

Abstract
Viruses cannot replicate autonomously, but must rely on the host cellular machinery to support their life cycle. Through natural selection, viruses have evolved strategies to co-opt the host organism to be a better site for their propagation. Some of these strategies are directed at the cellular machinery and involve complicated and ingenious solutions to optimize infection, replication, viral gene expression and new virion assembly and shedding. Other strategies are directed at the host’s innate and adaptive immune systems permitting the virus to evade clearance mechanisms. The more common pathogenic viral infections in nephrology, CMV, HIV-1, HCV, polyomavirus BK and parvovirus B19, all have acquired subversion strategies that benefit the virus, but because they interfere with normal cellular and immune processes, also have become pathogenic to the host. In addition, the highly prevalent viruses CMV, BK, and B19 only cause severe disease in the setting of immunosuppression, revealing the very delicate balance some viruses have achieved with their host’s immune system. Thus, selective pressure for survival drives both the evolution of more sophisticated viruses and also the host immune system as it evolves to combat the environment of adapting and emerging infectious agents. Understanding the molecular mechanisms of these viral subversion strategies may reveal new targets for the development of highly specific anti-viral therapies and also aide vaccine development.
Introduction
All viruses are obligate intracellular parasites and are unable to replicate without the support of the cellular machinery of the host. Because of this dependency, they are at the threshold of what is considered a living organism. For a virus to use a eukaryotic cell as a host, the virus encounters many barriers to completing its lifecycle and has evolved mechanisms to modify the host both at the cellular and the organismal level to ensure its survival. The goal of this article is to review host-virus interactions, from the perspective of the virus, focusing on the more common pathogenic viruses encountered in nephrology including cytomegalovirus (CMV), human immunodeficiency virus 1 (HIV-1), hepatitis C virus (HCV), polyomavirus BK, and parvovirus B19.
Viruses are some of the smallest and simplest living organisms consisting of only a piece of genetic material, either DNA or RNA, surrounded by a protein coat. All viral genomes contain non-coding sequences that control replication and transcription, as well as coding sequences which specify the viral proteins. How many proteins each virus encodes depends mostly on the physical size of the virion, since there are limitations on the amount of DNA or RNA that can be packaged within the protein coat. Viral proteins can be categorized into three general functions (1). The first function is to provide for the structural proteins of the coat; the second function is to ensure the replication of the genetic material, and the third is to modify the host cellular machinery to facilitate the first two functions. This latter function is typically unique to each virus and since it subverts normal cellular activities is the aspect of the virus that often leads to pathogenesis and disease.
All viruses encounter similar barriers when using a eukaryotic cell as a host (1). The first major barrier is entering the cell, and viruses typically exploit either a receptor-mediated mechanism or an endocytic process to cross the plasma membrane (Fig. 1). Once inside the cell, the virus encounters a second major barrier in that it must cross the nuclear membrane. Some viruses, such as parvovirus B19 (2), have evolved no specific mechanism to cross the nuclear membrane. Subsequently, these viruses can only replicate in actively dividing cells because they must wait for the cell to enter mitosis (where the nuclear envelop breaks down) to gain access to the critical replication and transcriptional machinery of the nucleus. After entering the nucleus, the subsequent steps of replication, transcription, translation of the viral genes, and assembly of new virions require extensive use of the cellular machinery. The final release of new virions can occur by budding, commonly used by the enveloped viruses like HIV-1, which preserves the host cell. Other viruses have evolved no mechanism to exit the cell and either must wait for the host cell to die or as in the case of parvovirus B19, induce the cell to die through the expression of the viral nonstructural protein NS1, thereby releasing progeny virions on cell rupture (3). Thus, for a virus to successfully use a cell type as a host, it must accommodate any restrictions it encounters in the host cell environment, or else, evolve a strategy to circumvent the barriers.
Figure 1.
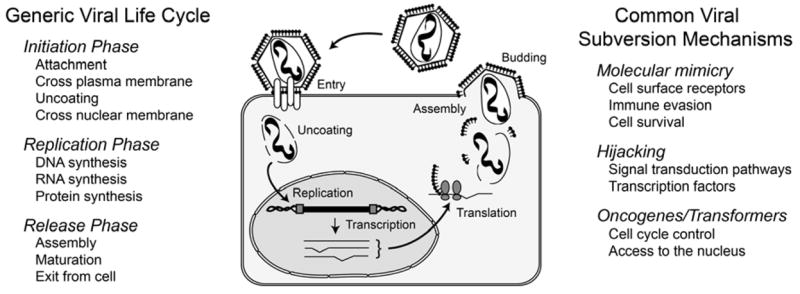
Eukaryotic cell barriers to the viral life cycle and types of subversion mechanism employed by the virus. Viruses require the use of the host cellular machinery to replicate their nucleic acid and produce viral proteins for the assembly of new virion. In addition, the virus, whether intracellular and extracellular, must escape the anti-viral defense mechanisms of the host’s immune system. For the virus to survive, it must evolve complex strategies to circumvent any cellular blocks to completing its life cycle and to evade the host immune system.
Subversion Mechanisms
There are several subversion strategies viruses use to support their replication in the host cell (Fig. 1). The strategies are typically deployed through the use of novel, non-structural “accessory” proteins which either evolve de novo or are co-opted from the host. These accessory genes will be retained in the viral genome if they provide an evolutionary selective advantage to the virus through improve viral fitness or replication capacity. However, since these accessory genes function to modify the host cell they also frequently interfere with normal cell functions leading to pathogenesis (see summary Table 1). The more common strategies include: molecular mimicry; hijacking host signaling pathways; and oncogenes or transforming proteins to modify the cell cycle.
Table 1.
Examples of viral genes and their roles in kidney-related host pathogenesis.
| Virus | Number of Viral Proteins | Role in host pathogenesis | Reference | |
|---|---|---|---|---|
| Total | Non-structural, accessory * | |||
| B19 | 4(9#) | 2(4#) |
Cytotoxic; induces IL-6 expression None identified |
3;57 |
| NS1
NS2 |
||||
| BK | 6 | 3 |
Blocks apoptosis, increases cell division None identified |
38 |
| T antigen
t antigen, agnoprotein |
||||
| CMV | ~160 | ~100 | Immunomodulatory | 4 |
| HCV | 11 | 3 |
Ablates TLR3 signaling: blocks dsRNA-induced antiviral response Activates STAT3, increases IL-8 production; Inhibits PKR: blocks dsRNA-induced response |
7;8 7;8 |
| NS3/NS4a complex
NS5a |
||||
| HIV-1 | 15 | 6 |
Mechanism unknown, possible contribution to disease in transgenic mice Hijacks Src-signaling (Stat3 & MAPK1,2 targets); Hijacks the cell cycle (Cyclin D1 expression & Rb phosphorylation) Mechanism unknown, contributes to disease in transgenic mice None identified |
13;23 15;34 22;23 |
| Tat
Nef Vpr Rev, Vif, Vpu |
||||
Non-structural proteins with enzymatic functions are not included.
Nine unique mRNA transcripts have been identified; expression and/or function have not been determined for all putative proteins.
Molecular Mimicry Strategies
Molecular mimicry is a strategy to imitate host proteins, and in many instances the viral homologues are genes directly pirated from the host genome through recombination events (4;5). As part of the viral lifecycle, many viruses will integrate within the host genome, and it is during these recombination events that host genes may be incorporated within the viral genome. As is typically of many of the DNA viruses, targets for molecular mimicry are frequently immunomodulatory proteins directed at neutralizing the host’s antiviral measures, or proteins that preserve infected cells from death by blocking apoptosis or cell-mediated killing activities.
The large β-herpes virus CMV is a classic example of a virus that uses molecular mimicry to modify the innate, adaptive and inflammatory immune responses of the host. CMV produces approximately 160 proteins, and of these, only 50–60 of the proteins are necessary for viral replication (6). The remaining 100 viral proteins have evolved to manipulate the host’s immune system through mimicking host cytokines and chemokines (virokines), cytokine and chemokines receptors (viroreceptors), and soluble cytokine binding proteins which function as cytokine scavengers (Table 1). These viral homologues have broad ranging effects on lymphocyte recruitment, cytokine activation, apoptosis and T and B cell functions ultimately resulting in the persistence of a lifelong infection with periodic reactivations.
A key immune evasion strategy of CMV is to modulate the major histocompatibility complexes (MHC) I and II (6). The CMV glycoproteins US2, US3, US6, and US11 function to post-translationally downregulate MHCI cell surface expression. US3, which is expressed in the endoplasmic reticulum early in infection, also prevents the loading of peptides into MHCI and disrupts intracellular MHCII invariant chain interactions. These viral proteins effectively function to elude effector and cytotoxic T cell responses, and thus prevent the normal host response to eliminate infected cells. CMV also produces many chemokines and chemokines receptor homologues that can function either as agonists or antagonists to cytokine and chemokine activity. For example, CMV produces an interleukin-10 homologue (vIL-10) that functions to suppress the production of other pro-inflammatory cytokines and the activity of macrophages (4). Alternatively, some virokines function to support virus dissemination, rather than immune evasion, by recruiting immune cells to the site of infection thereby providing additional new target cells for the virus to infect.
RNA viruses, such as the flavivirus Hepatitis C virus (HCV), also have evolved intricate immune evasion strategies (Table 1). Following infection, the appearance of the HCV RNA in the cytoplasm triggers the release of interferons (IFN), the key effector molecules of the immediate early response to viral infection (7). The HCV non-structural (NS) protein complex NS3/4a inactivates components of the Toll-like receptor (TLR) signaling complex blocking the initiating mechanism of the IFN antiviral mechanism. This blockade of a key innate immune response can result in the persistence of HCV beyond the acute infection phase, especially in individuals with a weakened or suppressed immune system (8).
Hijacking Strategies
As already discussed, viruses can modify the functions of cytokines and chemokines by producing viral homologues of these key immunomodulatory host proteins. However, another strategy for viruses to accomplish the same effect is to modulate the expression of the host cytokine and chemokine genes by altering the activity of host transcription factors. This “hijacking” of signal transduction or transcription factor activity is a second major subversion strategy employed frequently by RNA viruses to manipulate the host cell (5;9). Typically, viruses achieve these novel functions not by directly mimicking host protein function, but through the evolution of unique accessory proteins to interact with important intracellular signaling cascades.
In HIV-1 infection, the virus has evolved six non-structural accessory proteins (Table 1). The function of the accessory proteins in the viral life cycle is fairly well known (10), but their role in contributing to host pathogenesis is just beginning to emerge. In HIV-associated nephropathy (HIVAN), it is now accepted that a part of the pathologic process is the direct infection of renal epithelia which results in the expression of these accessory proteins within kidney cells (reviewed in 11;12). Extensive work with recombinant viruses and cell culture systems (13–17), and also studies with transgenic rodents (18–25) has shown that the expression of these accessory genes, independent of infection per se, is directly responsible for many aspects of HIVAN pathogenesis. In both rodent models and in infected patients, HIVAN is characterized by phenotypic changes in epithelial cell behavior including apoptosis, cellular dedifferentiation, and most significantly, excessive proliferation (26).
As an example of a hijacking strategy in HIVAN, HIV-1 is well known to interfere with the signal transduction pathway that activates the transcription factor NF-κB (27;28). NF-κB is the central regulator of most immune and inflammatory processes and controls the transcriptional of many host genes (29). As with all viruses, the expression of the viral genes is dependent on the available host transcription factors. HIV-1 has evolved to exploit NF-κB as it is a very potent transcriptional activator in T cells and macrophages, the primary targets for HIV-1 infection. NF-κB activation is controlled by phosphorylation and degradation of its inhibitor, IκB, which results in the removal of the inhibitor from NF-κB allowing it to translocate to the nucleus and activate transcription. HIV-1 infection of both immune cells (30;31) and kidney cells (32) results in the hijacking of the NF-κB activation process at the level of IκB phosphorylation, inducing a persistent degradation of IκB and subsequent persistent activation of NF-κB. This hijacking strategy ensures the viral genes are transcribed at a high rate; however, the disrupted NF-κB activation also alters the expression of many NF-κB-dependent host genes. In HIVAN, some of these host target genes include Fas and FasL (33) and Cyclin D1 (34). The HIV-1-induced dysregulation of these host genes leads to increased apoptosis and increased proliferation respectively in renal parenchymal cells, both contributing processes in HIVAN pathogenesis.
Oncogenes and Transformation Strategies
The increased proliferation of epithelial cells in HIVAN represents a significant difference in the pathogenic mechanism from the other more common forms of focal segmental glomerulosclerosis which are associated with overall loss of renal cell mass (35). This nonmalignant transformation of renal epithelial cells underlies both glomerular pseudocrescent and tubular cyst formation, each a significant pathological component of HIVAN. Using cell culture studies, this transformation includes both increased proliferation as well as loss of contact inhibition and appears to be caused by the function of the HIV accessory protein Nef (13) through its hijacking of the Src-dependent signal transduction pathways (15). Pharmaceutical suppression of this proliferative phenotype in a transgenic mouse model of HIVAN resulted in a significant improvement in kidney function and pathology (36;37). Although Nef appears to be a critical disease determinant in HIVAN pathogenesis, the exact causal mechanism is not known. Nonetheless future therapies directed at inhibiting Nef function may be useful in preventing or slowing the progression of HIVAN. Current studies in transgenic models have shown that limiting the effect of HIV on the cell cycle alone through the use of cyclin kinase inhibitors can improve renal function and histopathological changes demonstrating this may be a useful target for drug development (36;37).
Modifying the host cell cycle and oncogenic transformation is an important survival strategy for viruses that can only replicate in actively dividing cells, such as the polyomaviruses BK, JC and SV40. Some of the most potent transforming proteins are the polyomavirus tumor (T) antigens. T antigens are accessory proteins that are required for replication of the viral DNA and also hijack the cell cycle by blocking the function of p53 and the retinoblastoma (Rb) family members (38). The potency of T antigens to transform cells into a state of neoplastic proliferation is best characterized for the SV40 T antigen, which is frequently used in research applications to immortalize cell lines for their unlimited propagation in vitro. The polyomavirus BK, which naturally infects cells of the uroepithelium, produces a small t and large T antigen (Table 1), and a possible link between BK virus infection and cancer has been proposed (39). Although it is has been shown that BK viral sequences are present in cancers of the kidney and urinary tract, it is still debated as to a role for the virus in oncogenesis (40–42). BK infection alone may not induce cancer, since BK virus infects up to 90% of the population (39) and most infected persons do not develop cancer. Alternatively, it remains a possibility that the BK T antigen may be oncogenic and BK infection could be a contributing factor to urogenital and other cancers either in combination with another mutagenic event (either in the host cell or in the virus), or possibly in the setting of co-infection with another virus such as HIV-1 (43) or SV40 (44).
The Symbiosis of Host-Virus Interactions
In the best possible scenario, the interaction of a virus with its host is commensal, in which the virus benefits from exploiting the host to ensure its own propagation, but the host remains unharmed. The only evolutionary driving force for the virus is to replicate: to generate as many progeny virus as possible and to infect as many hosts as possible. Whereas some viruses establish a lifelong infection of the host while others move quickly from one host to the next, causing disease in the host is counterproductive to the virus’s biological success since a sick or dead host will only impede its ability to replicate.
Adaptation and Attenuation
An example of a virulence-defeating host-virus interaction is Ebola infection in humans (45). Although Ebola is highly transmissible between humans, it is also highly pathogenic resulting in almost immediate death of the host. Since Ebola is a recent infectious agent to appear in humans, the immune system has not had the evolutionary time need to establish the symbiotic balance between virus and host, in which both the virus adapts to the host and the human immune system devises a way to control the new infectious agent. Over time, the biological success of the virus (dependent on natural selection) improves in the host by both attenuation, a process where the virus becomes less pathogenic, and by an increased viral replication capacity.
Another emerging infectious disease, HIV-1, has similarly not had the evolutionary time to adapt and attenuate to humans (46). Simian immunodeficiency virus, SIV, now accepted to be the ancestor to HIV-1, has been infecting chimpanzees for likely 10,000 years or more (47). Within the past 100 years, SIV made a species jump to humans (48). A significant difference between SIV infection in chimps and HIV infection in humans is that chimps do not develop AIDS and die from their infections. A recent study has shown that the viral accessory protein Nef may play a significant role in this difference (49). Versions of the Nef protein are found in HIV-1, HIV-2 and the various SIV strains, and it functions to remove or prevent the plasma membrane presentation of receptors on the cell surface. One cell surface receptor efficiently downmodulated by the SIV Nefs is CD3, which through association with the T cell receptor initiates activation induced cell death (AICD), a normal and self-limiting process in the homeostasis of the immune system. The HIV-1 Nefs in human cells do not effectively downmodulate CD3. In the setting of a chronic infection, this failure to downmodulate CD3 results in robust AICD causing a depletion of effector T cell populations, which subsequently causes immunodeficiency and AIDS. Thus, SIV has adapted to the chimpanzee immune system over the past 10,000 years to be less pathogenic, in part by protecting the chimp’s immune system through CD3 downmodulation.
The Balance of Host-Virus Interactions and Immunosuppression
It is of interest to note that the more common viral infections related to chronic kidney disease include both viruses such as HIV-1 and HCV, which have symptomatic infections and a low prevalence (2% and 0.4% respectively in the U.S.), and other viruses such as B19, BK and CMV, which have a very high prevalence (80–90% of the U.S. population seroconverting in childhood) and individuals typically remain asymptomatic throughout life (50). Why does a virus that infects everyone cause disease in only a few, and typically only in the setting of immunosuppression? The obvious answer centers on the immunocompromised hosts, and underscores the delicate balance that exists between the host anti-viral mechanisms and the ability of the virus to replicate.
CMV is an example of a virus that has achieved a successful but delicate balance between the immune system and viral escape. CMV infection only causes severe disease in either the fetus or newborn infants, or in an immunocompromised adult, both situations where the immune system is not fully functional (6). Immunosuppression upsets this balance permitting the virus to escape the clearance mechanisms of the host. In both acute (51) and likely chronic graft dysfunction (52), CMV disease primarily involves the activation of cell-mediated immunity and is associated with inflammation and fibrosis in the graft.
As another example, B19 reactivations in immunocompromised patients (53) cause epo-resistant anemia from the direct infection and lysis of erythroid progenitor cells, as the B19 lifecycle requires the death of the host cell to release new virions (54). In addition, patient morbidity and graft injury results from a general, multi-organ inflammation (myocarditis, pneumonitis, hepatitis) and subsequent organ damage from scarring (55). Thus, in transplantation, the reactivations of CMV, BK and B19 generally result in a constant state of immune activation, with end organ damage occurring as a result of uncontrolled immune and inflammatory processes. Current therapies to control these reactivations have been most successful with the use of specific anti-virals exploiting a vulnerable point in the viral life cycle, such as the use ganciclovir to manage CMV infection and disease. A better understanding of the basic biology of these different viruses and how they escape latency will aide the future development of therapeutic agents to limit their replication in immunocompromised hosts.
In the setting of transplantation, several mechanisms have been proposed for the reactivation of both BK and CMV infections (56). Polyomaviruses have evolved no specific mechanism to cross the nuclear membrane and thus can only infect actively dividing cells. In the setting of transplantation, the frequently associated ischemic injury results in cell death of primarily tubular epithelia, a known target cell for BK infection. The normal repair response from ischemic injury includes the remaining tubular epithelial cells to proliferate to replace the lost cells. This reparative proliferation becomes an ideal environment for replication and propagation of BK virus in the kidney epithelium. Similarly, there are well known cellular responses to allogeneic transplantation such as the production of inflammatory cytokines TNF and IFN-γ, which and known to activate the host transcription factors NF-κB and AP-1. In CMV reactivation, the immediate-early (IE) gene set must be expressed to institute a new round of viral replication. Interestingly, the CMV major immediate early promoter has evolved to bind NF-κB and AP-1. Thus, the normal host responses to an allogeneic graft induce transcription factors that control the expression of the CMV IE genes, subsequently providing an environment to initiate a burst of new virus production.
Conclusions
Viruses are incapable of autonomous replication and must use the available cellular machinery of the host to support their life cycle. Because of this, viruses have evolved or borrowed from the host unique survival mechanisms to permit completion of their life cycle and include molecular mimicry, hijacking and oncogenic transformation. Host pathogenesis develops from both the host’s immune response to the infection, as well as the disruption of the normal cell cycle and cellular functions due to hijacking strategies employed by the virus. It is through survival-driven selective pressure and co-evolution of the virus with the host that shapes not only the virus but also the host and its immune system. In this co-evolution, the processes of adaptation and attenuation improve viral fitness as well as establish a more commensal symbiosis by making the virus less pathogenic to the host. By studying the adaptation and subversion strategies of “old” viral infections in humans may provide insight into how to treat “new” infections that are highly virulent and result in severe disease and mortality.
Acknowledgments
Dr. Bruggeman is supported by NIH grant DK61395 and is an investigator in the Case Center for AIDS Research supported by NIH grant AI36219.
References
- 1.Albrecht T, Fons M, Boldogn I, Rabson AS. Mechanisms of viral pathogenesis: effects on cell. In: Barns S, editor. Medical Microbiology. 4. Elsworth Mosby; 1996. [Google Scholar]
- 2.Heegaard ED, Brown KE. Human parvovirus B19. Clin Microbiol Rev. 2002;15:485–505. doi: 10.1128/CMR.15.3.485-505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yaegashi N, Niinuma T, Chisaka H, Uehara S, Moffatt S, Tada K, Iwabuchi M, Matsunaga Y, Nakayama M, Yutani C, Osamura Y, Hirayama E, Okamura K, Sugamura K, Yajima A. Parvovirus B19 infection induces apoptosis of erythroid cells in vitro and in vivo. J Infect. 1999;39:68–76. doi: 10.1016/s0163-4453(99)90105-6. [DOI] [PubMed] [Google Scholar]
- 4.Alcami A. Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol. 2003;3:36–50. doi: 10.1038/nri980. [DOI] [PubMed] [Google Scholar]
- 5.Murphy PM. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat Immunol. 2001;2:116–122. doi: 10.1038/84214. [DOI] [PubMed] [Google Scholar]
- 6.Mocarski ES., Jr Immune escape and exploitation strategies of cytomegaloviruses: impact on and imitation of the major histocompatibility system. Cell Microbiol. 2004;6:707–717. doi: 10.1111/j.1462-5822.2004.00425.x. [DOI] [PubMed] [Google Scholar]
- 7.Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 8.Lloyd AR, Jagger E, Post JJ, Crooks LA, Rawlinson WD, Hahn YS, Ffrench RA. Host and viral factors in the immunopathogenesis of primary hepatitis C virus infection. Immunol Cell Biol. 2007;85:24–32. doi: 10.1038/sj.icb.7100010. [DOI] [PubMed] [Google Scholar]
- 9.Sodhi A, Montaner S, Gutkind JS. Viral hijacking of G-protein-coupled-receptor signalling networks. Nat Rev Mol Cell Biol. 2004;5:998–1012. doi: 10.1038/nrm1529. [DOI] [PubMed] [Google Scholar]
- 10.Frankel AD, Young JA. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Herman ES, Klotman PE. HIV-associated nephropathy: Epidemiology, pathogenesis, and treatment. Semin Nephrol. 2003;23:200–208. doi: 10.1053/snep.2003.50018. [DOI] [PubMed] [Google Scholar]
- 12.Kimmel PL. HIV-associated nephropathy: virologic issues related to renal sclerosis. Nephrol Dial Transplant. 2003;18(Suppl 6):vi59–63. vi59–vi63. doi: 10.1093/ndt/gfg1062. [DOI] [PubMed] [Google Scholar]
- 13.Husain M, Gusella GL, Klotman ME, Gelman IH, Ross MD, Schwartz EJ, Cara A, Klotman PE. HIV-1 Nef induces proliferation and anchorage-independent growth in podocytes. J Am Soc Nephrol. 2002;13:1806–1815. doi: 10.1097/01.asn.0000019642.55998.69. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz EJ, Cara A, Snoeck H, Ross MD, Sunamoto M, Reiser J, Mundel P, Klotman PE. Human immunodeficiency virus-1 induces loss of contact inhibition in podocytes. J Am Soc Nephrol. 2001;12:1677–1684. doi: 10.1681/ASN.V1281677. [DOI] [PubMed] [Google Scholar]
- 15.He JC, Husain M, Sunamoto M, D’Agati VD, Klotman ME, Iyengar R, Klotman PE. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J Clin Invest. 2004;114:643–651. doi: 10.1172/JCI21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husain M, D’Agati VD, He JC, Klotman ME, Klotman PE. HIV-1 Nef induces dedifferentiation of podocytes in vivo: a characteristic feature of HIVAN. AIDS. 2005;19:1975–1980. doi: 10.1097/01.aids.0000191918.42110.27. [DOI] [PubMed] [Google Scholar]
- 17.Sunamoto M, Husain M, He JC, Schwartz EJ, Klotman PE. Critical role for Nef in HIV-1-induced podocyte dedifferentiation. Kidney Int. 2003;64:1695–1701. doi: 10.1046/j.1523-1755.2003.00283.x. [DOI] [PubMed] [Google Scholar]
- 18.Hanna Z, Kay DG, Rebai N, Guimond A, Jothy S, Jolicoeur P. Nef harbors a major determinant of pathogenicity for an AIDS-like disease induced by HIV-1 in transgenic mice. Cell. 1998;95:163–75. doi: 10.1016/s0092-8674(00)81748-1. [DOI] [PubMed] [Google Scholar]
- 19.Hanna Z, Kay DG, Cool M, Jothy S, Rebai N, Jolicoeur P. Transgenic mice expressing human immunodeficiency virus type 1 in immune cells develop a severe AIDS-like disease. J Virol. 1998;72:121–32. doi: 10.1128/jvi.72.1.121-132.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanna Z, Weng X, Kay DG, Poudrier J, Lowell C, Jolicoeur P. The pathogenicity of human immunodeficiency virus (HIV) type 1 Nef in CD4C/HIV transgenic mice is abolished by mutation of its SH3-binding domain, and disease development is delayed in the absence of Hck. J Virol. 2001;75:9378–9392. doi: 10.1128/JVI.75.19.9378-9392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanna Z, Priceputu E, Kay DG, Poudrier J, Chrobak P, Jolicoeur P. In vivo mutational analysis of the N-terminal region of HIV-1 Nef reveals critical motifs for the development of an AIDS-like disease in CD4C/HIV transgenic mice. Virology. 2004;327:273–286. doi: 10.1016/j.virol.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 22.Zuo Y, Matsusaka T, Zhong J, Ma J, Ma LJ, Hanna Z, Jolicoeur P, Fogo AB, Ichikawa I. HIV-1 genes vpr and nef synergistically damage podocytes, leading to glomerulosclerosis. J Am Soc Nephrol. 2006;17:2832–2843. doi: 10.1681/ASN.2005080878. [DOI] [PubMed] [Google Scholar]
- 23.Dickie P, Roberts A, Uwiera R, Witmer J, Sharma K, Kopp JB. Focal glomerulosclerosis in proviral and c-fms transgenic mice links Vpr expression to HIV-associated nephropathy. Virology. 2004;322:69–81. doi: 10.1016/j.virol.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 24.Kajiyama W, Kopp JB, Marinos NJ, Klotman PE, Dickie P. Glomerulosclerosis and viral gene expression in HIV-transgenic mice: role of nef. Kidney Int. 2000;58:1148–1159. doi: 10.1046/j.1523-1755.2000.00271.x. [DOI] [PubMed] [Google Scholar]
- 25.Ray PE, Liu XH, Robinson LR, Reid W, Xu L, Owens JW, Jones OD, Denaro F, Davis HG, Bryant JL. A novel HIV-1 transgenic rat model of childhood HIV-1-associated nephropathy. Kidney Int. 2003;63:2242–2253. doi: 10.1046/j.1523-1755.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 26.Barisoni L, Kopp JB. Modulation of podocyte phenotype in collapsing glomerulopathies. Microsc Res Tech. 2002;57:254–262. doi: 10.1002/jemt.10084. [DOI] [PubMed] [Google Scholar]
- 27.Santoro MG, Rossi A, Amici C. NF-kappaB and virus infection: who controls whom. EMBO J. 2003;22:2552–2560. doi: 10.1093/emboj/cdg267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowie AG, Zhan J, Marshall WL. Viral appropriation of apoptotic and NF-kappaB signaling pathways. J Cell Biochem. 2004;91:1099–1108. doi: 10.1002/jcb.20026. [DOI] [PubMed] [Google Scholar]
- 29.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 30.DeLuca C, Petropoulos L, Zmeureanu D, Hiscott J. Nuclear IkappaBbeta maintains persistent NF-kappaB activation in HIV-1-infected myeloid cells. J Biol Chem. 1999;274:13010–13016. doi: 10.1074/jbc.274.19.13010. [DOI] [PubMed] [Google Scholar]
- 31.McElhinny JA, MacMorran WS, Bren GD, Ten RM, Israel A, Paya CV. Regulation of I kappa B alpha and p105 in monocytes and macrophages persistently infected with human immunodeficiency virus. J Virol. 1995;69:1500–1509. doi: 10.1128/jvi.69.3.1500-1509.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinka S, Bruggeman LA. Persistent NF-{kappa}B activation in renal epithelial cells in mouse modelof HIV-associated nephropathy. Am J Physiol Renal Physiol. 2005;290:F657–F665. doi: 10.1152/ajprenal.00208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ross MJ, Martinka S, D’Agati VD, Bruggeman LA. NF-κB regulates Fas-mediated apoptosis in HIV-associated nephropathy. J Am Soc Nephrol. 2005;16:2403–2411. doi: 10.1681/ASN.2004121101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson PJ, Sunamoto M, Husain M, Gelman IH. HIV-1 expression induces cyclin D1 expression and pRb phosphorylation in infected podocytes: cell-cycle mechanisms contributing to the proliferative phenotype in HIV-associated nephropathy. BMC Microbiol. 2002;e2:26. doi: 10.1186/1471-2180-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albaqumi M, Soos TJ, Barisoni L, Nelson PJ. Collapsing glomerulopathy. J Am Soc Nephrol. 2006;17:2854–2863. doi: 10.1681/ASN.2006030225. [DOI] [PubMed] [Google Scholar]
- 36.Nelson PJ, D’Agati VD, Gries JM, Suarez JR, Gelman IH. Amelioration of nephropathy in mice expressing HIV-1 genes by the cyclin-dependent kinase inhibitor flavopiridol. J Antimicrob Chemother. 2003;51:921–929. doi: 10.1093/jac/dkg175. [DOI] [PubMed] [Google Scholar]
- 37.Gherardi D, D’Agati V, Chu TH, Barnett A, Gianella-Borradori A, Gelman IH, Nelson PJ. Reversal of collapsing glomerulopathy in mice with the cyclin-dependent kinase inhibitor CYC202. J Am Soc Nephrol. 2004;15:1212–1222. doi: 10.1097/01.asn.0000124672.41036.f4. [DOI] [PubMed] [Google Scholar]
- 38.Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006;25:5277–5285. doi: 10.1038/sj.onc.1209621. [DOI] [PubMed] [Google Scholar]
- 39.Reploeg MD, Storch GA, Clifford DB. Bk virus: a clinical review. Clin Infect Dis. 2001;33:191–202. doi: 10.1086/321813. [DOI] [PubMed] [Google Scholar]
- 40.Fioriti D, Videtta M, Mischitelli M, Degener AM, Russo G, Giordano A, Pietropaolo V. The human polyomavirus BK: Potential role in cancer. J Cell Physiol. 2005;204:402–406. doi: 10.1002/jcp.20300. [DOI] [PubMed] [Google Scholar]
- 41.Tognon M, Corallini A, Martini F, Negrini M, Barbanti-Brodano G. Oncogenic transformation by BK virus and association with human tumors. Oncogene. 2003;22:5192–5200. doi: 10.1038/sj.onc.1206550. [DOI] [PubMed] [Google Scholar]
- 42.Barbanti-Brodano G, Sabbioni S, Martini F, Negrini M, Corallini A, Tognon M. BK virus, JC virus and Simian Virus 40 infection in humans, and association with human tumors. Adv Exp Med Biol. 2006;577:319–41. doi: 10.1007/0-387-32957-9_23. [DOI] [PubMed] [Google Scholar]
- 43.Gorrill T, Feliciano M, Mukerjee R, Sawaya BE, Khalili K, White MK. Activation of early gene transcription in polyomavirus BK by human immunodeficiency virus type 1 Tat. J Gen Virol. 2006;87:1557–1566. doi: 10.1099/vir.0.81569-0. [DOI] [PubMed] [Google Scholar]
- 44.Li RM, Mannon RB, Kleiner D, Tsokos M, Bynum M, Kirk AD, Kopp JB. BK virus and SV40 co-infection in polyomavirus nephropathy. Transplantation. 2002;74:1497–1504. doi: 10.1097/00007890-200212150-00004. [DOI] [PubMed] [Google Scholar]
- 45.Peters CJ. Marburg and Ebola--arming ourselves against the deadly filoviruses. N Engl J Med. 2005;352:2571–2573. doi: 10.1056/NEJMp058109. [DOI] [PubMed] [Google Scholar]
- 46.Heeney JL, Dalgleish AG, Weiss RA. Origins of HIV and the evolution of resistance to AIDS. Science. 2006;313:462–466. doi: 10.1126/science.1123016. [DOI] [PubMed] [Google Scholar]
- 47.Brander C, Walker BD. Gradual adaptation of HIV to human host populations: good or bad news? Nat Med. 2003;9:1359–1362. doi: 10.1038/nm941. [DOI] [PubMed] [Google Scholar]
- 48.Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JF, Sharp PM, Shaw GM, Peeters M, Hahn BH. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science. 2006;313:523–526. doi: 10.1126/science.1126531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schindler M, Munch J, Kutsch O, Li H, Santiago ML, Bibollet-Ruche F, Muller-Trutwin MC, Novembre FJ, Peeters M, Courgnaud V, Bailes E, Roques P, Sodora DL, Silvestri G, Sharp PM, Hahn BH, Kirchhoff F. Nef-mediated suppression of T cell activation was lost in a lentiviral lineage that gave rise to HIV-1. Cell. 2006;125:1055–1067. doi: 10.1016/j.cell.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 50.Centers for Disease Control. Summary of notifiable disease -- United States, 2004. MMWR. 2006;53:6–39. [Google Scholar]
- 51.Helantera I, Koskinen P, Tornroth T, Loginov R, Gronhagen-Riska C, Lautenschlager I. The impact of cytomegalovirus infections and acute rejection episodes on the development of vascular changes in 6-month protocol biopsy specimens of cadaveric kidney allograft recipients. Transplantation. 2003;75:1858–1864. doi: 10.1097/01.TP.0000064709.20841.E1. [DOI] [PubMed] [Google Scholar]
- 52.Helantera I, Koskinen P, Finne P, Loginov R, Kyllonen L, Salmela K, Gronhagen-Riska C, Lautenschlager I. Persistent cytomegalovirus infection in kidney allografts is associated with inferior graft function and survival. Transpl Int. 2006;19:893–900. doi: 10.1111/j.1432-2277.2006.00364.x. [DOI] [PubMed] [Google Scholar]
- 53.Young NS, Brown KE. Parvovirus B19. N Engl J Med. 2004;350:586–597. doi: 10.1056/NEJMra030840. [DOI] [PubMed] [Google Scholar]
- 54.Azzi A, Cesaro S, Laszlo D, Zakrzewska K, Ciappi S, De SR, Fanci R, Pesavento G, Calore E, Bosi A. Human polyomavirus BK (BKV) load and haemorrhagic cystitis in bone marrow transplantation patients. J Clin Virol. 1999;14:79–86. doi: 10.1016/s1386-6532(99)00055-4. [DOI] [PubMed] [Google Scholar]
- 55.Cavallo R, Merlino C, Re D, Bollero C, Bergallo M, Lembo D, Musso T, Leonardi G, Segoloni GP, Ponzi AN. B19 virus infection in renal transplant recipients. J Clin Virol. 2003;26:361–368. doi: 10.1016/s1386-6532(02)00104-x. [DOI] [PubMed] [Google Scholar]
- 56.Hummel M, Abecassis MM. A model for reactivation of CMV from latency. J Clin Virol. 2002;25(Suppl 2):S123–36. doi: 10.1016/s1386-6532(02)00088-4. [DOI] [PubMed] [Google Scholar]
- 57.Moffatt S, Tanaka N, Tada K, Nose M, Nakamura M, Muraoka O, Hirano T, Sugamura K. A cytotoxic nonstructural protein, NS1, of human parvovirus B19 induces activation of interleukin-6 gene expression. J Virol. 1996;70:8485–8491. doi: 10.1128/jvi.70.12.8485-8491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]