

Abstract
The presumed role of an overactive Protein Arginine Deiminase 4 (PAD4) in the pathophysiology of rheumatoid arthritis (RA) suggests that PAD4 inhibitors could be used to treat an underlying cause of RA, potentially offering a mechanism to stop further disease progression. Thus, the development of such inhibitors is of paramount importance. Towards the goal of developing such inhibitors, we initiated efforts to characterize the catalytic mechanism of PAD4; and thereby identify important mechanistic features that can be exploited for inhibitor development. Herein we report the results of mutagenesis studies as well as our efforts to characterize the initial steps of the PAD4 reaction, in particular, the protonation status of Cys645 and His471 prior to substrate binding. The results indicate that Cys645, the active site nucleophile, exists as the thiolate in the active form of the free enzyme. pH studies on PAD4 further suggest that this enzyme utilizes a reverse protonation mechanism.
Rheumatoid Arthritis1 (RA) is a chronic and progressive autoimmune disorder of unknown etiology. It is the second most common type of arthritis, affecting ~1% of the adult US population and causing a mean reduction in life expectancy of 5 – 10 years (1, 2). Due to its idiopathic nature, the therapeutic options available for RA largely focus on disease management, i.e. treating its symptoms rather than treating an underlying cause(s) of disease (3). Over the last several years, however, serological, genetic, and biochemical studies (4–8) have suggested a role for a dysregulated Protein Arginine Deiminase 4 (PAD4) activity in the onset and progression of this autoimmune disorder – PAD4 catalyzes the post-translational conversion of peptidyl-Arg to peptidyl-citrulline (Cit) (Figure 1). For example, RA associated mutations have been identified in the PAD4 gene (4) and autoantibodies that recognize citrullinated proteins are specifically produced by RA patients (7, 8). Furthermore, the treatment of rodents with citrullinated collagen leads to a higher incidence and a faster rate of onset of collagen-induced arthritis in rodent models of RA (9, 10). On the basis of this information, we and others have suggested that the deiminating activity of PAD4 is up-regulated in RA patients, generating an aberrant immune response to citrullinated epitopes in the RA synovium (5, 11, 12). Thus, PAD4 inhibitors hold the promise of being effective therapeutics for RA. In addition to its presumed role in RA, dysregulated PAD4 activity and/or expression has recently been associated with the etiology of multiple sclerosis and cancer (13–15), thereby suggesting that the therapeutic value of PAD4 inhibitors could be broader than initially considered.
Figure 1.

Reaction catalyzed by PAD4.
PAD4 is predominantly expressed in blood lymphocytes and has been suggested to play roles in apoptosis and differentiation (4, 16–19). Additionally, PAD4 is known to be a calcium dependent nuclear enzyme that deiminates histones H2A, H3, and H4 and acts as a transcriptional corepressor for the estrogen receptor (16, 17, 20–22). However, and despite its importance as a therapeutic drug target, the physiological role(s) of PAD4 are incompletely defined and are only beginning to be deciphered. The recent development of potent and bioavailable PAD4 inhibitors and activity based protein profiling reagents (23–25) will undoubtedly be useful tools for obtaining a more complete description of the physiological role(s) of this enzyme.
Recent in vitro studies have generated significant data regarding the molecular details of PAD4 catalysis (although key gaps remain). For example, preliminary investigations have confirmed the identity and stoichiometry of the reaction products (11, 26) and demonstrated that solvent oxygen is incorporated into the product, i.e. peptidyl-Cit (11, 26). Also, initial pH rate profiles performed with low substrate concentrations, approximating kcat/Km conditions, suggested that two ionizable groups were critical for catalysis (11). And finally, several structures of PAD4, determined by X-ray crystallography, have confirmed that PAD4 is a member of the amidinotransferase superfamily of enzymes (27–29). On the basis of this homology (30–32), and preliminary site directed mutagenesis experiments (27), there are 4 key catalytic residues, including Asp350, His471, and Asp473 that contribute to rate enhancement by playing loosely defined roles in substrate binding (Asp350 and Asp473) and general acid/general base catalysis (His471). Cys645, the fourth key catalytic residue, most likely acts as a nucleophile to generate a covalent S-alkylthiouronium intermediate akin to the acyl enzyme intermediates observed in other cysteine hydrolases. Note that while a kinetically competent covalent intermediate has yet to be demonstrated for PAD4, the fact that F- and Cl-amidine, two haloacetamidine bearing mechanism based inactivators, irreversibly inactivate PAD4 by modifying Cys645 (24, 25) argues forcefully for a role for Cys645 as the active site nucleophile when combined with the abundance of evidence for covalent catalysis among other amidinotransferase family members (e.g. rapid quench kinetic studies on Arginine Deiminase (ADI) (33), mass spectrometry studies on dimethylarginine dimethylaminohydrolase (DDAH) (34), and crystal structures of the S-alkylthiouronium intermediate in ADI (31)).
While at least 4 different mechanisms have been proposed to explain the deiminating activity of the various amidinotransferase family members (11, 30–32, 35), we initially proposed a working model that involves a nucleophilic thiolate whose charge is stabilized via an ion pair with His471, analogously to the Cys-imidazolium ion pair observed in papain (36). Herein we report the results of site directed mutagenesis studies, pKa measurements on the active site thiol, pH rate profiles, and solvent isotope effects that support the existence of a nucleophilic active site thiolate. Further, the results indicate that at the pH optimum only a small fraction of the enzyme exists in the catalytically competent thiolate form and in total suggest that PAD4 utilizes a reverse protonation mechanism (37, 38).
EXPERIMENTAL PROCEDURES
Chemicals
Iodoacetamide and 2-chloroacetamidine were obtained from Oakwood Products (Columbia, SC). Dithiothreitol (DTT), iodoacetic acid, protease inhibitor cocktail (Cat#P8465), and Benzoyl L-arginine ethyl ester (BAEE) were acquired from Sigma-Aldrich (St. Louis, MO). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was obtained from Fluka. Dideuterium oxide was acquired from Cambridge Isotope Laboratories (Andover, MA).
Purification of PAD4
Recombinant human PAD4 was expressed and purified analogously to previously described methods (11, 39). Our optimized protocol is described in detail in the supplementary material.
pH Profile
The pH profile of PAD4 was constructed by measuring the steady state kinetic parameters for the deimination of BAEE over a pH range of 6.0—9.0. Reaction buffers consisted of 100 mM Bis-Tris (5.7–7.5), 100 mM Tris-HCl (7.5–8.5), or 100 mM CHES (8.5–9.0) plus 2 mM DTT, 10 mM CaCl2, 50 mM NaCl, and BAEE in various concentrations (0 to 20 mM in a final volume of 60 μL). Stock concentrations of BAEE were dissolved in 50 mM buffer at the desired pH. Enzyme assays were performed essentially as described in Kearney et al (11). The initial rates obtained from these experiments were fit to eq 1
| (1) |
using GraFit version 5.0.11 (40). The kcat and kcat/Km values obtained from this analysis were plotted as a function of pH and fit to eq 2
| (2) |
using GraFit version 5.0.11. Lim1 is the amount of activity observed at low pH, lim2 is pHopt, lim3 is the amount of activity observed at high pH.
Substrate Protection Experiments
Reaction mixtures containing 10 mM CaCl2, 100 mM Tris-HCl pH 7.6, 500 μM TCEP, 50 mM NaCl, and BAEE (2 mM or 10 mM) were pre-incubated with either 2-chloroacetamidine (5 mM) or iodoacetamide (1.25 mM) at 37 °C for ten minutes. Subsequently, PAD4 (0.2 μM final) was added and aliquots (60 μL) were removed at various time points (0–15 minutes) and residual activity assayed as described above. Samples incubated without chloroacetamidine or iodoacetamide were used as controls. The data obtained were fit to eq 3
| (3) |
using GraFit version 5.0.11. P is the amount of citrulline produced, vi is the initial velocity, kobs is the observed rate of inactivation, and t is the time.
Iodoacetamide Inactivation Kinetics
Inactivation reactions containing 500 μM TCEP, 10 mM CaCl2, and 100 mM of buffer (pH 6.5 Bis-Tris, pH 7.6–9.0 Tris) were pre-incubated with 2.0 μM PAD4 for 10 minutes at 37 °C (20 μL total volume). Varying concentrations of iodoacetamide (0–2.0 mM) were then added. Inactivation reaction were quenched with DTT (20 mM final) at various time points (0–30 minutes) and then immediately added to a reaction mixture to measure the residual activity of PAD4. These reaction mixtures, which contained 10 mM CaCl2, 50 mM NaCl, 100 mM Tris pH 7.6, 2 mM DTT, and 10 mM BAEE (60 μL total volume), were pre-incubated at 37 °C for ten minutes, before adding aliquots from the quenched inactivation mixture. Activity assays proceeded for 15 minutes at which point the reaction was stopped by flash freezing in liquid nitrogen. Residual enzymatic activity was then quantified using the methodology described above. The data obtained at each iodoacetamide concentration were fit to eq 4
| (4) |
using Grafit 5.0.1.1. v is the velocity, vo is the initial velocity, k is the pseudo first order rate constant for inactivation, and t is time. Due to a lack of inactivator saturation, the second order rate constant for enzyme inactivation, kinact/KI, was determined by plotting the observed inactivation rates (kobs) versus inactivator concentration and fitting the data to eq 5
| (5) |
using Grafit 5.0.1.1. kinact is the maximal rate of inactivation; KI is the concentration of inactivator that yields half-maximal inactivation, and [I] is the concentration of inactivator. The slopes thus obtained, i.e. kinact/KI, were plotted versus pH and subsequently fit to eq 6
| (6) |
using GraFit version 5.0.11. Lim1 is the minimum rate and Lim2 is the maximum rate.
Iodoacetamide versus Iodoacetic acid Inactivation Kinetics
Inactivation reactions containing 500 μM TCEP, 10 mM CaCl2, and 100 mM HEPES pH 7.6 were pre-incubated with 2.0 μM PAD4 at 37 °C (60 μL total volume). After ten minutes, iodoacetamide or iodoacetic acid (1.25 mM) was added to the mixture. The residual activity of PAD4 was measured as described above.
2-Chloroacetamidine Inactivation Kinetics
Inactivation mixtures containing 2 mM DTT, 10 mM CaCl2, and 100 mM of buffer (pH 6.5 MES, pH 7.0 to 9.0 Tris-HCl)) were preincubated with 2 μM PAD4 at 37 °C. After ten minutes, 2-chloroacetamidine (0–30 mM) was added to the mixture. Residual activity was measured by removing aliquots from the inactivation mix at various time points (0 to 30 minutes). These aliquots were diluted ten-fold into reaction buffer (10 mM CaCl2, 50 mM NaCl, 2 mM DTT, 100 mM Tris-HCl pH 7.6, and 10 mM BAEE). The data were processed using the methods described above.
Site directed mutagenesis
Catalytic mutants of PAD4 were generated using the Quik Change Site Directed Mutagenesis Kit™ (Stratagene). Forward and reverse primers (IDT DNA technologies) used for each mutagenesis experiment are listed in Table 1. Note that the entire open reading frame of each mutant was sequenced to ensure that the proper mutation had been incorporated and that no additional mutations had been introduced during the PCR reaction. Mutant PAD4 enzymes were purified using the methodology described above.
Table 1.
Sequences of forward and reverse primers for site directed mutagenesisa
| Mutant | Forward primer | Reverse primer |
|---|---|---|
| H471A | tggctgtccgtgggcgccgtggacgagttc | gaactcgtccacggcgcccacggacagcca |
| H471G | tggctgtccgtgggcggcgtggacgagttc | gctcaggaactcgccgcccacggacagcca |
| H471Q | ctgtccgtgggccaggtggacgagttcctg | caggaactcgtccacctggcccacggacag |
| D350A | gaccagtggatgcaggccgaaatggagatcggc | gccgatctccatttcggcctgcatccactggtc |
| D350E | gaccagtggatgcaggaagaaatggagatcggc | gccgatctccatttcttcctgcatccactggtc |
| D350N | gaccagtggatgcagaatgaaatggagatcggctac | gtagccgatctccatttcattctgcatccactggtc |
| D473A | gtgggccacgtggccgagttcctgagc | gctcaggaactcggccacgtggcccac |
| D473E | gtccgtgggccacgtggaagagttcctgagctttg | caaagctcaggaactcttccacgtggcccacggac |
| D473N | gtccgtgggccacgtgaatgagttcctgagctttg | caaagctcaggaactcattcacgtggcccacggac |
| C645S | ggggaggtgcactccggcaccaacgtgc | gcacgttggtgccggagtgcacctcccc |
| C645A | ggggaggtgcacgccggcaccaacgtgc | gcacgttggtgccggcgtgcacctcccc |
All primers are written 5′ to 3′.
Kinetic studies on mutant enzymes
Steady state kinetic parameters for PAD4 mutants were determined as previously described. Because the catalytic activity of all of the mutants described in this study was significantly impaired relative to wild type PAD4, enzyme assays were performed with higher amounts of enzyme (2.5 μM final) and for longer time periods (2 h) to achieve detectable amounts of citrulline.
Solvent Isotope Effects
Solvent Isotope Effects (SIE) were measured in reaction buffers containing 50 mM Bis-Tris (pL 5.75–7.00), 50 mM Tris-HCl (pL 7.0–8.5), or 50 mM CHES (pL 8.5–9.0) as well as 10 mM CaCl2, 2 mM DTT, 50 mM NaCl, PAD4, and various concentrations of BAEE (0–10 mM) in > 96% D2O. The kinetic parameters were determined using the methods described above. To control for possible solvent viscosity effects (SVE), the steady state kinetic parameters for the deimination of BAEE were determined under identical conditions at the pH optimum in assay buffer containing 10 % glycerol. The SVE and SIE on the calcium dependence of PAD4 were also assessed as a further control. For these studies, PAD4 activity was measured in assay buffers containing either > 96% D2O or 10% glycerol plus CaCl2 (0–10 mM), 50 mM Tris-HCl pH 7.6, 10 mM BAEE, 50 mM NaCl, 2 mM DTT, and PAD4. The K0.5 for calcium was determined using the methodology described in Kearney et al (11).
RESULTS
Site directed mutagenesis
To gain insights into the catalytic mechanism of PAD4, we generated a series of site specific mutations in the PAD4 gene at sites corresponding to Asp350, His471, Asp473, and Cys645, i.e. the 4 key catalytic residues involved in substrate binding (Asp350 and Asp473), nucleophilic catalysis (Cys645), and general acid/general base catalysis (His471). Initially, all four residues were individually mutated to Ala, the proteins purified, and estimates of the kinetic parameters determined (Table S1). In all cases, kcat/Km was decreased by greater than 7,900-fold, thereby indicating that all 4 residues are essential for catalysis, consistent with previous mutagenesis studies (27). Note that high concentrations of purified enzyme and extended incubation times were used to estimate kcat/Km and that partial proteolysis studies (see Figure S1), which are a sensitive indicator of PAD4 structure (11), were used to ensure that the lack of activity is not caused by a major structural perturbation, analogously to other systems (41–43).
To gain further insights into the roles of these residues in substrate binding and catalysis, a series of conservative mutations were generated. For example, a Cys → Ser mutant was generated for Cys645; Asp → Asn and Glu mutants were generated for Asp350 and Asp473; and a His → Gln mutant was generated for His471. The enzymatic activity of the Cys645Ser and His471Gln mutants was decreased by greater than 14,000- and 34,000-fold (Table S1), respectively, consistent with the essential roles of these residues in nucleophilic and general acid/base catalysis. Somewhat surprisingly, mutation of Asp350 and Asp473 to Asn and Glu led to very dramatic declines in PAD4 activity, on the order of those observed for the Asp → Ala mutations (Table S1). These results indicate that PAD4 activity is sensitive to even conservative mutations and further suggest that the correct positioning of the guanidinium group, with respect to the active site Cys, and charge neutralization are critical determinants of catalytic power. Overall, the lack of activity observed with these mutants may be due to a lack of synergy between these 4 key catalytic residues.
Note that the lack of activity was not due to a gross conformational change because all of the mutants behaved similarly to wild type enzyme in our partial proteolysis assay (Figure S1). Also note that the studies described herein are consistent with the fact that active site mutations in a related enzyme, Arginine Deiminase (ADI) from Pseudomonas aeruginosa, lead to similar decreases in deiminating activity (44). Finally, the specific effects of the mutations described above on each of the two half reactions, i.e. intermediate formation and intermediate hydrolysis, are unknown but will be the subject of future investigations.
pH Studies
To further probe the PAD4 reaction mechanism, pH rate profiles were determined for wild type enzyme. These studies were pursued because pH rate profiles can suggest the identity of catalytically important functional groups in the free enzyme (kcat/Km versus pH), the ES complex (kcat versus pH), or the substrate (kcat/Km versus pH) (45, 46). For these studies, pH profiles were constructed by measuring the steady state kinetic parameters for the deimination of BAEE over a range of pH’s (5.7–9.0). Note that BAEE is a synthetic substrate that is commonly used to study PAD activity. Also note that there is essentially no effect on the calcium dependence of PAD4 over this pH range and that PAD4 activity is linear with respect to time at all pH values used in this study (11).
The plots of log kcat/Km versus pH are bell-shaped and fit well to a model with two apparent pKa values and a limiting non-zero plateau (133 M−1s−1) for the ascending limb (Figure 3A). The apparent pKa’s of the ascending and descending limbs are 6.9±0.2 (slope of 0.68) and 9.0±0.1 (slope of −1.15), respectively. Note that because these pKa values are within 3 units they must be considered apparent pKa’s. Therefore, the corrected values, calculated according to the method of Segel (47), are 7.3 and 8.2 for the ascending and descending limbs, respectively. While not definitive, the presence of a non-zero plateau for the ascending limb could suggest that a second ionization form of the enzyme is active at low pH. The plots of kcat versus pH are similarly bell-shaped and fit well to a model with two apparent pKa values and limiting non-zero plateaus of 0.76 s−1 for the ascending limb and 0.24 s−1 for the descending limb (Figure 3B). The apparent pKa’s of the ascending and descending limbs are 7.0±0.3 (slope of 0.37) and 8.7±0.4 (slope of −0.60), respectively. The pKa values obtained from this analysis most likely represent the protonation states of His471 and Cys645 in the ES complex (see Figure 2); although considering the stepwise nature of the reaction mechanism it is difficult to definitively assign either of these values to a particular residue.
Figure 3.

(A) Plot of log kcat/Km versus pH. (B) Plot of log kcat versus pH.
Figure 2.

(A) Active site of PAD4. (B) Working model of PAD4 catalysis. A possible mechanism of PAD4 catalysis involves a nucleophilic thiolate (Step 1) and His 471 acts as a general acid, donating a proton to the departing amine during the collapse of the first tetrahedral intermediate (Step 2). This leads to the formation of an S-alkyl thiouronium intermediate. The exchange of a molecule of water for ammonia occurs and, as drawn, His471 acts as a general base to activate a water molecule for nucleophilic attack on the thiouronium intermediate (Step 3). This leads to the formation of the second tetrahedral intermediate that collapses to eliminate the Cys thiolate and in the process generate Cit. Asp473 is appropriately positioned to deprotonate the hydroxyl. Step 4 involves the exchange of product for substrate.
pKa measurements on the active site Cysteine (Cys645) by iodoacetamide inactivation kinetics
On the basis of the structure and proposed mechanism of PAD4, the simplest assumption is that the ascending limb in the kcat/Km versus pH plots corresponds to the formation of the active site thiolate, i.e. the deprotonation of Cys645, and that the descending limb corresponds to the deprotonation of His471, i.e. the pKa values of Cys645 and His471 are 7.3 and 8.2, respectively. However, an alternative possibility is the special case of a ‘reverse protonation’ mechanism (37, 38). In such a mechanism, the pKa assignments are the ‘reverse’ of the simplest assumption, i.e. the pKa values of Cys645 and His471 are 8.2 and 7.3, respectively.
To distinguish between these two possibilities, direct measurements of the Cys645 side chain pKa were made by determining the rates of iodoacetamide-induced enzyme inactivation over a range of pH’s (pH 6.5 to pH 9) and concentrations of iodoacetamide, similarly to related systems (48, 49). The percentage of activity remaining after iodoacetamide treatment was determined as a function of time and the resulting plots were fit to a single exponential to determine the pseudo first order rate constant for inactivation, i.e. kobs. Figure 4A depicts a representative plot of the data, obtained at pH 7.6. Plots of kobs versus iodoacetamide concentration were linear, indicating second order inactivation kinetics (Figure 4B). The second order rate constants of inactivation, i.e. kinact/KI, obtained at each of the indicated pH values were subsequently plotted against pH; and as depicted in Figure 4C the results indicate that the rate of PAD4 inactivation increases with rising pH. Graphical analyses are consistent with a single pKa value of 8.3±0.07. This pKa value is in excellent agreement with that obtained for the descending limb in the kcat/Km versus pH rate profiles, suggesting that PAD4 utilizes a ‘reverse protonation’ mechanism.
Figure 4.

Time and concentration dependent inactivation of PAD4 by iodoacetamide. (A) observed inactivation at pH 7.6 by different concentrations of iodoacetamide: 0 (◇), 250 (▼), 500 (●), 750 (▲), 1000 (○), and 1500 μM (■). (B) The pseudo first order rate constant of PAD4 inactivation is plotted versus iodoacetamide concentration and plots were fitted to equation 5 as described in materials and methods. (C) The pKa of C645. Second order rate constants were plotted versus pH and fit to equation 6 as described in materials and methods.
Note that two sets of control experiments were performed to confirm that the iodoacetamide induced inactivation of PAD4 was due to the modification of an active site residue, i.e. Cys645. First, the rates of iodoacetamide- and iodoacetic-induced PAD4 inactivation were compared. The results demonstrate that PAD4 is preferentially inactivated by iodoacetamide (Figure 5A). The fact that iodoacetic acid is a relatively poorer PAD4 inactivator is at least partially consistent with the modification of an active site residue because electrostatic repulsions between Asp350 and Asp473 and iodoacetic acid would be expected to inhibit the interaction between this compound and PAD4. Secondly, the rate of iodoacetamide-induced inactivation is higher at lower concentrations of substrate (Figure 5B). This result is also consistent with the modification of an active site residue because substrate binding would be expected to preclude inactivator binding.
Figure 5.

Inactivation of PAD4 with iodoacetamide. (A) Iodoacetamide is a better PAD4 inactivator than iodoacetic acid. Percent activity remaining was plotted versus time and fit to equation 4 as described in materials and methods. (B) Substrate protects against iodoacetamide-induced inactivation of PAD4. Plots of product formation versus time are depicted for PAD4 in the absence and presence of iodoacetamide (1.25 mM) at two different concentrations of BAEE (2 and 10 mM).
pKa measurements on the active site Cysteine (Cys645) by 2-chloroacetamidine inactivation kinetics
The pKa of Cys645 was also measured with 2-chloroacetamidine, a positively charged PAD4 inactivator, by determining the rates of enzyme inactivation over a range of pH’s (pH 6.5 to pH 9) and concentrations. As described above for the iodoacetamide-induced inactivation of PAD4, the percentage of activity remaining after 2-chloroacetamidine treatment was determined as a function of time and the resulting plots were fit to a single exponential to determine the pseudo first order rate constant for inactivation, i.e. kobs, at each concentration of inactivator (Figure 6A). Plots of kobs versus 2-chloroacetamidine concentration were linear within the concentration range used (Figure 6B) and gratifyingly the second order rate constant for PAD4 inactivation, 55 ± 6.0 M−1min−1 at pH 7.6, is quite close to that reported by the Fast group (35 M−1min−1) (39). Plots of kinact/KI versus pH (Figure 6C) reveal that the pKa of Cys645 is 7.9±0.16; in good agreement with both the descending limb of the kcat/Km versus pH rate profiles and that obtained via iodoacetamide inactivation kinetics. Furthermore, these results indicate that this positively charged inactivator has only minimal effects on the pKa of the Cys645 thiol. Note that substrate could protect against PAD4 inactivation by 2-chloroacetamidine (Figure 6D), consistent with the modification of an active site residue.
Figure 6.

Time and concentration dependent inactivation of PAD4 by 2-chloroacetamidine. (A) Observed inactivation at pH 7.6 by different concentrations of 2-chloroacetamidine: 0 (◇), 250 (●), 500 (▲), 1000 (○), and 1500 μM (■). (B) The pseudo first order rate constant of PAD4 inactivation is plotted versus 2-chloroacetamidine concentration and plots were fitted to equation 5 as described in materials and methods. (C) The pKa of C645. Second order rate constants were plotted versus pH and fit to equation 6 as described in materials and methods. (D) Substrate protects against 2-chloroacetamidine-induced inactivation of PAD4. Plots of product formation versus time are depicted for PAD4 in the absence or presence of 2-chloroacetamidine (5 mM) at two different concentrations of PAD4: 10 mM BAEE (●), 2 mM BAEE (■), 10 mM BAEE with 2-chloroacetamidine (○), and 2 mM BAEE with 2-chloroacetamidine (□).
Solvent Isotope Effects (SIE’s)
Because PAD4 catalyses a hydrolysis reaction that involves a nucleophilic thiolate, we evaluated the SIE by performing the reaction in D2O over a range of pL values. The SIE on kcat was normal with a kcatH/kcatD of 1.25 at the pL optimum, indicating that proton abstraction does not contribute significantly to the rate limiting step of the reaction. In contrast to the normal SIE on kcat, an inverse SIE was observed for kcat/Km over the entire pL range studied (Figure 7). At the pL optimum the ratio kcat/KmH/kcat/KmD was 0.43. Inverse SIE’s of this type have previously been used to support the existence of a thiolate•imidazolium ion pair (50–54).
Figure 7.

Solvent Isotope Effect (SIE). Plots of log kcat/Km versus pL in H2O (●) or D2O (○).
Note that under identical conditions, the concentration of calcium required for half maximal activity, i.e. the K0.5, was virtually unaffected (0.56 ± 0.04 for H2O (11) versus 1.0 ± 0.4 for D2O), thereby indicating that the inverse SIE is unlikely to be due to a change in the calcium dependence of the enzyme. Also, note that a normal SVE on both kcat (1.32) and kcat/Km (1.34) was observed when the steady state kinetic parameters were determined in the presence of 10% glycerol (w/v), a concentration of glycerol that closely matches the viscosity of D2O (55); thereby indicating that the inverse SIE is not due to an increase in the viscosity of buffers containing D2O.
DISCUSSION
On the basis of the structure of PAD4 and literature precedents from related systems (30–32), we initially proposed a working model of PAD4 catalysis that involved a nucleophilic thiolate (Figure 2; (11)) and hypothesized that this species was stabilized via the formation of an ion pair with His471, analogously to the thiolate-imidazolium ion pair observed in papain (36). To begin to test the validity of this hypothesis, we generated a series of semi-conservative and non-conservative active site mutants (Cys → Ala and Ser; His → Gly, Ala, Gln) designed to directly evaluate the roles of Cys645 and His471. While the activity associated with these mutant enzymes is negligible (kcat/Km ↓ by ≥ 7,900-fold), the results are nevertheless consistent with an essential role for these residues in PAD4 catalysis and are consistent with the proposed mechanism, i.e. that a nucleophilic thiolate and properly oriented imidazole group2 are required for nucleophilic and general acid/base chemistry, respectively. Similar results have been obtained for other amidinotransferase family members, e.g. ADI (44).
pH rate profiles (kcat/Km versus pH) were also constructed to identify and characterize the roles of catalytically important residues; and the results are consistent with the presence of two key ionizable groups with pKa’s of 7.3 and 8.2. On the basis of the proposed catalytic mechanism, these pKa values most likely correspond to the protonation states of His471 and Cys645 before the substrate has bound to the enzyme. To more definitively assign one of these pKa values to Cys645, pKa measurements were made on the active site thiol by determining the effect of pH on the rate of PAD4 inactivation by iodoacetamide and 2-chloroacetamidine. As described above, the rates of inactivation increased with pH for both compounds and in both cases the pKa determined for Cys645 is quite close to the value obtained for the descending limb of the kcat/Km versus pH rate profile, i.e. pH 8.2. The fact that the pKa of Cys645 corresponds to the descending limb of the kcat/Km versus pH rate profile is most consistent with a ‘reverse protonation’ mechanism (see above) and thereby suggests that the pKa of His471 is ~7.3.
The fact that the iodoacetamide and 2-chloroacetamidine inactivation kinetics yield similar pKa’s for Cys645 is inconsistent with a pure substrate assisted mechanism of thiol deprotonation. 3 This is the case because in a pure substrate assisted mechanism, binding to the thiol form of the enzyme (i.e., E-SH; where E is the enzyme, SH is the thiol; upper pathway in Scheme 1) is obligatory. Therefore, if 2-chloroacetamidine, a guanidinium analog, forms this initial encounter complex (i.e. E-SH•2-chloroacetamidine), a reasonable assumption given that 2-chloroacetamidine displays saturation kinetics at the pH optimum (39), the observed pKa should have shifted dramatically from the resting state by the factor α. While an effect on the kinact/KI titration curve may not be apparent if 2-chloroacetamidine does not form an initial encounter complex with E-SH or if the inactivator can bind to either the thiol or thiolate forms of the enzyme (i.e., binding to E-SH is not obligatory), the results obtained are most consistent with preferential binding of this positively charged inactivator to the thiolate form of the enzyme at the pH optimum via the lower pathway in Scheme 1, i.e. a reverse protonation mechanism.
Scheme 1.

Binding of inactivator to either the E-SH (upper pathway) or E-S− (lower pathway) complexes.
A reverse protonation mechanism is also supported by the fact that the decrease in kcat/Km at both pH extremes is largely driven by an increase in Km. While effects of pH on Km are often difficult to interpret, large changes in Km as a function of pH are consistent with the preferential binding of substrate to one form of the enzyme at the pH optimum (47). Thus when taken in combination, our results are most consistent with substrate binding to a form of the enzyme that consists of a negatively charged thiolate and a positively charged imidazolium ion, i.e. the ES−H+ form, where E is enzyme, S− is the thiolate, and H+ is the imidazolium ion. This appears to be the case despite the fact that only a small fraction of PAD4 (~15% of the enzyme) exists as ES−H+ at the pH optimum – this is the case because the titration curves for His471 and Cys645 are overlapping (Figure 8).
Figure 8.
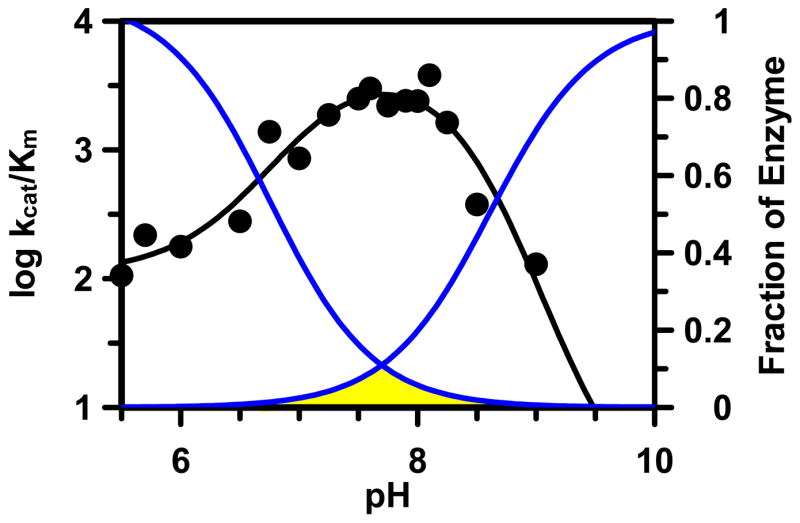
Fraction of enzyme in the active form (yellow) with C645 deprotonated and H471 protonated, i.e., E-S−H+.
If substrate binds preferentially to the ES−H+ form of PAD4 at the pH optimum, as our data suggests, it is likely that the negative charge of the thiolate is at least partially stabilized via the formation of an ion pair with the imidazolium ion. Such an interaction would be important because the PAD4 active site is highly anionic (Asp350 and Asp473 should be deprotonated), disfavoring thiolate formation; and His471 is the only positively charged residue within the vicinity of Cys645 that could serve this function. The inverse SIE on kcat/Km is consistent with the dissociation of a proton from a thiol with a fractionation factor of less than 1 and such inverse SIE’s have previously been used to support the existence of a thiolate•imidazolium ion pair in other Cys hydrolases (50–54). The inverse SIE on kcat/Km is also noteworthy because it is inconsistent with previously proposed mechanisms (27, 35, 56) that involve His471 acting as a general base to deprotonate the substrate guanidinium, which subsequently deprotonates the thiol, because if such a mechanism were operative, a normal SIE would be expected.
Although the distance between the imidazole nitrogen of His471 and the β-carbon of Cys645 is 6.9 Ǻ in the structure of the PAD4C645A•Calcium complex (the best available representation of active PAD4 before substrate has bound; PDB ID 1WD9), it is important to recognize that the true distance between these two residues is likely to be considerably shorter. For example, based on the length of a C—S bond (~1.6 Ǻ), the distance between this nitrogen and the thiolate is likely less than ~5.3 Ǻ. Furthermore at the pH used for protein crystallization (pH 8.0), and considering the reverse protonation mechanism described above, this structure represents less than 10 % of active PAD4; thus, the distance may appear large because this structure actually represents an average of inactive conformers.
Implications for other amidinotransferase family members
PAD4 belongs to a large superfamily of guanidinium modifying enzymes; and despite shared active site architectures differences in specific active site residues and kinetic properties exist between members of the amidinotransferase superfamily of enzymes. For example: (i) the pH-optimum of various family members varies significantly from < pH 5.5 for Pseudomonas aeruginosa ADI to pH 7.6 for PAD4 (11, 30, 57, 58); (ii) the active site Cys in DDAH appears to be deprotonated via a substrate assisted mechanism (59); iii) the distance between the imidazolium ion and the active site Cys is considerably longer in both ADI and DDAH; (iv) PAD4 requires high concentrations of DTT to maintain activity whereas other amidinotransferase family members do not (11, 39, 44); and (v) PAD4 has a His-Cys dyad; whereas ADI and DDAH possess a His-Cys-Glu catalytic triad. Thus, the ion pair mechanism proposed herein for PAD4 may not be universally utilized by other amidinotransferase family members.
In summary, our results indicate that in the free and active form of the enzyme Cys645 exists as the thiolate. Furthermore, pH studies and pKa measurements with iodoacetamide and 2-chloroacetamidine strongly suggest that PAD4 utilizes a reverse protonation mechanism. These studies have already aided our successful efforts to synthesize PAD4 inhibitors (23–25) and will undoubtedly serve as a guide to others interested in developing a PAD4 targeted RA pharmaceutical.
Supplementary Material
SUPPORTING INFORMATION AVAILABLE. Supplementary Methods, Supplementary Table 1, and Supplementary Figure 1. This material is available free of charge via the internet at http://pubs.acs.org.
ABBREVIATIONS
- PAD
protein arginine deiminase
- Cit
citrulline
- RA
rheumatoid arthritis
- BAEE
benzoyl L-arginine ethyl ester
- BAA
benzoyl L-arginine amide
- DTT
dithiothreitol
- GST
glutathione S-transferase
- TCEP
Tris(2-carboxyethyl)phosphine hydrochloride
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- ADI
Arginine Deiminase
- DDAH
Dimethylarginine Dimethylamino Hydrolase
- SIE
Solvent Isotope Effect
- SVE
Solvent Viscosity Effect
Footnotes
This work was supported in part by the start up funds from the University Of South Carolina Research Foundation (P.R.T) and in part by National Institutes of Health grant GM079357 to PRT.
PAD, protein arginine deiminase; Cit, citrulline; RA, rheumatoid arthritis; BAEE, benzoyl L-arginine ethyl ester; BAA, benzoyl L-arginine amide; DTT, dithiothreitol; GST, glutathione S-transferase; TCEP, Tris(2-carboxyethyl)phosphine hydrochloride; HEPES, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid).
The activity of neither the His471Gly nor the His471Ala mutant could be chemically rescued by the addition of a variety of imidazole- and guanidinium-containing compounds.
In a substrate assisted mechanism of thiol deprotonation, the proximity of the positively charged substrate guanidinium to a high pKa active site Cys promotes thiol deprotonation by either an unknown general base or by proton donation to solvent (59). The term pure is used to indicate that the substrate can only bind to the enzyme when Cys645 exists as the thiol (Scheme 1 upper pathway) and is used to differentiate between the possibility that the substrate can bind to the enzyme regardless of whether Cys645 exists as the thiol or thiolate, i.e. substrate can bind via either pathway in Scheme 1.
References
- 1.Akil M, Amos RS. ABC of Rheumatology: Rheumatoid arthritis I: Clinical features and diagnosis. Brit Med J. 1995;310:587–590. doi: 10.1136/bmj.310.6979.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finesilver AG. Newer approaches to the treatment of rheumatoid arthritis. Wisconsin Medical Journal. 2003;102:34–37. [PubMed] [Google Scholar]
- 3.Smolen JS, Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003;2:473–488. doi: 10.1038/nrd1109. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, Ohtsuki M, Furukawa H, Yoshino S, Yukioka M, Tohma S, Matsubara T, Wakitani S, Teshima R, Nishioka Y, Sekine A, Iida A, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 5.Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays. 2003;25:1106–1118. doi: 10.1002/bies.10357. [DOI] [PubMed] [Google Scholar]
- 6.Vossenaar ER, Van Venrooij WJ. Citrullinated proteins: sparks that may ignite the fire in rheumatoid arthritis. Arthritis Res Ther. 2004;6:107–111. doi: 10.1186/ar1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, van Venrooij WJ. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43:155–163. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Lundberg K, Nijenhuis S, Vossenaar ER, Palmblad K, van Venrooij WJ, Klareskog L, Zendman AJ, Harris HE. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res Ther. 2005;7:R458–R467. doi: 10.1186/ar1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006;116:961–973. doi: 10.1172/JCI25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kearney PL, Bhatia M, Jones NG, Luo Y, Glascock MC, Catchings KL, Yamada M, Thompson PR. Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry. 2005;44:10570–10582. doi: 10.1021/bi050292m. [DOI] [PubMed] [Google Scholar]
- 12.Thompson PR, Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem Biol. 2006;1:433–441. doi: 10.1021/cb6002306. [DOI] [PubMed] [Google Scholar]
- 13.Moscarello MA, Mastronardi FG, Wood DD. The Role of Citrullinated Proteins Suggests a Novel Mechanism in the Pathogenesis of Multiple Sclerosis. Neurochem Res. 2007;32:251–256. doi: 10.1007/s11064-006-9144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mastronardi FG, Wood DD, Mei J, Raijmakers R, Tseveleki V, Dosch HM, Probert L, Casaccia-Bonnefil P, Moscarello MA. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci. 2006;26:11387–11396. doi: 10.1523/JNEUROSCI.3349-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang X, Han J. Expression of peptidylarginine deiminase type 4 (PAD4) in various tumors. Mol Carcinog. 2006;45:183–196. doi: 10.1002/mc.20169. [DOI] [PubMed] [Google Scholar]
- 16.Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, Kouzarides T. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–553. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, Roeder RG, Clarke S, Stallcup MR, Allis CD, Coonrod SA. Human PAD4 Regulates Histone Arginine Methylation Levels via Demethylimination. Science. 2004;306:279–283. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- 18.Liu GY, Liao YF, Chang WH, Liu CC, Hsieh MC, Hsu PC, Tsay GJ, Hung HC. Overexpression of peptidylarginine deiminase IV features in apoptosis of haematopoietic cells. Apoptosis. 2006;11:183–196. doi: 10.1007/s10495-006-3715-4. [DOI] [PubMed] [Google Scholar]
- 19.Lee YH, Coonrod SA, Kraus WL, Jelinek MA, Stallcup MR. Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc Natl Acad Sci U S A. 2005;102:3611–3616. doi: 10.1073/pnas.0407159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakashima K, Hagiwara T, Yamada M. Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J Biol Chem. 2002;277:49562–49568. doi: 10.1074/jbc.M208795200. [DOI] [PubMed] [Google Scholar]
- 21.Hagiwara T, Nakashima K, Hirano H, Senshu T, Yamada M. Deimination of arginine residues in nucleophosmin/B23 and histones in HL-60 granulocytes. Biochem Biophys Res Comm. 2002;290:979–983. doi: 10.1006/bbrc.2001.6303. [DOI] [PubMed] [Google Scholar]
- 22.Hagiwara T, Hidaka Y, Yamada M. Deimination of Histone H2A and H4 at Arginine 3 in HL-60 Granulocytes. Biochemistry. 2005;44:5827–5834. doi: 10.1021/bi047505c. [DOI] [PubMed] [Google Scholar]
- 23.Luo Y, Knuckley B, Bhatia M, Thompson PR. Activity Based Protein Profiling Reagents for Protein Arginine Deiminase 4 (PAD4): Synthesis and in vitro Evaluation of a Fluorescently-labeled Probe. J Am Chem Soc. 2006;128:14468–14469. doi: 10.1021/ja0656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR. A Fluoro-Acetamidine Based Inactivator of Protein Arginine Deiminase 4 (PAD4): Design, Synthesis, and in vitro and in vivo Evaluation. J Am Chem Soc. 2006;128:1092–1093. doi: 10.1021/ja0576233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Thompson PR. Inhibitors and Inactivators of Protein Arginine Deiminase 4: Functional and structural characterization. Biochemistry. 2006;45:11727–11736. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hidaka Y, Hagiwara T, Yamada M. Methylation of the guanidino group of arginine residues prevents citrullination by peptidylarginine deiminase IV. FEBS Lett. 2005;579:4088–4092. doi: 10.1016/j.febslet.2005.06.035. [DOI] [PubMed] [Google Scholar]
- 27.Arita K, Hashimoto H, Shimizu T, Nakashima K, Yamada M, Sato M. Structural basis for Ca2+-induced activation of human PAD4. Nat Struct Mol Biol. 2004;11:777–783. doi: 10.1038/nsmb799. [DOI] [PubMed] [Google Scholar]
- 28.Arita K, Shimizu T, Hashimoto H, Hidaka Y, Yamada M, Sato M. Structural basis for histone N-terminal recognition by human peptidylarginine deiminase 4. Proc Natl Acad Sci U S A. 2006;103:5291–5296. doi: 10.1073/pnas.0509639103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirai H, Mokrab Y, Mizuguchi K. The guanidino-group modifying enzymes: structural basis for their diversity and commonality. Proteins. 2006;64:1010–1023. doi: 10.1002/prot.20863. [DOI] [PubMed] [Google Scholar]
- 30.Galkin A, Lu X, Dunaway-Mariano D, Herzberg O. Crystal structures representing the Michaelis complex and the thiouronium reaction intermediate of Pseudomonas aeruginosa arginine deiminase. J Biol Chem. 2005;280:34080–34087. doi: 10.1074/jbc.M505471200. [DOI] [PubMed] [Google Scholar]
- 31.Das K, Butler GH, Kwiatkowski V, Clark AD, Jr, Yadav P, Arnold E. Crystal structures of arginine deiminase with covalent reaction intermediates; implications for catalytic mechanism. Structure. 2004;12:657–667. doi: 10.1016/j.str.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 32.Murray-Rust J, Leiper J, McAlister M, Phelan J, Tilley S, Santa Maria J, Vallance P, McDonald N. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat Struct Biol. 2001;8:679–683. doi: 10.1038/90387. [DOI] [PubMed] [Google Scholar]
- 33.Lu X, Galkin A, Herzberg O, Dunaway-Mariano D. Arginine deiminase uses an active-site cysteine in nucleophilic catalysis of L-arginine hydrolysis. J Am Chem Soc. 2004;126:5374–5375. doi: 10.1021/ja049543p. [DOI] [PubMed] [Google Scholar]
- 34.Stone EM, Person MD, Costello NJ, Fast W. Characterization of a transient covalent adduct formed during dimethylarginine dimethylaminohydrolase catalysis. Biochemistry. 2005;44:7069–7078. doi: 10.1021/bi047407r. [DOI] [PubMed] [Google Scholar]
- 35.Shirai H, Blundell TL, Mizuguchi K. A novel superfamily of enzymes that catalyze the modification of guanidino groups. Trends Biochem Sci. 2001;26:465–468. doi: 10.1016/s0968-0004(01)01906-5. [DOI] [PubMed] [Google Scholar]
- 36.Lewis SD, Johnson FA, Shafer JA. Effect of cysteine-25 on the ionization of histidine-159 in papain as determined by proton nuclear magnetic resonance spectroscopy. Evidence for a his-159--Cys-25 ion pair and its possible role in catalysis. Biochemistry. 1981;20:48–51. doi: 10.1021/bi00504a009. [DOI] [PubMed] [Google Scholar]
- 37.Frankel BA, Kruger RG, Robinson DE, Kelleher NL, McCafferty DG. Staphylococcus aureus sortase transpeptidase SrtA: insight into the kinetic mechanism and evidence for a reverse protonation catalytic mechanism. Biochemistry. 2005;44:11188–11200. doi: 10.1021/bi050141j. [DOI] [PubMed] [Google Scholar]
- 38.Mock WL, Stanford DJ. Anisylazoformylarginine: a superior assay substrate for carboxypeptidase B type enzymes. Bioorg Med Chem Lett. 2002;12:1193–1194. doi: 10.1016/s0960-894x(02)00128-2. [DOI] [PubMed] [Google Scholar]
- 39.Stone EM, Schaller TH, Bianchi H, Person MD, Fast W. Inactivation of two diverse enzymes in the amidinotransferase superfamily by 2-chloroacetamidine: dimethylargininase and peptidylarginine deiminase. Biochemistry. 2005;44:13744–13752. doi: 10.1021/bi051341y. [DOI] [PubMed] [Google Scholar]
- 40.Leatherbarrow RJ. Erathicus Software. Staines, UK: 2004. [Google Scholar]
- 41.Thompson PR, Schwartzenhauer J, Hughes DW, Berghuis AM, Wright GD. The COOH terminus of aminoglycoside phosphotransferase (3′)-IIIa is critical for antibiotic recognition and resistance. J Biol Chem. 1999;274:30697–30706. doi: 10.1074/jbc.274.43.30697. [DOI] [PubMed] [Google Scholar]
- 42.Thompson PR, Boehr DD, Berghuis AM, Wright GD. Mechanism of aminoglycoside antibiotic kinase APH(3′)-IIIa: role of the nucleotide positioning loop. Biochemistry. 2002;41:7001–7007. doi: 10.1021/bi0256680. [DOI] [PubMed] [Google Scholar]
- 43.Boehr DD, Thompson PR, Wright GD. Molecular Mechanism of Aminoglycoside Antibiotic Kinase APH(3′)-IIIa. Roles of conserved active site residues. J Biol Chem. 2001;276:23929–23936. doi: 10.1074/jbc.M100540200. [DOI] [PubMed] [Google Scholar]
- 44.Lu X, Li L, Wu R, Feng X, Li Z, Yang H, Wang C, Guo H, Galkin A, Herzberg O, Mariano PS, Martin BM, Dunaway-Mariano D. Kinetic analysis of Pseudomonas aeruginosa arginine deiminase mutants and alternate substrates provides insight into structural determinants of function. Biochemistry. 2006;45:1162–11672. doi: 10.1021/bi051591e. [DOI] [PubMed] [Google Scholar]
- 45.Cleland WW. The use of pH studies to determine chemical mechanisms of enzyme-catalyzed reactions. Methods Enzymol. 1982;87:390–405. doi: 10.1016/s0076-6879(82)87024-9. [DOI] [PubMed] [Google Scholar]
- 46.Dixon M, Webb EC. Enzymes. Academic Press; New York: 1964. [Google Scholar]
- 47.Segel IH. Enzyme kinetics: Behavior and analysis of rapid equilibrium and steady-state enzyme systems. Wiley-Interscience; New York: 1975. [Google Scholar]
- 48.Zhang ZY, Dixon JE. Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402. Biochemistry. 1993;32:9340–9345. doi: 10.1021/bi00087a012. [DOI] [PubMed] [Google Scholar]
- 49.Lohse DL, Denu JM, Santoro N, Dixon JE. Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase PTP1. Biochemistry. 1997;36:4568–4575. doi: 10.1021/bi963094r. [DOI] [PubMed] [Google Scholar]
- 50.Quinn DM, Sutton LD. Theoretical basis and mechanistic utility of solvent isotope effects. In: Cook PF, editor. Enzyme Mechanism from Isotope Effects. CRC Press; Boca Raton, FL: 1991. pp. 73–126. [Google Scholar]
- 51.Wandinger A, Creighton DJ. Solvent isotope effects on the rates of alkylation of thiolamine models of papain. FEBS Lett. 1980;116:116–121. doi: 10.1016/0014-5793(80)80541-2. [DOI] [PubMed] [Google Scholar]
- 52.Brocklehurst K, Kowlessur D, Patel G, Templeton W, Quigley K, Thomas EW, Wharton CW, Willenbrock F, Szawelski RJ. Consequences of molecular recognition in the S1–S2 intersubsite region of papain for catalytic-site chemistry. Change in pH-dependence characteristics and generation of an inverse solvent kinetic isotope effect by introduction of a P1–P2 amide bond into a two-protonic-state reactivity probe. Biochem J. 1988;250:761–772. doi: 10.1042/bj2500761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polgar L. Mercaptide-imidazolium ion-pair: the reactive nucleophile in papain catalysis. FEBS Lett. 1974;47:15–18. doi: 10.1016/0014-5793(74)80415-1. [DOI] [PubMed] [Google Scholar]
- 54.Polgar L. Deuterium isotope effects on papain acylation. Evidence for lack of general base catalysis and for enzyme--leaving-group interaction. Eur J Biochem. 1979;98:369–374. doi: 10.1111/j.1432-1033.1979.tb13196.x. [DOI] [PubMed] [Google Scholar]
- 55.Karsten WE, Lai CJ, Cook PF. Inverse solvent isotope effects in the NAD-Malic enzyme reaction are the result of the viscosity difference between D2O and H2O: Implications for solvent isotope effect studies. J Am Chem Soc. 1995;117:5914–5918. [Google Scholar]
- 56.Galkin A, Kulakova L, Sarikaya E, Lim K, Howard A, Herzberg O. Structural insight into arginine degradation by arginine deiminase, an antibacterial and parasite drug target. J Biol Chem. 2004;279:14001–14008. doi: 10.1074/jbc.M313410200. [DOI] [PubMed] [Google Scholar]
- 57.Lu X, Li L, Wu R, Feng X, Li Z, Yang H, Wang C, Guo H, Galkin A, Herzberg O, Mariano PS, Martin BM, Dunaway-Mariano D. Kinetic Analysis of Pseudomonas aeruginosa Arginine Deiminase Mutants and Alternate Substrates Provides Insight into Structural Determinants of Function. Biochemistry. 2006;45:1162–1172. doi: 10.1021/bi051591e. [DOI] [PubMed] [Google Scholar]
- 58.Weickmann JL, Himmel ME, Smith DW, Fahrney DE. Arginine deiminase: demonstration of two active sites and possible half-of-the-sites reactivity. Biochem Biophys Res Commun. 1978;83:107–113. doi: 10.1016/0006-291x(78)90404-7. [DOI] [PubMed] [Google Scholar]
- 59.Stone EM, Costello AL, Tierney DL, Fast W. Substrate-assisted cysteine deprotonation in the mechanism of dimethylargininase (DDAH) from Pseudomonas aeruginosa. Biochemistry. 2006;45:5618–5630. doi: 10.1021/bi052595m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION AVAILABLE. Supplementary Methods, Supplementary Table 1, and Supplementary Figure 1. This material is available free of charge via the internet at http://pubs.acs.org.