

Abstract
Plasmodium falciparum causes the most severe form of human malaria, which kills ∼1.5–2.7 million people every year, but the molecular mechanisms underlying the clinical symptoms and the host–parasite interaction remain unclear. We show here that P. falciparum produces prostaglandins (PGs) D2, E2, and F2α. After incubation with 1 mM arachidonic acid (AA), cell homogenates of P. falciparum produced PGs as determined by enzyme immunoassay and gas chromatography–selected ion monitoring. PG production in the parasite homogenate was not affected by the nonsteroidal antiinflammatory drugs aspirin and indomethacin, and was partially heat resistant, whereas PG biosynthesis by mammalian cyclooxygenase was completely inhibited by these chemicals and by heat treatment. Addition of AA to the parasite cell culture markedly increased an ability of the parasite cell homogenate to produce PGs and of parasitized red blood cells to accumulate PGs in the culture medium. PGD2 and PGE2 accumulated in the culture medium at the stages of trophozoites and schizonts more actively than at the ring stage. These findings are the first evidence of the direct involvement of a malaria parasite in the generation of substances that are pyrogenic and injurious to the host defenses. We will discuss a possible contribution of the parasite-produced PGs to pathogenesis and host-parasite interaction of P. falciparum.
Keywords: Plasmodium falciparum, prostaglandin, malaria pathogenesis, tumor necrosis factor α, pyrogen
Malaria symptoms in an acute infection are general inflammatory responses, including periodic fever with shivering, headache, body pains, sleepiness, and loss of appetite, that are caused by the proliferating malaria parasite cells at the erythrocytic stage. During acute blood stage malaria, suppression of both T and B cell–mediated immune responses is a common occurrence (1–3), although the corresponding mechanism is not known, suggesting that malaria parasites have certain strategies for evasion of host defenses to establish the infection. Several papers have reported enhancement of the TNF-α level in malaria patient sera infected with Plasmodium falciparum and Plasmodium vivax, suggesting that TNF-α acts as an endogenous pyrogenic substance (4–6).
Prostaglandins (PGs) are metabolites of arachidonic acid (AA) via the cyclooxygenase pathway in mammals. They play versatile biological roles in maintaining the homeostasis of cell, tissue, and body. Among PGs, PGE2 is known to be a pyrogenic substance (7) as well as an immunosuppressor (8), and PGD2 is a somnogenic substance (9). They mediate inflammation and initiate physiological responses that are similar to the symptoms observed during malaria.
For many years, it has been thought that malaria symptoms are mostly, if not solely, mediated by PGs produced by host cells. We addressed the question whether or not P. falciparum produces PGs. Here we show the evidence that P. falciparum produces PGs in a way distinguishable from the mammalian system.
Materials and Methods
Culture and Preparation of Parasite Cells.
P. falciparum FCR3 and Honduras-1 strains were cultured as previously described (10) and modified by Sugiyama et al. (11) in a 5% O2 and 5% CO2 atmosphere with 3% (vol/vol) of type O RBCs in complete medium that consists of incomplete medium and 10% (vol/vol) heat inactivated type O human serum; incomplete medium is RPMI 1640 supplemented with 25 mM Hepes, 25 mM NaHCO3, 0.36 mM hypoxanthine, 3.4 mM glutamine, 10 μg/ml gentamycin, 100 U/ml penicillin, and 100 μg/ml streptomycin. Type O blood was freshly withdrawn into a tube containing citrate phosphate dextrose as anticoagulant. RBCs were washed three times with incomplete medium and cells were collected by centrifugation at 3,000 rpm for 15 min. White blood cells were not detected (<0.01%) in the prepared RBCs by visual microscopic inspection. The parasite cell culture was synchronized by sorbitol treatment as previously described (11). Infected RBCs were isolated by Percoll (Amersham Pharmacia Biotech Inc., Uppsala, Sweden) density centrifugation as described by Tosta et al. (12) with minor modifications. The cell suspension was overlaid on 3 vol of 63% (vol/vol) Percoll in PBS. After centrifugation at 1,800 g for 10 min at 25°C, the interphase that contained mainly schizonts and residual trophozoites was collected. The resulting cell fraction was diluted with 2 vol of PBS and centrifuged. The parasite cells were prepared by lysing the infected RBC membrane with 0.075% saponin (11). The resulting parasite cell preparation contained a small number of RBCs (0.5–0.9% of parasite cells), but white blood cells were not detectable (<0.01%). Infected RBCs and isolated parasite cells were suspended in 1.5 vol of PBS and frozen before homogenate preparation.
Preparation and Incubation of Parasite Cell Homogenates.
A suspension of 109 parasite cells in 0.3 ml of PBS was mixed with 5.7 ml of 100 mM sodium phosphate, pH 7.0, containing a protease inhibitor cocktail (Complete™; Boehringer Mannheim, Mannheim, Germany) and 600 mg of acid-washed glass beads (425– 600 μm; Sigma Chemical Co., St. Louis, MO) in a glass tube. The content of the tube was vigorously vortexed for 2 min and kept in an ice bath for 2 min. The cycle was repeated five times to disrupt the cells. The resulting suspension was used as the cell homogenates. The reaction mixture (500 μl) contained 100 mM sodium phosphate (pH 7.0), 2 mM hematin, 5 mM tryptophan, 1 mM AA, and 300 μl of cell homogenates. The mixture was incubated at 37°C for 30 min and the reaction was stopped by addition of 100 μl of 1 M HCl and 6 vol of cold ethyl acetate.
Extraction and Quantification of PGs.
3H-PGD2, 3H-PGE2, and 3H-PGF2α (60 Bq for each per assay; DuPont-NEN, Boston, MA) were added to each sample as tracer to determine the recovery of the following purification procedures. PGs produced in the homogenates (300 μl) and those in the culture medium (8 ml) were extracted three times with 6 vol of cold ethyl acetate and separated by HPLC as previously described (13). PGD2, PGE2, and PGF2α were quantified by enzyme immunoassay (EIA) by use of the respective EIA kits (Cayman Chemical Co., Ann Arbor, MI).
Gas Chromatography Mass Spectrometry Analysis.
Gas chromatography–selected ion monitoring (GC-SIM) analyses were run on a double focusing mass spectrometer (M-80B; Hitachi, Tokyo, Japan) equipped with a Van den Berg's solventless injector and a fused silica capillary column (Ultra#1; 25 m length, 0.32 mm internal diameter 280°C column temperature; Hewlett Packard, Palo Alto, CA). The parasite cells and the culture medium were prepared from the FCR3 strain grown in the medium containing 33 μM AA. The synchronized 150-ml culture at trophozoite and schizont stages was centrifuged at 1,800 g for 5 min at room temperature and washed with incomplete medium. Fresh complete medium containing 33 μM AA was added to the culture. After another 2-h incubation, the culture medium was taken and PGs were extracted. Then PGs in culture medium and PGs produced in the reaction by the parasite cell homogenates were fractionated by HPLC. The PGs obtained in the eluate were converted into their corresponding methyl ester–dimethylisopropylsilyl (ME-DMiPS) ether or ME-methoxime (MO)-DMiPS ether derivatives according to the method described previously, before applying on GC-SIM (14).
Results
Cell homogenates of RBCs infected with the FCR3 (Gambia) strain of P. falciparum and of the isolated parasite cells by saponin treatment produced significant amounts of PGD2, PGE2, and PGF2α after incubation with 1 mM AA at 37°C for 30 min (Fig. 1). PGD2 production in both homogenates is highest among three PGs. The amount of PGF2α produced by homogenate of isolated parasite cells was significantly more than that of infected RBCs. PG production in the parasite homogenates required exogenous addition of AA, i.e., no significant amounts of PGs were detected after incubation without AA or without incubation (Fig. 1). PG production was not observed in the homogenates of uninfected RBCs or with complete medium (Fig. 1).
Figure 1.

Production of PGs by homogenates of uninfected RBCs, infected RBCs, and parasite cells. Parasite cells and infected RBCs were isolated from trophozoite- and schizont-rich FCR3 cultures as described in Materials and Methods. Uninfected RBCs were from the same RBC source used for the parasite cultures. Each cell homogenate was incubated at 37°C for 30 min in the presence or absence of 1 mM AA. The amounts of PG produced by the cell homogenates were measured by EIA and standardized to 1 ml of packed cell volume. White, light gray, and dark gray bars represent PGD2, PGE2, and PGF2α, respectively.
To rule out the possibility that PG production is strain specific, PG production was examined in two parasite strains, FCR3 and Honduras-1. Fig. 2 a shows PG production was observed in both strains. In these parasite strains, the effect of exogenous addition of AA into culture medium was investigated. When parasite cells were grown in the medium supplemented with 33 μM AA for 48 h before harvest, PG production by the homogenates of FCR3 increased from 200 to 700 pg/108 cells for PGD2, and from 100 to 550 pg/108 cells for PGF2α whereas PGE2 production remained almost at the same level (120 pg/108 cells) (Fig. 2 a). The Honduras-1 cell homogenates also produced PGD2 and PGF2α (120 and 80 pg/108 cells, respectively), and the PG production increased to 630 and 330 pg/108 cells, respectively, by adding 33 μM AA to the culture.
Figure 2.

Effect of AA addition on the production of PG by P. falciparum strains. (a) PG production by cell homogenates. P. falciparum FCR3 and Honduras-1 strains were grown in medium with or without the addition of 33 μM AA for 48 h before harvesting. After incubation of the parasite homogenates with 1 mM AA at 37°C for 30 min, PGs produced were measured by EIA. The values shown are from three independent experiments with SE. In a typical experiment, FCR3 culture contained 0, 2.5, and 2.5% of RBCs infected with rings, trophozoites, and schizonts, respectively, and that of Honduras-1 contained 0.8, 3.3, and 1.4% of RBCs with rings, trophozoites, and schizonts, respectively. White, light gray, and dark gray bars represent PGD2, PGE2, and PGF2α, respectively. (b) PG release into the culture medium. FCR3 and Honduras-1 strain cultures initially contained 0.3% of RBCs infected with trophozoites and were cultivated for 48 h in medium (10 ml) with or without 33 μM AA. After 48 h, 0.06, 1.25, and 0.53% of RBCs in the FCR3 culture and 0, 1.1, and 0.1% of RBCs in the Honduras-1 culture were infected with rings, trophozoites, and schizonts, respectively. Cells were collected by centrifugation at 1,800 g for 5 min and washed twice with incomplete medium. Fresh medium containing 33 μM AA was added to the culture, and the parasite cells were incubated for another 2 h. The culture supernatant (8 ml) was then taken and centrifuged at 5,000 g for 30 min at 4°C to remove residual cells. The culture supernatants obtained were assayed for PGs by EIA.
Accumulation of PGs in the culture medium of P. falciparum was observed in both strains and this accumulation was also increased by the addition of AA to culture medium (Fig. 2 b). Culture supernatant of FCR3 and Honduras-1 strains cultured in the presence of 33 μM AA contained PGD2 (670 and 500 pg/ml, respectively) in the greatest amount, followed by PGE2 (370 and 330 pg/ml, respectively), and PGF2α (50 and 100 pg/ml, respectively) in lesser amounts. In the control reaction of uninfected RBCs preincubated with 33 μM AA for 48 h, PGs were not produced in the homogenates or in the medium (data not shown). These results indicate that the capacity to produce PGs of parasite cell was increased by cultivation with exogenous AA and that the produced PGs were accumulated in the culture medium.
To examine the stage-specificity of PG production, FCR3 strain cells were tightly synchronized with a life span of 4–6 h and the amounts of PG in culture medium were measured along with its development. Fig. 3 clearly shows that PGs accumulated in the medium at trophozoite and schizont stages more actively than at the ring stage.
Figure 3.
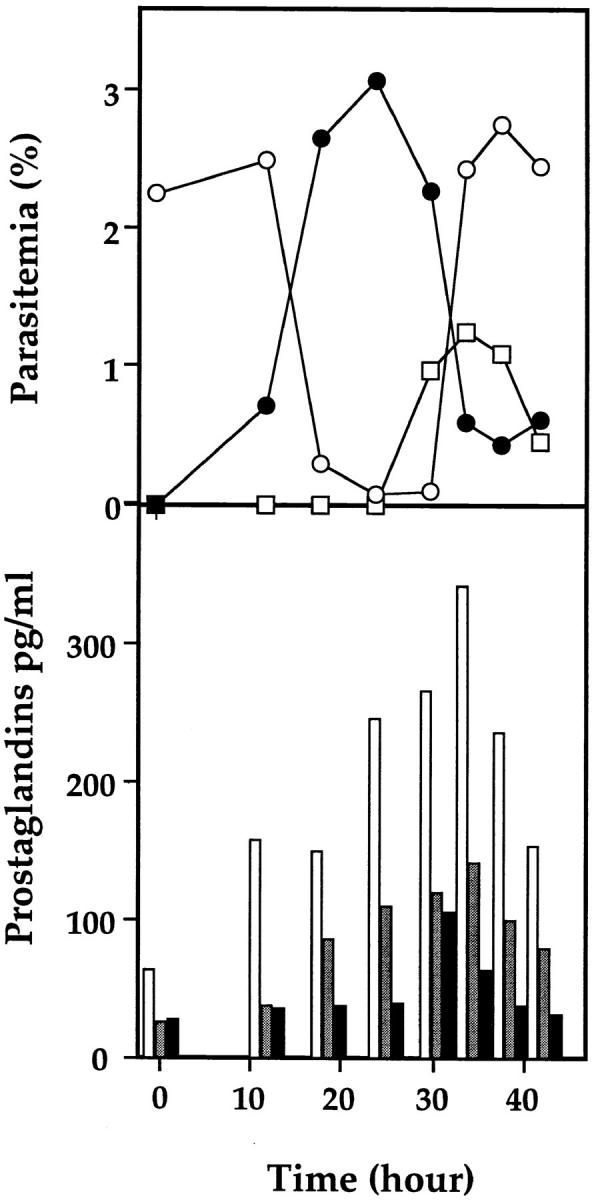
Time course of PG release from parasite cells. A ring-rich parasite cell culture (120 ml) of P. falciparum FCR3 strain was grown in complete medium containing 33 μM AA for 48 h. This culture was then treated with sorbitol and incubated for 30 h followed by a second sorbitol treatment to obtain synchronized early rings with a life span of 4–6 h after invasion of fresh RBCs. The synchronized 120-ml culture, supplemented with additional AA, was divided into 10 plastic plates. The time course was started after the last sorbitol treatment. The culture in each plate was centrifuged at 1,800 g for 5 min at room temperature at the indicated times and washed twice with incomplete medium. Fresh complete medium containing 33 μM AA was added to the culture, and the cultivation was continued for another 2 h. The culture supernatant (8 ml) was then collected by centrifugation at 5,000 g and stored at −80°C until PG quantification by EIA was performed. The Giemsa-stained parasite cells were counted microscopically. Open and filled circles and open squares represent ring, trophozoite, and schizont, respectively. White, light gray, and dark gray bars represent PGD2, PGE2, and PGF2α, respectively.
The molecular identity of parasite-produced PGs was supported by observations that each PG eluted at the same position as that of its authentic counterpart in HPLC used for purification of PGs before quantification by EIA and GC mass spectrometry (MS). In addition, parasite-produced PGs showed the exactly proportional titration curves in EIA to those of authentic PGs (data not shown). The molecular identities of PGs produced by the parasite were further confirmed by GC-SIM analysis. Analyses based on the use of SIM and the DMiPS ether derivatives revealed that all of the materials eluted from the HPLC had exactly the expected molecular masses (m/z 668 for PGF2α and 595 for PGD2 and PGE2) and their characteristic fragment ions (m/z 564, [M-31]+; 552, [M-43]+; and 524, [M-71]+ for PGD2 and PGE2; and m/z 625, [M-43]+; 597, [M-71]+; and 550, [M-118(DMiPSOH)]+ for PGF2α). Relative ion intensity ratios of individual derivatives of PGs in the eluate were identical to those of authentic standards. Fig. 4 shows representative selected ion recordings for the ME-MO-DMiPS ether derivatives of parasite-derived PGE2 compared with those of authentic PGE2. Taken together, we concluded that molecular structures of parasite-produced PGs were identical to those of authentic PGs.
Figure 4.

Selected ion recordings of PGE2 produced by the parasite cell homogenate. The HPLC-purified sample was dissolved in 15% EtOH and acidified with distilled HCl to pH 3. The resulting solution was subjected to Sep-Pak C18 cartridge (Waters Co., Milford, MA). The cartridge was subsequently washed with 15% EtOH and n-hexane, and then PGE2 was eluted with AcOEt. The resulting extract was methylated with freshly prepared ethereal diazomethane and then purified by means of silica gel column chromatography. Purified ME of PGE2 was converted to its MO-DMiPS ether derivative as previously described (14) and then subjected to GC-MS. Selected ion recordings monitoring its characteristic ions for PGE2, m/z 595, m/z 564, m/z 552, and m/z 524 are shown. The number indicates ion intensity for each peak. (A) Traces of the selected ion recordings of the ME-MO-DMiPS ether derivative of authentic PGE2. (B) Traces of the selected ion recordings of the ME-MO-DMiPS ether derivative of PGE2 produced in a reaction with parasite cell homogenate.
To exclude the possibility of contamination with mammalian cyclooxygenase, characteristics of the catalyst for production of PGs in the parasite were examined. Table 1 shows that the activity from parasite cells was only partially inhibited by treatment at 100°C for 10 min, and it was completely abolished by heating at 121°C for 90 min. Furthermore, PG production by parasite cell homogenates was not affected by 3 mM of aspirin or 42 μM of indomethacin. In contrast, PG production catalyzed by the cyclooxygenase preparation from sheep seminal vesicle was completely abolished by heat treatment (at 100°C for 10 min) and by these chemicals (Table 1). These results indicate that the catalytic substance(s) for PG production in P. falciparum is clearly different from the well-known enzymes involved in the mammalian arachidonate cascade system.
Table 1.
Effects of Nonsteroidal Antiinflammatory Drugs and Heat on PG Production by P. falciparum FCR3
| Enzyme source | Treatment | PGD2 | PGE2 | PGF2α (pg/assay) | ||||
|---|---|---|---|---|---|---|---|---|
| P.f. | None | 454 | 105 | 328 | ||||
| P.f. | Aspirin | 420 | 92 | 292 | ||||
| P.f. | Indomethacin | 408 | 100 | 279 | ||||
| P.f. | 100°C, 10 min | 272 | 79 | 213 | ||||
| P.f. | 121°C, 90 min, 1.1 atm | ND | ND | ND | ||||
| Sheep cox | None | 930 | 1150 | 180 | ||||
| Sheep cox | Aspirin | 48 | 53 | 12 | ||||
| Sheep cox | Indomethacin | 30 | 46 | 15 | ||||
| Sheep cox | 100°C, 10 min | ND | ND | ND |
The parasite cells (P.f.) were prepared from P. falciparum FCR3 strain grown in a medium containing 33 μM AA as described in Materials and Methods. The cyclooxygenase preparation of sheep seminal vesicle (sheep cox) was obtained from Eldan-Tech Ltd. (Israel). The parasite homogenates (5 × 107 cells) and sheep cyclooxygenase (300 ng protein) were incubated at 37°C for 30 min with 1 mM AA in the presence or absence of 3 mM aspirin or 42 μM indomethacin. Heat treatment was carried out at 100°C for 10 min and 121°C for 90 min before incubation with 1 mM AA. The amounts of PGs produced were measured by EIA. Detection limits were 25, 11, and 10 pg in each assay for PGD2, PGE2, and PGF2α, respectively. ND, not detected.
When homogenates of P. falciparum cultured with 33 μM AA were incubated with various concentrations of AA, PG production increased in a concentration-dependent manner (Fig. 5). However, the concentration-dependent curves are kinetically distinctive among PGs. PGD2 production almost linearly increased to 700 pg/108 cells with 1 mM AA, giving an apparent Km value for AA >1 mM, whereas PGE2 and PGF2α formation reached almost the maximum at ∼0.25 mM AA (180 and 550 pg/108 cells, respectively) and increased slightly with the higher concentrations, giving an apparent Km value for AA of ∼50 μM. Since the Km values for AA of the human cyclooxygenase-1 and -2 are reportedly 5–6 μM (15, 16), the catalytic substance(s) for PG production in P. falciparum is clearly different from the mammalian enzyme.
Figure 5.

AA dependency of PG formation in the cell homogenates of P. falciparum. P. falciparum FCR3 strain was grown in medium with the addition of 33 μM AA for 48 h before harvesting. The parasite homogenates were incubated at 37°C for 30 min with various concentrations of AA added exogenously. The amounts of PGs produced were quantified as described under Materials and Methods. Triangles, circles, and squares represent PGD2, PGE2, and PGF2α, respectively.
Discussion
We presented the evidence for the first time that P. falciparum produces PGD2, PGE2, and PGF2α in a way that is clearly distinguishable from the PG biosynthesis by mammalian cyclooxygenase. We also certified the molecular identity of P. falciparum produced PGs by GC-SIM. The finding of the production of PGs in a protozoan would shed light on the evolution of PGs and the possibility of novel pathways for their metabolism.
Since the catalysis for PG production in the parasite cell was partially heat stable, the catalytic mechanism may not involve the enzymatic activity. However, it is important to notice that the capacity of PG production was increased several-fold after cultivating the parasites with exogenously added AA (33 μM) in a concentration comparable to AA in blood stream (10 μM). Almost all of the parasite-produced PGs accumulated in the culture medium but not in the cell, because we could not detect PGs in the cell homogenate without incubation with exogenous AA. It was reported that arachidonic acid content in phospholipids of P. falciparum–infected RBC plasma membrane is much lower than that of normal RBC (7.85 and 17.36%, respectively) (17). Since Plasmodium is incapable of de novo biosynthesis of fatty acids (18), P. falciparum directly or indirectly uses an exogenous source of arachidonic acid.
The amount of PGF2α produced by parasite homogenates was higher than that of PGE2. However, culture medium of both strains contained low levels of PGF2α in comparison to those of PGE2. PGF2α might be degraded by parasite cells because after incubation of 3H-PGs (PGD2, PGE2, and PGF2α) with parasite cell homogenates, a recovered amount of 3H-PGF2α was decreased and a peak of unidentified degradation product was produced from only 3H-PGF2α (data not shown).
The concentration-dependent curves for AA of in vitro reaction with P. falciparum homogenates showed that productions of PGF2α and PGE2 were kinetically different from that of PGD2. Two distinct pathways may be involved in PG formation in P. falciparum homogenates, i.e., one produces PGF2α and PGE2; the other, PGD2. If any, the Km values for AA (∼50 μM and >1 mM, respectively) were much higher than those of the mammalian cyclooxygenase-1 and -2 (5–6 μM) (15, 16). Another possibility is that P. falciparum homogenates contain both production and degradation systems for PGs. In this case, the generation system may produce mainly PGE2 and PGF2α, and PGD2 only weakly, whereas PGE2 and PGF2α may be more actively degenerated than PGD2. However, we could not detect the degradation product of PGE2 by HPLC after incubation of the homogenates with [3H]-PGE2 (data not shown).
The malaria fever is coordinated with the schizont rupture associated with increased TNF-α level in the blood stream. It was shown that a glycosylphosphatidylinositol-like substance from malaria parasites stimulates the host TNF-α production (19). The malaria fever was, therefore, supposed to be caused by TNF-α after the induction of host PGE2 production. Our observation that parasite-produced PGs were accumulated in the culture medium at trophozoite and schizont stages raises a possibility that parasite-produced PGE2 and PGD2 directly contribute to fever and sleepiness as well.
Why does P. falciparum produce PGs? The most intriguing possibility is that the parasite-produced PGs modulates the host defense mechanism against malaria infection. For instance, an increased level of TNF-α was reported to suppress the parasite growth in vivo (20, 21). Lowering the host TNF-α production by parasite produced PGE2 would be beneficial to the parasite, since PGE2 regulates the level of TNF-α (22, 23). In the other parasitic organism, it was reported that microfilariae of Brugia malayi, intravascular nematode, generates and releases prostacyclin and PGE2 from arachidonic acid (24, 25). The reported PG synthesis is sensitive to indomethacin, unlike P. falciparum. These prostanoids may influence host immune and other cellular responses to ease the parasitism of microfilariae.
Further investigation into the contribution of parasite-produced PGs and catalysts in the clinical manifestation of this disease would provide a new means for controlling malaria.
Acknowledgments
We are grateful to Dr. J.E. Hyde for critical reading of the manuscript.
This work was supported by the Grant-in-Aid for Scientific Research on Priority Areas “Molecular Basis for Malaria Control” (08281104) from the Ministry of Education, Science, Sports, and Culture of Japan, and partly supported by Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation.
References
- 1.Ho M, Webster HK, Looareesuwan S, Supanaranond W, Phillips RE, Chanthavanich P, Warrell DA. Antigen-specific immunosuppression in human malaria due to Plasmodium falciparum. . J Infect Dis. 1986;153:763–771. doi: 10.1093/infdis/153.4.763. [DOI] [PubMed] [Google Scholar]
- 2.Riley EM, Andersson G, Otoo LN, Jepsen S, Greenwood BM. Cellular immune responses to Plasmodium falciparum antigens in Gambian children during and after an acute attack of falciparummalaria. Clin Exp Immunol. 1988;73:17–22. [PMC free article] [PubMed] [Google Scholar]
- 3.Whittle HC, Brown J, Marsh K, Blackman M, Jobe O, Shenton F. The effects of Plasmodium falciparummalaria on immune control of B lymphocytes in Gambian children. Clin Exp Immunol. 1990;80:213–218. doi: 10.1111/j.1365-2249.1990.tb05236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark, I.A., K.A. Rockett, and W.B. Cowden. 1992. TNF and malaria. In Tumor Necrosis Factors: The Molecules and Their Emerging Role in Medicine. B. Beutler, editor. Raven Press, Ltd., New York. 303–328.
- 5.Karunaweera ND, Grau GE, Gamage P, Carter R, Mendis KN. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivaxmalaria. Proc Natl Acad Sci USA. 1992;89:3200–3203. doi: 10.1073/pnas.89.8.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwiatkowski D, Cannon JG, Manogue KR, Cerami A, Dinarello CA, Greenwood BM. Tumour necrosis factor production in falciparummalaria and its association with schizont rupture. Clin Exp Immunol. 1989;77:361–366. [PMC free article] [PubMed] [Google Scholar]
- 7.Dinarello CA. Interleukins, tumor necrosis factors (cachectin), and interferons as endogenous pyrogens and mediators of fever. Lymphokines. 1987;14:1–31. [Google Scholar]
- 8.Goodwin JS, Ceuppens J. Regulation of the immune response by prostaglandins. J Clin Immunol. 1983;3:295–315. doi: 10.1007/BF00915791. [DOI] [PubMed] [Google Scholar]
- 9.Hayaishi O, Matsumura H. Prostaglandins and sleep. Adv Neuroimmunol. 1995;5:211–216. doi: 10.1016/0960-5428(95)00010-y. [DOI] [PubMed] [Google Scholar]
- 10.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 11.Sugiyama T, Suzue K, Okamoto M, Inselburg J, Tai K, Horii T. Production of recombinant SERA proteins of Plasmodium falciparum in Escherichia coliby using synthetic genes. Vaccine. 1996;14:1069–1076. doi: 10.1016/0264-410x(95)00238-v. [DOI] [PubMed] [Google Scholar]
- 12.Tosta CE, Sedegah M, Henderson DC, Wedderburn N. Plasmodium yoelii and Plasmodium berghei: isolation of infected erythrocytes from blood by colloidal silica gradient centrifugation. Exp Parasitol. 1980;50:7–15. doi: 10.1016/0014-4894(80)90003-x. [DOI] [PubMed] [Google Scholar]
- 13.Ujihara M, Urade Y, Eguchi N, Hayashi H, Ikai K, Hayaishi O. Prostaglandin D2formation and characterization of its synthetases in various tissues of adult rats. Arch Biochem Biophys. 1988;260:521–531. doi: 10.1016/0003-9861(88)90477-8. [DOI] [PubMed] [Google Scholar]
- 14.Miyazaki H, Ishibashi M, Yamashita K, Nishikawa Y, Katori M. Dimethylisopropylsilyl ether derivatives in gas chromatography mass spectrometry of prostaglandins and thromboxane B2. Biomed Mass Spectrom. 1981;8:521–526. [Google Scholar]
- 15.Barnett J, Chow J, Ives D, Chiou M, Mackenzie R, Osen E, Nguyen B, Tsing S, Bach C, Freire J, et al. Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochim Biophys Acta. 1994;1209:130–139. doi: 10.1016/0167-4838(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 16.Laneuville O, Breuer DK, Dewitt DL, Hla T, Funk CD, Smith WL. Differential inhibition of human prostaglandin endoperoxide H synthase-1 and -2 by nonsteroidal anti-inflammatory drugs. J Pharmacol Exp Ther. 1994;271:927–934. [PubMed] [Google Scholar]
- 17.Hsiao LL, Howard TJ, Aikawa M, Taraschi TF. Modification of host cell membrane lipid composition by the intra-erythrocytic human malaria parasite Plasmodium falciparum. . Biochem J. 1991;274:121–132. doi: 10.1042/bj2740121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holz GG., Jr Lipids and the malarial parasite. Bull WHO. 1977;55:237–248. [PMC free article] [PubMed] [Google Scholar]
- 19.Bate CA, Taverne J, Roman E, Moreno C, Playfair JH. Tumour necrosis factor induction by malaria exoantigens depends upon phospholipid. Immunology. 1992;75:129–135. [PMC free article] [PubMed] [Google Scholar]
- 20.Clark IA, Hunt NH, Butcher GA, Cowden WB. Inhibition of murine malaria (Plasmodium chabaudi) in vivoby recombinant interferon-gamma or tumor necrosis factor, and its enhancement by butylated hydroxyanisole. J Immunol. 1987;139:3493–3496. [PubMed] [Google Scholar]
- 21.Taverne J, Sheikh N, de Souza JB, Playfair JH, Probert L, Kollias G. Anaemia and resistance to malaria in transgenic mice expressing human tumour necrosis factor. Immunology. 1994;82:397–403. [PMC free article] [PubMed] [Google Scholar]
- 22.Kunkel SL, Spengler M, May MA, Spengler R, Larrick J, Remick D. Prostaglandin E2regulates macrophage-derived tumor necrosis factor gene expression. J Biol Chem. 1988;263:5380–5384. [PubMed] [Google Scholar]
- 23.Renz H, Gong J-H, Schmidt A, Nain M, Gemsa D. Release of tumor necrosis factor-alpha from macrophages. Enhancement and suppression are dose-dependently regulated by prostaglandin E2and cyclic nucleotides. J Immunol. 1988;141:2388–2393. [PubMed] [Google Scholar]
- 24.Liu LX, Serhan CN, Weller PF. Intravascular filarial parasites elaborate cyclooxygenase-derived eicosanoids. J Exp Med. 1990;172:993–996. doi: 10.1084/jem.172.3.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu LX, Weller PF. Arachidonic acid metabolism in filarial parasites. Exp Parasitol. 1990;71:496–501. doi: 10.1016/0014-4894(90)90076-o. [DOI] [PubMed] [Google Scholar]