

Abstract
CD4+ T lymphocyte depletion in human immunodeficiency virus type 1 (HIV-1)–infected humans underlies the development of acquired immune deficiency syndrome. Using a model in which rhesus macaques were infected with chimeric simian–human immunodeficiency viruses (SHIVs), we show that both the level of viremia and the structure of the HIV-1 envelope glycoprotein ectodomains individually contributed to the efficiency with which CD4+ T lymphocytes were depleted. The envelope glycoproteins of recombinant SHIVs that efficiently caused loss of CD4+ T lymphocytes exhibited increased chemokine receptor binding and membrane-fusing capacity compared with those of less pathogenic viruses. These studies identify the HIV-1 envelope glycoprotein ectodomains as determinants of CD4+ T lymphocyte loss in vivo and provide a foundation for studying pathogenic mechanisms.
Keywords: simian–human immunodeficiency virus, Rhesus macaques, envelope glycoprotein, CD4+ T lymphocyte depletion, pathogenesis
Human immunodeficiency virus type 1 (HIV-1) is the cause of acquired immune deficiency syndrome (AIDS) in humans (1, 2). The related simian immunodeficiency viruses (SIVs)1 cause AIDS-like illness in Old World monkeys (3). Acute infection with the primate immunodeficiency viruses is typically characterized by a high level of viremia and a transient loss of CD4+ T lymphocytes (4, 5). This phase of the infection is followed by a reduction in the viral load and an increase in CD4+ T lymphocytes, sometimes to near normal levels. In the absence of antiretroviral therapy, a gradual decline in CD4+ T cells and eventual rise in viremia occurs, ultimately resulting in immunosuppression in the chronically infected subject. The mechanisms underlying the selective depletion of CD4+ T cells during acute and chronic HIV-1 infection are not well understood. However, studies of virus and cell turnover in HIV-1–infected individuals suggest that active viral gene expression is required for the rapid destruction of infected cells (6, 7). Direct viral cytopathic effects and cytotoxic antiviral immune responses have been suggested as potential mechanisms for CD4+ T lymphocyte destruction in HIV-1–infected humans.
A variety of in vitro cell culture systems have been used to study HIV-1–induced cytopathic effects, which consist of the formation of multinucleated giant cells (syncytia) and the lysis of single cells. The HIV-1 envelope glycoproteins have been shown to be responsible for both forms of cytopathic effects (8–11). The HIV-1 envelope glycoproteins, gp120 and gp41, support virus entry by binding to the viral receptors, CD4, and chemokine receptors on the target cell (12–20) and by fusing the viral and the target cell membranes. HIV-1 envelope glycoproteins expressed on the infected cell surface fuse these cells with receptor-bearing, uninfected cells, leading to the formation of lethal syncytia (8, 9). Intracellular envelope glycoprotein-receptor complexes are capable of mediating membrane fusion reactions that lead to the lysis of single cells (11).
The identification of HIV-1 genes that are important for CD4+ T lymphocyte depletion in the infected host could provide insights into pathogenic mechanisms. Theoretically, at least two factors contribute to pathogenic outcome in vivo: virus replication and a property herein called intrinsic pathogenicity. Each of these factors could be influenced by viral genetic polymorphism. Adequate levels of virus replication must be achieved to sustain CD4+ lymphocyte depletion in vivo (21–25). Decreases in the replication of primate immunodeficiency viruses in human or animal hosts have resulted from genetic alterations in the virus or host and from antiviral therapy, with a consequent reduction in the appearance or progression of disease. Since there are numerous ways by which virus replication can be reduced (25), identification of viral determinants of replicative ability does not necessarily provide clues to the mechanism of CD4+ T lymphocyte destruction. Intrinsic pathogenicity describes the ability of a virus to cause pathogenic consequences at a given level of virus replication. In this instance, two primate immunodeficiency viruses that differ in intrinsic pathogenicity would deplete CD4+ T lymphocytes to different degrees, even though virus replication is identical. Genetic alterations in a virus that affect intrinsic pathogenicity would thus alter the relationship between virus replication and CD4+ T lymphocyte depletion. The identification of HIV-1 genetic determinants of intrinsic pathogenicity should provide clues to the mechanism(s) underlying CD4+ T cell destruction.
In vivo studies of HIV-1 pathogenesis require suitable animal models, such as SIV infection of rhesus macaques (3, 23, 26) and HIV-1 infection of chimpanzees (27). Studies using rhesus macaques infected with SIVmac239 have provided several insights into lentivirus pathogenesis, but this model does not allow an analysis of the role of HIV-1–encoded gene products. The use of chimpanzees presents a problem due to the endangered status and expense of these animals.
To study the contribution of HIV-1 genes to in vivo infection and pathogenicity, simian–human immunodeficiency virus chimeras (SHIVs) have been constructed. SHIVs containing the HIV-1 tat, rev, vpu, and env genes in an SIVmac239 background have been shown to establish persistent infection in a number of Old World monkey species, including the readily available rhesus macaque (28–31). We have previously shown that SHIV-89.6, which contains the envelope glycoproteins of a primary HIV-1 isolate, replicated to high levels in rhesus macaques (32). Despite this high level of replication, CD4+ T lymphocyte counts remained normal and no disease was induced in the infected animals. To derive a pathogenic variant of SHIV-89.6, we subjected the virus to animal-to-animal passage (33). The resulting virus, SHIV-89.6P, caused rapid and severe depletion of CD4+ T lymphocytes in rhesus macaques within 2 wk after inoculation; furthermore, SHIV-89.6P–infected animals often exhibited AIDS-like illness within 1 yr of infection (33). Of interest was the observation that the in vivo viremia in SHIV-89.6P–infected animals during the first weeks of infection was not dramatically increased compared with that seen in SHIV-89.6–infected animals. This observation suggested the possibility that animal passage had resulted in an increase in the intrinsic pathogenicity of the virus.
To study the genetic basis of the increased pathogenicity of SHIV-89.6P, molecular proviral clones were obtained and analyzed. One SHIV-89.6P proviral clone, designated KB9, generated viruses that caused CD4+ T cell depletion in all animals inoculated (34). Compared with the parental SHIV-89.6, SHIV-KB9 contained nucleotide changes in the LTR and coding changes in the tat and env genes (34). Some of the env changes resulted in single amino acid changes in the gp120 and gp41 envelope glycoprotein ectodomains. In addition, a large env deletion resulted in a gp41 cytoplasmic tail containing elements of both HIV-1 and SIV gp41 cytoplasmic domains (34).
The availability of molecularly cloned nonpathogenic and pathogenic SHIVs presents an ideal opportunity to identify HIV-1 determinants of rapid CD4+ lymphocyte depletion in a setting where potentially confounding, specific antiviral immune responses are minimal. Here, chimeras between SHIV-89.6 and SHIV-KB9 were evaluated in rhesus monkeys for replication and in vivo CD4+ T cell– depleting ability during acute infection. Viral genetic determinants of replication and intrinsic pathogenicity were independently defined, and in vitro phenotypes associated with the identified segments were characterized.
Materials and Methods
Cells and Viruses.
Ficoll-enriched rhesus PBMCs were stimulated with 1 μg/ml phytohemagglutinin (Murex Diagnostics Ltd., Dartfield, UK) for 48–72 h and incubated with human IL-2 (10% IL-2; Hemagen Diagnostics Inc., Columbia, MD) beginning 24 h before infection. Rhesus macrophages were cultured as previously described (35).
The molecularly cloned parental virus, SHIV-89.6, and the in vivo–passaged variant, SHIV-KB9, have been described elsewhere (32, 34). Infectious virus was produced by ligation of the SphI-digested 5′ and 3′ proviral halves, followed by transfection of CEM×174 cells, as described previously (28). For this study, three additional 3′ proviral halves were constructed as follows. For the KB9ct 3′ proviral plasmid, the KpnI–XhoI fragment of SHIV-89.6 was cloned into the KB9 3′ proviral plasmid. For the 89.6* 3′ proviral plasmid, the KpnI–NcoI fragment of SHIV-89.6 was cloned into the KB9 3′ proviral plasmid to produce the LT plasmid. Then the KpnI–BamHI fragment of KB9ct was cloned into LT. For the KB9ecto 3′ proviral plasmid, the KpnI–BamHI fragment of KB9 was cloned into the 89.6* 3′ proviral plasmid. For generation of infectious SHIV-89.6 virus, the SHIV-89.6 3′ half was ligated to the SIVmac239 wt 5′ half. For the 89.6*, KB9ct, KB9ecto, and KB9 viruses, the appropriate 3′ proviral halves were ligated to the SIVmac239R 5′ proviral half. The SIVmac239R 5′ proviral half contains a single passage-associated nucleotide change in the R region of the LTR when compared with the SIVmac239 5′ proviral clone (34). Virus-containing supernatants from transfected CEM×174 cells were normalized according to reverse transcriptase (RT) activity and frozen for subsequent in vitro and in vivo infections.
Animal Inoculations.
Rhesus monkeys (Macaca mulatta) were inoculated intravenously with SHIV-KB9, SHIV-KB9ct, SHIV-KB9ecto, and SHIV-89.6* viruses using supernatants from transfected CEM×174 cells normalized for equivalent RT activity. The supernatants were harvested at peak replication in the CEM×174 culture and 1 ml of supernatant, ∼106 RT units, was used in each inoculation. The rhesus monkeys used in this study were maintained in accordance with the guidelines of the Committee on Animals for the Harvard Medical School and the Guide for the Care and Use of Laboratory Animals (NIH 85-23). Monkeys were anesthetized with ketamine-HCl for all blood sampling and inoculations, and with Telazol for biopsies.
Flow Cytometry.
Peripheral blood lymphocytes obtained from the infected animals were phenotyped for CD3/CD4 and CD3/ CD8, as previously described (36). Absolute lymphocyte counts in blood were determined on an automated hematology analyzer (T540; Coulter Corp., Hialeah, FL) that provided a partial differential count.
SIVmac p27 Assay.
The concentration of SIVmac p27 core antigen in heparinized plasma from SHIV-infected animals, or from rhesus macrophage cultures infected in vitro, was determined by using a commercial kit (SIV Core [p27] Antigen EIA kit; Coulter Corp.).
Plasma Viral RNA Quantitation.
Quantitative assays for the measurement of SHIV RNA were performed by Chiron Corp. (Emeryville, CA), using a branched DNA signal amplification assay for the SIV pol region similar to the Quantiplex HIV-RNA branched DNA assay (37). The assay results were quantified by comparison with purified and quantitated in vitro–transcribed SIV pol RNA.
In Situ Hybridization for SHIV RNA in Lymph Nodes.
In situ hybridization of lymph nodes obtained on days 10 and 21 after virus inoculation was performed, using two previously described permeabilization procedures (26, 38). Cells with at least 20 silver grains, which corresponded to a sixfold increase in silver grains over the background level, were scored as viral RNA–positive. By using epiluminescent illumination, viral RNA–positive cells in 10 standard areas (450 × 700 μm) of the T cell–dependent zones of the lymph nodes were counted with an ×20 objective and the means were calculated.
Immunohistochemistry of Lymph Node Sections.
Pieces of lymph nodes were embedded in OCT compound medium and snap-frozen in liquid nitrogen. Cryostat sections were cut under RNase-free conditions, fixed in 100% acetone for 30 min, and stored at −70°C until use. Before immunohistochemistry, the sections were postfixed in 2% paraformaldehyde (15 min at room temperature), washed in PBS, and incubated either with a pool of CD4 antibodies consisting of 3 μl Leu3a (Becton Dickinson, Heidelberg, Germany), 7.5 μl OKT4 (Ortho Diagnostic System, Neckargemünd, Germany), and 3.5 μl NCL CD4-1F6 (Newcastle-upon-Tyne, UK) or with Dako-CD68 (1:70 dilution; Dakopatts, Copenhagen, Denmark) for macrophages. Binding of primary antibodies was visualized by the alkaline phosphatase/anti–alkaline phosphatase technique using New Fuchsin as chromogen, and the sections were subjected to in situ hybridization to detect SIV RNA. The hybridizations on the frozen sections were performed as described above, with the exception that the permeabilization procedure was omitted and the exposure time was shorter (3 d at 4°C).
Statistics.
The goal of the statistical analysis was to identify plausible models for describing the role of viral replication and viral genetic determinants as predictors of CD4+ T cell depletion during the acute infection. As a measure of viral replication, the cumulative p27 antigenemia for days 0–21 was derived by estimating the area under the p27 versus time curve using the trapezoidal method. As a measure of CD4+ T lymphocyte levels, the median value of the absolute peripheral blood CD4+ T cell counts recorded from day 14 to 36 after inoculation was calculated. A two-by-two factorial design characterized the experiment. Two animals had cumulative p27 values below detectable limits (these animals were confirmed to be infected by their seropositivity). These animals were considered ineligible and were omitted from the analysis. Partial F tests were used to compare nested models within the same family. Because each animal's measurements were used in two models of CD4+ T lymphocyte counts and cumulative antigenemia, a Bonferroni correction was made to the type I error, and a two-sided significance level of 0.025 was used. The adjusted R2 statistic was used to characterize the proportion of variability explained by each model (39).
Plasmids Expressing Envelope Glycoproteins.
The pSVIIIenv plasmid expressing the HIV-1 89.6 envelope glycoproteins has been described elsewhere (40). An envelope expressor plasmid, designated pSVIIIenv-89.6tail, containing the modified COOH terminus of the KB9 gp41 envelope glycoprotein, was constructed using the BamHI–NcoI (blunted) fragment of KB9 cloned into the BamHI–XhoI (blunted)–digested pSVIIIenv-89.6 plasmid. The KpnI–BamI fragment of the 3′ proviral halves was cloned into pSVIIIenv-89.6tail (KB9, KB9ct) or into pSVIIIenv-89.6 (KB9ecto) to create the corresponding envelope expressor plasmids. These were designated: pSVIIIenv-KB9, pSVIIIenv-KB9ct, and pSVIIIenv-KB9ecto. For expression of gp120 in Drosophila Schneider 2 cells, sequences encoding the 89.6 and KB9 gp120 were PCR amplified and cloned into the pMt vector (41), introducing a stop codon that terminated the protein at the natural gp120/gp41 proteolytic cleavage site. The gp120 glycoproteins were produced and purified from stably transfected Drosophila cells, as previously described (19, 41).
In Vitro Infections.
Equivalent amounts (50,000 RT units) of virus from transfected CEM×174 cells were added to 3 × 106 cells (for CEM×174 and PBMCs) or to each well (for macrophages) and incubated overnight. The virus was washed out 1 d after the infection and cultures were monitored for RT activity or p27 production for the duration of the experiment.
Syncytium Formation Assays.
COS-1 cells were cotransfected with the pSVIIIenv plasmids and with a plasmid expressing the HIV-1 Tat protein. A portion of the cells was used to control for envelope glycoprotein expression by labeling with [35S]cysteine overnight and precipitating the cell lysates with serum from an HIV-1–infected individual. For the syncytium formation assays, envelope glycoprotein-expressing COS-1 cells were detached using 50 mM EDTA in PBS and plated at 150,000 cells/well (in 0.5 ml RPMI 1640 plus FBS) with 1.5 × 106 target CEM×174 cells (in 0.5 ml RPMI 1640 plus FBS) in duplicate in 48-well plates. Wells were scored for syncytia after 6 h of cocultivation at 37°C.
ELISA for Binding of Human Soluble CD4 to Monomeric gp120.
96-well plates were coated with 100 ng per well of human soluble (s)CD4 in 100 μl of 100 mM sodium carbonate/bicarbonate buffer, pH 9.6. Plates were incubated at 4°C for 24 h, washed five times with PBS containing 0.2% Tween (PBS/ Tween), and incubated an additional 24 h at 4°C with 300 μl blocking buffer (PBS containing 2% nonfat dried milk and 5% heat inactivated FBS). After removal of the blocking buffer, the plates were incubated for 1 h at room temperature with twofold serial dilutions of 89.6 or KB9 gp120 envelope glycoprotein in PBS/Tween, starting at a concentration of 9.4 × 10−8 M in a total volume of 100 μl. Plates were washed 10 times and incubated for 1 h at room temperature with primary antibody (rabbit anti-ΔV1/V2/V3 serum, pool III, diluted 1:1,000 in blocking buffer (Wyatt, R., unpublished observations). The plates were washed and incubated with anti–rabbit biotin (Amersham Biotech, Arlington Heights, IL), diluted 1:2,000 in blocking buffer overnight at 4°C. This was followed by washes and incubation with streptavidin–horseradish peroxidase (Pierce Chemical Co., Rockford, IL), diluted 1:2,000 in PBS/Tween for 30 min at room temperature. Plates were washed and incubated with 100 μl/well TMB substrate (Bio-Rad, Hercules, CA) and the reaction was stopped with 100 μl 180 mM HCl per well. The plates were read at 450 nm.
Binding of Human and Rhesus sCD4 to Oligomeric Envelope Glycoproteins.
COS-1 cells were transfected with pSVIIIenv-89.6, pSVIIIenv-KB9, or pSVIIIenv-368 D/R as described above. The pSVIIIenv-368 D/R plasmid encodes an HIV-1 envelope glycoprotein that is defective for CD4 binding (42) and was included in these experiments as a control for nonspecific binding. The envelope glycoprotein surface expression was analyzed by incubation of cells with serum from an HIV-1–infected individual, followed by washing, cell lysis, and precipitation with protein A–Sepharose. For the sCD4 binding assays, supernatants containing metabolically labeled human or rhesus soluble CD4 were produced in COS-1 cells and incubated with envelope glycoprotein–expressing cells. After washing, bound and unbound sCD4 were immunoprecipitated from lysed cells or supernatants, respectively, using a polyclonal antiserum against CD4.
Env Complementation Assay and Analysis of Coreceptor Usage.
Approximately 10,000 RT units of recombinant HIV-1 virions containing the different HIV-1 envelope glycoproteins were produced in COS-1 cells and incubated for 3 h at 37°C with Cf 2Th cells expressing human or rhesus CD4 and chemokine receptors, as previously described (16).
Chemokine Receptor–binding Experiments.
Binding of 89.6 and KB9 gp120 to human CCR5 was analyzed using L1.2 cells stably expressing human CCR5. Relative binding constants for the gp120 glycoproteins were determined by incubating iodinated macrophage inflammatory protein (MIP)-1β with the CCR5-L1.2 cells in the presence of increasing concentrations of unlabeled envelope glycoprotein competitor, as previously described (19). The YU2Δ1/2/3 glycoprotein was included as a negative control in the competition experiments (30), and unlabeled MIP-1β (R&D Systems, Minneapolis, MN) was included as a positive control.
Neutralization Assays with Antibodies, sCD4, or Plasma.
COS-1 cell supernatants containing recombinant virions (5,000 RT units) expressing different envelope glycoproteins were incubated with increasing concentrations of antibodies or sCD4 for 1 h at 37°C before the addition of CEM×174 cells. Medium was added 1 d after infection and cultures were incubated another 48 h before harvesting and analysis. Plasma samples obtained from infected animals were heat-inactivated for 15 min at 55°C and used at a 1:50 dilution for neutralization experiments.
Seroconversion.
Cell lysates from radiolabeled COS-1 cells transfected with pSVIIIenv-89.6, pSVIIIenv-KB9ct, pSVIIIenv-KB9ecto, and pSVIIIenv-KB9 were immunoprecipitated with 8 μl heat-inactivated plasma from infected animals obtained at different time points after virus inoculation. Emergence of antibodies recognizing the homologous envelope glycoproteins was detected by SDS-PAGE and autoradiography.
SHIV-specific Cytotoxic Effector Cell Responses.
Virus-specific cytolytic effector cell activity was determined as previously described (43), using autologous B lymphoblastic target cells infected with recombinant vaccinia viruses expressing the HIV-1 89.6 envelope glycoproteins (Dr. Dennis Panicali, Therion Biologics Corporation, Cambridge, MA).
Results
CD4+ T lymphocyte Counts and Viral Replication in Monkeys Infected with SHIV Variants.
To examine the role of specific envelope glycoprotein determinants in the rapid CD4+ T lymphocyte depletion seen in SHIV-KB9–infected monkeys, the in vivo replication and CD4+ T cell-depleting ability of four SHIV variants were studied. The starting virus for these studies was SHIV-89.6*, which contains only the passage-associated LTR and tat changes compared with the nonpathogenic SHIV-89.6 virus. The other three SHIV variants differed from SHIV-89.6* only in the sequences of the envelope glycoproteins (Fig. 1). SHIV-KB9 has both the passage-associated single amino acid changes in the gp120/gp41 ectodomains and the gp41 cytoplasmic tail change compared with SHIV-89.6*. SHIV-KB9ecto has only the ectodomain changes, whereas SHIV-KB9ct has only the gp41 cytoplasmic tail change.
Figure 1.

Genomic organization of SHIV variants. The SHIV proviral genome is shown on the top. Sequences from SIVmac239 are shown in gray boxes and sequences from HIV-1 are shown in white (47, 52). Enzyme restriction sites relevant for cloning of the SHIV variants are shown. The proviral structures of the SHIV variants used in this study are depicted beneath. SHIV-89.6* is identical to the parental SHIV-89.6, except that it contains the passage-associated tat and LTR changes (34). The other three variants differ from SHIV-89.6* only in the sequence of the envelope glycoproteins. SHIV-KB9 has the passage-associated amino acid changes in the gp120/gp41 ectodomains and the gp41 cytoplasmic tail change compared with SHIV-89.6*. SHIV-KB9ct has only the gp41 cytoplasmic tail change, and SHIV-KB9ecto has only the gp120/gp41 ectodomain changes.
A total of 28 monkeys (7 with each of the four SHIV variants) were inoculated intravenously with virus. The absolute numbers of CD4+ and CD8+ T lymphocytes and CD20+ B lymphocytes in the peripheral blood were determined at regular intervals after infection. As has been previously observed in SIV- and SHIV-infected monkeys (26, 33), most of the animals exhibited a transient, modest lymphopenia that involved all of the lymphocyte subsets examined (data not shown). Between wk 1 and 2 after infection, the CD8+ T lymphocyte and CD20+ B lymphocyte counts returned to normal levels. The CD4+ T lymphocyte counts exhibited different patterns, depending upon the infecting SHIV (Fig. 2 A). Consistent with previously published results (34), all monkeys inoculated with SHIV-KB9 exhibited severely depressed CD4+ lymphocyte levels, ranging from 30 to 220 cells per μl of blood by 14 d after infection. Animals inoculated with SHIV-89.6* showed no persistent loss of CD4+ T cells, indicating that the passage-associated tat and LTR changes are not sufficient to cause CD4+ T lymphocyte depletion. Inoculation with SHIV-KB9ct resulted in variable levels of CD4+ T cell depletion: three out of seven animals (13898, 18390, and 15655) maintained normal CD4+ T cell levels; moderate depletion was observed in two animals (15865 and 18284); and more severe depletion was seen in two animals (11796 and 13939). Inoculation with SHIV-KB9ecto resulted in less variability; all animals except two (14122 and 15669) exhibited severe CD4+ T cell depletion within 14 d of infection.
Figure 2.


Absolute CD4+ T cell counts and plasma p27 antigenemia in infected monkeys. (A) The absolute CD4+ T cell counts in the peripheral blood of animals infected with SHIV-89.6*, SHIV-KB9ct, SHIV-KB9ecto, and SHIV-KB9 during the first 36 d of infection are shown. (B) The p27 antigen levels in the plasma of SHIV-infected animals are shown.
To examine the in vivo replicative abilities of the four SHIV variants, the level of plasma p27 antigenemia during acute infection was examined (Fig. 2 B). The animals infected with SHIV-89.6* exhibited the lowest levels of replication (<0.55 ng/ml at peak viremia), whereas peak viremia in the SHIV-KB9ct-, SHIV-KB9ecto-, and SHIV-KB9-infected animals ranged between 0.3 and 6.0 ng/ml, 0.11 and 4.0 ng/ml, and 0.78 and 10.0 ng/ml, respectively. In 16 of the animals (4 from each group), the level of viremia on day 10 after infection was confirmed by measurement of plasma viral RNA. The latter values correlated almost precisely with those obtained using the p27 assay (Spearman r = 0.98, P < 0.0001).
The relationship between virus replication and CD4+ T lymphocyte depletion was examined. Within a group of monkeys infected with the same virus, the observed levels of replication helped to explain the degree of CD4+ T lymphocyte decline in the monkeys. For example, among the monkeys infected with SHIV-KB9ct, the two animals (11796 and 13939) that exhibited the most severe depletion of CD4+ T cells also exhibited the highest levels of replication. In the group infected with SHIV-KB9ecto, the two monkeys (14122 and 15669) that maintained normal CD4+ lymphocyte counts exhibited the lowest levels of replication. However, CD4 depletion in monkeys infected with different viruses often could not be explained simply by accounting for the level of virus replication. For example, all of the animals inoculated with SHIV-KB9 exhibited severe CD4+ T cell depletion, even those monkeys (13921, 15823, and 13950) in which the peak level of viremia was relatively low (between 0.5 and 2 ng/ml on day 10). Several animals (13916, 13945, 16155, and 15487) with low CD4+ T cell counts and low levels of replication were also present in the SHIV-KB9ecto group. In contrast, the animals in the SHIV-KB9ct group that replicated the virus in this range (animals 15865, 18390, 18284, and 15655) did not exhibit severe loss of CD4+ T cells. These data suggested the possibility that viruses with the KB9 envelope glycoprotein ectodomains might deplete CD4+ T cells more efficiently than a comparable number of viruses lacking the passage-associated changes in the envelope glycoprotein ectodomains.
The hypothesis that specific env determinants equip the SHIV variants with different intrinsic pathogenicity (i.e., the ability to deplete CD4+ T lymphocytes) was tested using statistical analysis. The contributions of the two viral envelope glycoprotein determinants, the ectodomain changes and the cytoplasmic tail change, were analyzed. To this end, several plausible models were investigated. These included a nonlinear exponential decay model with three covariates, a linear transformation of the exponential decay model, an inverse model, and a nonlinear hyperbolic model. Based on the F statistics, which describe goodness of fit, and the adjusted R2 value, which represents the proportion of variability in CD4+ T lymphocyte levels explained by the model, a nonlinear exponential decay model with parameters for cumulative p27 antigenemia, envelope glycoprotein changes (yes/no), and the interaction between cumulative p27 antigenemia and envelope glycoprotein changes was selected as “best”. Although extensive examination of residuals could not be undertaken due to the small sample size, a visual assessment of residuals did not reveal serious violations of model assumptions. The relationship between cumulative p27 antigenemia and CD4+ T lymphocyte counts for viruses that differ in either envelope glycoprotein ectodomain or cytoplasmic tail sequences is shown in Fig. 3, panels A and B, respectively. The curves shown are based on a nonlinear exponential decay model. Partial F tests revealed that ectodomain changes and their associated interaction contributed significantly to the model's explanatory power (P < 0.001), whereas the passage-associated cytoplasmic tail modification did not (P = 0.0532). Thus, for a given level of antigenemia, viruses with KB9 envelope glycoprotein ectodomains deplete CD4+ T lymphocytes more efficiently than viruses with wild-type 89.6 ectodomain sequences. In contrast, the KB9 cytoplasmic tail does not contribute to the intrinsic ability of the viruses to cause CD4+ T cell decline. Thus, although the presence of KB9 sequences in both envelope glycoprotein domains contributes to an increase in virus replication in vivo, only the passage-associated ectodomain changes increase the efficiency with which CD4+ T lymphocytes are depleted.
Figure 3.


Relationship between viremia and CD4+ T lymphocyte counts for SHIV variants with different envelope glycoprotein domains. A “set-point” value for the absolute CD4+ T lymphocyte count in each animal (the median of the CD4 counts recorded on days 14–36 after infection) was plotted against the cumulative p27 antigenemia (area under the p27 versus time curves in Fig. 2 B). The data were grouped according to the sequences (either wild-type 89.6 or containing the KB9 passage- associated changes) of the viral envelope glycoprotein ectodomains (A) or cytoplasmic tails (B). The curves are fitted to the data using a nonlinear exponential decay model.
Analysis of Virus-infected Cells in Lymph Nodes.
Previous studies demonstrated that at the peak of viremia in monkeys acutely infected by SIV or SHIV a large number of cells expressing viral RNA are present in the T cell–dependent areas of the lymph nodes (26, 32). This number diminishes rapidly so that by day 21 after infection, when the viremia has cleared, only a few infected cells can be identified in lymphoid organs. To examine the lymphatic tissues in the monkeys infected with the SHIV variants, lymph node sections obtained at days 10 and 21 after infection were probed for viral RNA expression by in situ hybridization. These studies focused on the T cell–dependent zones in the lymph nodes, since these areas harbor the majority of potentially infectable T lymphocytes. As was previously seen for SIV- or SHIV-infected monkeys (26, 32), the number of viral RNA–positive cells was significantly higher on day 10 than on day 21 for all of the SHIV variants (data not shown). Fig. 4 shows that there was a strong inverse correlation between the number of viral RNA–positive cells in the lymph nodes at day 10 and the peripheral blood CD4+ T lymphocyte counts. These results suggest that the lymph node sections sampled in this study were reasonably representative, and that CD4+ T lymphocyte depletion in this model is strongly influenced by the number of infected cells in the lymphoid organs.
Figure 4.

Relationship between infected lymph node cells and CD4+ T lymphocyte counts. The number of viral RNA–positive cells in the T cell zone of lymph nodes, which were taken from the animals at day 10 after inoculation, was compared with the “set point” values for the peripheral blood CD4+ T lymphocyte counts. The animal identification numbers for three of the data points discussed in the text are provided.
The number of CD4+ cells in the T cell–dependent zones of the lymph nodes from infected monkeys was determined by immunohistochemistry. There was a very strong correlation (Spearman r = 0.87, P < 0.001) between the number of CD4+ cells in the lymph node sections and the peripheral blood CD4+ lymphocyte counts at day 21 (data not shown). This observation is consistent with studies demonstrating that by day 14 after infection with uncloned SHIV-89.6P, CD4+ lymphocytes were depleted in all of the lymphoid organs examined at necropsy (Reimann, K., and N. Letvin, unpublished observations).
By combining immunohistochemistry with in situ hybridization for viral RNA, we attempted to estimate the percentage of virus-infected CD4+ cells in the lymph nodes of two SHIV-KB9–infected monkeys, animals 13921 and 13970 (Table 1). In both animals, the number of viral RNA-positive cells in a lymph node section was higher on day 10 after infection than on day 21. Some of the viral RNA–positive cells were CD4− and probably represent cells that had intrinsically low levels of CD4 expression or that had downregulated CD4 after infection. A technical limitation of the methodology is that, when viral RNA signals are very high, definitive assignment of the cell to either the CD4+ or CD4− subset is not possible. Even if one assumes that these uncharacterized cells were CD4+, only ∼4–5% of the CD4+ cells in the lymph node of monkey 13921 were viral RNA-positive 10 d after infection. By day 21, a continued decrease in the number of CD4+ cells in the T cell–dependent region of the lymph node was observed, and the percentage of CD4+ cells that were viral RNA–positive decreased to 0.4%. In monkey 13970, the number of CD4+ cells in the lymph nodes was only 10% of that seen in uninfected animals by day 10 after infection, and declined even further by day 21. Assuming that all of the cells with extremely high viral RNA signals were CD4+, up to 19% of the CD4+ cells in the lymph node section were viral RNA–positive at day 10 after infection. This value decreased to <3% by day 21 after infection. Thus, a minority of CD4+ cells in the lymph nodes of SHIV-KB9–infected animals were detectably viral RNA-positive at the time points examined, even when CD4+ lymphocyte depletion was extensive.
Table 1.
Expression of Viral RNA and CD4 in Cells in the T Cell–dependent Zone of Lymph Nodes
| Animal | Days after infection | Total CD4+ cells | CD4+ viral RNA+ cells | CD4− viral RNA+ cells | Uncharacterized cells with very high viral RNA | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 13921 | 10 | 267 (206–311) | 10.8 (8–16) | 1.8 (1–2) | 1.0 (0–2) | |||||
| 21 | 120 (93–154) | 0.5 (0–1) | 0.5 (0–1) | 0 | ||||||
| 13970 | 10 | 38 (13–82) | 2.0 (1–3) | 9.5 (6–14) | 5 (1–8) | |||||
| 21 | 17 (9–27) | 0.4 (0–1) | 1.4 (0–3) | 0 | ||||||
| Uninfected control | – | 409 (381–447) | ND | ND | ND | |||||
| Uninfected control | – | 386 (349–421) | ND | ND | ND |
The number of cells positive for viral RNA or CD4 expression in a standard field is reported. The numbers in parentheses represent the range of values observed.
Infectivity and Syncytium-forming Ability of SHIV Variants.
The in vitro replicative abilities of the SHIV variants were examined in primary rhesus monkey PBMCs and macrophages. SHIV-KB9 exhibited the most efficient replication in activated PBMC, closely followed by SHIV-KB9ecto (Fig. 5 A). SHIV-KB9ct showed increased levels of replication compared with the parental SHIV-89.6 and SHIV-89.6*, which replicated with identical efficiencies. The SHIV-KB9ct virus replicated less efficiently than either SHIV-KB9 or SHIV-KB9ecto. Thus, changes in both the gp120/gp41 ectodomains and the gp41 cytoplasmic domain of the passaged virus contribute to the increased replication of KB9 in cultured rhesus PBMCs. When the ability of the SHIV variants to replicate in rhesus macrophages was assessed, a modest increase was observed for SHIV-KB9 and SHIV-KB9ecto compared with the other variants. However, these levels of replication were barely detectable and were ∼10-fold lower than that observed for SIVmac316, a macrophage-tropic SIV included in the experiments as a positive control (data not shown).
Figure 5.
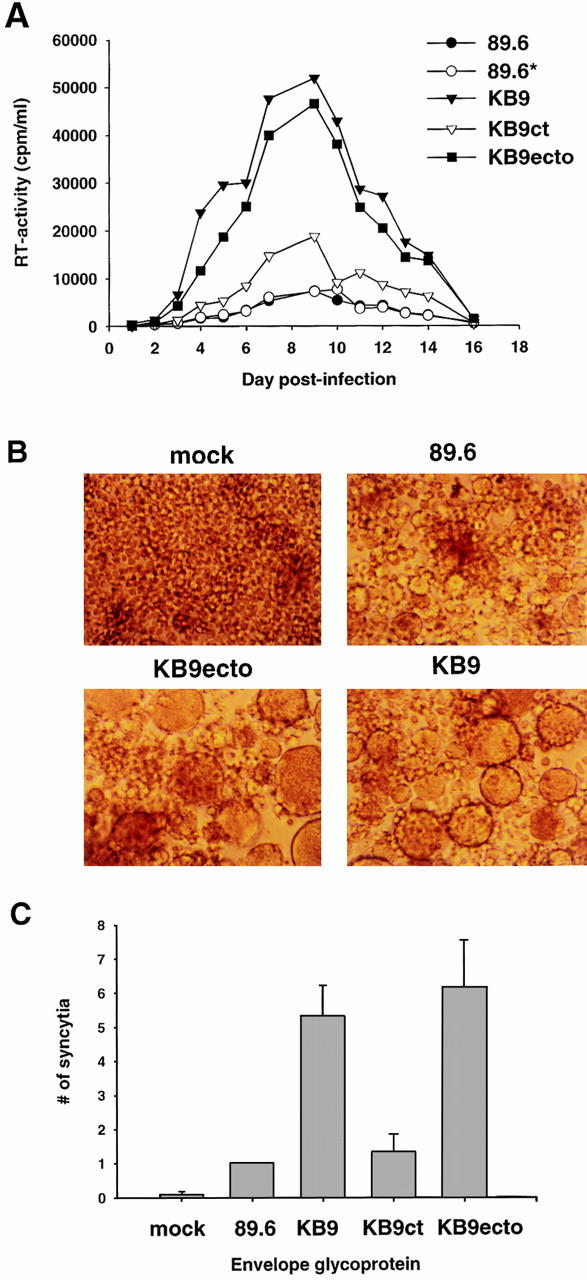
Replication and syncytium-forming ability of SHIV variants. (A) Rhesus PBMCs were PHA-stimulated and infected with equal amounts of SHIV-89.6, SHIV-89.6*, SHIV-KB9ct, SHIV-KB9ecto, and SHIV-KB9. Cells were maintained in the presence of IL-2 for the duration of the experiment and an aliquot of the culture medium was removed every day for RT analysis. (B) Uninfected CEM×174 cultures (mock) were compared with cultures infected with SHIV-89.6, SHIV-KB9ecto, and SHIV-KB9 for the presence of syncytia. (C) COS-1 cells, transiently expressing the 89.6, KB9ct, KB9ecto, or KB9 envelope glycoproteins, were cocultivated with CEM×174 cells for 6 h at 37°C. The number of syncytia was scored and normalized to that observed for the parental 89.6 envelope glycoproteins. The mean values and SE derived from three independent experiments are shown.
The replication of the SHIV variants in a human cell line, CEM×174, was also examined. Interestingly, SHIV-KB9 did not exhibit a replication advantage in this cell line (data not shown). In fact, the parental SHIV-89.6 replicated more efficiently than the other variants in CEM×174 cells. Apparently some of the replication phenotypes associated with the changes in SHIV-KB9 are influenced by host cell type. A consistent and striking increase in the number and size of syncytia was seen in the cultures infected with SHIV-KB9 and SHIV-KB9ecto, even when compared with cultures infected with the more efficiently replicating SHIV-89.6 (Fig. 5 B). In contrast, the syncytia in cultures infected with SHIV-89.6* and SHIV-KB9ct were similar in appearance to those in the SHIV-89.6-infected cells (data not shown). These observations suggested that the syncytium-forming ability of the viruses with the KB9 envelope glycoprotein ectodomains might be better than that of viruses with wild-type 89.6 ectodomains.
To analyze the fusogenic activity of the envelope glycoproteins in a more controlled fashion, syncytium formation assays were performed by cocultivation of COS-1 cells expressing the viral envelope glycoproteins with target CEM×174 cells. All of the envelope glycoproteins exhibited comparable processing of the gp160 envelope glycoprotein precursor, gp120-gp41 association and cell surface expression in transfected COS-1 cells (data not shown). Both the KB9 and KB9ecto envelope glycoproteins induced the formation of syncytia with approximately fivefold higher efficiency than the parental 89.6 envelope glycoproteins (Fig. 5 C). The KB9ct envelope glycoproteins mediated syncytium formation at an efficiency comparable to that seen for the 89.6 envelope glycoproteins. Thus, the KB9 envelope glycoprotein ectodomain sequences specify an increased fusogenic capacity.
Receptor Binding of Envelope Glycoproteins of SHIV Variants.
Two different assays were used to determine whether the KB9 envelope glycoproteins bind CD4 more efficiently than do the parental 89.6 envelope glycoproteins. First, binding of the monomeric gp120 envelope glycoproteins to sCD4 was compared using an ELISA. In brief, sCD4 was coated onto the ELISA plate and serially diluted 89.6 or KB9 gp120 was bound to the CD4, followed by detection with an anti-gp120 rabbit serum. The two gp120 glycoproteins bound sCD4 with comparable affinity (Fig. 6 A). To analyze the interaction of soluble CD4 with the oligomeric 89.6 and KB9 envelope glycoproteins, binding of metabolically labeled sCD4 to cells expressing the envelope glycoproteins was performed. In this assay, both human and rhesus monkey soluble CD4 bound the oligomeric 89.6 and KB9 envelope glycoproteins with similar affinity (data not shown). Thus, neither a generic increase in CD4-binding affinity nor a specific adaptation to rhesus CD4 appears to account for the enhanced fusogenic activity of the KB9 envelope glycoproteins.
Figure 6.


Receptor-binding of the 89.6 and KB9 gp120 envelope glycoproteins. (A) The binding of the 89.6 and KB9 gp120 glycoproteins to human sCD4 absorbed onto the surface of an ELISA plate is shown. Values represent the mean and standard deviations from two independent experiments, each containing four replicate samples. (B) The ability of the 89.6 and KB9 gp120 glycoproteins to compete with MIP-1β binding to mouse cells expressing human CCR5 was assessed in the presence of 100 nM sCD4. The results shown are representative of two independent experiments, each performed with duplicate samples at each concentration of competitor. Relative inhibitory constants for MIP-1β (positive control), 89.6 gp120, and KB9 gp120 were 0.20 nM, 39.0 nM, and 4.4 nM, respectively. The YU2ΔV1/2/3 glycoprotein, which lacks the V3 loop and thus is unable to bind CCR5 (19), was included as a negative control.
To assess whether SHIV-KB9 exhibited an altered ability, either qualitative or quantitative, to initiate infection via human and rhesus chemokine receptors, compared with SHIV-89.6, an env-complementation assay (16) was used. Recombinant HIV-1 viruses were produced by cotransfecting COS-1 cells with two plasmids, pHXBH10ΔenvCAT and pSVIIIenv. The pHXBH10ΔenvCAT plasmid contains an env-defective HIV-1 provirus that encodes chloramphenicol acetyltransferase (CAT); the pSVIIIenv plasmids encode either the 89.6 or KB9 envelope glycoproteins. Virus-containing COS-1 supernatants were normalized for RT activity and used for infection of Cf2Th canine thymocytes transiently expressing human or rhesus CD4, in conjunction with either human or rhesus monkey chemokine receptors. For all of the receptor combinations tested (human CD4 and either human CXCR4 or CCR5; rhesus CD4 and either rhesus CXCR4 or CCR5), viruses with the KB9 envelope glycoproteins were more efficient at mediating entry than were viruses with the 89.6 envelope glycoproteins (data not shown). Under conditions in which a relatively brief (3-h) incubation of virus with target cells was used, viruses with the KB9 envelope glycoproteins were approximately threefold more efficient than viruses with the 89.6 envelope glycoproteins. No evidence of specific adaptation to rhesus monkey receptors was observed.
Since the 89.6 envelope glycoproteins can use coreceptors in addition to CCR5 and CXCR4 (16, 18), the ability of the KB9 envelope glycoproteins to use a wider panel of seven transmembrane proteins was tested. This panel included coreceptors that support SIV but not HIV-1 entry (14, 44). Recombinant viruses with KB9 envelope glycoproteins were more efficient than viruses with the 89.6 envelope glycoproteins at infecting cells expressing CD4 and the human CCR2b, CCR3, and apj proteins (data not shown). The magnitude of this increase in efficiency was comparable to that seen with the CCR5 and CXCR4 coreceptors. Viruses with the KB9 and 89.6 envelope glycoproteins did not infect cells expressing CD4 and the known SIV coreceptors (STLR33, gpr15, and gpr1). Thus, the changes in the KB9 envelope glycoproteins specify a more efficient usage of several coreceptors used by the 89.6 envelope glycoproteins, but no qualitative changes in coreceptor usage were observed.
The increase in the efficiency with which the KB9 envelope glycoproteins, compared with the 89.6 envelope glycoproteins, mediate infection of cells bearing specific coreceptors might be due to a relative increase in binding to the coreceptors. To test this, the ability of purified 89.6 and KB9 gp120 glycoproteins to bind human CCR5 expressed on the surface of murine L1.2 cells was examined. An indirect competition assay (19, 20) was used, since this assay does not require iodination of the gp120 glycoproteins, which might introduce artifactual changes in affinity for chemokine receptors. Iodinated MIP-1β, a CCR5 ligand, was bound to L1.2-CCR5 cells in the presence of increasing concentrations of the KB9 and 89.6 gp120 glycoproteins. Since gp120 binding to CCR5 is greatly enhanced by prior CD4 binding (19, 20), an excess (100 nM) of sCD4 was included in these experiments. Unlabeled MIP-1β was included as a positive control, and the YU2ΔV1/2/3 protein as a negative control. The latter protein is a gp120 derivative that binds CD4 efficiently but is deficient in CCR5 binding due to deletion of the V3 loop (19). Fig. 6 B shows that, as expected, the unlabeled MIP-1β but not the YU2ΔV1/2/3 protein competed for the binding of radiolabeled MIP-1β to the L1.2-CCR5 cells. The KB9 gp120 competed more efficiently than did the 89.6 gp120. The inhibitory (K i) constants were 4.4 nM for the KB9 gp120 and 39.0 nM for the 89.6 gp120. Thus, the KB9 gp120 exhibited an approximately ninefold apparent increase in affinity for CCR5 compared with the parental 89.6 gp120.
Sensitivity of SHIV Variants to Neutralization.
Two of the three in vivo passages of SHIV-89.6 that gave rise to the pathogenic SHIV-89.6P were performed relatively late, at 12 and 14 wk after the infection (33). Therefore, some of the passage-associated amino acid substitutions in the KB9 envelope glycoproteins may have arisen in response to immune pressure from the hosts. The neutralization sensitivity of viruses with either the parental 89.6, the KB9 or the KB9ct envelope glycoproteins was tested. Since, like most primary HIV-1 isolates, viruses with the parental 89.6 envelope glycoproteins are relatively resistant to neutralization (40), a small panel of antibodies known to neutralize the 89.6 virus was tested. These included IgG1b12, an antibody directed against the CD4-binding site (45), and AG1121, a V3 loop–directed antibody. The sensitivity of the three viruses to neutralization by sCD4 was also examined. As shown in Fig. 7, viruses bearing the KB9 envelope glycoproteins were less sensitive to neutralization by both IgG1b12 and AG1121 antibodies, compared with viruses bearing the 89.6 or the KB9ct glycoproteins. All three viruses exhibited comparable sensitivity to neutralization by sCD4, indicating that HIV-1 can evolve to resist antibodies directed against receptor-binding regions of gp120 without significantly decreasing affinity for CD4.
Figure 7.


Neutralization of viruses with the 89.6, KB9ct and KB9 envelope glycoproteins. Recombinant viruses encoding CAT and bearing either the 89.6, KB9ct, or KB9 envelope glycoproteins were incubated with different concentrations of IgG1b12 (A), AG1121 (B), or sCD4 (C). The viruses were then incubated with CEM×174 cells. CAT activity in the CEM×174 cells was assessed 3 d later and is expressed as the percentage of CAT activity seen in the absence of antibody or sCD4. The results shown are representative of those obtained in at least two independent experiments.
Antiviral Immune Responses in SHIV-infected Animals.
To characterize the antibody responses in the infected monkeys, plasma samples from multiple time points after infection were used to precipitate the homologous envelope glycoproteins from radiolabeled COS-1 cell lysates. The development of a strong antibody response against the gp120 and/or gp160 envelope glycoproteins was observed in the majority of the animals ∼14–17 d after inoculation (data not shown). The monkeys with the most severe decline in CD4+ lymphocytes (13970, 13916, and 15431) failed to seroconvert during the first 71 d after infection. Neutralizing activity against recombinant viruses with the homologous envelope glycoproteins could be detected in the plasma of most infected animals by days 22–45 after inoculation (data not shown). Some of the monkeys (13950 and 13945) that seroconverted did not raise neutralizing antibodies. As expected, no neutralizing activity was detected in the plasma of monkeys that failed to seroconvert.
The cytotoxic T cell responses against the 89.6 envelope glycoproteins were assessed in four monkeys infected with SHIV-KB9ct and four monkeys infected with SHIV-KB9. Virus-specific cytotoxic T cell responses were measurable in three animals (two SHIV-KB9ct–infected monkeys and one SHIV-KB9–infected monkey) by 2 wk after inoculation and in all eight animals by 3 wk after inoculation (data not shown). There was no discernible difference in the magnitude of the response between the two groups of animals.
Discussion
The availability of molecularly cloned SHIVs that exhibit profound differences in the ability to induce CD4+ T lymphocyte loss and AIDS, while differing only moderately in proviral sequence (34), allowed an investigation of the molecular basis of the in vivo viral phenotypes. To this end, it was useful to distinguish viral genetic determinants of in vivo replicative ability from those viral elements that influence intrinsic pathogenicity, i.e., CD4+ T lymphocyte–depleting capacity. The rather modest differences in levels of viremia between the pathogenic SHIV-89.6P virus and the parental virus SHIV-89.6, combined with the large variation in virus replication in different monkeys inoculated with the identical virus, suggested that genetic determinants of both replication and intrinsic pathogenicity were operative in this system and could be defined.
In the pathogenic SHIV-KB9, changes both in the envelope glycoprotein ectodomains and cytoplasmic tail contributed to modest and additive increases in virus replication during the first weeks of infection in vivo. The envelope glycoprotein ectodomain changes associated with the pathogenic virus specify an increase in virus replication in cultured rhesus monkey PBMC. This increase is reflected in an increase in the efficiency with which the chemokine receptors used by the parental SHIV-89.6 virus are used by SHIV-KB9. No qualitative changes in chemokine receptor usage were observed between SHIV-89.6 and SHIV-KB9. At least part of the increased efficiency of virus entry associated with the SHIV-KB9 envelope glycoprotein ectodomains can be attributed to an increase in the affinity of gp120–CD4 complexes for the chemokine receptors. Although we could document this increased affinity in the only available quantitative binding assay, which uses CCR5 as a target (19), the increased efficiency of SHIV-KB9 entry into target cells expressing CXCR4, CCR3, and CCR2b raises the possibility that the KB9 gp120 exhibits an increased affinity for other coreceptors besides CCR5.
The gp41 cytoplasmic tail change associated with the pathogenic SHIV-KB9 is dramatic, involving the replacement of HIV-1 envelope glycoprotein sequences with those of SIV (34). This change apparently contributes to virus replication in vivo, and to modest increases in replication in cultured rhesus monkey PBMCs. The basis for these increases is uncertain. One obvious possibility is that the modified KB9 gp41 cytoplasmic tail is more compatible with the SIV matrix protein in the SHIV chimera. In viruses produced in CEM×174 cells, we did not observe any difference in the efficiency with which envelope glycoproteins containing KB9 cytoplasmic tails were incorporated into SHIV virions (Karlsson, G., and J. Sodroski, unpublished observations), an expected consequence of improved matrix–gp41 compatibility. It remains possible that such phenotypes are subtle and/or dependent on the virus-producing cell types.
The risk of clinical sequelae of HIV-1 infection in humans is highly dependent upon the absolute CD4+ T lymphocyte levels (46, 47). Likewise, SHIV-infected monkeys that develop AIDS-like illness and die within 12 mo of infection uniformly exhibit rapid and profound decreases in the number of CD4+ T lymphocytes in the peripheral blood and tissues in the first 3 wk after infection (33). Our results demonstrate that the structure of the SHIV envelope glycoprotein ectodomains determines the efficiency of CD4+ T lymphocyte depletion, beyond any effects on the overall pattern or level of viremia. Thus, HIV-1 envelope glycoprotein ectodomain sequences can apparently influence the intrinsic pathogenicity of SHIVs. In this study we identified two properties specified by the KB9 ectodomain sequences: an increase in membrane fusogenic capacity and a decreased sensitivity to antibody neutralization. The increase in fusogenic activity is probably closely related to the increase in virus entry discussed above and is at least in part a result of increased chemokine receptor binding. That more fusogenic envelope glycoprotein ectodomains arose in conjunction with the generation of a virus with improved capability of depleting CD4+ T lymphocytes in vivo is intriguing. The ability of the HIV-1 envelope glycoproteins to mediate membrane fusion has been strongly associated with both forms of viral cytopathic effects, single cell lysis and syncytium formation (8, 9, 11). Thus, at least in tissue culture systems, both the destruction of the infected cell and of receptor-bearing, uninfected cells is dependent on the efficiency with which the HIV-1 envelope glycoproteins fuse membranes.
Further work will be required to define precisely the mechanisms by which the envelope glycoprotein ectodomains modulate the efficiency of CD4+ T cell depletion. An understanding of pathogenic mechanisms in the SHIV-macaque model would benefit from knowledge of the relative involvement of infected and uninfected CD4+ T lymphocytes in T cell loss. Based on the resolution of the antigenemia, the average in vivo life span of cells producing any of the SHIV variants must be <3–4 d, consistent with values observed in HIV-1–infected humans and SIV-infected monkeys (6, 7, 48). Thus, although envelope glycoprotein ectodomain sequences could influence the rate of infected cell destruction, the eventual outcome of infection and virus production is cell death. This is consistent with the observation that all of the SHIV variants used in this study retain some degree of cytopathic potential and can elicit cytotoxic T cells, which will eventually destroy virus-producing cells not eliminated by viral killing mechanisms.
Our observation that similar degrees of CD4 T lymphocyte depletion occur in animals with a wide range of viral RNA–positive cells in the lymph nodes suggests that uninfected CD4+ T lymphocytes are destroyed in some SHIV-infected monkeys. If so, our data imply that a direct or indirect interaction with virus-producing cells drives this process or, if free virions contribute, that the envelope glycoprotein ectodomain sequences determine the efficiency of cell killing. A potentially relevant observation is that even when comparable numbers of viral RNA-positive cells were present in the lymph nodes, an animal (13930) infected with a KB9 ectodomain-containing virus exhibited much lower CD4+ T lymphocyte counts than animals (15655 and 18431) infected with viruses containing wild-type 89.6 ectodomains (Fig. 4). Perhaps under conditions where only low percentages of CD4+ T lymphocytes are infected, as in this example, the increased potential of envelope glycoproteins containing KB9 ectodomains to mediate the destruction of uninfected cells is manifest.
Since envelope glycoprotein structure could influence the generation of immune responses that might modulate the longevity of the infected cell, the cytotoxic T cell and antibody responses to the envelope glycoproteins in SHIV-infected animals were examined. The efficiencies with which Env-specific cytotoxic T lymphocytes were generated did not significantly differ among animals infected with SHIV variants differing in the envelope glycoprotein ectodomains. Since more monkeys infected with SHIVs containing the KB9 envelope glycoprotein ectodomains exhibited lower CD4+ lymphocyte counts, neutralizing antibody responses in this group were in some cases delayed or undetectable. Thus, an increase in the efficiency with which the envelope glycoproteins with KB9 ectodomains elicit immune responses is not the explanation for the in vivo properties related to CD4+ lymphocyte destruction specified by these sequences.
SHIVs with the KB9 envelope glycoprotein ectodomains are more resistant to neutralizing antibodies than are viruses with the wild-type 89.6 ectodomains. Although this ectodomain-specified property is likely to be important for the maintenance of a chronic infection and eventual disease induction, it seems less likely to contribute to the efficiency of acute CD4+ T lymphocyte depletion. Antibodies directed against the viral envelope glycoprotein were not detectable in the SHIV-infected monkey plasma until 2–3 wk after infection, after the course of CD4+ T lymphocyte decline was established. Neutralizing antibodies were not detected in any of the animals until well after CD4+ lymphocyte counts had reached their nadir. In monkeys exhibiting the greatest CD4+ T lymphocyte decline, virus-specific antibody responses were delayed or, in some cases, undetectable.
The studies reported herein implicate the HIV-1 exterior envelope glycoprotein domains in determining the efficiency of CD4+ T lymphocyte destruction in SHIV-89.6P– infected monkeys. These ectodomain sequences modulate the relationship between virus load and CD4+ T lymphocyte numbers in a manner distinct from that seen for viral sequences (e.g., the gp41 cytoplasmic tail) that influence only virus replication. This model system and approach could be used to address the contribution of other HIV-1 or SIV genes to CD4+ T lymphocyte destruction. A definition of genetic determinants of intrinsic pathogenicity is the foundation for an understanding of pathogenic mechanisms.
Acknowledgments
We thank Gudrun Großschupff and Birgit Raschdorff for excellent technical assistance. We thank Drs. David Camerini, Hyeryun Choe, Michael Farzan, Lijun Wu, Kathleen Martin, Paul Ponath, Dennis Burton, Qing Ma, Timothy Springer, Raymond Sweet, and Ronald Desrosiers for reagents, and Drs. Keith Reimann, Wolfgang Hofmann, and Carlo Rizzuto for helpful discussions. We thank Ms. Sheri Farnum and Yvette McLaughlin for manuscript preparation.
This work was supported by National Institutes of Health grants AI-20729, CA-50139, CA-06516, AI-33832, RR-07000, RR-00163, and RR-00168, and Center for AIDS Research Grant AI-28691; a grant from the German Ministry of Education and Research (BMBF 01 Kl 9714/6); support from the G. Harold and Leila Mathers Foundation; support from the Friends 10; and a gift from the late William F. McCarty-Cooper. Gunilla Karlsson was supported by a fellowship from Douglas and Judi Krupp.
Abbreviations used in this paper
- CAT
chloramphenicol acetyltransferase
- MIP
macrophage inflammatory protein
- RT
reverse transcriptase
- sCD4
human soluble CD4
- SHIV
simian–human immunodeficiency virus
- SIV
simian immunodeficiency virus
References
- 1.Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vezinet-Brun F, Rouzioux C, et al. Isolation of a T lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, Palker TJ, Redfield R, Oleske J, Safai B, et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science. 1984;224:500–503. doi: 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- 3.Letvin NL. Animal models for the study of human immunodeficiency virus infections. Curr Opin Immunol. 1992;4:481–485. doi: 10.1016/s0952-7915(06)80043-9. [DOI] [PubMed] [Google Scholar]
- 4.Daar ES, Moudgil T, Meyer RD, Ho DD. Transient high levels of viremia in patients with primary human immunodeficiency virus type 1 infection. N Engl J Med. 1991;324:961–964. doi: 10.1056/NEJM199104043241405. [DOI] [PubMed] [Google Scholar]
- 5.Clark SJ, Saag MS, Decker D, Campbell-Hill S, Robertson JL, Veldkamp PJ, Kappes JC, Hahn BH, Shaw GM. High titers of cytopathic virus in plasma of patients with symptomatic primary HIV-1 infection. N Engl J Med. 1991;324:954–960. doi: 10.1056/NEJM199104043241404. [DOI] [PubMed] [Google Scholar]
- 6.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 7.Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 8.Sodroski J, Goh WC, Rosen C, Campbell K, Haseltine WA. Role of the HTLV-III/LAV envelope in syncytium formation and cytopathicity. Nature. 1986;322:470–474. doi: 10.1038/322470a0. [DOI] [PubMed] [Google Scholar]
- 9.Lifson J, Feinberg M, Reyes G, Rabin L, Banapour B, Chakrabarti S, Moss B, Wong-Staal F, Steimer K, Engelman E. Induction of CD4-dependent cell fusion by the HTLV-III/LAV envelope glycoprotein. Science. 1986;323:725–728. doi: 10.1038/323725a0. [DOI] [PubMed] [Google Scholar]
- 10.Koga Y, Sasaki M, Nakamura K, Kimura G, Nomoto K. Intracellular distribution of the envelope glycoprotein of human immunodeficiency virus and its role in the production of cytopathic effect in CD4+ and CD4− human cell lines. J Virol. 1990;64:4661–4671. doi: 10.1128/jvi.64.10.4661-4671.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao J, Park I-W, Cooper A, Sodroski J. Molecular determinants of acute single-cell lysis by human immunodeficiency virus type 1. J Virol. 1996;70:1340–1354. doi: 10.1128/jvi.70.3.1340-1354.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984;312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 13.Dalgleish AG, Beverley PCL, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 14.Deng H, Unutmaz D, Kewal VN, Ramani, Littman DR. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature. 1997;388:296–300. doi: 10.1038/40894. [DOI] [PubMed] [Google Scholar]
- 15.Feng Y, Broder C, Kennedy P, Berger E. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein–coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 16.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, MacKay CR, Larosa G, Newman W, et al. The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 17.Alkhatib G, Combadiere C, Broder CC, Feng Y, Murphy PM, Berger E. CC-CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 18.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. A dual-tropic primary HIV-1 isolate that uses fusin and the β-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 19.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardins E, Newman W, et al. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 20.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. CD4-independent, antibody-sensitive interaction between HIV-1 and its co-receptor CCR-5. Nature. 1996;384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 21.Chakrabarti L, Cumont MC, Montagnier L, Hurtrel B. Variable course of simian immunodeficiency virus infection in lymph nodes: relation to disease progression. J Virol. 1994;64:6634–6642. doi: 10.1128/jvi.68.10.6634-6643.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mellors JW, Rinaldo C, Gupta P, White R, Todd J, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 23.Ruprecht RM, Baba T, Rasmussen R, Hu Y, Sharma P. Murine and simian retrovirus models: the threshold hypothesis. AIDS (London) 1996;10:S33–40. doi: 10.1097/00002030-199601001-00005. [DOI] [PubMed] [Google Scholar]
- 24.Lifson J, Nowak M, Goldstein S, Rossio J, Kinter A, Vasquez G, Wiltrout T, Brown C, Schneider D, Wahl L, et al. The extent of early viral replication is a critical determinant of the natural history of simian immunodeficiency virus infection. J Virol. 1997;71:9508–9514. doi: 10.1128/jvi.71.12.9508-9514.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desrosiers RC, Lifson J, Gibbs J, Czajak S, Howe A, Arthur L, Johnson RP. Identification of highly attenuated mutants of simian immunodeficiency virus. J Virol. 1998;72:1431–1437. doi: 10.1128/jvi.72.2.1431-1437.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reimann KA, Tenner-Racz K, Racz P, Montefiori DC, Yasutomi Y, Lin W, Ransil BJ, Letvin NL. Immunopathogenic events in acute infection of rhesus monkeys with simian immunodeficiency virus macaques. J Virol. 1994;68:2362–2370. doi: 10.1128/jvi.68.4.2362-2370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novembre FJ, Saucier M, Anderson DC, Klumpp SA, O'Neil SP, Brown CR, Jr, Hart CE, Guenthner PC, Swenson RB, McClure HM. Development of AIDS in a chimpanzee infected with human immunodeficiency virus type 1. J Virol. 1997;71:4086–4091. doi: 10.1128/jvi.71.5.4086-4091.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Lord CI, Haseltine W, Letvin NL, Sodroski J. Infection of cynomolgus monkeys with chimeric HIV-1/SIVmac virus that expresses the HIV-1 envelope glycoproteins. J Acquir Immune Defic Syndr. 1992;5:639–646. [PubMed] [Google Scholar]
- 29.Igarashi T, Shibata R, Hasebe F, Ami Y, Shinohara K, Komatsu T, Stahl-Hennig C, Petry H, Hunsmann G, Kuwata T, et al. Persistent infection with SIVmac chimeric virus having tat, rev, vpu, env, and nef of HIV type 1 in macaque monkeys. AIDS Res Hum Retrovirus. 1994;10:1021–1029. doi: 10.1089/aid.1994.10.1021. [DOI] [PubMed] [Google Scholar]
- 30.Luciw PA, Pratt-Lowe E, Shaw KES, Levy JA, Cheng-Mayer C. Persistent infection of rhesus macaques with T-cell-line–tropic and macrophage-tropic clones of simian/human immunodeficiency viruses (SHIV) Proc Natl Acad Sci USA. 1995;92:7490–7494. doi: 10.1073/pnas.92.16.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li JT, Halloran M, Lord CI, Watson A, Ranchalis J, Fung M, Letvin NL, Sodroski JG. Persistent infection of macaques with simian-human immunodeficiency virus. J Virol. 1995;69:7061–7071. doi: 10.1128/jvi.69.11.7061-7067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reimann KA, Li JT, Voss G, Lekutis C, Tenner-Racz K, Racz P, Lin W, Montefiori DC, Lee-Parritz DE, Lu Y, et al. An envgene derived from a primary human immunodeficiency virus type 1 isolate confers high in vivo replicative capacity to a chimeric simian/human immunodeficiency virus in rhesus monkeys. J Virol. 1996;70:3198–3206. doi: 10.1128/jvi.70.5.3198-3206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reimann KA, Li JT, Veazey R, Halloran M, Park I-W, Karlsson GB, Sodroski J, Letvin NL. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. J Virol. 1996;70:6922–6928. doi: 10.1128/jvi.70.10.6922-6928.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karlsson GB, Halloran M, Li J, Park I-W, Gomila R, Reimann K, Axthelm MK, Iliff SA, Letvin NL, Sodroski J. Characterization of molecularly cloned simian-human immunodeficiency viruses causing rapid CD4+ lymphocyte depletion in rhesus monkeys. J Virol. 1997;71:4218–4225. doi: 10.1128/jvi.71.6.4218-4225.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephens EB, Mukherjee S, Sahni M, Zhuge W, Raghavan R, Singh DK, Leung K, Atkinson B, Li Z, Joag SV, et al. A cell-free stock of simian-human immunodeficiency virus that causes AIDS in pig-tailed macaques has a limited number of amino acid substitutions in both SIVmac and HIV-1 regions of the genome and has altered cytotropism. Virology. 1997;231:313–321. doi: 10.1006/viro.1997.8534. [DOI] [PubMed] [Google Scholar]
- 36.Reimann KA, Waite BCD, Lee-Parritz DE, Lin W, Uchanska-Ziegler B, O'Connell MJ, Letvin NL. Use of human leukocyte-specific monoclonal antibodies for clinically immunophenotyping lymphocytes of rhesus monkeys. Cytometry. 1994;17:102–108. doi: 10.1002/cyto.990170113. [DOI] [PubMed] [Google Scholar]
- 37.Pachl C, Todd JA, Kern DG, Sheridan PJ, Fong SJ, Stempien M, Hoo B, Besemer D, Yeghiazarian T, Irvine B, et al. Rapid and precise quantification of HIV-1 RNA in plasma using a branched DNA signal amplification assay. J Acquir Immune Defic Syndr. 1995;8:446–454. doi: 10.1097/00042560-199504120-00003. [DOI] [PubMed] [Google Scholar]
- 38.Tenner-Racz K, Stellbrink H-J, van Lunzen J, Schneider C, Jacobs H-P, Raschdorff B, Großschupff G, Steinman RM, Racz P. The enlarged lymph nodes of HIV-1–infected asymptomatic patients with high CD4 T cell counts are sites for virus replication and CD4 T cell proliferation. The impact of highly active antiretroviral therapy. J Exp Med. 1998;187:949–959. doi: 10.1084/jem.187.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamilton, L. 1992. Regression with Graphics: A Second Course in Applied Statistics. Duxbury Press, Belmont, CA. 41.
- 40.Sullivan N, Sun Y, Li J, Hofmann W, Sodroski J. Replicative function and neutralization sensitivity of envelope glycoproteins from primary and T-cell line–passaged human immunodeficiency virus type 1 isolates. J Virol. 1995;69:4413–4422. doi: 10.1128/jvi.69.7.4413-4422.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Culp JS, Johansen H, Hellmig B, Beck J, Matthews TJ, Delers A, Rosenberg M. Regulated expression allows high level production and secretion of HIV-1 gp120 envelope glycoprotein in Drosophila Schneider cells. Biotechnology. 1991;9:173–177. doi: 10.1038/nbt0291-173. [DOI] [PubMed] [Google Scholar]
- 42.Olshevsky U, Helseth E, Furman C, Li J, Haseltine WA, Sodroski J. Identification of individual human immunodeficiency virus type 1 gp120 amino acids important for CD4 receptor binding. J Virol. 1990;64:5701–5707. doi: 10.1128/jvi.64.12.5701-5707.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voss G, Li J, Manson K, Wynand M, Sodroski J, Letvin NL. Human immunodeficiency virus type 1 envelope glycoprotein–specific cytotoxic T lymphocytes in simian-human immunodeficiency virus-infected rhesus monkeys. Virology. 1995;208:770–775. doi: 10.1006/viro.1995.1209. [DOI] [PubMed] [Google Scholar]
- 44.Farzan M, Choe H, Martin K, Marcon L, Hofmann W, Karlsson G, Sun Y, Barrett P, Marchand N, Sullivan N, et al. Two orphan seven-transmembrane segment receptors which are expressed in CD4-positive cells support simian immunodeficiency virus infection. J Exp Med. 1997;186:405–411. doi: 10.1084/jem.186.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burton DR, Pyati J, Koduri R, Sharp SJ, Thornton GB, Parren PWHI, Sawyer LSW, Hendry RM, Dunlop N, Nara PL, et al. Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science. 1994;266:1024–1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 46.Vanhems P, Allard R, Toma E, Cyr L, Beaulieu R. Prognostic value of the CD4+T cell count for HIV-1–infected patients with advanced immunosuppression. Int J STD AIDS. 1996;7:495–501. doi: 10.1258/0956462961918572. [DOI] [PubMed] [Google Scholar]
- 47.Mellors JW, Munoz A, Giorgi J, Margolick J, Tassoni C, Gupta P, Kingsley L, Todd J, Saah A, Detels R, et al. Plasma virus load and CD4+lymphocytes as prognostic markers of HIV-1 infection. Ann Intern Med. 1997;126:946–954. doi: 10.7326/0003-4819-126-12-199706150-00003. [DOI] [PubMed] [Google Scholar]
- 48.Nowak M, Lloyd A, Vasquez G, Wiltrout T, Wahl L, Bischofberger N, Williams J, Kinter A, Fauci A, Hirsch V, Lifson J. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J Virol. 1997;71:7518–7525. doi: 10.1128/jvi.71.10.7518-7525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]