

Abstract
Analysis of T regulatory cells (Treg) and T effector cells (Teff) in experimental autoimmune encephalomyelitis is complicated by the fact that both cell types express CD4 and CD25. We demonstrate that encephalitogenic T cells, following antigen recognition, up regulate cell surface expression of CD4. The CD4high sub-population contains all of the antigen response as shown by proliferation and cytokine secretion, and only these cells are capable of transferring EAE to naive animals. On the other hand, a FACS separable CD25+ sub-population of cells displayed consistent levels of CD4 prior to and after antigen stimulation. These cells displayed characteristics of Treg, such as expressing high levels of the Foxp3 gene and the ability to suppress mitogenic T cell responses.
Keywords: Experimental autoimmune encephalomyelitis, CD4, CD25, T cells, cellular proliferation, FACS, Treg
1. Introduction
Major effort to understand the etiology and mechanisms of multiple sclerosis (MS) has utilized experimental autoimmune encephalomyelitis (EAE) as an animal model. Although EAE is not a complete model, it does share many of the immunological features seen in MS (Swanborg 1995). EAE is characterized by infiltration of mononuclear cells into the CNS and demyelination of nerve cells (Traugott, McFarlin, & Raine 1986;Traugott, Raine, & McFarlin 1985). This inflammatory reaction is initiated by CD4+ T cells that recognize myelin autoantigens such as MBP (Sakai et al. 1988;Zamvil et al. 1985;Zamvil et al. 1986), MOG (Amor et al. 1994;Johns et al. 1995), and myelin proteolipid protein (PLP) (Amor et al. 1993;Trotter et al. 1987;Tuohy et al. 1989). In mice, EAE can be induced by active immunization with neuroantigen emulsified in complete Freund's adjuvant (CFA) (Traugott, Raine, & McFarlin 1985) or by passive transfer of neuroantigen-primed and in vitro-activated lymph node cells into naive recipients (Bernard 1976;Mokhtarian, McFarlin, & Raine 1984a;Pettinelli & McFarlin 1981). The initiation phase of the disease is a typical Th1 response involving production of gamma-interferon and IL-12 (Powell et al. 1990;Segal, Dwyer, & Shevach 1998). Recent studies implicate IL-23 and IL-17 in the propagation of the disease (Cua et al. 2003;Komiyama et al. 2006;Langrish et al. 2005;Sutton et al. 2006).
The nature of the effector T cells that mediate disease is still unclear. One of the difficulties in studying effector cell functions is the paucity of such cells, even after priming. We previously discovered that T cells up-regulated their cell surface expression of CD4 following antigen recognition and that antigen-specific effector cells, as analyzed by antigen-induced proliferation and limiting dilution assays, were all confined to a CD4high cell subpopulation isolated by flow cytometry (Ridgway, Fasso, & Fathman 1998). This observation had been confirmed in autoimmune disease models such as non-obese diabetic (NOD) mice (Lejon & Fathman 1999) and experimental autoimmune myasthenia gravis (Standifer, Kraig, & Infante 2003). The use of the CD4high marker should allow enrichment of antigen-specific effector T cells for further functional analysis.
Recent studies have implicated a population of regulatory T cells (Treg) in the maintenance of tolerance as well as in the control of autoimmune diseases (Sakaguchi et al. 2006). These cells typically also express the cell surface molecules CD4 and CD25 (Asano et al. 1996;Sakaguchi et al. 1995). Subsequent studies showed that CD5, OX40, CD45RBlo, CD62L, cytotoxic T-lymphocyte antigen-4 (CTLA-4) and glucocorticoid-induced tumor necrosis factor receptor (GITR) are other markers associated with Treg cells (McHugh et al. 2002;Shimizu et al. 2002). As these cell surface markers are also up-regulated in activated T cells that mediate effector functions, it has been difficult to separate these two populations for further studies. In recent years, a transcription factor of the forkhead/winged-helix family called forkhead box protein 3 (Foxp3) has been identified specifically in Treg cells (Fontenot, Gavin, & Rudensky 2003;Khattri et al. 2003). Mice deficient in Foxp3 do not have Treg cells and develop lymphoproliferative diseases (Lahl et al. 2007). Scurfy mice, which have deletion of the forkhead domain of the scurfin protein Foxp3 also develop a T cell mediated X-linked autoimmune disorder (Chang et al. 2005). Foxp3 thus provides a useful marker to identify Treg cells.
Since their initial description by Sakaguchi and colleagues (Sakaguchi, Sakaguchi, Asano, Itoh, & Toda 1995), CD4+CD25+ Treg cells have been reported to be involved in several models of autoimmunity, such as diabetes, EAE, colitis, gastritis and collagen-induced arthritis (Asano, Toda, Sakaguchi, & Sakaguchi 1996;Green, Choi, & Flavell 2002;Groux et al. 1997;Huehn et al. 2004;Kohm et al. 2002). In EAE, Kohm et al. isolated CD4+CD25+ cells from the lymph nodes of naïve unprimed C57BL/6 mice and demonstrated that transfer of these cells into naive syngeneic recipient mice three days prior to active induction of EAE with MOG35-55 significantly protected the animals from development of clinical EAE (Kohm, Carpentier, Anger, & Miller 2002). By pretreating B10.S mice, which are resistant to EAE induction with PLP139-151, with anti-CD25 antibodies (clone PC61) prior to immunization with PLP139-151, Reddy et al. demonstrated that a percentage of the mice became susceptible to EAE development, thus implying that EAE resistance might be maintained by “natural” CD25+ cells(Reddy et al. 2004). Both studies involve activities of pre-existing Treg cells in naïve unprimed animals and are referred to as “natural Treg” cells. It is not clear whether Treg cells in antigen-primed animals have different characteristics and mechanisms of actions. Unfortunately, as pointed out earlier, both effector T cells and Treg cells express very similar cell surface molecules. A method that would separate these two cell populations for further functional studies is obviously needed.
In this report, we demonstrate that effector T cells and Treg display different levels of the CD4 antigen. We make use of this difference to enrich for effector T cells and Treg cells, respectively, in mice with EAE. We first demonstrate that CD4high cells isolated from CNS-antigen primed mice indeed contain the bulk of the antigen-specific T cell proliferative responses in vivo and in vitro. In addition, CD4high cells, and not CD4norm cells, cause EAE after adoptive transfer to naïve recipients. Based on expression of CD4 and CD25, we next identify two separable cell populations of CD4+ cells in antigen activated LN cultures, CD4highCD25+ and CD4normalCD25+. The CD4high cells proliferate in response to specific antigen and do not express Foxp3, while the CD4normal cell population does not proliferate to antigen stimulation, expresses the Foxp3 gene and are able to suppress mitogen induced T cell responses. Lastly, we show that CD4high, but not CD4normal cells express the inflammatory cytokines IFN-γ and IL-17. Taken together, we demonstrate that high expression of CD4 is a reliable marker of antigen-specific EAE effector T cells and can be used to separate this cell population from Treg cells from in vitro cultures.
2. Materials and Method
2.1. Mice
B10.PL and C57BL/6 (B6) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained in the Stanford Medical Center Department of Comparative Medicine or the Wayne State University Department of Laboratory Animal Research. Mice were used between 6 and 12 weeks of age. All study related protocols were approved either by the Stanford University or Wayne State University animal investigation committees prior to performing the studies.
2.2. Antigens
MOG peptide p35-55 (MEVGWYRPSFSRVVHLYRNGK) and MBP peptide Ac1-11 (AcASQKRPSQRHG) was synthesized by Genemed Synthesis (South San Fransisco CA) and purity confirmed by HPLC.
2.3. Immunization
Groups of 3-5 mice were immunized subcutaneously at four sites in the flanks with 50 μl (each site) of an emulsion containing IFA plus 10 mg/ml heat killed Mycobacterium tuberculosis, H37RA (Difco Laboratories Inc, Detroit MI) plus 200 μg/mouse antigen (MOG35-55 or MBPAc1-11) suspended in an equal volume of Dulbecco's PBS.
2.4. In vitro culture
Eight to ten days after immunization, draining inguinal and axillary lymph nodes were removed and single cell suspensions prepared. LN cells were activated with the priming antigen (50 ug/ml), 8 x 106 cells/well in 24-well flat-bottom plates in EAE medium at 37° C supplemented with 5% CO2. EAE media consisted of RPMI 1640 supplemented with 2 mM L-Glutamine, penicillin/streptomycin, nonessential amino acids, sodium pyruvate, and 10 mM hepes buffer (Gibco Laboratories, Grand Island, NY), 50 mM 2-ME (Sigma Chemical Co., St. Louis MO), and 10% FCS. For long term cultures, cells were allowed to “rest” for 10 days without media addition. Cells were then restimulated as follows: T cells were harvested and cultured (2 x 106 cells) with APC's (2.5 x 107 syngeneic irradiated spleen cells) and antigen (50 ug/ml), in T25 flasks in 10 ml EAE media.
2.5. FACS analysis
For studies in vitro, aliquots of lymph node cells were taken from culture, washed with FACS buffer (Dulbecco's PBS with 2% FCS), stained for 1 or 2 color flow cytometric analysis with fluorescein and phycoerythrin antibodies at a predetermined optimal concentration for 20 minutes at 4° C, washed just prior to analysis. Anti-CD4 (clone GK1.5), anti-CD25 (clone 7D4) fluoresceinated antibodies were obtained from BD-Pharmingen, Inc. (San Diego, CA ). Anti-CD4 and CD25 (same clones)-phycoerythrin antibodies were also obtained from BD-Pharmingen. Just prior to analysis, propidium iodide (P.I.-1ug/ml) was added to the cells. Positive P.I. staining was used to gate dead cells from the samples. 2 x 105 cells were analyzed by two color flow cytometry on a Becton-Dickinson Facscan cytometer. The data was analyzed using WinMDI 2.8 software.
2.6. FACS sorting and proliferation assays
Single cell suspensions were obtained either from 4 day cultures or directly from the lymph nodes of immunized animals (in both cases, harvested 8-10 days after immunization, as above). The cells were stained with anti-CD4 antibody (and in some experiments also with anti-CD25 antibodies). The cultured cells were analyzed by FACS for determination of appropriate gates, sterile sorted into the indicated populations for further analysis. For proliferation assays, cells were plated into 96 well plates at the given cell number, pulsed with [3H] thymidine for 18 hours, and then counted on a Beta plate.
2.7. Adoptive transfer of cell populations for determination of encephalitogenicity
Cell populations were tested for encephalitogenicity using the adoptive transfer and challenge protocol of Shaw et. al. (Shaw et al. 1992). Briefly, donor B6 mice were immunized with MOG35-55 peptide as described above. Ten days later draining LN cells were activated by in vitro culture with the same antigen for four days. After FACS sorting or in vitro culture, cells were washed and resuspended in PBS. Viable cells were assayed by tryphan blue exclusion counting. Cells were then resuspended to appropriate volumes for transfer to naïve irradiated (500R) recipient mice, which received the indicated number of viable cells by tail vein injection in 200 ul sterile PBS. Recipient mice were examined daily for clinical signs of disease and were graded according to the following scale: 0-no abnormality; 1-loss of tail tonicity; 2-paralysis in a single hind limb; 3-dual hind limb paralysis; 4-paralysis involving the forelimbs; 5-moribund; 6-death. Subcutaneous injections of normal saline were administered to all animals losing significant body weight (> 20%). EAE was induced in B10.PL mice in a similar manner except that the antigen used was MBP Ac1-11 and recipient mice were not irradiated. After thirty days those mice that did not succumb to disease were challenged with a sub-encephalitogenic dose of antigen (50 ug/mouse) in CFA, and the mice examined for signs of disease for the next sixty days. Disease was charted using the following criteria, 1) average disease grade: average disease of those mice which succumbed to disease; 2) average day of onset: average day of onset of those mice which succumbed to disease; 3) incidence of disease: number of mice with disease/total mice treated.
2.8. Intracellular staining of LN cells for IFN-g, and IL-17
Antigen activated LN cultures were stained for intracellular cytokine expression using the Cytofix/Cytoperm Plus Kit (BD Pharmingen) according to the manufacturer's instructions. Protein transport inhibitor was added 4 hours prior to staining. Anti IFN-γ (clone XMG1.2) and anti-IL-17 (clone TC11-18H10) were both purchased from BD Pharmingen and used at predetermined optimal concentrations. Specificity of staining was determined in control experiments by pre-staining with unconjugated cytokine antibodies.
2.9. Intracellular staining for FoxP3
Antigen stimulated LN cultures were stained for intracellular FoxP3 expression using the mouse Treg staining kit (eBioscience) according to manufacturers instructions.
2.9. Real time PCR measurement of gene expression
Day 3 LN cultures of B6, MOG peptide stimulated cells were harvest and FACS sorted as described above to isolate CD4highCD25+ and CD4norm CD25+ cells. Immediately following sorting, total RNA was isolated from the sorted cells using the RNeasy, RNA isolation kit (Qiagen, Valencia, CA) following the manufacturer's suggested protocol. Total RNA was quantitated by measuring A260/A280 absorbance. cDNA was produced from 1 ug total RNA using a cDNA synthesis kit (Gibco BRL) following the manufactures instructions. Real time PCR was performed in a Smartcycler (Cephied) thermocycler using taqman primer and probe sets specific for FoxP3, IFN-g and IL-17 and the housekeeping gene ubiquitin. For each cDNA, both ubiquitin and specific primers were used in separate tubes. As well, duplicate samples were run for each DNA/primer set. cDNA was subject to PCR amplification under the following conditions: 50°C for 2 min, 95°C for 5 mins, followed by 40 cycles of 15s at 95°C, 1 min at 60°C. Primer sequences are as follows:
IL-17 sense, 5′ CCCTCTGTGATCTGGGAAGC;
IL-17 antisense, 5′- TTTCCCTCCGCATTGACAC;
IL-17 taqman probe 5'- CAGTGCCGCCACCAGCGC;
IFN-γ sense, 5'- CATTGAAAGCCTAGAAAGTCTGAATAAC,
IFN-γ antisense, 5'- TGGCTCTGCAGGATTTTCATG;
IFN-γ taqman probe 5'-TCACCATCCTTTTGCCAGTTCCTCCAG;
FoxP3 sense GGCCCTTCTCCAGGACAGA;
FoxP3 antisense GCTGATCATGGCTGGGTTGT;
FoxP3 taqman probe 5'- TexRed-ACTTCATGCATCAGCTCTCCACTGTGGAT-BHQ2;
ubiquitin sense GCAAGCAGCTGGAAGATGG,
ubiquitin antisense GACCAGGTGGAGGGTGGA;
ubiquitin taqman probe 5'-Tex Red-CGGACGCTGTCAGACTAC.
Amplification of RNA (without reverse transcription) confirmed that no contaminating genomic DNA was present in the samples. All reactions were performed using the TaqMan Gold RT-PCR kit according the manufacturer's recommendation (PE Applied Biosystems). Normalization to ubiquitin expression was performed for each sample using the ΔΔCT method (Pagliarulo et al. 2004).
2.10 Treg suppression of CD3 stimulated T cells
Naïve CD4+CD25− T cells (responder cells- Miltenyi CD4 bead isolated) were cultured in 96-well round bottom plates at 1×104 cells/well with 2 × 104 irradiated (3000R) syngeniec T depleted (Thy 1 Miltenyi bead isolated) spleen cells (as APC) in the presence of varying amounts of CD4normalCD25+ T cells (containing Tregs). Cell cultures were stimulated with 2μg/ml anti-CD3 Ab (Clone 2G3) for four days with the addition of 1μCi [3H]thymidine (Amersham) for the final 16h of culture. Cells were harvested with an automatic cell harvester and uptake of radioactivity was measured in a β-plate liquid scintillation counter (Wallac). The suppressive capacity of Tregs towards responder cells in co-culture (Tresp-Treg ratio 1:0 to 1:8) for each cell ratio was compared by expressing proliferation as a stimulation index (si=specific proliferation-cpm/background cpm), and comparing the si of each cell ratio to proliferation of stimulated Teff alone.
3. Results
3.1 MBP and MOG-specific T cells up regulate CD4 expression
Prompted by the fact that T cells responding to conventional antigens up regulate expression of CD4 (Ridgway, Fasso, & Fathman 1998), we questioned whether T cells responding to CNS autoantigens could be similarly phenotyped. To test this possibility, MOG p35-55 mice were immunized with an encephalitogenic peptide of MOG (p35-55) at four sites in the flanks. Eight days after immunization, draining lymph nodes (LN) were harvested and the cells were stained with anti-CD4 antibodies and analyzed by flow cytometry. As shown in Figure 1A, a distinct population of cells, comprising less than 25% of total lymph node cells expressed the CD4 molecule. A small subpopulation in each sample, representing 1.2% of the total cells, can also be discerned to express a higher level of CD4. The LN cells were then cultured in vitro with the priming peptide for four days. On each subsequent day, aliquots of the cells were analyzed by FACS. The percentage of CD4high cells in the MOGp35-55 activated culture increased steadily throughout the culture period, from 1.2% of live cells initially to almost 12% of the cells by 96 hours in culture (Fig. 1B). This increased percentage could be the result of death of other cells over time, as well as an absolute increase in the number of CD4high cells due to antigen stimulation. In similar experiments we also observed the up regulation of CD4 expression in LN cells from B10.PL mice immunized and cultured with the dominant epitope of MBP Ac1-11. In this case, the CD4high population of cells was less discernable in the uncultured LN cells, constituting only 0.7% of total cells (Figure 1C). However, upon culture for 4 days, a prominent CD4high population was again observed accounting for 8.4% of total cells (Figure 1D). In similar experiments we have detected up regulation of CD4 in response to a number encephalitogenic CNS autoantigens. B6 mice immunized with PLPp171-191 (Tompkins et al. 2002)or MBPp60-80 (Shaw et al. 1996), SJL mice immunized with MBP (whole molecule) and Balb/c mice immunized with PLPp180-199 (Lyons et al. 2002) all display discrete populations of CD4high T cells (data not shown). The fact that CD4 up regulation occurs in all of these mouse strains immunized with different encephalitogenic peptides suggests that this is a general phenomenon of CD4 T cell reactivity to CNS autoantigens.
Figure 1. FACS staining of LN cells demonstrates the presence of CD4high cells.

LN cells from MOG p35-55 primed B6 mice (panel A-B), or LN cells from MBP Ac1-11 primed B10.PL mice (panel C-F) were stained with CD4 antibodies. In panels A and B the CD4 antibody is FITC conjugated. In panels C-F, the CD4 antibody is PE conjugated. For the sake of clarity, the cells are plotted against an empty channel (x axis-no stain) in a density plot. The percentage CD4high cells are determined by the indicated gates. Panel A shows LN cells isolated from a MOG35-55 primed B6 mouse without in vitro culture; Panel B: shows the same LN cells after in vitro culture with MOG peptide for four days; Panel C shows LN cells from a MBP Ac1-11 primed B10.PL mouse without culture; Panel D shows the same LN cells after four days in vitro culture with MBP peptide; Panel E shows the same LN cells after 6 rounds of in vitro culture with MOG peptide and syngeneic APC's, 4 days after stimulation; Panel F shows the same cells as in Panel E except 11 days after stimulation.
By extension, we anticipated that long term T cell lines, which are more homogeneous in their antigen specificity, would be more uniformly CD4high. To test this hypothesis, MBPAc1-11-primed and activated LN cells were carried in vitro for two months by repeated stimulation with antigen and syngeneic APC's at 10 day intervals. Upon FACS analysis, the CD4+ cells were found to be greater than 87 % CD4high (Figure 1E). The remaining CD4+, but CD4norm cells are likely still viable cells derived from the APC's used for stimulation. To confirm this fact, the cultured cells were allowed to rest for an additional 7 days (during which time the irradiated APC's die and can be gated by positive propidium iodide staining) and reanalyzed. In a resting culture, the percentage of CD4high cells is 98% (Figure 1F). Interestingly, this indicates that the CD4high phenotype is persistent, and not dependant upon the recent activation of the T cells.
3.2 CD4high cells contain the bulk of the proliferative response
To test the proliferative capacity of the different CD4 cell populations, total CD4, CD4high and CD4norm cells from 72-hour cultures of MOGp35-55-primed B6 LN cells or MBP Ac1-11 primed B10.PL LN cells were sorted by flow cytometry. The sorted cells (1 × 105) were re-cultured for 18 hours and were pulsed with tritiated thymidine during this culture period. As shown in Table 1, the CD4high cell population exhibited vigorous proliferation while the CD4norm cells had minimal proliferation. Whole lymph node cell response to antigen from the same mice was also tested. Since CD4high cell constituted only a fraction of the total lymph node cells, a multiplier (percent of total LN cells) was used to normalize the responses for comparison. When the proliferation data are corrected by such a multiplier, it is seen that the majority of the proliferative responses to MOG35-55 and MBP1-11 are contained in the CD4high population (Table I). Little proliferation was contributed by the CD4norm cell fraction and that seen is probably the result of sorting error. (0.5-1.5% as determined by post sort analysis)
Table 1. The CD4high sub-population contains all of the proliferative response.
Three days after in vitro culture, encephalitogenic peptide primed and in vitro activated LN cells were sterile sorted into CD4high and CD4norm populations. Unsorted LN cells were used for comparison. The cells, 1 × 105 cells of each type, were plated and pulsed immediately with 3H-thimidine for 18 hours before scintillation counting. Results are shown as raw data and corrected for the percentage of each CD4 sub-population represented in the original culture (as determined by sort gating). The percentage of CD4high and CD4norm cells for each experiment is indicated as the multiplier.
| Mouse strain antigen combination |
Cells type cultured (1 × 105) |
CPM | Multiplier (% of total CD4 cells in culture) |
Corrected CPM |
|
|---|---|---|---|---|---|
| C57BL/6 mice immunized with MOG35-55 |
Total CD4 Cells CD4norm Cells CD4high Cells |
31,579 2,411 220,932 |
1.0 0.858 0.142 |
(100%) (85.8%) (14.2%) |
31,579 2,067 31,482 |
| B10.PL mice immunized with MBPAc1-11 |
Total CD4 Cells CD4norm Cells CD4high Cells |
14,200 888 165,993 |
1.0 0.919 0.081 |
(100%) (91.9%) (8.1%) |
14,200 816 13,445 |
3.3 CD4high cells are encephalitogenic
Since the bulk of the antigen responsiveness was contained in CD4high T cells, we next tested if the cells were also encephalitogenic in a series of adoptive transfer experiments. LN cells from MOGp35-55-primed B6 donor mice were cultured in vitro with antigen for 4 days. Viable cells were then separated into CD4high and CD4normal populations by FACS. The cells were transferred into naive syngeneic recipients at the indicated cells numbers (Table 2). Unsorted cells were also transferred to serve as controls. As is shown, transfer of 6.3 × 106 CD4high cells elicited EAE in recipient mice by day 15 post cell transfer, demonstrating their encephalitogenic potential. As expected, unseparated LN cells also transferred disease, with similar kinetics. We noticed a slight difference in disease grade, which may be due to the fact that an equivalent number of CD4high cell were not transferred. Due to limitations in FACS sorting we were only able to collect sufficient cells to transfer 82% the unsorted equivalent. Alternatively, the difference in disease grade could also be due to non-CD4+ cells present in the unsorted cell population. More importantly, CD4norm cells were unable to transfer disease even though 5 × 107 cells were transferred to recipients.
Table 2. Adoptive transfer of EAE in B6 mice using isolated CD4high cells.
The indicated CD4 cell populations were sterile sorted from day 4 cultures of MOG activated B6 LN cells. Cells were transferred i.v. to naïve syngeneic mice. EAE is graded as described in methods.
| Disease Incidence |
Average Day of Onset |
Average Disease Grade |
|
|---|---|---|---|
| Unsorted Cells (5 × 107) |
5/5 | 12.2 | 3.0 |
| CD4norm Cells (5 × 107) |
0/3 | -- | -- |
| CD4high Cells (6.3 × 106) |
3/3 | 15.0 | 1.0 |
In a second experiment, CD4high and CD4norm cells from MBP Ac1-11 primed B10.PL mice were similarly transferred to naïve recipient mice. In this case however, only adoptive transfer of the unsorted cell population could induce disease in naïve recipients (Table 3A). The fact that none of the sorted cell populations produced EAE in host animals was not completely unexpected, since the threshold number of cells necessary to elicit disease under these conditions is unknown. We previously reported that low encephalitogenicity in B6 and SJL mice could be enhanced to detectable levels by challenging the recipients with antigen (Shaw, Kim, Ho, Lisak, & Tse 1992;Shaw et al. 2007). The antigenic challenge apparently served to boost the activities of the transferred cells since challenge prior to cell transfer or depletion of the donor cells prior to challenge did not elicit EAE (Shaw, Li, Ho, Hoa, Lisak, & Tse 2007). To further test the encephalitogenicity of the sorted cell populations, the groups of recipients that did not succumb to EAE were immunized with a sub-encephalitogenic dose of MBPAc1-11 at four sites in the flanks. As shown in Table 3B, antigenic challenge resulted in disease development only in mice that received CD4high cells. In addition, onset of clinical disease correlated with the graded number of cells transferred. Mice that received 4 × 106 CD4high cells succumbed to EAE in less than five days. Mice that received only 1 × 105 sorted CD4high cells developed EAE in fifteen days post challenge. Mice that received high numbers of CD4norm cells, on the other hand, did not develop EAE even after antigenic challenge. As a control, naïve B10.PL mice were immunized with the same MBP peptide emulsion used for challenge. None of these mice developed EAE. These experiments strongly support the concept that in EAE, antigen-specific encephalitogenic T cells primarily reside in the CD4high subpopulation, similar to that reported for type I diabetes and EAMG (Lejon & Fathman 1999;Standifer, Kraig, & Infante 2003).
Table 3A/B. Adoptive transfer of EAE in B10.PL mice by sorted CD4 cell subsets.
A: Sorted CD4 cell subsets do not induced EAE by adoptive transfer only. B10.PL mice were immunized with MBP Ac1-11 and LN cells activated in vitro with the same peptide. After a four day culture the cells were FACS sorted into CD4 subpopulations and transferred to naïve recipients at the numbers indicated. Mice were observed for clinical signs of EAE for 30 days. B: Clinical EAE in B10.PL mice after antigenic challenge. After 30 days, those mice that had not displayed clinical EAE were immunized with a sub-encephalitogenic dose of MBP Ac1-11 (50 ug/mouse). Naive animals, immunized with MBP Ac1-11, served as controls, demonstrating that the dosage used is sub-encephalitogenic.
| Table 3A | |||
|---|---|---|---|
| Disease Incidence (pre-challenge) |
Average Day of Onset |
Average Disease Grade |
|
| Unsorted Cells (5 × 107) |
3/3 | 11.0 | 3.0 |
| CD4norm Cells (1 × 107) |
0/3 | -- | -- |
| CD4high Cells (1.5 × 105) |
0/3 | -- | -- |
| CD4high Cells (1.5 × 106) |
0/3 | -- | -- |
| CD4high Cells (4 × 106) |
0/3 | -- | -- |
| Table 3B | |||
| Disease Incidence (post-challenge) |
Average Day of Onset |
Average Disease Grade |
|
| Unsorted Cells (5 × 107) |
na | na | na |
| CD4norm Cells (1 × 107) |
0/3 | -- | -- |
| CD4high Cells (1.5 × 105) |
3/3 | 15.3 | 1.0 |
| CD4high Cells (1.5 × 106) |
3/3 | 8.7 | 3.0 |
| CD4high Cells (4 × 106) |
3/3 | 4.7 | 3.0 |
| Naïve mice immunized with 50 ug/ml MBP Ac1-11 |
0/6 | NA | NA |
3.4 CD4highCD25+ effector cells can be distinguished from CD4normCD25+ regulatory T cells
Since their initial description by Sakaguchi and colleagues (Sakaguchi, Sakaguchi, Asano, Itoh, & Toda 1995), CD4+CD25+ Treg cells have been reported to be involved in several models of autoimmunity, such as diabetes, EAE, colitis, gastritis and collagen-induced arthritis (Asano, Toda, Sakaguchi, & Sakaguchi 1996;Green, Choi, & Flavell 2002;Groux, O'Garra, Bigler, Rouleau, Antonenko, de Vries, & Roncarolo 1997;Huehn, Siegmund, Lehmann, Siewert, Haubold, Feuerer, Debes, Lauber, Frey, Przybylski, Niesner, de la, Schmidt, Brauer, Buer, Scheffold, & Hamann 2004;Kohm, Carpentier, Anger, & Miller 2002). These cells typically also express the cell surface molecules CD4 and CD25 (Kuniyasu et al. 2000). As the CD25 marker is also up-regulated in activated T cells that mediate effector functions, it has been difficult to separate these two populations for further studies. Based on our observation that encephalitogenic T cells up regulate their CD4 expression upon immunization of the animals with neuro-antigens, we speculated that this feature may differentially distinguish antigen-specific effector cells from regulatory T cells. To test this hypothesis, B6 mice were immunized with MOG35-55 peptide. Ten days after immunization, draining lymph node cells were isolated and cultured with antigen for 4 days. At the end of the culture period, cells were stained with anti-CD4 and anti-CD25 antibodies and analyzed on FACS. As can be seen in the upper right quadrant of Figure 2, two distinct populations of CD4+/CD25+ cells can be identified, one with intermediate level and one with high level of CD4 expression. These two populations are referred to as, CD4normalCD25+ (gate R2) and CD4highCD25+ (gate R3) respectively.
Figure 2. FACS analysis of MBP-primed lymph node cells from B6 mice after a four-day culture and stained with anti-CD4 and anti-CD25 antibodies.
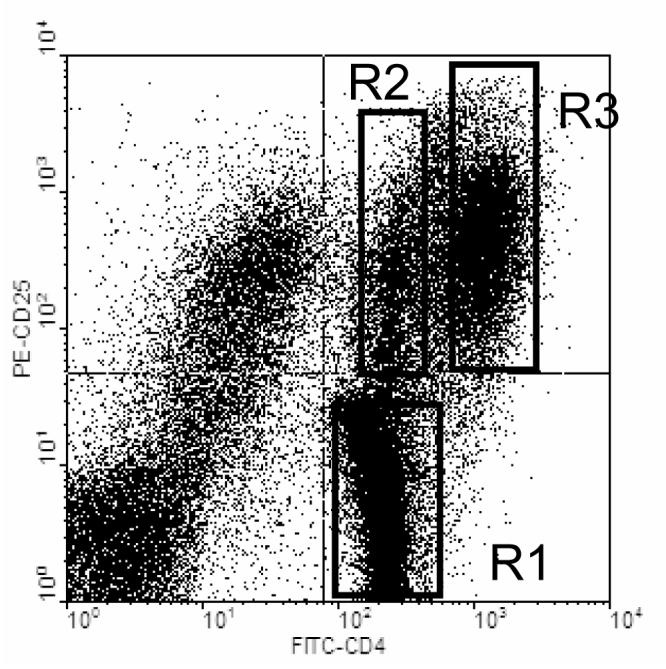
LN cells from MOG primed B6 mice were stimulated in vitro with the priming peptide for four days. FACS analysis of CD4 (FITC) and CD25 (PE) expression reveals two distinct CD4+/CD25+ cell populations. CD4normalCDC25+ cells (putative Treg) are delineated by gate R2 while CD4highCD25+ cells (putative Teff) are delineated by gate R3. FACS staining is typical of all cultured LN cells tested.
To differentiate the function of these cell populations, we next examined the expression of genes associated with both Treg and T effector cells in EAE. We expected that the R2 population of cells would contain Treg cells which would express high levels of FoxP3. Alternatively, we expected that the R3 population of cells contained effector T cells which would produce the inflammatory cytokines IFN-γ and IL-17, (Ando et al. 1989;Langrish, Chen, Blumenschein, Mattson, Basham, Sedgwick, McClanahan, Kastelein, & Cua 2005). To test this hypothesis, B6 mice were immunized with MOG35-55 and draining LN cells cultured with the peptide. Three days later the cells were harvested and stained for FACS analysis of intracellular FoxP3. As can be seen in Figure 3, the vast majority of the CD4normalCD25+ cells (R2 gate) stained positive for FoxP3, while the CD4high cells (R3 gate) were uniformly negative.
Figure 3. FoxP3 is only is contained primarily in CD4normal CD25+ cells.

LN cells from MOG p35-55 primed mice were cultured for four days with the priming peptide followed by analysis of intracellular FoxP3 expression. LN cells were stained with antibodies to CD4 (FITC) and CD25 (APC) then fixed and stained for intracellular expression of FoxP3 (PE) using a Treg staining kit (eBioscience). Cell populations defined by gates R2 and R3 in the density plot were further analyzed for FoxP3 expression in the histograms at right. Staining is representative of at least three experiments.
In similar experiments we stained for inflammatory cytokines, and found that the CD4high population of cells contained the bulk of the cytokine producing cells (Figure4). Nearly half of the CD4high cells from these animals were found to produce IFN-γ (9.9% total CD4high, 4.1% CD4high/IFN-γ+--Figure 4, upper panel). In separate experiments we found that expression of IL-17 was also contained solely in the CD4high sub-population, but fewer cells produced IL-17. Only 15% of the CD4high cells were positive for IL-17 expression (Figure 4, lower panel). To further quantitate the levels of expression of these genes in the different cells populations, B6 mice were immunized with MOG peptide, draining LN cells were cultured for four days and CD4normalCD25+ (gate R2) and CD4highCD25+ (gate R3) cells FACS sorted. The yield for R2 was 0.7 × 106 cells per mouse and the yield for R3 was nearly 2 × 106 cells per mouse. IL-17 and IFN-g expression was analyzed by real time PCR. For comparison, gene expression was compared to that of unsorted LN cells using the ΔΔCt method (Pagliarulo, George, Beil, Groshen, Laird, Cai, Willey, Cote, & Datar 2004). As shown in Figure 5, the CD4normalCD25+ population had greater than 300 fold increased expression of Foxp3 compared to unsorted LN cells. On the other hand, IFN-γ and IL-17 expression was highly enriched in sorted CD4high cells, with 52 and 187 fold increases in gene expression respectively.
Figure 4. Inflammatory cytokine expression is contained in the CD4high cell population.
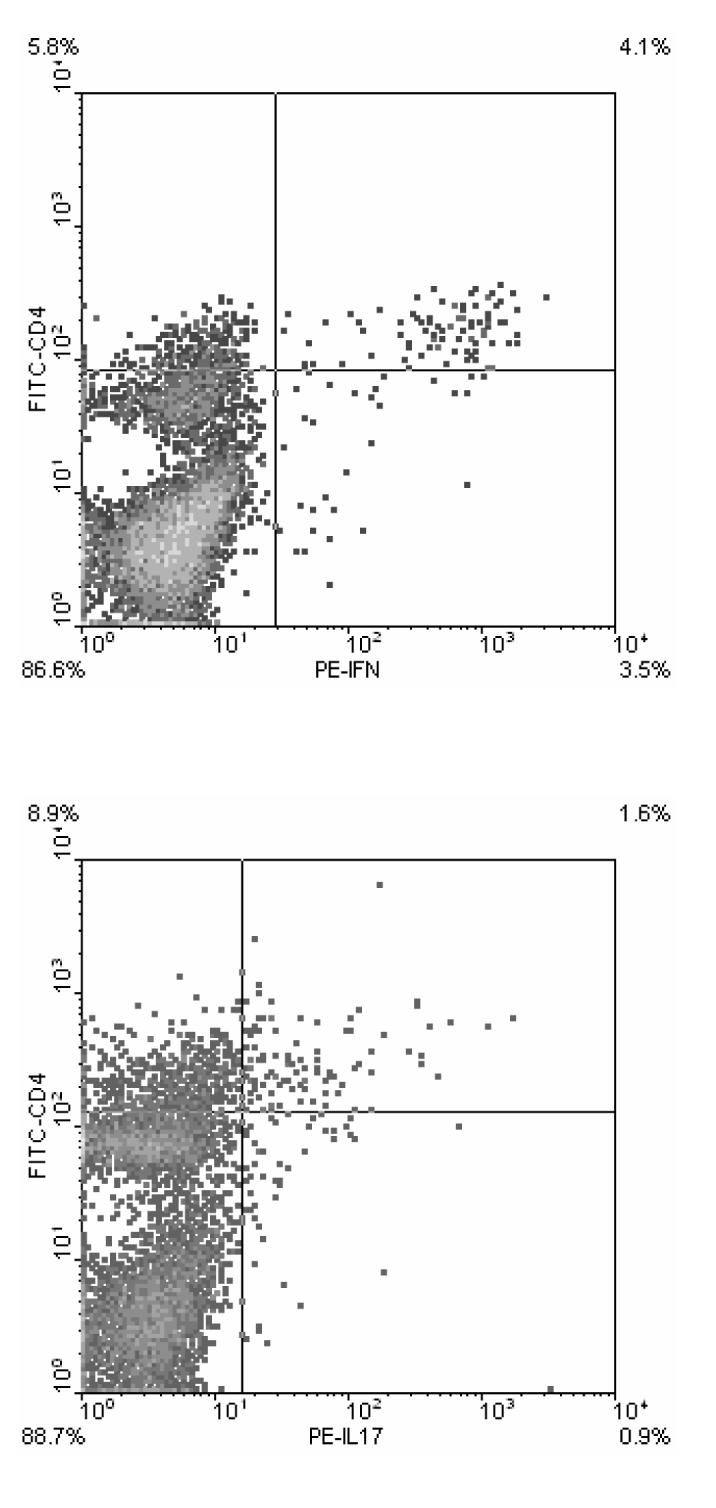
LN cells from MOG p35-55 primed mice were cultured for four days with the priming peptide. Cells were then stained for analysis of CD4 (FITC) expression and intracellular IFN-γ and IL-17 expression (both PE) as described in methods. Quadrant gates are set to differentiate CD4high cells from CD4+ cells (set arbitrarily), and cytokine producing cells from non-producing cells (as determined by analysis of unstained control samples). Staining is representative of at least two experiments.
Figure 5. Inflammatory cytokine and FoxP3 gene in sorted cell subsets.

MOG peptide primed and in vitro stimulated LN cells were stained with CD4 and CD25 antibodies as described in methods. CD4high Teff cells and CD4normalCD25+ Treg cells were sorted using gating similar to that shown in Figure 3. Gene expression from sorted cells (8 × 104 cells) was analyzed by real time PCR.
Since the bulk of the FoxP3 expression was confined to the CD4normalCD25+ cell population, these cells were tested for their ability to inhibit the proliferative responses of anti-CD3 stimulated naïve CD4+CD25− spleen cells (Pace, Pioli, & Doria 2005). Figure 6 shows that CD4normalCD25+ cells are extremely effective in suppressing the proliferative responses even at Treg:target (Effector) cell ratio of 1:8. CD4normalCD25+ Treg cells by themselves did not respond to anti-CD3 stimulation (data not shown). These results show clearly that the CD4normalCD25+ cell population contains the bulk of Treg cells. Thus, this approach has achieved separation of encephalitogenic effector T cells (CD4highCD25+) and Treg cells (CD4normalCD25+) in mice primed with a neuroantigen ten days earlier.
Figure 6. CD4normalCD25+ cells can suppress mitogenic T cell responses.

CD4normalCD25+ (Treg) cells were isolated from 4-day cultures of MOG35-55-immunized B6 MOG p35-55 lymph nodes cells by sterile sorting using gates similar to that depicted in Figure 3. Naïve CD4 positive T cells (1 × 104) were cultured for 3 days in round-bottom 96-well plates with the sorted cells and 2 × 104 APCs (T-cell-depleted splenocytes) per well in triplicate in the presence of 2.0 ug/ml of anti-CD3 antibody (clone 145-2C11) in ratios of Treg/E as indicated above. Sixteen hours before harvesting, cells were pulsed with 3H-Thymidine. Cells were harvested and incorporation of radioactive thymidine was counted in a beta- counter. Results are representative of at least two experiments.
4. Discussion
The results presented here demonstrate that CNS-antigen-specific T cells up regulate CD4 expression both in vitro and in vivo. The entire proliferative response to an autoantigen was found within this population of cells as shown by thymidine incorporation assays as well as by limiting dilution analysis of precursor frequency (data not shown). We have also demonstrated that the encephalitogenic response to CNS autoantigens was contained in the CD4high population of cells by adoptive transfer of disease using this sub-population of cells. Moreover, CD4high cells exclusively produce two key inflammatory cytokines implicated in EAE pathogenesis, namely IFN-γ and IL-17.
The frequency of T cells specific for an auto-antigen is extremely low, on the order of 1:50,000-300,000 T cells in unprimed mice, and approximately 1:1000-10,000 in primed mice (Ford & Burger 1983;Gebel et al. 1983;Kojima et al. 1988). Although several methods have been developed to study autoreactive T cell responses in vivo (e.g. adoptive transfer of T cell clones and TCR transgenic mice) (Mendel et al. 2004;Mokhtarian, McFarlin, & Raine 1984b;Zamvil, Nelson, Trotter, Mitchell, Knobler, Fritz, & Steinman 1985), the characterization of autoantigen-specific T cell responses requires manipulation of the normal physiology of the immune response. T cell clones and T cells from TCR transgenic mice utilize a single TCR αβ heterodimer and these T cells generate a homogenous, single affinity response to antigen. The methods used to isolate T cell clones, and also the T cells from which TCR transgenic mice are derived, have used culture conditions predisposed to selecting only the best growing T cells from culture; the T cells with the proliferative response “most favorable” for growth. Thus, although these methods have provided important insights into autoreactive T cell responses in vivo, they share some drawbacks in studies of conventional immune response.
We find that CD4high effector cells expressed both IFN-γ and IL-17, the major inflammatory mediators implicated in EAE development. Our experiments indicate that CD4high effector cells express IFN-γ more than twice as frequently as they did IL-17. This is consistent with previous reports in SJL and B6 mice in which IFN-γ producing cells outnumbered IL-17 producing cells (Harrington et al. 2005;Langrish, Chen, Blumenschein, Mattson, Basham, Sedgwick, McClanahan, Kastelein, & Cua 2005), and indicates that the CD4high phenotype encompasses all EAE effector cell types. Interestingly, we found no T cells expressing both cytokines simultaneously. This is in contrast to a recent report by Suryani and Sutton, who found that the majority of IL-17 producing cells recovered from the CNS of B6 mice with active EAE also, expressed IFN-γ (Asano, Toda, Sakaguchi, & Sakaguchi 1996). This difference may due to the cell source since our studies concentrated on cells from the draining LN's not the CNS. The difference seen could also be due to the length of the in vitro activation. Suryani found that cytokine double positive cells became single positive IL-17+ producing cells within 30 hours after in vitro activation. It is not known if this phenotype change is an artifact of ex vivo antigenic stimulation. We have found that encephalitogenic CD4high T cells can be detected in the CNS of mice with active EAE (data not shown) and are currently determining the cytokine expression profile of these cells. The use of the CD4high phenotype should allow us to examine these cells without the need for ex vivo stimulation.
Recent studies have implicated a population of regulatory T cells (Treg) in the maintenance of tolerance as well as controlling EAE. Kohm et al. (Kohm, Carpentier, Anger, & Miller 2002) isolated CD4+CD25+ cells from the lymph nodes of naïve unprimed C57BL/6 mice and demonstrated that transfer of these cells into naive syngeneic recipient mice three days prior to active induction of EAE with MOG35-55 significantly protected the animals from development of clinical EAE. The difficulty in isolating Treg cells from in vitro cultures of antigen stimulated LN cells lies in the fact that both cell types express CD4 and CD25. We have demonstrated that CD4+CD25+ Treg cells can be differentiated from CD4+ effector cells on the basis of differential CD4 expression. CD4high effector cells were found to be uniformly negative for the transcription factor FoxP3, a marker for Treg cells. Treg cells were contained in a distinct population of cells expressing intermediate levels of CD4 (CD4normal) and high levels of CD25 after three to four days of in vitro culture with antigen. These cells expressed high levels of FoxP3 and did not express the inflammatory cytokines IFN-γ or IL-17. Nearly all of these cells expressed FoxP3 indicating that the majority of the cells in this population were Treg. The use of antigen-specific tetramers (Korn et al. 2007) should allow for more specific study of antigen-specific Treg without resorting to the use of genetically manipulated mice such as FoxP3-GFP knock-in animals (Korn, Reddy, Gao, Bettelli, Awasthi, Petersen, Backstrom, Sobel, Wucherpfennig, Strom, Oukka, & Kuchroo 2007).
Acknowledgments
The authors would like to acknowledge the expert technical assistance of Ms. Cariel Taylor and the invaluable administrative assistance of Ms Robin Kizer and Ms Lynette Rae. The authors would also like to thank Dr. Jeannette Thai for review and thoughtful discussion of the manuscript.
Grant Support
This work was sponsored by the National Institutes of Health grants DK39959 and DK44837 and National Multiple Sclerosis Society Grant RG 3112-A-2. Michael K. Shaw was a fellow of the National Multiple Sclerosis Society for a portion of this work.
Footnotes
Disclosures
The authors have no financial conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amor S, Baker D, Groome N, Turk JL. Identification of a major encephalitogenic epitope of proteolipid protein (residues 56-70) for the induction of experimental allergic encephalomyelitis in Biozzi AB/H and nonobese diabetic mice. J Immunol. 1993;150(12):5666–5672. [PubMed] [Google Scholar]
- Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, Matthieu JM, Baker D. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol. 1994;153(10):4349–4356. [PubMed] [Google Scholar]
- Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124(1):132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J.Exp.Med. 1996;184(2):387–396. doi: 10.1084/jem.184.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard CC. Experimental autoimmune encephalomyelitis in mice: genetic control of susceptibility. J Immunogenet. 1976;3(4):263–274. doi: 10.1111/j.1744-313x.1976.tb00583.x. [DOI] [PubMed] [Google Scholar]
- Chang X, Gao JX, Jiang Q, Wen J, Seifers N, Su L, Godfrey VL, Zuo T, Zheng P, Liu Y. The Scurfy mutation of FoxP3 in the thymus stroma leads to defective thymopoiesis. J.Exp.Med. 2005;202(8):1141–1151. doi: 10.1084/jem.20050157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421(6924):744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat.Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Ford D, Burger D. Precursor frequency of antigen-specific T cells: effects of sensitization in vivo and in vitro. Cell Immunol. 1983;79(2):334–344. doi: 10.1016/0008-8749(83)90075-8. [DOI] [PubMed] [Google Scholar]
- Gebel HM, Scott JR, Parvin CA, Rodey GE. In vitro immunization to KLH. II. Limiting dilution analysis of antigen-reactive cells in primary and secondary culture. J Immunol. 1983;130(1):29–32. [PubMed] [Google Scholar]
- Green EA, Choi Y, Flavell RA. Pancreatic lymph node-derived CD4(+)CD25(+) Treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity. 2002;16(2):183–191. doi: 10.1016/s1074-7613(02)00279-0. [DOI] [PubMed] [Google Scholar]
- Groux H, O'Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389(6652):737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat.Immunol. 2005;6(11):1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, Niesner U, de la RM, Schmidt CA, Brauer R, Buer J, Scheffold A, Hamann A. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J.Exp.Med. 2004;199(3):303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns TG, Kerlero de RN, Menon KK, Abo S, Gonzales MF, Bernard CC. Myelin oligodendrocyte glycoprotein induces a demyelinating encephalomyelitis resembling multiple sclerosis. J.Immunol. 1995;154(10):5536–5541. [PubMed] [Google Scholar]
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat.Immunol. 2003;4(4):337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J.Immunol. 2002;169(9):4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- Kojima M, Cease KB, Buckenmeyer GK, Berzofsky JA. Limiting dilution comparison of the repertoires of high and low responder MHC-restricted T cells. J Exp.Med. 1988;167(3):1100–1113. doi: 10.1084/jem.167.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J.Immunol. 2006;177(1):566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat.Med. 2007;13(4):423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuniyasu Y, Takahashi T, Itoh M, Shimizu J, Toda G, Sakaguchi S. Naturally anergic and suppressive CD25(+)CD4(+) T cells as a functionally and phenotypically distinct immunoregulatory T cell subpopulation. Int.Immunol. 2000;12(8):1145–1155. doi: 10.1093/intimm/12.8.1145. [DOI] [PubMed] [Google Scholar]
- Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J.Exp.Med. 2007;204(1):57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J.Exp.Med. 2005;201(2):233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejon K, Fathman CG. Isolation of self antigen-reactive cells from inflamed islets of nonobese diabetic mice using CD4high expression as a marker. J Immunol. 1999;163(10):5708–5714. [PubMed] [Google Scholar]
- Lyons JA, Ramsbottom MJ, Trotter JL, Cross AH. Identification of the encephalitogenic epitopes of CNS proteolipid protein in BALB/c mice. Journal of Autoimmunity. 2002;19(4):195–201. doi: 10.1006/jaut.2002.0619. [DOI] [PubMed] [Google Scholar]
- McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16(2):311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- Mendel I, Natarajan K, Ben Nun A, Shevach EM. A novel protective model against experimental allergic encephalomyelitis in mice expressing a transgenic TCR-specific for myelin oligodendrocyte glycoprotein. J Neuroimmunol. 2004;149(12):10–21. doi: 10.1016/j.jneuroim.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Mokhtarian F, McFarlin DE, Raine CS. Adoptive transfer of myelin basic protein-sensitized T cells produces chronic relapsing demyelinating disease in mice. Nature. 1984a;309(5966):356–358. doi: 10.1038/309356a0. [DOI] [PubMed] [Google Scholar]
- Mokhtarian F, McFarlin DE, Raine CS. Adoptive transfer of myelin basic protein-sensitized T cells produces chronic relapsing demyelinating disease in mice. Nature. 1984b;309(5966):356–358. doi: 10.1038/309356a0. [DOI] [PubMed] [Google Scholar]
- Pace L, Pioli C, Doria G. IL-4 modulation of CD4+CD25+ T regulatory cell-mediated suppression. J.Immunol. 2005;174(12):7645–7653. doi: 10.4049/jimmunol.174.12.7645. [DOI] [PubMed] [Google Scholar]
- Pagliarulo V, George B, Beil SJ, Groshen S, Laird PW, Cai J, Willey J, Cote RJ, Datar RH. Sensitivity and reproducibility of standardized-competitive RT-PCR for transcript quantification and its comparison with real time RT-PCR. Mol.Cancer. 2004;3:5. doi: 10.1186/1476-4598-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinelli CB, McFarlin DE. Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2- T lymphocytes. J.Immunol. 1981;127(4):1420–1423. [PubMed] [Google Scholar]
- Powell MB, Mitchell D, Lederman J, Buckmeier J, Zamvil SS, Graham M, Ruddle NH, Steinman L. Lymphotoxin and tumor necrosis factor-alpha production by myelin basic protein-specific T cell clones correlates with encephalitogenicity. Int.Immunol. 1990;2(6):539–544. doi: 10.1093/intimm/2.6.539. [DOI] [PubMed] [Google Scholar]
- Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, Sobel RA, Wucherpfennig KW, Kuchroo VK. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc.Natl.Acad.Sci.U.S.A. 2004;101(43):15434–15439. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway W, Fasso M, Fathman CG. Following antigen challenge, T cells up-regulate cell surface expression of CD4 in vitro and in vivo. J Immunol. 1998;161(2):714–720. [PubMed] [Google Scholar]
- Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol.Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J.Immunol. 1995;155(3):1151–1164. [PubMed] [Google Scholar]
- Sakai K, Zamvil SS, Mitchell DJ, Lim M, Rothbard JB, Steinman L. Characterization of a major encephalitogenic T cell epitope in SJL/J mice with synthetic oligopeptides of myelin basic protein. J Neuroimmunol. 1988;19(12):21–32. doi: 10.1016/0165-5728(88)90032-x. [DOI] [PubMed] [Google Scholar]
- Segal BM, Dwyer BK, Shevach EM. An interleukin (IL)-10/IL-12 immunoregulatory circuit controls susceptibility to autoimmune disease. J.Exp.Med. 1998;187(4):537–546. doi: 10.1084/jem.187.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MK, Kim C, Hao HW, Chen F, Tse HY. Induction of myelin basic protein-specific experimental autoimmune encephalomyelitis in C57BL/6 mice: mapping of T cell epitopes and T cell receptor V beta gene segment usage. J Neurosci.Res. 1996;45(6):690–699. doi: 10.1002/(SICI)1097-4547(19960915)45:6<690::AID-JNR5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Shaw MK, Kim C, Ho KL, Lisak RP, Tse HY. A combination of adoptive transfer and antigenic challenge induces consistent murine experimental autoimmune encephalomyelitis in C57BL/6 mice and other reputed resistant strains. J Neuroimmunol. 1992;39(12):139–149. doi: 10.1016/0165-5728(92)90183-l. [DOI] [PubMed] [Google Scholar]
- Shaw MK, Li J, Ho P, Hoa H, Lisak RP, Tse HY. Induction of Myelin Basic Protein-Specific Adoptive Experimental Autoimmne Encephalomyelitis in C57BL/6 Mice: Longevity of Donor T Cells and Lack of Disease Relapses. In: Broglio P, editor. Neuroimmunology Research Focus. Nova Science Publishers, Inc.; 2007. [Google Scholar]
- Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat.Immunol. 2002;3(2):135–142. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- Standifer NE, Kraig E, Infante AJ. A hierarchy of T cell receptor motifs determines responsiveness to the immunodominant epitope in experimental autoimmune myasthenia gravis. J.Neuroimmunol. 2003;145(12):68–76. doi: 10.1016/j.jneuroim.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J.Exp.Med. 2006;203(7):1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanborg RH. Experimental autoimmune encephalomyelitis in rodents as a model for human demyelinating disease. Clin Immunol.Immunopathol. 1995;77(1):4–13. doi: 10.1016/0090-1229(95)90130-2. [DOI] [PubMed] [Google Scholar]
- Tompkins SM, Padilla J, Dal Canto MC, Ting JP, Van KL, Miller SD. De novo central nervous system processing of myelin antigen is required for the initiation of experimental autoimmune encephalomyelitis. J.Immunol. 2002;168(8):4173–4183. doi: 10.4049/jimmunol.168.8.4173. [DOI] [PubMed] [Google Scholar]
- Traugott U, McFarlin DE, Raine CS. Immunopathology of the lesion in chronic relapsing experimental autoimmune encephalomyelitis in the mouse. Cell Immunol. 1986;99(2):395–410. doi: 10.1016/0008-8749(86)90248-0. [DOI] [PubMed] [Google Scholar]
- Traugott U, Raine CS, McFarlin DE. Acute experimental allergic encephalomyelitis in the mouse: immunopathology of the developing lesion. Cell Immunol. 1985;91(1):240–254. doi: 10.1016/0008-8749(85)90047-4. [DOI] [PubMed] [Google Scholar]
- Trotter JL, Clark HB, Collins KG, Wegeschiede CL, Scarpellini JD. Myelin proteolipid protein induces demyelinating disease in mice. J.Neurol.Sci. 1987;79(12):173–188. doi: 10.1016/0022-510x(87)90271-1. [DOI] [PubMed] [Google Scholar]
- Tuohy VK, Lu Z, Sobel RA, Laursen RA, Lees MB. Identification of an encephalitogenic determinant of myelin proteolipid protein for SJL mice. J.Immunol. 1989;142(5):1523–1527. [PubMed] [Google Scholar]
- Zamvil S, Nelson P, Trotter J, Mitchell D, Knobler R, Fritz R, Steinman L. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature. 1985;317(6035):355–358. doi: 10.1038/317355a0. [DOI] [PubMed] [Google Scholar]
- Zamvil SS, Mitchell DJ, Moore AC, Kitamura K, Steinman L, Rothbard JB. T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature. 1986;324(6094):258–260. doi: 10.1038/324258a0. [DOI] [PubMed] [Google Scholar]