

Abstract
The transcription factor NF-κB is sequestered in the cytoplasm by the inhibitor proteins of the IκB family. Each member of the IκB exhibits structural and biochemical similarities as well as differences. In an effort to address the functional redundancy of two closely related IκB molecules, IκBα and IκBβ, we generated knock-in mice by replacing the IκBα gene with the IκBβ gene. The knock-in mice do not express IκBα, but express a T7-tagged IκBβ under the promoter and regulatory sequence of ikba. Unlike the IκBα-deficient mice, which display severe postnatal developmental defects and die by postnatal day 8, homozygous knock-in mice survive to adulthood, are fertile, and exhibit no apparent abnormalities. Furthermore, thymocytes and embryonic fibroblasts from the knock-in animals exhibit an inducible NF-κB response similar to that of wild-type animals. These results indicate that IκBα and IκBβ share significant similarities in their biochemical activity, and that they acquired their different functions from divergent expression patterns during evolution.
Keywords: nuclear factor κB, IκB, transgenic mice, knockout mice, hematopoiesis
Nuclear factor κB (NF-κB)1 plays an important role in regulating genes involved in inflammatory and immune responses. In vertebrates, NF-κB consists of homo- or heterodimers of the subunits p50, p52, RelA, RelB, and cRel. These subunits share a highly conserved NH2-terminal sequence termed the Rel-homology domain (RHD), which is required for DNA binding, dimerization, nuclear localization, and interaction with the inhibitor IκB molecules (for review see references 1–5). In resting cells, NF-κB is sequestered in the cytoplasm by the inhibitor IκBs. Cytoplasmic retention is achieved via interaction between a conserved sequence motif known as the ankyrin repeats in the IκBs and the RHDs of NF-κB subunits. Upon cell stimulation by a variety of agents, specific serine residues on the IκBs are phosphorylated, signaling for ubiquitination which in turn targets IκBs for proteasome-mediated degradation. NF-κB released from the inhibitor translocates into the nucleus and activates transcription of target genes, which include those involved in inflammatory, immune, and acute phase responses.
In mammalian cells, the IκB family consists of IκBα, IκBβ, IκBε, Bcl-3, p105, and p100 (for review see references 2–4). Knockout mice studies have revealed the critical roles that some of these IκB molecules play in development and immune responses. For example, the knockout mice of IκBα have increased basal NF-κB activity in hematopoietic organs. Extensive granulopoeisis, dermatitis, and death by postnatal day 8–10 are observed in these mutant animals (5, 6). In contrast, p100-deficient mice displayed gastric hyperplasia and an impaired proliferative response in lymphocytes (7). Bcl-3–deficient mice develop normally but are incapable of antigen-specific antibody response (8, 9). These differences in phenotypes suggest that each IκB family member plays a unique and nonredundant role in regulating NF-κB activity in the hematopoietic system.
Among the IκB members, IκBα and IκBβ are the most prominent and have been extensively characterized. IκBα and IκBβ were first identified as two fractions from HeLa extracts that had similar kinetics of activities (10). In addition to the ankyrin motif, IκBα and IκBβ also have similar phosphoacceptor sites in their NH2 termini that mediate signal-induced degradation and a similar specificity of interaction with the RelA and cRel complexes (11, 12). Given these similarities, it seemed surprising that IκBβ could not compensate for IκBα in the IκBα-deficient mice. Two potential explanations are: first, in spite of the similarities in structure, these two proteins have different in vivo biochemical activities and, therefore, they cannot replace each other's function. Alternatively, they are in vivo biochemically equivalent, but differential expression patterns have made them functionally incapable of compensating for one another. In an attempt to discriminate between these two possibilities, we have replaced IκBα with IκBβ using homologous recombination in embryonic stem cells in order to determine whether IκBα can be functionally replaced by IκBβ. The targeting event brought the integrated IκBβ gene under the control of the ikba promoter and at the same time introduced a null mutation in ikba. Here we report that the “knock-in” mice develop to adulthood without apparent abnormalities, are fertile, have no increase in their basal NF-κB activity, and can elicit NF-κB responses. These observations contrast strongly with those obtained with the IκBα-deficient mice, which present extensive granulopoeisis, dermatitis, and death by postnatal day 8–10 (5, 6). Thus, our data demonstrate that in vivo IκBα can be functionally replaced by IκBβ, and that these two molecules have acquired different functions by differential tissue pattern expressions and responses to NF-κB inducers.
Materials and Methods
Generation of the IκBα Knock-in Mice.
To generate mice with the replacement of IκBα by IκBβ, the targeting vector pPNT-abki was constructed using the pPNT vector (13). Murine ikba and ikbb genomic clones were used for vector construction. A 5′ 4.7-kb NotI–SalI and a 3′ 9.5-kb HindIII–NotI restriction fragment of ikba genomic sequence were cloned into the respective sides of the neo cassette in the pPNT targeting vector. A pair of oligonucleotide linkers containing the bacterial phage T7 Tag, and a 9.5-kb NcoI–SalI genomic restriction fragment containing the entire coding sequence of ikbb, were inserted downstream of the translation initiation of ikba. To ensure efficient translation initiation from the T7 Tag sequence, the endogenous translational initiation sequence of ikbb was deleted. Additionally, to ensure that the presence of the pgk-neo cassette would not interfere with transcription of the recombined ikbb, loxp sites were inserted flanking the 5′ and 3′ ends of the pgk-neo cassette to allow cyclic AMP responsive element–mediated excision of this marker. Since our studies (see below) indicated that transcription of the knock-in allele was not affected, the pgk-neo cassette was not removed subsequently. The resulting vector pPNT-abki was linearized by NotI and electroporated into murine CJ7 embryonic stem (ES) cells. Neomycin- and gancyclovir-resistant cells were selected and screened by Southern blot analysis using a 3′ external probe of the ikba gene. DNA from a homologously recombined locus yields an 11-kb EcoRV fragment, whereas the wild-type DNA yields a 15-kb fragment. In addition, hybridization using several internal probes, including neomycin, confirmed that the entire genomic region of the IκBβ gene was integrated into the IκBα locus. Correctly targeted ES cells were injected into blastocysts or aggregated with morula of ICR mice. Male chimeras were mated with ICR females to obtain germline transmission of the mutated allele.
Western Blot Analysis, Electrophoretic Mobility Shift Assay, and Immunoprecipitation.
Single cell suspensions of thymocytes or splenocytes were prepared from 4–6-wk-old mice according to standard procedures (14) in RPMI 1640 containing 10% heat-inactivated FCS. Cells were incubated with 20 ng/ml of PMA and 1 μg/ml of PHA, or 5 ng/ml mouse TNF at 37°C for the indicated periods of time before harvest. Preparation of cytoplasmic and nuclear extracts was performed as previously described (15). For immunoprecipitation, a total of 5 × 105 ES cells or 6 × 106 thymocytes were incubated with 0.5 mCi/ml of [35S]methionine in methionine-free DMEM containing 10% dialyzed FCS for 8 h at 37°C. Cells were lyzed in radioimmunoprecipitation assay buffer and immunoprecipitation was performed as previously described (16). Western blot analysis using equal amounts of cytoplasmic extracts was carried out using standard protocols. For electrophoretic mobility shift assay (EMSA), nuclear extracts were tested for binding to the palindromic κB-specific probe (17) or an octamer-specific probe (18).
Histology and Flow Cytometry Analysis.
Mouse tissues were immersion fixed in 10% buffered formalin and embedded in paraffin blocks. Sections were stained with hematoxylin and eosin. Flow cytometry analysis with single cell suspension from thymus, spleen, and bone marrow was performed using commercially available antibodies with a flow cytometer (Becton Dickinson, San Jose, CA). 3 × 105 cells were first incubated with 1 μg of the antibodies and then incubated with PE- or FITC-conjugated antibodies.
Results
IκBα Knock-in Mice.
The murine IκBα gene was replaced by the IκBβ gene in ES cells by homologous recombination with the gene-targeting vector pPNT-abki (Fig. 1 A). The targeting vector deleted the entire coding sequence of the IκBα gene and replaced it with the genomic sequence encoding the murine IκBβ gene. To distinguish the IκBβ introduced by homologous recombination from the endogenous IκBβ, a 34-bp nucleotide sequence encoding the bacterial phage T7-Tag was placed in front of the ATG start codon of the IκBβ gene in the targeting vector. This targeting vector was transfected into CJ7 ES cells, and ES cell clones carrying the replacement of the endogenous IκBα gene were identified by Southern blot analysis (Fig. 1 B). The resulting knock-in allele was abbreviated as +/ki or ki/ki for heterozygotes or homozygotes, respectively.
Figure 1.
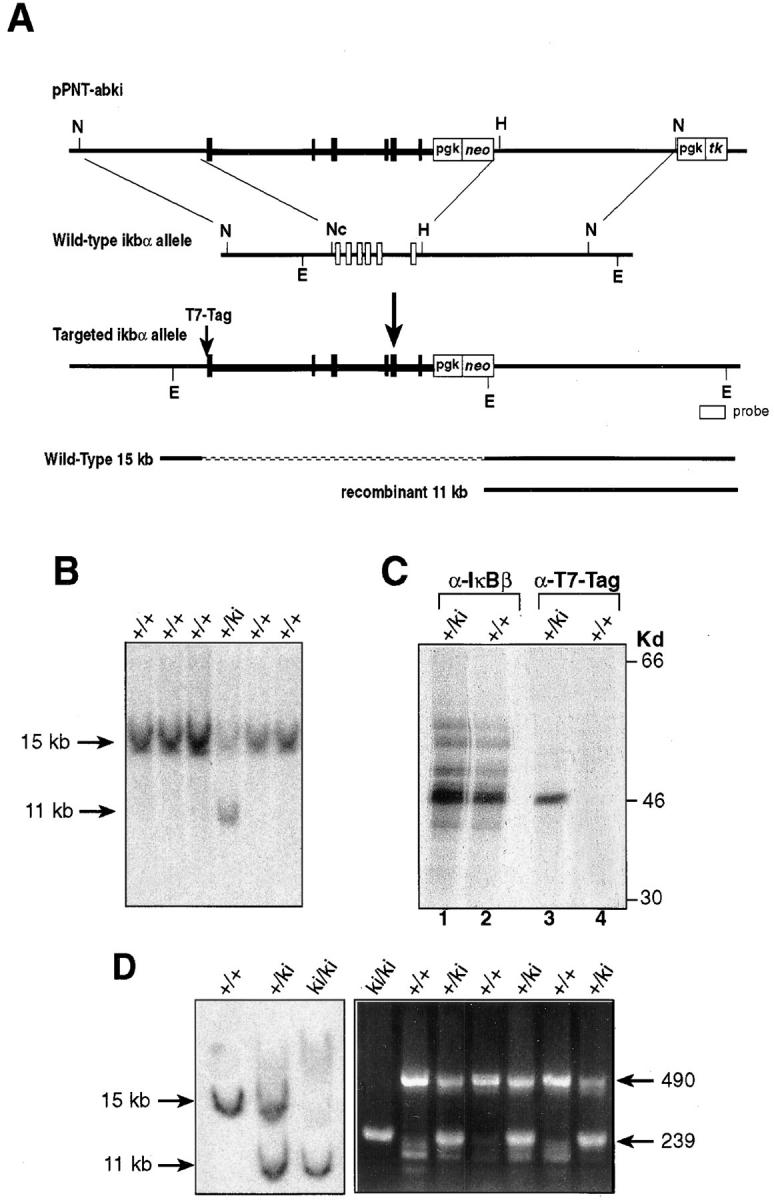
Targeted replacement of IκBα with the IκBβ gene. (A) The targeting vector pPNT-abki is shown at the top. Black boxes and thickened lines represent IκBβ exons and introns. Open boxes and thin lines represent IκBα exons and introns. Homologous recombination takes place between the 5′ and 3′ flanking regions of the ikba gene, resulting in replacement of the entire IκBα locus by IκBβ. Additionally, the ATG start codon of the IκBβ was also replaced with the T7-Tag sequence. N, NotI; Nc, NcoI; H, HindIII; E, EcoRV. (B) Southern blot analysis of DNA isolated from ES cell lines. EcoRV-digested DNA from cells that have undergone homologous recombination yielded an 11-kb signal when hybridized to an external probe. (C) Immunoprecipitation analysis of recombinant knock-in ES cells. Equal numbers of wild-type and +/ki ES cells were labeled with [35S]methionine and immunoprecipitated with antiserum against IκBβ (lanes 1 and 2) or T7-Tag (lanes 3 and 4). (D) Analysis of homozygous mouse tail DNA. (Left) Southern blot analysis of wild-type, heterozygous, and homozygous mouse tail DNA; (right) PCR analysis of tail DNA using IκBα primers and neomycin primers. Amplification of IκBα yields a 490-bp PCR product, whereas amplification of the neomycin gene yields a 239-bp PCR product.
To ensure that the homologous recombination resulted in replacement of the IκBα gene with a functional IκBβ gene, an immunoprecipitation experiment was performed in the ES cells. Extracts from [35S]methionine labeled wild-type (+/+) and recombined (+/ki) ES cells were immunoprecipitated with antiserum against IκBβ (Fig. 1 C, lanes 1 and 2), and then with antiserum against the T7-Tag epitope (Fig. 1 C, lanes 3 and 4). The T7-Tag antiserum precipitated a protein that is similar in molecular weight to the IκBβ protein in the recombinant ES cells but not in the wild-type cells, indicating that the introduced IκBβ was expressed and can be distinguished from the endogenous IκBβ using antibodies against the T7-Tag epitope.
The Knock-in Animals Develop Normally and Present no Changes in NF-κB Activity.
Aggregation chimeras giving germline transmission were obtained from two targeted ES cell lines. Both lines of heterozygous animals (+/ki) were crossed to obtain homozygous mice (ki/ki). Homozygous ki/ki mice were born in normal Mendelian ratio, indicating that replacement of IκBα with IκBβ did not affect embryonic development (Fig. 1 D).
In contrast to the previously described IκBα-deficient mice, which have severe developmental defects and die by postnatal day 8 (5, 6), both lines of the ki/ki mice developed to adulthood. Examination of 5-wk-old ki/ki mice revealed that the phenotype was both grossly and histologically similar to that of wild-type mice. Additionally, peripheral blood cells and serum biochemical analysis were also similar to those from wild-type animals. In particular, there was no evidence of granulopoiesis or epidermal dysplasia with hyperkeratosis as reported in the IκBα-deficient mice (data not shown). The lack of granulopoiesis was further confirmed by flow cytometric analysis of bone marrow from wild-type and knock-in animals using the granulocyte/macrophage-specific surface markers Mac-1 and Gr-1. Additionally, a normal distribution of B and T lymphocyte markers (CD4, CD8, TCRab, Thy1.2, CD25, B220, and IgM) and erythroid marker (Ter119) was observed, indicating that the lymphoid and erythroid compartments of these animals are also normal (data not shown).
To determine if replacement of IκBα by IκBβ affected the overall patterns of IκB and NF-κB gene expression, Western blot analysis was performed. Splenocytes from wild-type (+/+), heterozygous (+/ki), and homozygous (ki/ki) animals were analyzed. No differences in the steady-state levels of p105, p100, p50, and RelA were found between wild-type and knock-in splenocyte extracts (Fig. 2 A). As expected, IκBα protein was absent in the homozygous ki/ki animals, whereas the T7-Tag antibody detected a protein that migrated to the identical position as the IκBβ protein in +/ki and ki/ki splenocyte extracts. Consistent with the presence of extra IκBβ molecules in the heterozygous and homozygous animals, an increase was observed in the level of IκBβ protein in these animals compared with wild-type animals.
Figure 2.

Expression of NF-κB/IκB proteins and NF-κB binding activity in the knock-in mice. (A) Western blot analysis of splenocytes from wild-type, heterozygous, and homozygous mice with antisera against members of the NF-κB and IκB family. (B) Immunoprecipitation of cRel- and RelA-associated IκB proteins. Homozygous (ki/ki) and wild-type (+/+) thymocytes were labeled with [35S]methionine and immunoprecipitated with RelA and cRel antiserum in nondenaturing conditions. The immunoprecipitates were then denatured and sequentially reprecipitated with IκBα, T7-Tag, and IκBβ antisera. (C) EMSA for basal NF-κB activity in thymus, spleen, brain, and MEFs of the knock-in mice. Whole cell extracts were incubated with a palindromic κB-specific probe.
To demonstrate that the introduced IκBβ could complex with NF-κB subunits, coimmunoprecipitations were performed (Fig. 2 B). Wild-type and knock-in thymocytes were labeled with [35S]methionine and immunoprecipitated with antiserum against RelA and cRel under native conditions. The immunoprecipitates were denatured and then sequentially immunoprecipitated with IκBα, T7-Tag, and IκBβ antiserum. Fig. 2 B shows that in wild-type cells a large amount of IκBα was associated with RelA and/or cRel, whereas only a small amount of IκBβ was associated with these subunits. On the other hand, in homozygous knock-in cells, a large amount of T7-Tag-IκBβ was associated with RelA and/or cRel. The amount of T7-Tag–IκBβ in homozygous cells was comparable to that of IκBα in wild-type cells. This indicates that the IκBβ molecule introduced by homologous recombination was expressed at a similar level to IκBα in the wild-type animals, and that the introduced IκBβ is capable of interacting with the NF-κB subunits.
In IκBα-deficient animals, a prominent increase in the basal level of NF-κB activity was observed in spleen and thymus, emphasizing the importance of IκBα in controlling the basal NF-κB activity in hematopoietic organs (5, 6, 18). Thus, we determined the basal levels of NF-κB activity in thymus, spleen, brain, and embryonic fibroblasts from control and mutant mice using EMSA. Basal NF-κB activity in ki/ki animals remained relatively similar to that in the wild-type animals in all cell types examined (Fig. 2 C), indicating that the introduced IκBβ has replaced the role of IκBα in controlling basal levels of NF-κB activity. Furthermore, NF-κB activity in nonhematopoietic cells was not significantly altered, indicating that the presence of extra IκBβ molecules did not affect the basal machinery controlling NF-κB in these cells.
Signal-dependent NF-κB Response in the Thymus.
A significant feature that distinguishes IκBα and IκBβ is the inducibility of IκBα expression by NF-κB. After stimulation with NF-κB inducers, NF-κB accumulates in the nucleus and stimulates ikba transcription. Transcriptional stimulation of the IκBα gene is mediated by several κB enhancer elements present in its 5′ flanking region (19, 20) and leads to rapid accumulation of IκBα molecules, which in turn inhibit NF-κB response. It has been proposed that this regulatory loop is responsible for the transient induction of NF-κB activity. In contrast, the 5′ upstream region of the IκBβ gene does not contain functional κB enhancers, and its transcription is not regulated by NF-κB (11). This fundamental difference in regulation might be part of the functional differences that exist between IκBα and IκBβ. Since the IκBβ gene introduced by homologous recombination was placed under the control of the ikba promoter, it should become inducible by NF-κB. To test this inducibility and assess the functional consequence of this induction, we stimulated thymocytes from wild-type and ki/ki animals with PMA and PHA and analyzed NF-κB activity by EMSA (Fig. 3 A). Similar to the wild-type thymocytes, ki/ki thymocytes exhibited a rapid increase in NF-κB activity 15 min after PMA/PHA stimulation. The increase persisted for 6 h in wild-type and ki/ki cells, indicating that the knock-in thymocytes were capable of eliciting signal-dependent NF-κB response. Western blot analysis revealed that, similar to IκBα in the wild-type, the T7-Tag–IκBβ in ki/ki thymocytes reaccumulated 60 min after stimulation. The time course of reappearance for T7-Tag–IκBβ is similar to that for IκBα in the wild-type, indicating that the IκBβ gene placed downstream of the IκBα promoter is inducible by NF-κB (Fig. 3 B). However, in spite of the increased level of IκBβ protein in basal condition and after induction, signal-dependent NF-κB response in the knock-in thymocytes remained relatively similar to the wild-type.
Figure 3.

Signal dependent NF-κB activation in ki/ki thymocytes. (A) NF-κB binding activity from wild-type (+/+) and ki/ki thymocytes stimulated with PMA/PHA. Cells were stimulated for the indicated periods of time and EMSA was performed using the palindromic κB sequence with 5 μg of nuclear extracts. Oct-1 binding activity was used as control. (B) Western blot analysis of +/+ and ki/ki thymocytes stimulated with PMA/PHA. Similar to IκBα, T7-Tag–IκBβ is induced and reaccumulates after NF-κB stimulation. 20 μg of cytoplasmic extracts were used per lane and a sister blot was probed with antilactic dehydrogenase (LDH) antibodies as control for loading.
Signal-dependent NF-κB Response in Mouse Embryo Fibroblasts.
A tissue-specific difference in function has been proposed between IκBα and IκBβ. Based on the relative abundance of these two proteins, and on the fact that disruption of IκBα specifically affected NF-κB activity in hematopoietic cells, it has been postulated that IκBα plays a more important role in hematopoietic cells, whereas IκBβ is more important in nonhematopoietic cells (18). To assess the functional consequence of the replacement in nonhematopoietic cells, we tested NF-κB response in fibroblasts from 15-d-old embryos (Fig. 4). Wild-type and ki/ki primary mouse embryo fibroblasts (MEFs) were treated with TNF-α for various periods of time and harvested for EMSA and Western blot analysis. After 30 min of TNF-α treatment, NF-κB was activated in both ki/ki and wild-type MEFs (Fig. 4 A). The level of NF-κB induction was similar, indicating that the knock-in fibroblasts are capable of eliciting a normal NF-κB response. However, NF-κB activity in ki/ki cells diminished to near basal level 3 h after stimulation, whereas NF-κB activity in the wild-type remained prominent at the same time point. A Western blot analysis showed that the T7-Tag–IκBβ in ki/ki cells was induced after TNF-α stimulation, resulting in accumulation of a significantly higher amount of IκBβ in the knock-in cells than in the wild-type cells (Fig. 4 B). The time at which the T7-Tag–IκBβ accumulates dramatically in the cell correlates to the time point at which NF-κB activity becomes noticeably lower in the ki/ki fibroblasts.
Figure 4.


Signal dependent NF-κB activation in ki/ki embryo fibroblasts. (A) NF-κB binding activty of wild-type (+/+) and ki/ki fibroblasts stimulated with TNF-α for the indicated periods of time. EMSA was performed using 5 μg of nuclear extracts. Oct-1 binding was used in parallel as control. (B) Western blot analysis of +/+ and ki/ki fibroblasts stimulated with TNF-α. The T7-Tag–IκBβ reaccumulates in ki/ki fibroblasts after TNF-α stimulation. IκBα, T7-Tag–IκBβ, and IκBβ levels were determined on blots with 20 μg of cytoplasmic extracts.
IκBβ Is Capable of Postinduction Repression.
Studies using primary fibroblasts from IκBα-deficient embryos have demonstrated the importance of IκBα in postinduction repression. NF-κB activity in fibroblasts lacking IκBα persisted for >2 h after TNF-α removal, whereas NF-κB activity in wild-type fibroblasts quickly returned to basal level (within 30 min after stimulation; references 5, 6). To determine if the IκBβ introduced by homologous recombination was capable of inhibiting the NF-κB activity after induction, we performed postinduction experiments using the ki/ki fibroblasts (Fig. 5). Wild-type and ki/ki primary embryo fibroblasts were treated for 30 min with TNF-α, after which TNF-α was removed, and the cells were harvested 60, 120, and 240 min later. In the wild-type MEFs, NF-κB activity returned to basal levels nearly 60 min after TNF-α removal (Fig. 5 A). In ki/ki MEFs, the return of NF-κB to basal levels occurs on or after 120 min. A Western blot analysis revealed that, similar to IκBα, the T7-Tag–IκBβ was upregulated after induction. However, the time at which maximal T7-Tag–IκBβ accumulated was delayed compared to IκBα (60 min versus 2 h; Fig. 5 B). Peak T7-Tag–IκBβ accumulation corresponded to the time at which NF-κB activity diminished to basal level. Therefore, in the context of the IκBα promoter, IκBβ can be induced by NF-κB and is competent in postinduction repression, although the time course of repression is slower than that of IκBα in the wild-type cells.
Figure 5.
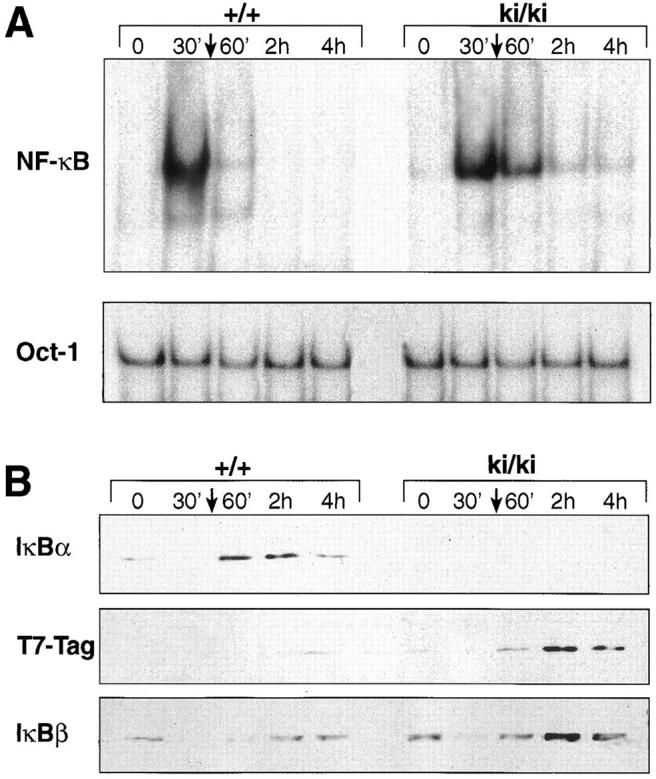
Postinduction repression of NF-κB in ki/ki embryo fibroblasts. (A) βF-κB binding activity of wild-type (+/+) and ki/ki fibroblasts treated with TNF-α for 30 min, after which cells were washed and medium without TNF-α was added to the cells for the indicated period. Nuclear extracts were prepared at the indicated periods after removal of TNF-α. A loading control using the Oct-1 probe was also performed. (B) Western blot analysis of +/+ and ki/ki fibroblasts treated with TNF-α. 20 μg each of the total cell extracts were analyzed for levels of IκBα, T7-Tag–IκBβ, and the endogenous IκBβ proteins.
Discussion
Using the knock-in approach, we have generated mice that carry a replaced IκBα gene. A T7-Tag–modified IκBβ gene was expressed under the promoter and the 5′ regulatory sequence of the IκBα gene. As a result, the ikba allele was inactivated, and the introduced ikbb was expressed and regulated like ikba. This approach allowed us to investigate the functional similarity of IκBα and IκBβ under physiological conditions. The knock-in mice survived to adulthood and did not display any of the abnormalities that are present in the IκBα-deficient mice. Basal levels of NF-κB activity in the hematopoietic organs of the knock-in mice were not elevated like those in the IκBα-deficient mice. Furthermore, signal-induced NF-κB response in thymocytes was also normal. These results indicate that IκBβ can functionally compensate for the absence of IκBα.
The notion that IκBα and IκBβ are biochemically equivalent is unexpected. Although many similarities exist between these two molecules, significant biochemical differences have also been reported. For example, IκBα degradation is induced by a wide variety of NF-κB inducers, but only a subset of these inducers effects IκBβ degradation (11, 21). The kinetics of degradation is also different between the two molecules. In general, IκBα has a more rapid degradation than IκBβ. Differences in the basal phosphorylation state, degradation mechanism (21), and affinity to RelA (22) have also been shown for these two molecules. These observations formed the basis of hypotheses that the biochemical differences of IκBα and IκBβ contribute to functional differences for these two molecules.
One prominent functional difference that has been proposed for the IκBs is the unique “chaperone” role of IκBβ (23). IκBβ molecules resynthesized after stimulation have been found to be hypophosphorylated. The hypophosphorylated IκBβ molecules can bind to NF-κB complexes, but do not mask the nuclear localization signals of NF-κB. Thus, they serve as chaperones for NF-κB complexes, protecting them from inhibition by the resynthesized IκBα molecules and, therefore, maintaining the persistent activation of NF-κB. However, recent reports have provided evidence against this model. For example, Miyamoto et al. have reported that inhibition of IκBβ degradation by proteasome inhibitors does not have any effect on the constitutive activity of NF-κB in WEHI cells (21). Prolonged nuclear localization of NF-κB complexes also is not necessarily associated with long-term depletion of IκBβ (24). Moreover, mice deficient in IκBβ have been also generated (Attar, R.M., manuscript in preparation). NF-κB responses elicited by various extracellular stimuli, along with various immune responses in these mice, are indistinguishable from those in wild-type mice, suggesting that persistent NF-κB activation can take place in the absence of IκBβ. Furthermore, NF-κB activity in thymocytes remained relatively unchanged in the knock-in mice, indicating that the resynthesized IκBβs did not chaperone more NF-κB complexes into the nucleus, although a large excess of T7-Tag–IκBβ is produced after stimulation (Fig. 3). It is surprising that such a large amount of IκBβ did not affect NF-κB activity. It appears as if the NF-κB complexes in the knock-in cells are protected from the resynthesized T7-Tag–IκBβ just as the NF-κB complexes in the wild-type cells are protected from the resynthesized IκBα. Possibly, a separate mechanism exists that is independent of the phosphorylation status of IκBβ for maintaining the persistent activation of NF-κB.
In light of the fact that the mice lacking IκBα but expressing IκBβ controlled by the ikba promoter regulatory elements appear healthy, we conclude that the two IκB molecules are biochemically equivalent, and that certain biochemical features reported on IκBβ, such as hypophosphorylation or slow degradation, do not contribute significantly to the functional difference between them. We postulate that the difference in relative level of expression between these two molecules is a primary component of their functional difference. It has been reported that these two IκB molecules are expressed differently in different tissues: IκBα mRNA is highly abundant in the spleen, whereas IκBβ mRNA is mostly abundant in the testis (11). IκBα protein expression is also known to be higher in the hematopoietic organs, whereas IκBβ protein is distributed equally between the hematopoietic and nonhematopoietic tissues (5). Consistent with these observations, we are able to immunoprecipitate a much larger amount of IκBα than IκBβ molecules in our thymocyte preparation (Fig. 2 B and data not shown). Additionally, our Western blots also show that the IκBβ introduced by homologous recombination is expressed at a higher level than the endogenous IκBβ molecules in thymocytes (Fig. 3 B). On the other hand, the relative increase in IκBβ level does not appear to be as high in the knock-in fibroblasts (Fig. 4 B). Although we can not rule out that the differences in stability may have contributed to some of the difference in steady state expression level, our observations suggest that the ikba promoter is more active in the hematopoietic system compared with nonhematopoietic systems.
In addition to tissue-specific differences in expression, the autoregulatory feature is also a fundamental difference between the two molecules. It has been proposed that NF-κB–inducible regulation of IκBα gene transcription is important for terminating NF-κB response in nonhematopoietic cells. By performing a postinduction repression experiment similar to that performed in the study of IκBα-deficient mice, we found that the T7-Tag–IκBβ molecule can serve as an inhibitor similar to IκBα in postinduction repression. This again supports the notion that IκBα and IκBβ are biochemically equivalent, and that regulatory κB enhancer elements upstream of the genes are crucial components of the functional differences between these IκBs. A more recently identified member of the IκB family, IκBε, also has this autoregulatory feature (25, 26). The fact that some, but not all, members of the IκB family share this property further implicates the importance of specific regulatory sequences in conferring different functions of these molecules. This auto-regulatory feature appears to be closely linked to tissue-specific difference in expression, as resynthesis of excess T7-Tag–IκBβ is associated with a different effect in fibroblasts than in thymocytes.
Despite the increase in understanding NF-κB function, the specificity and physiological relevance of the different Rel/NF-κB and IκB proteins remains unclear. The knockouts and transgenic animals have provided important information on the physiological roles of the individual members (7, 27–35), but functional redundancy probably has masked the full importance of each member. Besides knowledge provided from single and double knockout studies, the knock-in approach provides a powerful tool for analysis of redundancy and the physiological function of individual members. The knock-in approach could be used to dissect the importance of specific domains or structures within a molecule in conferring specificity. Similar to this study, the knock-in approach has been used to reveal redundancy of two other transcription factor families, the Engrailed family (En-1 and En-2; reference 36), and the MyoD family (Myogenin and Myf-5; reference 37). In both cases, replacement of a similar member of the same family resulted in complete rescue of the phenotype from single knockout. Together, these data demonstrate that many closely related members of a gene family acquire different functions in evolution through divergence of gene expression, rather than through divergence in biochemical function. Because overlapping gene function is likely to be prevalent in mammals, such approaches are critical for clarifying the unique and overlapping function of members of a family, and for determining the complete repertoire of functions of individual genes.
Acknowledgments
We thank Chery Rizzo for cell culture assistance, Mavis Swerdel and Alice Lee for chimera aggregations, Kenneth Class for flow cytometry, and the staff of Veterinary Sciences at Bristol-Myers Squibb for their excellent support. We also thank Donald Hawken for support and critical reading of the manuscript.
Abbreviations used in this paper
- EMSA
electrophoretic mobility shift assay
- ES
embryonic stem
- MEF
mouse embryo fibroblast, NF-κB, nuclear factor κB
References
- 1.Gilmore TD. Introduction: the Rel/NF-κB signal transduction pathway. Cancer Biol. 1997;8:61–62. doi: 10.1006/scbi.1997.0056. [DOI] [PubMed] [Google Scholar]
- 2.Whiteside ST, Israël S. IκB proteins: structure, function and regulation. Cancer Biol. 1997;8:75–82. doi: 10.1006/scbi.1997.0058. [DOI] [PubMed] [Google Scholar]
- 3.Israel A. A role for phosphorylation and degradation in the control of NF-κB activity. Trends Genet. 1995;11:203–205. doi: 10.1016/s0168-9525(00)89045-9. [DOI] [PubMed] [Google Scholar]
- 4.Beg AA, Baldwin AS., Jr The IκB proteins: multifunctional regulators of Rel/NF-κB transcription factors. Genes Dev. 1993;7:2064–2070. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 5.Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- 6.Klement JF, Rice NR, Car BD, Abbondanzo SJ, Powers GD, Bhatt H, Chen C-H, Rosen CA, Stewart CL. IκBα deficiency results in a sustained NF-κB response and severe widespread dermatitis in mice. Mol Cell Biol. 1996;16:2341–2349. doi: 10.1128/mcb.16.5.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishikawa H, Carrasco D, Claudio E, Ryseck R-P, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J Exp Med. 1997;186:999–1014. doi: 10.1084/jem.186.7.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Shacter E, Leonardi A, Anver M, et al. Critical roles for the Bcl-3 oncoprotein in T cell–mediated immunity, splenic microarchitecture and germinal center reactions. Immunity. 1997;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- 9.Schwarz EM, Krimpenfort P, Berns A, Verma IM. Immunological defects in mice with a targeted disruption in Bcl-3. Genes Dev. 1997;11:187–197. doi: 10.1101/gad.11.2.187. [DOI] [PubMed] [Google Scholar]
- 10.Zabel U, Baeuerle PA. Purified human IκB can rapidly dissociate the complex of the NF-κB transcription factor with its cognate DNA. Cell. 1990;61:255–265. doi: 10.1016/0092-8674(90)90806-p. [DOI] [PubMed] [Google Scholar]
- 11.Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. IκB-β regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995;80:573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 12.Chu Z-L, Mckinsey TA, Liu L, Qi X, Ballard DW. Basal phosphorylation of the PEST domain in IκBβ regulates its functional interaction with the c-rel proto-oncogene product. Mol Cell Biol. 1996;16:5974–5984. doi: 10.1128/mcb.16.11.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tybulewicz VLJ, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 14.Coligan, J.E., A.M. Kruisbeek, D.H. Margulies, E.M. Shevach, and W. Strober. 1992. Isolation and fractionation of mononuclear cell populations. In Current Protocols in Immunology. J.W. Sons, editor. Greene Publishing Associates & Wiley-Interscience, New York. 3.0.1–3.0.5.
- 15.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucl Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa H, Ryseck R-P, Bravo R. Characterization of ES cells deficient for the p105 precursor (NF-κB1): role of p50 NLS. Oncogene. 1996;13:255–263. [PubMed] [Google Scholar]
- 17.Dobrzanski P, Ryseck R-P, Bravo R. Both N- and C-terminal domains of RelB are required for full transactivation: role of the N-terminal leucine zipper-like motif. Mol Cell Biol. 1993;13:1572–1582. doi: 10.1128/mcb.13.3.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–169. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 19.de Martin R, Vanhove B, Cheng Q, Hofer E, Csizmadia V, Winkler H, Bach FH. Cytokine-inducible expression in endothelial cells of an IκBα-like gene is regulated by NFκB. EMBO (Eur Mol Biol Organ) J. 1993;12:2773–2779. doi: 10.1002/j.1460-2075.1993.tb05938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Bail O, Schmidt-Ullrich R, Israel A. Promoter analysis of the gene encoding the IκB-α/MAD3 inhibitor of NF-κB: positive regulation by members of the rel/ NF-κB family. EMBO (Eur Mol Biol Organ) J. 1993;12:5043–5049. doi: 10.1002/j.1460-2075.1993.tb06197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyamoto S, Seufzer BJ, Shumway SD. Novel IκBα proteolytic pathway in WEHI231 immature B cells. Mol Cell Biol. 1998;18:19–29. doi: 10.1128/mcb.18.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Attar RM, Macdonald-Bravo H, Raventos-Suarez C, Durham SK, Bravo R. Expression of constitutively active IκBβ in T cells of transgenic mice: persistent NF-κB activity is required for T-cell immune responses. Mol Cell Biol. 1998;18:477–487. doi: 10.1128/mcb.18.1.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suyang H, Phillips R, Douglas I, Ghosh S. Role of unphosphorylated, newly synthesized IκBβ in persistent activation of NF-κB. Mol Cell Biol. 1996;16:5444–5449. doi: 10.1128/mcb.16.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalli K, Huntoon C, Bell M, McKean DJ. Mechanism responsible for T-cell antigen receptor– and CD28- or interleukin 1 (IL-1) receptor–mediated regulation of IL-2 gene expression by NF-κB. Mol Cell Biol. 1998;18:3140–3148. doi: 10.1128/mcb.18.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whiteside ST, Epinat J-C, Rice NR, Israël A. IκBε, a novel member of the IκB family, controls RelA and cRel NF-κB activity. EMBO (Eur Mol Biol Organ) J. 1997;16:1413–1426. doi: 10.1093/emboj/16.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Nabel GJ. A new member of the IκB protein family, IκBε, inhibits RelA (p65)-mediated NF-κB transcription. Mol Cell Biol. 1997;17:6184–6190. doi: 10.1128/mcb.17.10.6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attar RM, Caamano J, Carrasco D, Iotsova V, Ishikawa H, Ryseck R-P, Weih F, Bravo R. Genetic approaches to study Rel/NF-κB/IκB function in mice. Semin Cancer Biol. 1997;8:93–101. doi: 10.1006/scbi.1997.0060. [DOI] [PubMed] [Google Scholar]
- 28.Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat Med. 1997;3:1285–1289. doi: 10.1038/nm1197-1285. [DOI] [PubMed] [Google Scholar]
- 29.Ishikawa H, Claudio E, Dambach D, Raventos-Suarez C, Ryan C, Bravo R. Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-κB1) but expressing p50. J Exp Med. 1998;187:985–996. doi: 10.1084/jem.187.7.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrasco D, Cheng J, Lewin A, Warr G, Yang H, Rizzo C, Rosas F, Snapper C, Bravo R. Multiple hemopoietic defects and lymphoid hyperplasia in mice lacking the transcriptional activation domain of the c-Rel protein. J Exp Med. 1998;187:973–984. doi: 10.1084/jem.187.7.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, Bravo R. Nuclear factor (NF)-κB2 (p100/p52) is required for normal splenic microarthitecture and B cell–mediated immune responses. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinberg A, Tran T, Scharton-Kersten T, Anver M, Love P, Brown K, Siebenlist U. Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck R-P, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/ Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 34.Sha WC, Liou H-C, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 35.Kontgen F, Frumont RJ, Strasser A, Metcalf D, Li R, Targlinton D, Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 36.Hanks M, Wurst W, Anson-Cartwright L, Auerbach AB, Joyner AL. Rescue of the En-1 mutant phenotype by replacement of En-1 with En-2. . Science. 1995;269:679–682. doi: 10.1126/science.7624797. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Schnegelsberg PNJ, Dausman J, Jaenisch R. Functional redundancy of the muscle-specific transcription factors Myf5 and myogenin. Nature. 1996;379:823–825. doi: 10.1038/379823a0. [DOI] [PubMed] [Google Scholar]