. Am Rev Respir Dis. 1989;139:1401–1406. doi: 10.1164/ajrccm/139.6.1401. [DOI] [PubMed] [Google Scholar]
- 7.Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, Enander I, Venge P, Ahlstedt S, Simony-Lafontaine J, Godard P, Michel FB. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 8.Azzawi M, Bradley B, Jeffrey PK, Frew AJ, Wardlaw AJ, Knowles G, Asoufi B, Collins JV, Durham S, Kay AB. Identification of activated T lymphocytes and eosinophils in bronchial biopsies in stable asthmatics. Am Rev Respir Dis. 1990;142:1407–1413. doi: 10.1164/ajrccm/142.6_Pt_1.1407. [DOI] [PubMed] [Google Scholar]
- 9.Djukanovic R, Wilson JW, Britten KM, Wilson SJ, Walls AF, Roche WR, Howarth PH, Holgate ST. Quantitation of mast cells and eosinophils in the bronchial mucosa of symptomatic atopic asthmatics and healthy control subjects using immunohistochemistry. Am Rev Respir Dis. 1990;142:863–871. doi: 10.1164/ajrccm/142.4.863. [DOI] [PubMed] [Google Scholar]
- 10.Broide DH, Gleich GJ, Cuomo AJ, Coburn DA, Federman EC, Schwartz LB, Wasserman SI. Evidence of ongoing mast cell and eosinophil degranulation in symptomatic asthma airway. J Allergy Clin Immunol. 1991;88:637–648. doi: 10.1016/0091-6749(91)90158-k. [DOI] [PubMed] [Google Scholar]
- 11.Dahl R, Venge P, Olsson I. Variations of blood eosinophils and eosinophil cationic protein in serum in patients with bronchial asthma. Allergy. 1978;33:211–215. doi: 10.1111/j.1398-9995.1978.tb01536.x. [DOI] [PubMed] [Google Scholar]
- 12.Corrigan CJ, Hartnell A, Kay AB. T lymphocyte activation in acute severe asthma. Lancet. 1988;1:1129–1132. doi: 10.1016/s0140-6736(88)91951-4. [DOI] [PubMed] [Google Scholar]
- 13.Walker C, Virchow J, Bruijnzeel PLB, Blaser K. T cell subsets and their soluble products regulate eosinophilia in allergic and nonallergic asthma. J Immunol. 1991;146:1829–1835. [PubMed] [Google Scholar]
- 14.Ying S, Durham SR, Barkans J, Masuyama K, Jacobson M, Rak S, Lowhagen O, Moqbel R, Kay AB, Hamid QA. T cells are the principal source of interleukin-5 mRNA in allergen-induced rhinitis. Am J Respir Cell Mol Biol. 1993;9:356–360. doi: 10.1165/ajrcmb/9.4.356. [DOI] [PubMed] [Google Scholar]
- 15.Maggi E, Romagnani S. Role of T cells and T-cell-derived cytokines in the pathogenesis of allergic diseases. Ann NY Acad Sci. 1994;725:2–12. doi: 10.1111/j.1749-6632.1994.tb39784.x. [DOI] [PubMed] [Google Scholar]
- 16.Umetsu DT, DeKruyff RH. Th1 and Th2 CD4+T cells in the pathogenesis of allergic diseases. Proc Soc Exp Biol Med. 1997;215:11–20. doi: 10.3181/00379727-215-44109. [DOI] [PubMed] [Google Scholar]
- 17.Coffman RL, Seymour BWP, Hudak S, Jackson J, Rennick D. Antibody to interleukin-5 inhibits helminth-induced eosinophilia in mice. Science. 1989;245:308–310. doi: 10.1126/science.2787531. [DOI] [PubMed] [Google Scholar]
- 18.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin-5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kopf M, Brombacher F, Hodgkin PD, Ramsay AJ, Milbourne EA, Dai WJ, Ovington KS, Behm CA, Kohler G, Young IG, et al. IL-5 deficient mice have a development defect in CD5+ B-1 cells and lack eosinophils but have normal antibody and cytotoxic T cell responses. Immunity. 1996;4:15–24. doi: 10.1016/s1074-7613(00)80294-0. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi Y, Suda T, Suda J, Eguchi M, Miura Y, Harada N, Kitamura S, Tominaga A, Takatsu K. Purified interleukin-5 supports the terminal differentiation and proliferation of murine eosinophilic precursors. J Exp Med. 1988;167:43–56. doi: 10.1084/jem.167.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopez AF, Sanderson CJ, Gamble JR, Campbell HD, Young IG, Vadas MA. Recombinant human interleukin 5 is a selective activator of human eosinophil function. J Exp Med. 1988;167:219–224. doi: 10.1084/jem.167.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clutterbuck EJ, Hirst EM, Sanderson CJ. Human interleukin-5 (IL-5) regulates the production of eosinophils in human bone marrow cultures: comparisons and interactions with IL-1, IL-3, IL-6 and GM-CSF. Blood. 1989;73:1504–1512. [PubMed] [Google Scholar]
- 23.Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ. Cooperation between interleukin-5 and the chemokine eotaxin to induce eosinophil accumulation in vivo. J Exp Med. 1995;182:1169–1174. doi: 10.1084/jem.182.4.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baldwin AS., Jr The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 25.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 26.Siebenlist U, Franzoso G, Brown K. Structure, regulation and function of NF-κB. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 27.Blackwell TS, Christman JW. The role of nuclear factor–kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol. 1997;17:3–9. doi: 10.1165/ajrcmb.17.1.f132. [DOI] [PubMed] [Google Scholar]
- 28.Barnes PJ. Nuclear factor-kappa B. Int J Biochem Cell Biol. 1997;29:867–870. doi: 10.1016/s1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- 29.Sha WC. Regulation of immune responses by NF-κB/Rel transcription factors. J Exp Med. 1998;187:143–146. doi: 10.1084/jem.187.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-κB. EMBO (Eur Mol Biol Organ) J. 1991;10:3805–3817. doi: 10.1002/j.1460-2075.1991.tb04950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang S-M, Tran A-C, Grilli M, Lenardo MJ. NF-κB subunit regulation in nontransformed CD4+ T lymphocytes. Science. 1992;256:1451–1456. doi: 10.1126/science.1604322. [DOI] [PubMed] [Google Scholar]
- 32.Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-kB-mediated inhibition. Nature. 1992;359:339–342. doi: 10.1038/359339a0. [DOI] [PubMed] [Google Scholar]
- 33.Ray P, Yang L, Zhang D-H, Ghosh K, Ray A. Selective upregulation of cytokine-induced RANTES gene expression in lung epithelial cells by overexpression of IκBR. J Biol Chem. 1997;272:20191–20197. doi: 10.1074/jbc.272.32.20191. [DOI] [PubMed] [Google Scholar]
- 34.Ishikawa H, Claudio E, Dambach D, Raventos-Suarez C, Ryan C, Bravo R. Chronic inflammation and susceptibility to bacterial infection in mice lacking the polypeptide p105 precursor (NF-κB1) but expressing p50. J Exp Med. 1998;187:985–996. doi: 10.1084/jem.187.7.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 36.Sha WC, Liou H-C, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 37.Snapper CM, Zelazowski P, Rosas FR, Kehry MR, Tian M, Baltimore D, Sha WC. B cells from p50/ NF-kB knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription and Ig class switching. J Immunol. 1996;156:183–191. [PubMed] [Google Scholar]
- 38.Nakajima H, Iwamoto I, Tomoe S, Matsumura R, Tomioka H, Takatsu K, Yoshida S. CD4+T-lymphocytes and interleukin-5 mediate antigen-induced eosinophil infiltration into the mouse trachea. Am Rev Respir Dis. 1992;146:374–377. doi: 10.1164/ajrccm/146.2.374. [DOI] [PubMed] [Google Scholar]
- 39.Gavett SH, Chen X, Finkelman F, Wills-Karp M. Depletion of murine CD4+T lymphocytes prevents antigen-induced airway hyperreactivity and pulmonary eosinophilia. Am J Respir Cell Mol Biol. 1994;10:587–593. doi: 10.1165/ajrcmb.10.6.8003337. [DOI] [PubMed] [Google Scholar]
- 40.Corry DB, Folfesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, Locksley RM. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med. 1996;183:109–117. doi: 10.1084/jem.183.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cohn L, Homer RJ, Marinov A, Rankin J, Bottomly K. Induction of airway mucus production by T helper 2 (Th2) cells: a critical role for interleukin 4 in cell recruitment but not mucus production. J Exp Med. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kung TT, Jones H, Adams GK, III, Umland SP, Kreutner W, Egan RW, Chapman RW, Watnick AS. Characterization of a murine model of allergic pulmonary inflammation. Int Arch Allergy Immunol. 1994;105:83–90. doi: 10.1159/000236807. [DOI] [PubMed] [Google Scholar]
- 43.Ray P, Tang WL, Wang P, Homer R, Kuhn C, III, Flavell RA, Elias JA. Regulated overexpression of interleukin 11 in the lung: use to dissociate development-dependent and -independent effects. J Clin Invest. 1997;100:2501–2511. doi: 10.1172/JCI119792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong S, Guerder S, Visintin I, Reich E, Swenson KE, Flavell RA, Janeway CA., Jr Expression of the co-stimulator molecule B7-1 in pancreatic B cells accelerates diabetes in the NOD mouse. Diabetes. 1995;44:326–329. doi: 10.2337/diab.44.3.326. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalo JA, Lloyd CM, Kremer L, Finger E, Martinez AC, Siegelman MH, Cybulsky M, Gutierrez-Ramos JC. Eosinophil recruitment to the lung in a murine model of allergic inflammation. The role of T cells, chemokines, and adhesion receptors. J Clin Invest. 1996;98:2332–2345. doi: 10.1172/JCI119045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siegel MD, Zhang D-H, Ray P, Ray A. Activation of the interleukin-5 promoter by cAMP in murine EL-4 cells requires the GATA-3 and CLE0 elements. J Biol Chem. 1995;270:24548–24555. doi: 10.1074/jbc.270.41.24548. [DOI] [PubMed] [Google Scholar]
- 47.Zhang D-H, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. 1997;272:21597–21603. doi: 10.1074/jbc.272.34.21597. [DOI] [PubMed] [Google Scholar]
- 48.Zhang D-H, Yang L, Ray A. Differential responsiveness of the interleukin-5 (IL-5) and IL-4 genes to transcription factor GATA-3. J Immunol. 1998;161:3817–3821. [PubMed] [Google Scholar]
- 49.Liou H-C, Nolan GP, Ghosh S, Fujita T, Baltimore D. The NF-κB precursor, p105, contains an internal IκB-like inhibitor that preferentially inhibits p50. EMBO (Eur Mol Biol Organ) J. 1992;11:3003–3009. doi: 10.1002/j.1460-2075.1992.tb05370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inoue J-I, Kerr LD, Kakizuka A, Verma IM. IκBγ, a 70kd protein identical to the C-terminal half of p110 NF-κB: a new member of the IκB family. Cell. 1992;68:1109–1120. doi: 10.1016/0092-8674(92)90082-n. [DOI] [PubMed] [Google Scholar]
- 51.Iademarco MF, McQuillan JJ, Rosen GD, Dean DC. Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1) J Biol Chem. 1992;267:16323–16329. [PubMed] [Google Scholar]
- 52.Neish AS, Williams AJ, Palmer HJ, Whitley MZ, Collins T. Functional analysis of the human vascular cell adhesion molecule 1 promoter. J Exp Med. 1992;176:1583–1593. doi: 10.1084/jem.176.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen CC, Rosenbloom CL, Anderson DC, Manning AM. Selective inhibition of E-selectin, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 expression by inhibitors of I kappa B-alpha phosphorylation. J Immunol. 1995;155:3538–3545. [PubMed] [Google Scholar]
- 54.Read MA, Neish AS, Luscinskas FW, Palombella VJ, Maniatis T, Collins T. The proteasome pathway is required for cytokine-induced endothelial-leukocyte adhesion molecule expression. Immunity. 1995;2:493–506. doi: 10.1016/1074-7613(95)90030-6. [DOI] [PubMed] [Google Scholar]
- 55.Roebuck KA, Rahman A, Lakshminarayanan V, Janakidevi K, Malik AB. H2O2 and tumor necrosis factor-alpha activate intercellular adhesion molecule 1 (ICAM-1) gene transcription through distinct cis-regulatory elements within the ICAM-1 promoter. J Biol Chem. 1995;270:18966–18974. doi: 10.1074/jbc.270.32.18966. [DOI] [PubMed] [Google Scholar]
- 56.Shu HB, Agranoff AB, Nabel EG, Leung K, Duckett CS, Neish AS, Collins T, Nabel GJ. Differential regulation of vascular cell adhesion molecule 1 gene expression by specific NF-κB subunits in endothelial and epithelial cells. Mol Cell Biol. 1993;13:6283–6289. doi: 10.1128/mcb.13.10.6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahmad M, Marui N, Alexander RW, Medford RM. Cell type-specific transactivation of the VCAM-1 promoter through an NF-κB enhancer motif. J Biol Chem. 1995;270:8976–8983. doi: 10.1074/jbc.270.15.8976. [DOI] [PubMed] [Google Scholar]
- 58.Ledebur HC, Parks TP. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-κB site and p65 homodimers. J Biol Chem. 1995;270:933–943. doi: 10.1074/jbc.270.2.933. [DOI] [PubMed] [Google Scholar]
- 59.McCarty JM, Yee EK, Deisher TA, Harlan JM, Kaushansky K. Interleukin-4 induces endothelial vascular cell adhesion molecule-1 (VCAM-1) by an NF-κB-independent mechanism. FEBS Lett. 1995;372:194–198. doi: 10.1016/0014-5793(95)00976-g. [DOI] [PubMed] [Google Scholar]
- 60.Taub D, Conlon K, Lloyd AR, Oppenheim J, Kelvin D. Preferential migration of activated CD4+ and CD8+T cells in response to MIP-1α and MIP-1β. Science. 1993;260:355–358. doi: 10.1126/science.7682337. [DOI] [PubMed] [Google Scholar]
- 61.Vanguri P, Farber JM. Identification of CRG-2: an interferon-inducible mRNA predicted to encode a murine monokine. J Biol Chem. 1990;265:15049–15057. [PubMed] [Google Scholar]
- 62.MacLean JA, Ownbey R, Luster AD. T cell– dependent regulation of eotaxin in antigen-induced pulmonary eosinophilia. J Exp Med. 1996;184:1461–1469. doi: 10.1084/jem.184.4.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187:875–883. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1998;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 65.Gerber BO, Zanni MP, Uguccioni M, Loetscher M, Mackay CR, Pichler WJ, Yawalkar N, Baggiolini M, Moser B. Functional expression of the eotaxin receptor CCR3 in T lymphocytes co-localizing with eosinophils. Curr Biol. 1997;7:836–843. doi: 10.1016/s0960-9822(06)00371-x. [DOI] [PubMed] [Google Scholar]
- 66.Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, Sinigaglia F. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med. 1998;187:129–134. doi: 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Daugherty BL, Siciliano SJ, DeMartino J, Malkowitz L, Sirontino A, Springer MS. Cloning, expression and characterization of the human eotaxin receptor. J Exp Med. 1996;183:2349–2354. doi: 10.1084/jem.183.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ponath PD, Qin S, Post TW, Wang J, Wu L, Gerard NP, Newman W, Gerard C, Mackay CR. Molecular cloning and characterization of a human eotaxin receptor expressed selectively on eosinophils. J Exp Med. 1996;183:2437–2448. doi: 10.1084/jem.183.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gao J-L, Sen A, Tiffany L, Yoshi O, Rothenberg M, Murphy P, Luster A. Identification of a mouse eosinophil-selective receptor for the CC chemokine eotaxin. Biochem Biophys Res Commun. 1996;223:679–684. doi: 10.1006/bbrc.1996.0955. [DOI] [PubMed] [Google Scholar]
- 70.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 71.Jose PJ, Adcock IM, Griffiths-Johnson DA, Berkman N, Wells TNC, Williams TJ, Power CA. Eotaxin: cloning of an eosinophil chemoattractant cytokine and increased mRNA expression in allergen-challenged guinea-pig lungs. Biochem Biophys Res Commun. 1994;205:788–794. doi: 10.1006/bbrc.1994.2734. [DOI] [PubMed] [Google Scholar]
- 72.Rothenberg ME, Luster AD, Leder P. Murine eotaxin: an eosinophil chemoattractant inducible in endothelial cells and in interleukin 4-induced tumor suppression. Proc Natl Acad Sci USA. 1995;92:8960–8964. doi: 10.1073/pnas.92.19.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ponath PD, Qin S, Ringler DJ, Clark-Lewis I, Wang J, Kassam N, Smith H, Shi X, Gonzalo JA, Newman W, et al. Cloning of the human eosinophil chemoattractant eotaxin. Expression, receptor binding, and functional properties suggest a mechanism for the selective recruitment of eosinophils. J Clin Invest. 1996;97:604–612. doi: 10.1172/JCI118456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garcia-Zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med. 1996;2:449–456. doi: 10.1038/nm0496-449. [DOI] [PubMed] [Google Scholar]
- 75.Gonzalo JA, Jia GQ, Aguirre V, Friend D, Coyle AJ, Jenkins NA, Lin GS, Katz H, Lichtman A, Copeland N, et al. Mouse eotaxin expression parallels eosinophil accumulation during lung allergic inflammation but it is not restricted to a Th2-type response. Immunity. 1996;4:1–14. doi: 10.1016/s1074-7613(00)80293-9. [DOI] [PubMed] [Google Scholar]
- 76.Rothenberg ME, Ownbey R, Mehlhop PD, Loiselle PM, van de Rijn M, Bonventre JV, Oettgen HC, Leder P, Luster AD. Eotaxin triggers eosinophil-selective chemotaxis and calcium flux via a distinct receptor and induces pulmonary eosinophilia in the presence of interleukin 5 in mice. Mol Med. 1996;2:334–348. [PMC free article] [PubMed] [Google Scholar]
- 77.Sanz M-J, Ponath PD, Mackay CR, Newman W, Miyasaka M, Tamatani T, Flanagan BF, Lobb RR, Williams TJ, Nourshargh S, Williams TJ. Human eotaxin induces α4 and β2 integrin-dependent eosinophil accumulation in rat skin in vivo: delayed generation of eotaxin in response to IL-4. J Immunol. 1998;160:3569–3576. [PubMed] [Google Scholar]
- 78.Garcia-Zepeda EA, Rothenberg ME, Weremowicz S, Sarafi MN, Morton CC, Luster AD. Genomic organization, complete sequence, and chromosomal location of the gene for human eotaxin (SCYA11), an eosinophil-specific CC chemokine. Genomics. 1997;41:471–476. doi: 10.1006/geno.1997.4656. [DOI] [PubMed] [Google Scholar]
- 79.Humbles AA, Conroy DM, Marleau S, Rankin SM, Palframan RT, Proudfoot AE, Wells TN, Li D, Jeffery PK, Griffiths-Johnson DA, et al. Kinetics of eotaxin generation and its relationship to eosinophil accumulation in allergic airways disease: analysis in a guinea pig model in vivo. J Exp Med. 1997;186:601–612. doi: 10.1084/jem.186.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mould AW, Matthaei KI, Young IG, Foster PS. Relationship between interleukin 5 and eotaxin in regulating blood and tissue eosinophilia in mice. J Clin Invest. 1997;99:1064–1071. doi: 10.1172/JCI119234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Swain SL. Generation and in vivo persistence of polarized Th1 and Th2 cells. Immunity. 1994;1:553–552. doi: 10.1016/1074-7613(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 82.Sewell WA, Mu HH. Dissociation of production of interleukin-4 and interleukin-5. Immunol Cell Biol. 1996;74:274–277. doi: 10.1038/icb.1996.48. [DOI] [PubMed] [Google Scholar]
- 83.Doi S, Gemou-Engesaeth V, Kay AB, Corrigan CJ. Polymerase chain reaction quantification of cytokine messenger RNA expression in peripheral blood mononuclear cells of patients with acute exacerbations of asthma: effect of glucocorticoid therapy. Clin Exp Allergy. 1994;24:854–867. doi: 10.1111/j.1365-2222.1994.tb01808.x. [DOI] [PubMed] [Google Scholar]
- 84.Tendler CL, Burton JD, Jaffe J, Danielpour D, Charley M, McCoy JP, Pittelkow MR, Waldmann TA. Abnormal cytokine expression in Sezary and adult T-cell leukemia cells correlates with the functional diversity between these T-cell malignancies. Cancer Res. 1994;54:4430–4435. [PubMed] [Google Scholar]
- 85.Jung T, Schauer U, Rieger C, Wagner K, Einsle K, Neumann C, Heusser C. Interleukin-4 and interleukin-5 are rarely co-expressed by human T cells. Eur J Immunol. 1995;25:2413–2416. doi: 10.1002/eji.1830250843. [DOI] [PubMed] [Google Scholar]
- 86.Zheng W-p, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 87.Lee HJ, O'Garra A, Arai K-I, Arai N. Characterization of cis-regulatory elements and nuclear factors conferring Th2-specific expression of the IL-5 gene: a role for a GATA-binding protein. J Immunol. 1998;160:2343–2352. [PubMed] [Google Scholar]
- 88.Ho I-C, Hodge MR, Rooney JW, Glimcher LH. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996;85:973–983. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- 89.Rao A, Luo C, Hogan PG. Transcription factors of the NF-AT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 90.Ray P, Zhang D-H, Elias JA, Ray A. Cloning of a differentially expressed IκB-related protein. J Biol Chem. 1995;270:10680–10685. doi: 10.1074/jbc.270.18.10680. [DOI] [PubMed] [Google Scholar]