

Abstract
Connexin 35 (Cx35) is a major component of electrical synapses in the central nervous system. Many gap junctions containing Cx35 are regulated by dopamine receptor pathways that involve protein kinase A (PKA). To study the mechanism of PKA regulation, we analyzed direct phosphorylation of Cx35 by PKA in vitro, and studied the regulation of Neurobiotin tracer coupling in HeLa cells expressing Cx35 or Cx35 mutants that lack phosphorylation sites. In Cx35-transfected cells, application of the PKA activator Sp-8-cpt-cAMPS caused a significant decline in coupling, while a PKA inhibitor, Rp-8-cpt-cAMPS, significantly increased tracer coupling. In vitro phosphorylation and mutagenic analysis showed that PKA phosphorylates Cx35 directly at two major sites, Ser110 in the intracellular loop and Ser276 in the carboxyl terminus. In addition, a minor phosphorylation site in the C-terminus was identified by truncation of the last 7 amino acids at Ser298. The mutations Ser110Ala or Ser276Ala significantly reduced regulation of coupling by the PKA activator, while a combination of the two eliminated regulation. Truncation at Ser298 reversed the regulation such that the PKA activator significantly increased and the PKA inhibitor significantly decreased coupling. The activation was eliminated in the S110A,S276A,S298ter triple mutant. We conclude that PKA regulates Cx35 coupling in a complex manner that requires both major phosphorylation sites. Furthermore, the tip of the C-terminus acts as a “switch” that determines whether phosphorylation will inhibit or enhance coupling. Reliance on the combined states of three sites provides fine control over the degree of coupling through Cx35 gap junctions.
Keywords: Connexin36, phosphorylation, cAMP, protein kinase, amacrine cell
1. INTRODUCTION
Electrical synapses, consisting of gap junctions between two neurons, occur frequently throughout the central nervous system. With the identification of connexin 35 (Cx35) [35,36] and its mammalian homologue Cx36 [5,44] as major neuronal connexins, the understanding of electrical synapses has increased rapidly. Many, if not most, gap junctions among neurons in the brain and spinal cord contain Cx35/36 [39,42,43]. In the retina, Cx36 is a critical component of the rod visual pathway, forming the gap junctions of AII amacrine cells with each other and with cone ON bipolar cells [11,29]. Cx35/36 is also an important component of cone-cone and rod-cone gap junctions [10,23,37].
Disruption of the Cx36 gene in mice impaired firing synchrony [7] and gamma-frequency oscillations [17] in the neocortex, and led to anomalous patterns of sharp-wave bursts and ripple oscillations in the hippocampus [25,38]. Cx36-null mice also showed deficits in visual transmission through the rod pathway [8,15], consistent with disruption of AII amacrine-cone ON bipolar cell and rod-cone gap junctions.
In many retinal neurons, the degree of electrical coupling varies with light adaptation state. Cone-cone coupling has been observed to increase with light adaptation in the turtle [6], as did rod-cone coupling in salamander [51]. This effect of light was partially mimicked by dopamine D2/D4-like receptor activation [20], which results in reduction of adenylyl cyclase activity and cytoplasmic cAMP concentration [4,34]. On the other hand, light adaptation [1] or dopamine application [16] reduced coupling in AII amacrine cells, which respond to dopamine through D1 dopamine receptors and elevation of cytoplasmic cAMP.
In both photoreceptors and AII amacrine cells, gap junctions contain Cx35/Cx36 [10,11,23,29,37]. Thus it is expected that Cx35/36 is regulated by pathways that alter cAMP levels and PKA activity. We have previously found that activation of PKA in HeLa cells results in uncoupling of Cx35 gap junctions, and that Cx35 is phosphorylated by PKA in vitro [37]. In this study, we examine the mechanism by which PKA regulates coupling in Cx35 gap junctions in more detail. We find that PKA regulation is complex, requiring several phosphorylation events that likely involve both direct and indirect pathways.
2. MATERIALS AND METHODS
Fusion protein constructs
Portions of the perch Cx35 cDNA coding for intracellular domains were cloned separately into bacterial expression vectors. The intracellular loop domain, amino acids 102-178, was cloned previously into pET15b [36]. The resulting construct codes for an 11.5 kDa fusion protein with an N-terminal 6x His tag. The carboxyl-terminal domain, consisting of amino acids 254-304, was subcloned into the EcoRI and HindIII sites of pET42c (Novagen, Madison, WI) from a pGexKG clone described previously [37]. This construct coded for a 38.5 kDa fusion protein containing amino-terminal glutathione-S-transferase and 6x His tags. A pET42 control clone was made by cutting pET42a with HindIII and EcoRV, filling the HindIII end with Klenow, and resealing the vector. This construct codes for a 31.7 kDa protein containing GST and the pET42 His-tag and multiple cloning site up to a point very close to the position into which the Cx35 CT was cloned.
Mutations in the connexin intracellular domains were made with a PCR-based site-directed mutagenesis protocol [12] using Pfu polymerase (Stratagene, La Jolla, CA). Mutations were designed to eliminate a putative phosphorylation site by converting the phosphorylatable serine or threonine residue to alanine. All constructs were sequenced on both strands to confirm mutations and screen for the presence of PCR-introduced mutations.
Fusion proteins were expressed in E. coli strain BL21(DE3). Intracellular loop proteins were purified by binding to Ni-NTA magnetic beads (Qiagen, Valencia, CA) using the manufacturer’s procedures. Proteins were left bound to the beads and stored in 25 mM Tris-Cl, pH 7.5, 5 mM MgCl2, and 50% glycerol. Due to lability of the C-terminal fusion proteins, different procedures were used to minimize proteolytic degradation. Proteins were expressed by induction with 1mM IPTG at 30°C for 1 hour. Cells were pelleted, suspended in water plus 1 mg/ml lysozyme (Sigma) for 3 minutes at RT, and then supplemented with 5 μl/ml of a protease inhibitor cocktail (Sigma). Cell suspensions were sonicated on ice for 30 sec, and brought to final solute concentrations of 50 mM Tris-Cl, pH 8.0, 0.3 M NaCl, 10 mM Imidazole by addition of a concentrated buffer stock. Fusion proteins were then purified from the soluble extract by binding to a Ni-NTA resin in spin columns (Active Motif, Carlsbad, CA) using the manufacturer’s protocol. Proteins were stored following elution in 25 mM Tris-Cl, pH 8.0, 150 mM NaCl, 125 mM imidazole, and 50% glycerol. Fusion protein concentration was estimated by densitometry of Coomassie blue stained bands compared to a series of BSA standards run in the same minigels. In the case of the C-terminal fusion protein preparations that contain proteolytic cleavage products, only the intact protein band was used for the protein determination.
In Vitro Phosphorylation Assays
Fusion proteins (200 or 400 ng protein per reaction) were incubated with 0.15 units PKA catalytic subunit (mouse α isoform; New England Biolabs, Beverly, MA) and γ32P-ATP or γ33P-ATP (ICN, Irvine, CA) for 40 min. at 37°C. For intracellular loop fusion proteins, the final solution contained 50 mM Tris-Cl (pH 7.5), 10 mM MgCl2, 20 μM ATP, 46 mM NaCl, 1 mM KCl, 4 mM Na2HPO4, and 27% glycerol. For C-terminal fusion proteins, the final solution contained 66 mM Tris-Cl (pH 7.7), 10 mM MgCl2, 20 μM ATP, 94 mM NaCl, 78 mM imidazole, and 31% glycerol. Reactions were stopped by addition of SDS sample buffer, heated to 70°C for 10 min., and electrophoresed on SDS polyacrylamide gels. Gels were blotted onto PVDF or nitrocellulose membranes for imaging onto X-ray film (Kodak). Estimates of phosphorylation were made by densitometry of the developed films using an Alpha Imager 2000 system (Alpha Innotech, San Leandro, CA).
Phosphoamino Acid Analysis
Phosphorylated proteins bound to PVDF membranes were excised from the membrane and hydrolyzed in 5.7M HCl at 110°C for 1 hr. The hydrolyzed proteins were dried, mixed with phosphoamino acid standards (Fluka, Buchs, Switzerland), and spotted onto cellulose plates (EM Sciences, Gibbstown, NJ) for one-dimensional thin layer electrophoresis [18]. Phosphoamino acid standards were visualized with ninhydrin and compared to 32P-phosphoamino acid residues detected by autoradiography.
Expression of connexins in HeLa cells
The full coding sequence of perch Cx35 was sub-cloned into pcDNA 3.1 Zeo (Invitrogen, Carlsbad, CA) from a previously-described pBluescript cDNA clone [36]. The Cx35 cDNA was excised with AluI and XhoI and cloned into the EcoRV and XhoI sites of pcDNA. HeLa cells (ATCC clone CCL2) were transfected with 2 μg of plasmid using GenePorterII reagent (Gene Therapy Systems, San Diego, CA) and grown under zeocin selection (100 μg/ml) for 6 weeks. Resistant colonies were isolated and amplified under selection. Clones were tested for connexin expression by immunostaining and RT-PCR as described previously [37] (data not shown). The Cx35 wild type cell line has been characterized previously [37].
To prepare mutated connexin constructs, we used the same primers and cycling conditions as used to make mutations in the fusion protein constructs. Wild-type Cx35 pcDNA clone was used as the template. For multiple mutants, appropriate singly or doubly-mutant Cx35 pcDNA clones were used as the templates. All mutants were confirmed by sequencing on both strands. Transfection was performed in the same manner. Stable cell lines were selected for each of the singly mutated connexin clones and used for tracer coupling assays. For analysis of double and triple mutants, transient transfection assays were utilized (see below). Transfection was performed as described above except that 5 μg of the mutant plasmid DNA was used per transfection. The wild-type Cx35 plasmid construct was used as a control.
Tracer Coupling Experiments
Connexin-transfected HeLa cells were maintained under zeocin selection until plating onto coverslips for tracer coupling experiments. One to two days prior to experiments, cells were fed with fresh medium, trypsinized, and plated at approximately 50% confluence onto glass cell culture coverslips (Fisher Scientific, Houston, TX). Cells were fed again 2 to 12 hours prior to experimentation. For transient transfection assays, non-transfected HeLa cells were plated onto coverslips as described above. 24-36 hours after plating, the cells were transfected with 5 μg of wild-type or mutant connexin plasmid in 1 ml of medium as described above, and grown for 24 hours without selective antibiotic. The cells were then fed with fresh medium and scrape-loading experiments were performed as with the stably-transfected cell lines.
Tracer coupling within Cx35 wild type and mutant cell lines was analyzed by scrape loading. HeLa cells were incubated at 25°C in oxygenated modified Ringer’s medium (containing in mM: 150 NaCl, 6.2 KCl, 1.2 NaH2PO4, 1.2 MgSO4, 2.5 CaCl2, 10 Hepes, 10 glucose, pH 7.4). The medium was supplemented with 0.05 % Neurobiotin (Vector Laboratories, Burlingame, CA) and 0.05% Alexa-488 dextran (Molecular Probes, Eugene, OR), and cells were scraped with a 25-guage needle. Incubation was continued for 10 minutes at 25°C to allow for loading and diffusion. Cells were then fixed with 4% formaldehyde in 0.1 M phosphate buffer for 1 hour, and visualized with streptavidin-Cy3 (Jackson ImmunoResearch). Fluorescence signals were photographed on a Zeiss fluorescence microscope with a 12-bit Hamamatsu Orca-100 digital camera using Simple PCI software (Compix, Cranberry Township, PA).
Drugs were applied by exchanging the bathing solution with oxygenated incubation medium plus drug for 10 minutes prior to scrape-loading. Cells were exposed continuously to drugs thereafter without further supplementation or washout. The PKA activator, Sp-8-cpt-cAMPS and PKA inhibitor, Rp-8-cpt-cAMPS, were from Alexis (San Diego, CA). Purity and concentration of cAMP analogs was assessed by reversed phase liquid chromatography using a BioCAD/Sprint system (Applied Biosystems, Framingham, MA) and a Poros20 HC column with a 20-70% acetonitrile gradient.
All tracer coupling experiments were performed on two to six separate days resulting from different platings of the HeLa cell lines. In the case of HeLa-Cx35S298ter, two independent clones were examined and the data combined. One clone was studied for the other Cx35 mutants analyzed. For all experiments, control and drug-treated conditions were examined on the same day with the same batch of cells. Due to some day-to-day variation in the level of tracer coupling, the diffusion data for some experiments were normalized to the mean diffusion coefficient (see below) for each cell line under control (no drug) conditions measured on the same day of experiments.
Data analysis
DNA and protein sequence analyses were performed with GeneTool 1.0 and Peptool 2.0 software (Biotools, Inc., Edmonton, AB). Additional analyses were done with PhosphoBase online [19], and basic statistical analyses were performed with Microsoft Excel.
For tracer coupling analyses, fluorescence intensities of coupled cells were used to calculate diffusion coefficients in a manner similar to that described in Mills and Massey [28], and extended in O’Brien et al. [37]. This procedure utilizes a linear 25-compartment diffusion model of the type described by Zimmerman and Rose [52] in which the movement of tracer between adjacent compartments (cells) is described by a series of 25 differential equations that are solved for tracer flux given the total amount of diffusion time and a diffusion coefficient, k. The diffusion coefficient, k, represents the proportion of tracer that diffuses from one compartment to another per second. All k values larger than 0 represent measurable tracer coupling. Optimal fits of intensity data to the model were determined in the program MatLab (Mathworks, Natick, MA) by systematically varying k and another parameter, bo, the bolus loading rate. The parameter bo defines the rate of addition of tracer to the initial compartment for the loading period, which was assumed to be 1 minute for scrape-loading experiments, and was set to zero thereafter. The value of this parameter is determined by the total amount of tracer in all the compartments and is insensitive to the diffusion rate between the cells [28]. Data fits were determined by plotting cell intensities on a log intensity axis and determining the diffusion coefficient k that best fit the rate of decline with distance, and the rate of delivery, bo, that fit the overall tracer concentration. An iterative procedure that minimized the sum of squared errors between the model and data was used to achieve these fits.
Since the model used is linear and assumes that a single compartment is loaded, we limited the data analyzed to narrow clusters of cells extending perpendicular to a loaded cell. In practice, analysis of scrape-loading data with this model yielded data with the same relative responses but larger absolute k’s compared to data obtained by microinjection with the same cell lines and drug treatments [37]. The larger k values in scrape-loading data could be explained entirely by tracer loading from multiple sources along the scrape rather than a single point source (data not shown). A more complicated 2-dimensional model with multiple loaded cells was not used for calculations due to the computational intensity of these models and the large number of repetitions required for iterative fitting to the data. Since relative changes in k were the important parameters in this study, the linear model was found to provide robust analysis of the data.
3. RESULTS
Cx35 gap junctions are regulated by PKA activity
To study the functional regulation of Cx35 gap junctions, we used stably transfected HeLa cell lines that express this connexin [37]. HeLa cells transfected with Cx35 support the transfer of Neurobiotin (figure 1A), but not Lucifer Yellow or Alexa-488 dextran (not shown), to adjacent cells through gap junctions. The diffusion of Neurobiotin could be measured by the fluorescence intensity of Cy3-streptavidin labeling in cells adjacent to the scrape. Fluorescence intensity declined with distance in either direction perpendicular to the scrape (figure 1D and E), and could be fit with the linear diffusion model described in the methods. The diffusion coefficients (k) measured in either direction from the scrape were similar: 0.0013 cells2/sec and 0.0012 cells2/sec. Since no diffusion was observed with Lucifer Yellow or Alexa-488 dextran, the diffusion coefficients for these dyes were 0 cells2/sec.
Figure 1. Effects of PKA activation and inhibition on tracer coupling in Cx35-HeLa cells measured by scrape-loading.
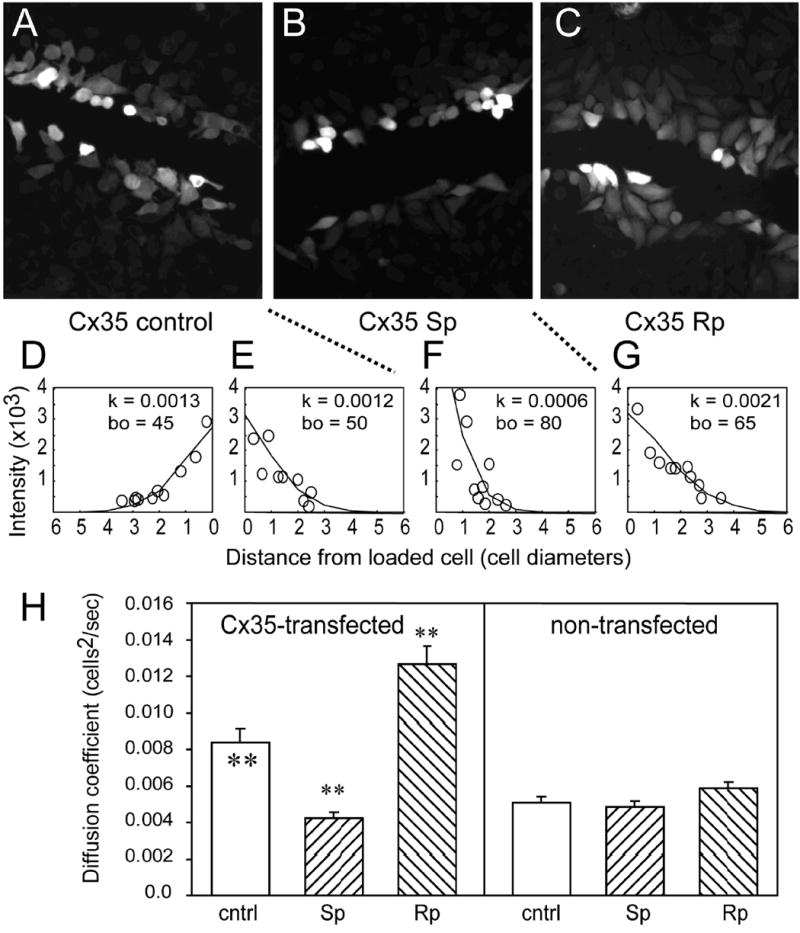
Neurobiotin loaded into cells along a scrape passes through gap junctions in HeLa cells expressing Cx35 (A). The concentration of Neurobiotin, as measured by the fluorescent intensity following fixation and staining with Cy3-streptavidin (see methods), declines with distance below (D) or above (E) the scrape. Diffusion coefficients (k) measured by fitting a linear diffusion model to the data (see methods) were 0.0013 cells2/s below and 0.0012 cells2/s above the scrape. Bath application of the PKA activator Sp-8-cpt-cAMPS (20 μM) reduced the spread of tracer (B) and reduced the diffusion coefficient (F) to 0.0006 cells2/s in Cx35-HeLa. Application of the PKA inhibitor Rp-8-cpt-cAMPS (20 μM) increased the spread of tracer (C) and increased the diffusion coefficient (G) to 0.0021 cells2/s. (H) Summary data for scrape-loading in HeLa-Cx35 and non-transfected HeLa cells. For Cx35-HeLa cells, the reduction in k relative to control (cntrl) caused by 20 μM Sp-8-cpt-cAMPS (Sp) and the increase in k relative to control caused by 20 μM Rp-8-cpt-cAMPS (Rp) were significant at the p < 0.01 level using a t-test. For non-transfected HeLa cells, neither drug treatment changed k significantly. Data shown are means of 14-53 measurements; error bars are +1 SEM. Significance of statistical comparisons is indicated by * for p < 0.05 and ** for p < 0.01. Significance symbols within the control condition bar represent comparison of the Cx35 cell line to the non-transfected cell line under control conditions.
We have previously observed using microinjection protocols that activators and inhibitors of PKA modulated tracer transfer in Cx35-transfected cells [37]. In scrape-loading experiments, tracer coupling was also inhibited by bath application of 20 μM Sp-8-cpt-cAMPS, a membrane-permeant PKA activator (figure 1B and F). The diffusion coefficient measured in this example was 0.0006 cells2/sec. 20 μM of the membrane-permeant PKA inhibitor Rp-8-cpt-cAMPS caused an increase in tracer coupling to 0.0021 cells2/sec (figure 1C and G).
Figure 1H shows the quantitative tracer coupling analysis of non-transfected and Cx35-transfected HeLa cells exposed to PKA activators and inhibitors. Under control conditions, Cx35-transfected cells transferred Neurobiotin with a diffusion coefficient of 0.0084 ± 0.0008 cells2/s (mean ± SEM; n = 32). This was significantly larger than the diffusion coefficient for Neurobiotin transfer in non-transfected HeLa cells (0.0051 ± 0.0003 cells2/s, n = 43; t = 4.09, p < 0.01). 20 μM Sp-8-cpt-cAMPS significantly reduced coupling in Cx35-transfected cells to 0.0042 ± 0.0003 cells2/s (n = 14; t = -5.93, p < 0.01), while 20 μM Rp-8-cpt-cAMPS significantly increased coupling to 0.0127 ± 0.0010 (n = 24; t = 3.92, p < 0.01). Neither drug caused a significant change in coupling in non-transfected HeLa cells (Sp vs. control: t = -0.61, p > 0.05, n = 46 for Sp; Rp vs. control: t = 1.89, p > 0.05, n = 53 for Rp). These data indicate that activation of PKA in HeLa cells causes uncoupling of Cx35 gap junctions, while inhibition of PKA increases coupling.
Cx35 contains predicted PKA phosphorylation sites
The observed regulation of gap junctional coupling by protein kinase A could occur through either direct or indirect pathways. We have previously observed that PKA catalytic subunit can phosphorylate Cx35 intracellular domains [37], suggesting that regulation of coupling may be a direct effect. To characterize this pathway further, we performed a phosphorylation motif analysis of the Cx35 sequence and those of its gene homologues mouse Cx36 and skate Cx35. Figure 2 shows that perch Cx35 contains several predicted PKA phosphorylation sites. Serines 110 and 128 in the intracellular loop, and threonine 265 and serine 276 in the carboxyl terminal tail were identified as consensus sites for PKA phosphorylation. The sequence surrounding Ser110 is conserved in mouse Cx36, and differs in skate Cx35 by only one amino acid (Gln109) that disrupts the predicted PKA recognition motif. In the C-terminus, the consensus PKA phosphorylation motif centered on Ser276 is completely conserved among perch, mouse, and skate connexins. The other two sites predicted in perch Cx35 are not conserved in the other connexin homologues.
Figure 2. Amino acid alignment of perch Cx35 with its homologues.
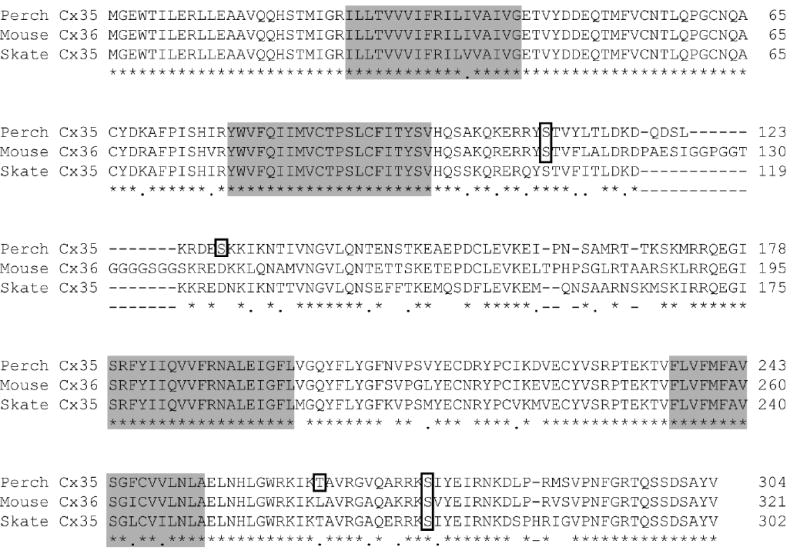
Perch Cx35 is highly similar to its homologues, mouse Cx36 and skate Cx35. In this diagram, amino acid identity is designated by an asterisk (*) and similarity by a period (.) below the alignment. Transmembrane domains (M1-M4) are shown with shaded boxes. Several consensus sequences for PKA phosphorylation are present (outlined residues). Two of these are conserved across species: serine 110 in the intracellular loop and serine 276 (numbering from perch Cx35) in the C-terminus. Perch Cx35 also has non-conserved sites at serine 128 and threonine 265.
In addition to predicted PKA phosphorylation motifs, the C-terminus contains a region rich in residues that may be phosphorylated. The last 7 amino acids of the CT contain 3 serine and one tyrosine residues with high potential for phosphorylation. Although not contained within a PKA consensus phosphorylation sequence, this region has substantial similarity to the consensus, and contains predicted phosphorylation sites for other kinases (analysis not shown).
PKA phosphorylates Cx35 at several sites
The predicted PKA phosphorylation sites contained both serine and threonine target residues. To examine whether all of these were phosphorylated by PKA, we performed phosphoamino acid analyses of in vitro phosphorylated fusion proteins containing the Cx35 intracellular loop (IL) and carboxyl terminal (CT) domains. Figure 3 shows that the only radioactive spots detected were those that co-migrate with the phosphoserine standard. No phosphothreonine was detected in our analyses, indicating that the predicted threonine target sites were negligibly phosphorylated.
Figure 3. Phosphoamino acid analysis of in vitro phosphorylated perch Cx35 I-loop and C-terminal fusion proteins.
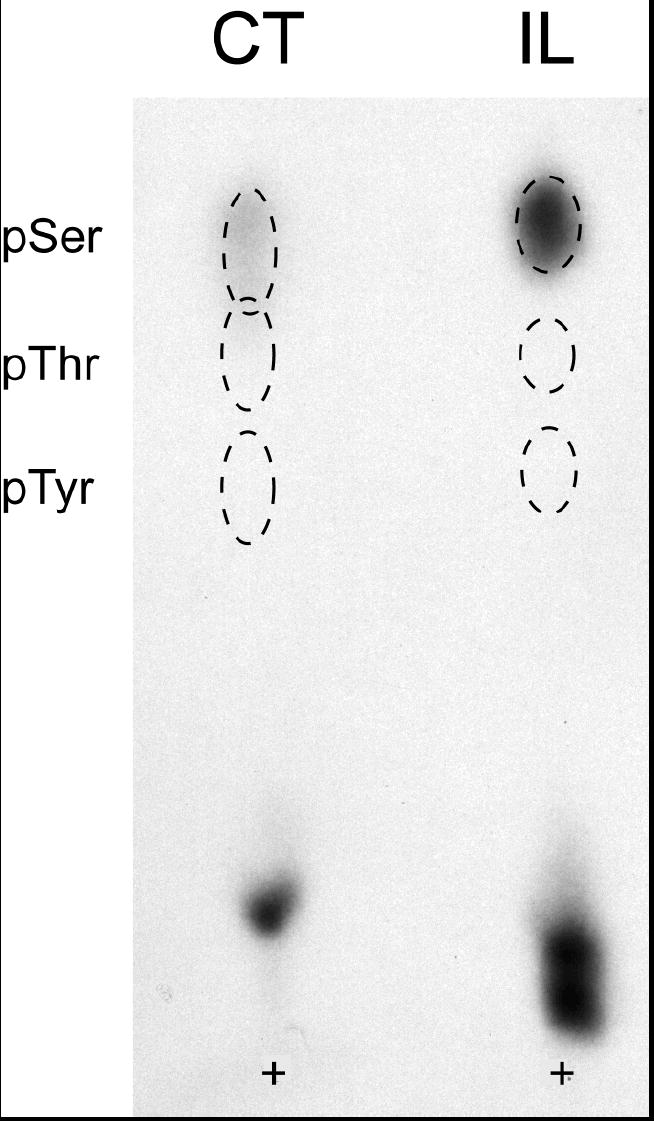
Cx35CT and Cx35IL fusion proteins phosphorylated in vitro with PKA were subjected to acid hydrolysis and one-dimensional thin layer electrophoresis with phospho-amino acid standards. Phospho-amino acid standards were detected with ninhydrin and phosphorylated residues by autoradiography. Positions of the phospho-amino acid standards are shown by dashed outlines, and the sample origin is labeled with a +. In both CT and IL fusion proteins, only phosphoserine was detected.
To determine which phosphorylation sites were used, we made mutants in the predicted phosphorylation sites that converted the acceptor serine or threonine to alanine. Mutant fusion proteins were expressed in bacteria and used for in vitro phosphorylation assays. Within the IL, two sites were predicted. Figure 4A shows that mutation of Ser110 (S110A) reduced phosphorylation by approximately 90%, while mutation of Ser128 (S128A) had little effect compared to the wild type control. The Ser110 mutant did not completely abolish phosphorylation, suggesting that a minor phosphorylation site exists in the IL. However, the conserved Ser110 site was the major site phosphorylated in this domain.
Figure 4. Mutational analysis of PKA phosphorylation in Cx35 IL and CT domains.
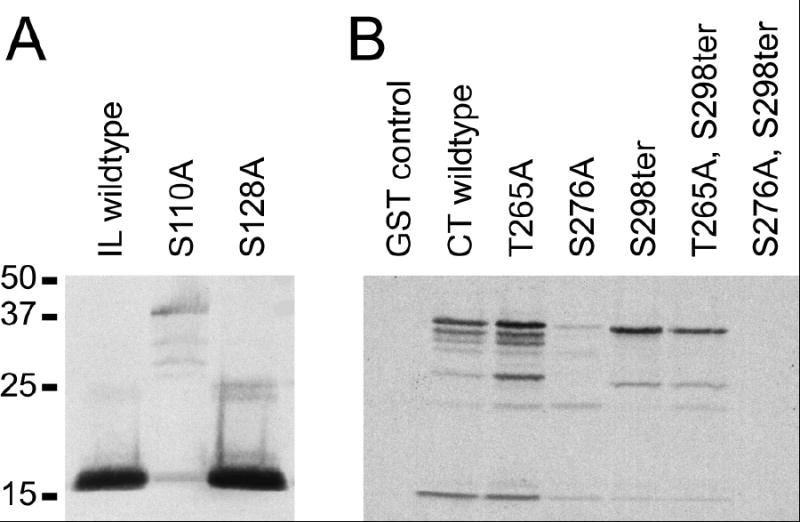
A. A 6x-His tagged construct of the Cx35 I-loop was expressed in bacteria and purified either in wild-type form, or with single mutations in each of the two predicted PKA recognition sites. Autoradiograph of the in vitro phosphorylation analysis shows that the mutation Ser110Ala eliminated most of the phosphorylation in this domain. Phosphorylation reactions contained 400 ng protein each.
B. Similar analysis of the Cx35 CT domain fused to GST. Note that the CT construct was prone to proteolytic degradation in bacteria. The topmost band represents non-degraded fusion protein, and protein content in the phosphorylations was normalized to this band (200 ng protein per reaction). The single mutation Ser276Ala had a much larger effect on phosphorylation than either Thr265Ala or Ser298ter, eliminating about 90% of phosphorylation. The double mutation, Ser276Ala, Ser298ter eliminated all phosphorylation, indicating that the minor phosphorylation site is in the final 7 amino acids of the C-terminus.
Within the CT, a serine and a threonine site were predicted to be phosphorylated, although no phosphothreonine was detected in the amino acid analysis. We nonetheless examined mutations in both Thr265 (T265A) and Ser276 (S276A). Figure 4B shows that mutation of Ser276 eliminated most of the phosphorylation, while mutation of Thr265 had little effect. Since the remaining phosphorylation was likely to be on serine, we made a mutation that truncated the CT at position 298 (labeled S298ter), eliminating the last 7 amino acids including 3 potentially phosphorylated serine residues. The S298ter mutation had little effect on total phosphorylation either alone or in combination with T265A. However, combination of S298ter with S276A completely eliminated phosphorylation (figure 4B). Thus PKA phosphorylated the CT at two sites, the major conserved consensus site (Ser276), and a minor site that does not strictly adhere to the consensus motif. The latter site, however, is completely conserved among the Cx35/36 homologues.
Mutation of phosphorylation sites reveals complex regulation by PKA
In order to study the contribution of each of the identified phosphorylation sites to regulation of Cx35 gap junctions, we made single mutants in the full-length connexin and generated stably-transfected HeLa cell lines expressing these mutants. The mutants examined were point mutations of the two major phosphorylation sites (S110A and S276A), and the truncation mutant in the minor C-terminal site (S298ter). We examined tracer coupling in these cell lines using the scrape-loading technique. Figure 5 shows the summary data for these experiments. These data have been normalized to the control condition for each cell line on each day of experimentation to account for differences in expression level of the connexins in the different cell lines. The Cx35 wild type data are the same as shown in figure 1H, being derived from 14 to 33 measurements under control conditions (cntrl) and after treatment with the PKA activator Sp-8-cpt-cAMPS (Sp) and PKA inhibitor Rp-8-cpt-cAMPS (Rp).
Figure 5. Effects of single mutations in PKA phosphorylation sites on regulation of coupling by PKA.
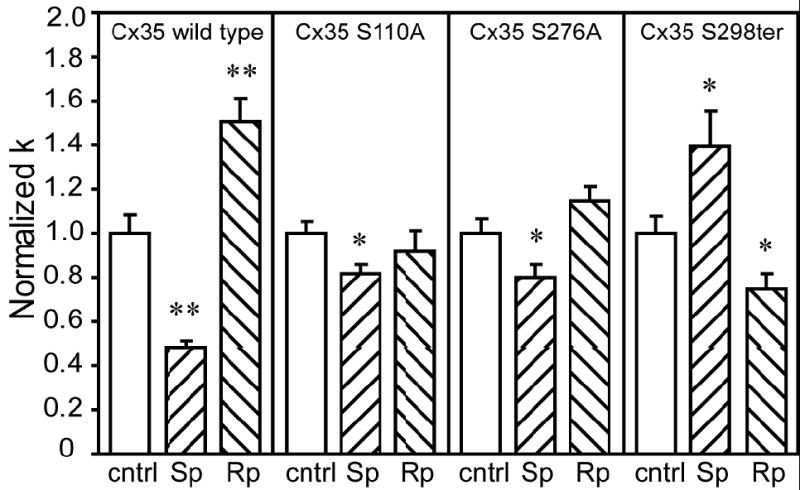
Neurobiotin tracer coupling was measured using the scrape-loading technique in HeLa cells stably transfected with Cx35 or phosphorylation-deficient Cx35 mutants. Treatments with the PKA activator Sp-8-cpt-cAMPS (designated Sp) and the PKA inhibitor Rp-8-cpt-cAMPS (designated Rp) were the same as in figure 1. Data shown are normalized to the no drug condition (cntrl) for each cell line examined, and data for Cx35 wild type are the same as in figure 1H. The single mutations Ser110Ala or Ser276Ala significantly reduced the effect of the PKA activator, and prevented the effect of the PKA inhibitor. In the truncation mutant, Ser298ter, the PKA activator increased coupling and the PKA inhibitor decreased coupling. Data shown are means of 14 to 39 independent measurements; error bars are +1 SEM. Significance at the p<0.05 level (using a t-test) is shown by *; significance at the p<0.01 level is shown by **.
Mutation of either of the two major PKA phosphorylation sites (S110A and S276A) reduced the effects of PKA activation or inhibition. Activation of PKA with 20 μM Sp-8-cpt-cAMPS caused significant reduction in coupling in both mutants (S110A: cntrl = 1.0 ± 0.06, n = 36, Sp = 0.82 ± 0.04, n = 34, t = -2.59, p < 0.05; S276A: cntrl = 1.0 ± 0.06, n = 39, Sp = 0.80 ± 0.06, n = 30, t = -2.38, p < 0.05), however, the reductions in coupling were significantly less than in wild type (S110A Sp vs. Cx35 Sp: t = 6.18, p < 0.01; S276A Sp vs Cx35 Sp: t = 4.61, p < 0.01). The changes in coupling caused by 20 μM Rp-8-cpt-cAMPS were not significant for either of these mutants. These data suggest that direct phosphorylation at both Ser110 and Ser276 is important for regulation; phosphorylation of either alone produces significantly reduced regulation.
Truncation of the last 7 amino acids of Cx35 resulted in anomalous reversal of the pattern of regulation by PKA (figure 5, right panel). Activation of PKA with 20 μM Sp-8-cpt-cAMPS resulted in significant increase in coupling (cntrl = 1.0 ± 0.07, n = 24, Sp = 1.40 ± 0.16, n = 23; t = 2.27, p < 0.05) while inhibition of PKA with 20 μM Rp-8-cpt-cAMPS resulted in significant decrease in coupling (Rp = 0.75 ± 0.07, n = 16; t = -2.49, p < 0.05). This effect was observed in two independent stable cell lines (data combined in figure 5), and so is not a result of unusual positional effects caused by insertion of the mutated clone into the genomic DNA. While the tip of the C-terminus was only a minor PKA phosphorylation site, these results suggest that the state of this region acts as an important “switch” in determining the regulation of coupling through Cx35 channels.
To examine further the dependence of Cx35 tracer coupling on the combined states of regulatory sites we prepared multiple mutants lacking both major phosphorylation sites, Ser110 and Ser276, and mutants lacking all three sites identified. We used transient transfection experiments to evaluate the effects of these mutations on tracer coupling. Figure 6, left panel, shows that Neurobiotin tracer coupling in HeLa cells transiently transfected with wild-type Cx35 was regulated by PKA activators and inhibitors in a manner qualitatively similar to that seen in the Cx35 stably-transfected cell line. 20 μM Sp-8-cpt-cAMPS significantly reduced coupling (cntrl = 1.0 ± 0.08, n = 24; Sp = 0.58 ± 0.06, n = 23, t = -4.45, p < 0.01), as was seen in the stable Cx35 cell line. 20 μM Rp-8-cpt-cAMPS did not produce a significant increase in coupling (Rp = 1.27 ± 0.12, n = 22; t = 1.88, p = 0.07), although the diffusion coefficient measured could not be distinguished from that of the Cx35 cell line under the same conditions (t = -1.43, p > 0.1). These results indicate that the transient transfection assay can be used to evaluate the effects of mutations on coupling through Cx35 gap junctions.
Figure 6. Effects of combined mutations on regulation of coupling by PKA.
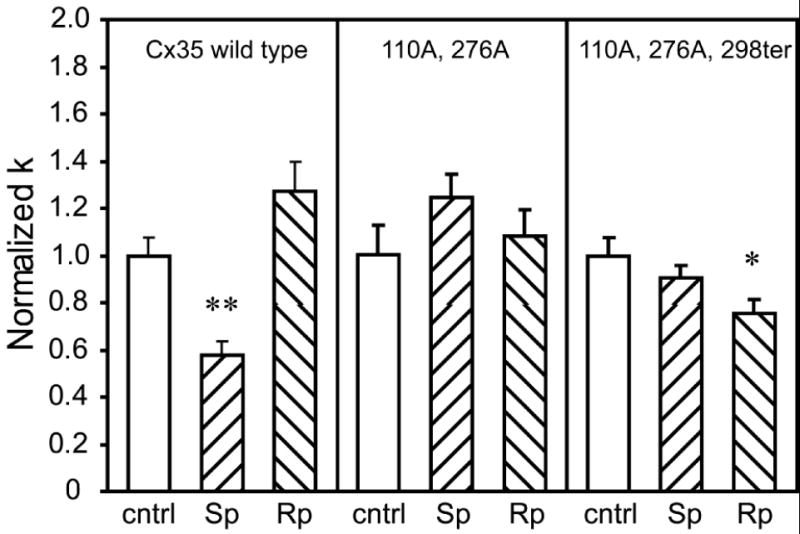
Neurobiotin tracer coupling was measured by scrape loading in HeLa cells transiently transfected with wild-type Cx35 or with the multiple mutants Cx35-S110A, S276A and Cx35- S110A, S276A, S298ter. Drug treatments were the same as shown in figure 5. Cells transfected with the Cx35 wild-type construct showed a similar pattern of regulation to that seen in the stably-transfected cell line (figure 5). The S110A, S276A double mutant did not show any statistically significant responses to the PKA activator or inhibitor, while the S110A, S276A, S298ter triple mutant showed no response to the PKA activator (Sp) and a small significant reduction in coupling with the PKA inhibitor (Rp). Data are means of 16-24 measurements; error bars are +1 SEM. Significance is displayed as in figure 5.
The double mutant, Cx35-S110A, S276A lacks both major PKA phosphorylation sites. Figure 6, center panel, shows that this mutant transiently transfected into HeLa cells showed no significant changes in coupling upon application of either Sp-8-cpt-cAMPS or Rp-8-cpt-cAMPS compared to control conditions (Sp vs. cntrl: t = 1.50, p > 0.05, n = 20 cntrl, n = 19 Sp; Rp vs. cntrl: t = 0.48, p > 0.05, n = 20 Rp). However, when compared to the stably-transfected S110A and S276A cell lines, the 20 μM Sp-8-cpt-cAMPS treatment resulted in a significantly elevated normalized diffusion coefficient in the double mutant (normalized k = 1.24 ± 0.10; double vs. S110A: t = 3.93, p < 0.01; double vs. S276A: t = 3.85, p < 0.01). No significant differences were found in treatments with 20 μM Rp-8-cpt-cAMPS (normalized k = 1.08 ± 0.11; double vs. S110A: t = 1.11, p > 0.1; double vs. S276A: t = -0.52, p > 0.1). These data confirm that the combined phosphorylation of both Ser110 and Ser276 is essential for the reduction of coupling observed when PKA is activated. Phosphorylation of either site alone results in moderate regulation of coupling that can be seen in the single-mutant cell lines.
To shed more light on the mechanism of the “switch” revealed by the S298ter truncation mutant, we examined the triple mutant, Cx35-S110A, S276A, S298ter (figure 6, right panel). Activation of PKA with 20 μM Sp-8-cpt-cAMPS did not change coupling significantly compared to control conditions (cntrl = 1.0 ± 0.08, n = 16, Sp = 0.90 ± 0.06, n = 18, t = -1.01, p > 0.1). This contrasts with the S298ter single mutant cell line, which displayed significantly elevated coupling when PKA was activated (figure 5, right panel). The difference in coupling between the triple mutant and the S298ter single mutant was statistically significant (t = -2.94, p < 0.01), suggesting that the elevated coupling when PKA is activated was due to phosphorylation at the Ser110 and/or Ser276 sites. Thus the S298ter mutation does switch the major phosphorylation sites from inhibitory to activating. Curiously, inhibition of PKA with Rp-8-cpt-cAMPS produced a small but significant decrease in coupling in the triple mutant (Rp = 0.75 ± 0.06, n = 18, t = -2.55, p < 0.05), which was indistinguishable from the response of the S298ter single mutant (t = 0.06, p > 0.9). Thus a small component of the regulatory response of Cx35 is not eliminated even in the triple mutant.
4. DISCUSSION
Gap junctional coupling is a highly dynamic property of cellular networks. Many processes influence the degree of coupling including gene transcription, plaque assembly, channel gating, and connexon degradation. Protein phosphorylation has an influence on many of these processes, and is a central element of gap junction regulation [22]. The neuronal gap junction protein connexin35/36 is regulated by the activity of protein kinase A in both cell culture systems [37] and intact neurons [27]. In the present study, we found that this regulation occurs via direct phosphorylation of the connexin, and not through indirect effects on synthetic or degradative pathways.
The pathway that regulates Cx35 is complex. We identified two major PKA phosphorylation sites, Ser110 and Ser276, by in vitro phosphorylation and mutagenesis. Both of these sites proved to be important for regulation of Cx35-mediated tracer coupling, since mutation of either site reduced the observed changes in tracer coupling. While either of these sites can confer some regulatory response to the connexin, our data suggest that both sites must be phosphorylated to achieve the degree of regulation observed in wild type channels.
Mitropoulou and Bruzzone [30] recently reported that perch Cx35 hemichannels were regulated by cAMP analogs in Xenopus oocytes. They found that conversion of the PKA recognition site associated with the Ser110 phosphorylation site to the sequence found in skate Cx35 eliminated regulation by cAMP analogs. They further found that conversion of skate Cx35 to the perch sequence in this region conferred cAMP regulation to the connexin, which was not otherwise responsive to cAMP analogs. Their findings are in agreement with the results of our study in that disruption of phosphorylation at Ser110 significantly reduced regulation by PKA. Thus regulation of hemichannel activity reflects the regulation in intact gap junction channels, as has been reported previously by DeVries and Schwartz [9] for fish horizontal cell gap junctions. Given this relationship between hemichannel and intercellular channel regulation, we would predict that similar results would also be obtained by mutation of the PKA recognition site associated with the Ser276 phosphorylation site.
Both Ser110 and Ser276 are conserved in all Cx35 homologues for which sequences have been deposited in GenBank, and only the skate Cx35 so far lacks the PKA consensus recognition sequence at one of these sites. This suggests that regulation by phosphorylation at these two sites is likely to be conserved in the Cx35 homologues. In the case of skate Cx35, it is unclear whether or not a skate isoform of PKA might recognize the modified recognition sequence and confer the expected regulation in skate neurons. It is also possible that other protein kinases recognize this sequence with higher efficiency and thus may accomplish regulation by phosphorylating Ser110 in conjunction with PKA (or other kinase) phosphorylation of Ser276.
The most unusual finding of this study is of the presence of an apparent “switch” located in the tip of the C-terminus. Truncation of the last 7 amino acids of Cx35 resulted in reversal of the effect of phosphorylating Ser110 and Ser276, and this unusual effect could be blocked by eliminating the latter two phosphorylation sites. These findings suggest that regulation of Cx35 channels by PKA depends on a set of interdependent sites.
The scheme of regulation we have described in this study is summarized in the model in figure 7. The effects of phosphorylation on Ser110 and Ser276 are well described. However, the nature of the “switch” mechanism is unclear and may depend either on direct effects of PKA on Cx35 or on indirect effects mediated by other proteins. The C-terminal site was phosphorylated very weakly by purified PKA catalytic subunit in our in vitro assays. For this reason, we consider it unlikely that direct phosphorylation by PKA accounts for the regulatory capacity of this site. However, it is possible that PKA alters the activity of a different protein kinase or phosphatase that influences the level of phosphorylation at this site. Such a scheme predicts that inhibition of the appropriate kinase or phosphatase or expression of Cx35 in a cell line lacking the protein would prevent reversal of regulation when the C-terminal truncation is present.
Figure 7. Regulation of Cx35 by protein kinase A.
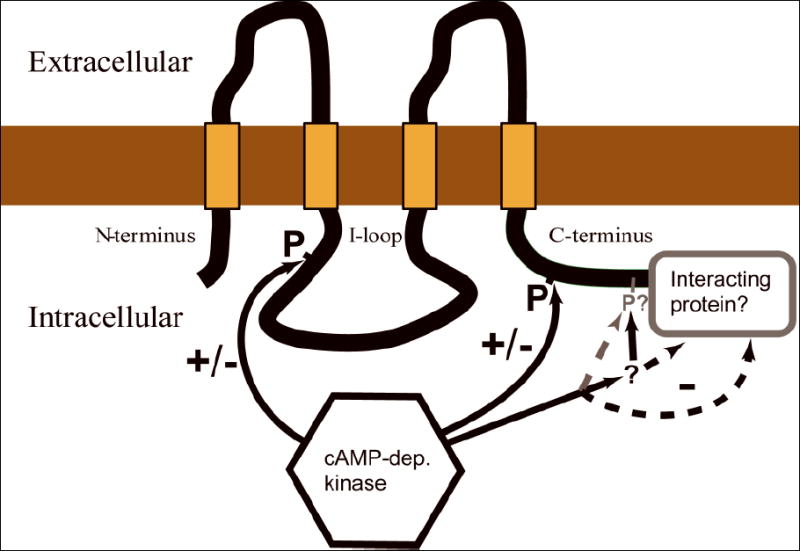
Schematic diagram of Cx35 showing the regulatory sites identified in this study. One major PKA phosphorylation site is present in the intracellular loop (Ser110) and one in the C-terminus (Ser276). Phosphorylation at both of these sites is required for the full inhibitory effect observed by activating PKA in HeLa cells. The domain containing the last 7 amino acids of the C-terminus has an important role in the regulatory response since truncation of this domain causes PKA phosphorylation at the other two sites to increase coupling. This constitutes a molecular “switch.” Candidates for the control of this site include: a) PKA phosphorylation at a minor site within the domain, b) phosphorylation/dephosphorylation by other protein kinases or phosphatases that act in this domain, c) direct physical interaction with a protein that binds this domain, or d) signaling via proteins assembled in a complex by a binding partner associated with the domain. The activity of PKA may change the properties of any of these signaling pathways.
A second possibility is that the regulatory capacity of the C-terminal site depends on a binding interaction with another protein. The mutation we used to identify this site removed the last 7 amino acids of the Cx35 C-terminal domain. Such a mutation may disrupt a binding interaction that involves the tip of the C-terminus. Several connexins have been shown to interact with proteins via the C-terminal domains [13,21,32,33,45], with the scaffolding protein ZO1 being the most prevalent partner. Indeed Cx36 has recently been found to interact with ZO1 at this site [24]. It is possible that such an association may have influence on dynamic channel regulation, either through physical effects on gating or through assembly of a signaling complex that acts upon the connexin.
Several other connexin types are regulated by cAMP and the cAMP-dependent protein kinases (PKA). For example, junctional coupling in cells containing Cx43 is generally increased within a few minutes by activation of PKA [2,14]. Increases in coupling have also been observed in T84 cells, which express Cx32 [3]. Conversely, Cx45 transfected into HeLa cells was weakly uncoupled by 1 mM 8-Br-cAMP [48].
The effects of cAMP on gap junction coupling may occur through several mechanisms. There are indirect effects of cAMP on connexin transcription and trafficking. For example, cAMP upregulates transcription of Cx43 15-40 fold in a rat hepatoma cell line [26], and Traub et al. [46] observed a 20% increase in amount of the 26 kD gap junction protein in mouse hepatocytes in 15 minutes in response to 1 mM dibutyryl-cAMP. However, there are also direct effects of phosphorylation on gap junction coupling. Moreno et al. [31] found that dephosphorylation of Cx43 with alkaline phosphatase led to a shift in the single channel conductance to larger values, but also reduced the number of active channels. On the other hand, 8-Br-cAMP caused a shift toward larger subconductance states in Cx40 channels expressed in SkHep1 cells [47].
In the mammalian retina, gap junctional coupling in AII amacrine cells, which express Cx36 [11,29], is closely regulated by cAMP levels. Treatment with dopamine D1 receptor agonists or forskolin plus IBMX, both of which elevate cAMP level, reduces coupling [16]. More specific PKA activators and inhibitors have the same effect [27], suggesting that this is due to PKA activity. Cx36 is highly homologous to Cx35 [36], and the PKA phosphorylation sites identified in this study are completely conserved (see figure 2). Thus it is likely that PKA regulation of coupling, as seen in Cx35, is a feature common to Cx36 gap junctions.
Many areas of the central nervous system express Cx35 or Cx36. The regulation of these channels by cAMP levels and PKA may have profound influence on the magnitude of electrical coupling. A few examples of this have been observed, such as in rat hippocampal neurons, where dopamine D1 receptor pathways reduce coupling and spikelet amplitude [49]. Interestingly, in the goldfish Mauthner cell mixed synapse, which has been shown recently to use Cx35 [39], electrical transmission is enhanced by dopamine, postsynaptic cAMP, and postsynaptic PKA activity [40,41,50]. Based on the results of our study, we would expect that the state of the “switch” at the C-terminus of Cx35 is different in this neuron upon D1 receptor activation than it is in other neurons that have been studied. Thus regulation of coupling through Cx35 by PKA activity in a neuronal system can be either positive of negative. The characteristics of this regulation will depend on the condition of all of the regulatory sites found in this study.
Acknowledgments
The authors wish to thank Dr. Alice Chuang for invaluable assistance in programming MatLab routines to calculate diffusion coefficients, and Dr. Stephen Mills for insightful discussions. This research was supported by grants from the National Eye Institute (EY12857 and EY10608) and Research to Prevent Blindness. VMW was supported in part by a student fellowship (SF01037) from the Fight for Sight research division of Prevent Blindness America.
References
- 1.Bloomfield SA, Xin D, Osborne T. Light-induced modulation of coupling between AII amacrine cells in the rabbit retina. Vis Neurosci. 1997;14:565–76. doi: 10.1017/s0952523800012220. [DOI] [PubMed] [Google Scholar]
- 2.Burghardt RC, Barhoumi R, Sewall TC, Bowen JA. Cyclic AMP induces rapid increases in gap junction permeability and changes in the cellular distribution of connexin43. J Membr Biol. 1995;148:243–53. doi: 10.1007/BF00235042. [DOI] [PubMed] [Google Scholar]
- 3.Chanson M, White MM, Garber SS. cAMP promotes gap junctional coupling in T84 cells. Am J Physiol. 1996;271:C533–9. doi: 10.1152/ajpcell.1996.271.2.C533. [DOI] [PubMed] [Google Scholar]
- 4.Cohen AI, Todd RD, Harmon S, O’Malley KL. Photoreceptors of mouse retinas possess D4 receptors coupled to adenylate cyclase. Proc Natl Acad Sci U S A. 1992;89:12093–7. doi: 10.1073/pnas.89.24.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Condorelli DF, Parenti R, Spinella F, Salinaro AT, Belluardo N, Cardile V, Cicirata F. Cloning of a new gap junction gene (Cx36) highly expressed in mammalian brain neurons. Eur J Neurosci. 1998;10:1202–8. doi: 10.1046/j.1460-9568.1998.00163.x. [DOI] [PubMed] [Google Scholar]
- 6.Copenhagen DR, Green DG. Spatial spread of adaptation within the cone network of turtle retina. J Physiol (Lond) 1987;393:763–76. doi: 10.1113/jphysiol.1987.sp016852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deans MR, Gibson JR, Sellitto C, Connors BW, Paul DL. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron. 2001;31:477–85. doi: 10.1016/s0896-6273(01)00373-7. [DOI] [PubMed] [Google Scholar]
- 8.Deans MR, Volgyi B, Goodenough DA, Bloomfield SA, Paul DL. Connexin36 is essential for transmission of rod-mediated visual signals in the mammalian retina. Neuron. 2002;36:703–12. doi: 10.1016/s0896-6273(02)01046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeVries SH, Schwartz EA. Hemi-gap-junction channels in solitary horizontal cells of the catfish retina. J Physiol (Lond) 1992;445:201–30. doi: 10.1113/jphysiol.1992.sp018920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feigenspan A, Janssen-Bienhold U, Hormuzdi S, Monyer H, Degen J, Sohl G, Willecke K, Ammermuller J, Weiler R. Expression of Connexin36 in Cone Pedicles and OFF-Cone Bipolar Cells of the Mouse Retina. J Neurosci. 2004;24:3325–3334. doi: 10.1523/JNEUROSCI.5598-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feigenspan A, Teubner B, Willecke K, Weiler R. Expression of Neuronal Connexin36 in AII Amacrine Cells of the Mammalian Retina. J Neurosci. 2001;21:230–239. doi: 10.1523/JNEUROSCI.21-01-00230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher CL, Pei GK. Modification of a PCR-based site-directed mutagenesis method. Biotechniques. 1997;23:570–1. 574. doi: 10.2144/97234bm01. [DOI] [PubMed] [Google Scholar]
- 13.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–4. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- 14.Godwin AJ, Green LM, Walsh MP, McDonald JR, Walsh DA, Fletcher WH. In situ regulation of cell-cell communication by the cAMP-dependent protein kinase and protein kinase C. Molecular & Cellular Biochemistry. 1993;127-128:293–307. doi: 10.1007/BF01076779. [DOI] [PubMed] [Google Scholar]
- 15.Guldenagel M, Ammermuller J, Feigenspan A, Teubner B, Degen J, Sohl G, Willecke K, Weiler R. Visual transmission deficits in mice with targeted disruption of the gap junction gene connexin36. J Neurosci. 2001;21:6036–44. doi: 10.1523/JNEUROSCI.21-16-06036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hampson EC, Vaney DI, Weiler R. Dopaminergic modulation of gap junction permeability between amacrine cells in mammalian retina. Journal of Neuroscience. 1992;12:4911–22. doi: 10.1523/JNEUROSCI.12-12-04911.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hormuzdi SG, Pais I, LeBeau FE, Towers SK, Rozov A, Buhl EH, Whittington MA, Monyer H. Impaired electrical signaling disrupts gamma frequency oscillations in connexin 36-deficient mice. Neuron. 2001;31:487–95. doi: 10.1016/s0896-6273(01)00387-7. [DOI] [PubMed] [Google Scholar]
- 18.Jelinek T, Weber MJ. Optimization of the resolution of phosphoamino acids by one-dimensional thin-layer electrophoresis. Biotechniques. 1993;15:628–630. [PubMed] [Google Scholar]
- 19.Kreegipuu A, Blom N, Brunak S, Brunak S. PhosphoBase a database of phosphorylation sites: release 2.0. Nucl Acids Res. 1999;27:237–239. doi: 10.1093/nar/27.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krizaj D, Gabriel R, Owen WG, Witkovsky P. Dopamine D2 receptor-mediated modulation of rod-cone coupling in the Xenopus retina. J Comp Neurol. 1998;398:529–38. [PMC free article] [PubMed] [Google Scholar]
- 21.Laing JG, Manley-Markowski RN, Koval M, Civitelli R, Steinberg TH. Connexin45 interacts with zonula occludens-1 and connexin43 in osteoblastic cells. J Biol Chem. 2001;276:23051–5. doi: 10.1074/jbc.M100303200. [DOI] [PubMed] [Google Scholar]
- 22.Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Arch Biochem Biophys. 2000;384:205–15. doi: 10.1006/abbi.2000.2131. [DOI] [PubMed] [Google Scholar]
- 23.Lee EJ, Han JW, Kim HJ, Kim IB, Lee MY, Oh SJ, Chung JW, Chun MH. The immunocytochemical localization of connexin 36 at rod and cone gap junctions in the guinea pig retina. Eur J Neurosci. 2003;18:2925–34. doi: 10.1046/j.1460-9568.2003.03049.x. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Olson C, Lu S, Kamasawa N, Yasumura T, Rash JE, Nagy JI. Neuronal connexin36 association with zonula occludens-1 protein (ZO-1) in mouse brain and interaction with the first PDZ domain of ZO-1. Eur J Neurosci. 2004;19:2132–46. doi: 10.1111/j.l460-9568.2004.03283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maier N, Guldenagel M, Sohl G, Siegmund H, Willecke K, Draguhn A. Reduction of high-frequency network oscillations (ripples) and pathological network discharges in hippocampal slices from connexin 36-deficient mice. J Physiol. 2002;541:521–8. doi: 10.1113/jphysiol.2002.017624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehta PP, Yamamoto M, Rose B. Transcription of the gene for the gap junctional protein connexin43 and expression of functional cell-to-cell channels are regulated by cAMP. Mol Biol Cell. 1992;3:839–50. doi: 10.1091/mbc.3.8.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills SL, Massey SC. Differential properties of two gap junctional pathways made by AII amacrine cells. Nature. 1995;377:734–7. doi: 10.1038/377734a0. [DOI] [PubMed] [Google Scholar]
- 28.Mills SL, Massey SC. The kinetics of tracer movement through homologous gap junctions in the rabbit retina. Vis Neurosci. 1998;15:765–77. doi: 10.1017/s0952523898154159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills SL, O’Brien JJ, Li W, O’Brien J, Massey SC. Rod pathways in the mammalian retina use connexin36. J Comp Neurol. 2001;436:336–350. [PMC free article] [PubMed] [Google Scholar]
- 30.Mitropoulou G, Bruzzone R. Modulation of perch connexin35 hemi-channels by cyclic AMP requires a protein kinase A phosphorylation site. J Neurosci Res. 2003;72:147–57. doi: 10.1002/jnr.10572. [DOI] [PubMed] [Google Scholar]
- 31.Moreno AP, Saez JC, Fishman GI, Spray DC. Human connexin43 gap junction channels. Regulation of unitary conductances by phosphorylation. Circ Res. 1994;74:1050–7. doi: 10.1161/01.res.74.6.1050. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen PA, Baruch A, Shestopalov VI, Giepmans BN, Dunia I, Benedetti EL, Kumar NM. Lens Connexins {alpha}3Cx46 and {alpha}8Cx50 Interact with Zonula Occludens Protein-1 (ZO-1) Mol Biol Cell. 2003;14:2470–2481. doi: 10.1091/mbc.E02-10-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nielsen PA, Beahm DL, Giepmans BN, Baruch A, Hall JE, Kumar NM. Molecular cloning, functional expression, and tissue distribution of a novel human gap junction-forming protein, connexin-31.9. Interaction with zona occludens protein-1. J Biol Chem. 2002;277:38272–83. doi: 10.1074/jbc.M205348200. [DOI] [PubMed] [Google Scholar]
- 34.Nir I, Harrison JM, Haque R, Low MJ, Grandy DK, Rubinstein M, Iuvone PM. Dysfunctional light-evoked regulation of cAMP in photoreceptors and abnormal retinal adaptation in mice lacking dopamine D4 receptors. J Neurosci. 2002;22:2063–73. doi: 10.1523/JNEUROSCI.22-06-02063.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Brien J, al-Ubaidi MR, Ripps H. Connexin 35: a gap-junctional protein expressed preferentially in the skate retina. Molecular Biology of the Cell. 1996;7:233–43. doi: 10.1091/mbc.7.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Brien J, Bruzzone R, White TW, Al-Ubaidi MR, Ripps H. Cloning and expression of two related connexins from the perch retina define a distinct subgroup of the connexin family. J Neurosci. 1998;18:7625–37. doi: 10.1523/JNEUROSCI.18-19-07625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Brien J, Nguyen HB, Mills SL. Cone photoreceptors in bass retina use two connexins to mediate electrical coupling. J Neurosci. 2004;24:5632–5642. doi: 10.1523/JNEUROSCI.1248-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pais I, Hormuzdi SG, Monyer H, Traub RD, Wood IC, Buhl EH, Whittington MA, LeBeau FE. Sharp wave-like activity in the hippocampus in vitro in mice lacking the gap junction protein connexin 36. J Neurophysiol. 2002:00549.2002. doi: 10.1152/jn.00549.2002. [DOI] [PubMed] [Google Scholar]
- 39.Pereda A, O’Brien J, Nagy JI, Bukauskas F, Davidson KG, Kamasawa N, Yasumura T, Rash JE. Connexin35 mediates electrical transmission at mixed synapses on Mauthner cells. J Neurosci. 2003;23:7489–503. doi: 10.1523/JNEUROSCI.23-20-07489.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pereda A, Triller A, Korn H, Faber DS. Dopamine enhances both electrotonic coupling and chemical excitatory postsynaptic potentials at mixed synapses. Proc Natl Acad Sci U S A. 1992;89:12088–92. doi: 10.1073/pnas.89.24.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pereda AE, Nairn AC, Wolszon LR, Faber DS. Postsynaptic modulation of synaptic efficacy at mixed synapses on the Mauthner cell. J Neurosci. 1994;14:3704–12. doi: 10.1523/JNEUROSCI.14-06-03704.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rash JE, Staines WA, Yasumura T, Patel D, Furman CS, Stelmack GL, Nagy JI. Immunogold evidence that neuronal gap junctions in adult rat brain and spinal cord contain connexin-36 but not connexin-32 or connexin-43. Proc Natl Acad Sci U S A. 2000;97:7573–8. doi: 10.1073/pnas.97.13.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rash JE, Yasumura T, Davidson KG, Furman CS, Dudek FE, Nagy JI. Identification of cells expressing Cx43, Cx30, Cx26, Cx32 and Cx36 in gap junctions of rat brain and spinal cord. Cell Adhes Commun. 2001;8:315–20. doi: 10.3109/15419060109080745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sohl G, Degen J, Teubner B, Willecke K. The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Lett. 1998;428:27–31. doi: 10.1016/s0014-5793(98)00479-7. [DOI] [PubMed] [Google Scholar]
- 45.Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem. 1998;273:12725–31. doi: 10.1074/jbc.273.21.12725. [DOI] [PubMed] [Google Scholar]
- 46.Traub O, Look J, Paul D, Willecke K. Cyclic adenosine monophosphate stimulates biosynthesis and phosphorylation of the 26 kDa gap junction protein in cultured mouse hepatocytes. Eur J Cell Biol. 1987;43:48–54. [PubMed] [Google Scholar]
- 47.van Rijen HV, van Veen TA, Hermans MM, Jongsma HJ. Human connexin40 gap junction channels are modulated by cAMP. Cardiovasc Res. 2000;45:941–51. doi: 10.1016/s0008-6363(99)00373-9. [DOI] [PubMed] [Google Scholar]
- 48.van Veen TA, van Rijen HV, Jongsma HJ. Electrical conductance of mouse connexin45 gap junction channels is modulated by phosphorylation. Cardiovasc Res. 2000;46:496–510. doi: 10.1016/s0008-6363(00)00047-x. [DOI] [PubMed] [Google Scholar]
- 49.Velazquez JL, Han D, Carlen PL. Neurotransmitter modulation of gap junctional communication in the rat hippocampus. Eur J Neurosci. 1997;9:2522–31. doi: 10.1111/j.1460-9568.1997.tb01681.x. [DOI] [PubMed] [Google Scholar]
- 50.Wolszon LR, Faber DS. The effects of postsynaptic levels of cyclic AMP on excitatory and inhibitory responses of an identified central neuron. J Neurosci. 1989;9:784–97. doi: 10.1523/JNEUROSCI.09-03-00784.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang XL, Wu SM. Modulation of rod-cone coupling by light. Science. 1989;244:352–4. doi: 10.1126/science.2711185. [DOI] [PubMed] [Google Scholar]
- 52.Zimmerman AL, Rose B. Permeability properties of cell-to-cell channels: kinetics of fluorescent tracer diffusion through a cell junction. J Membr Biol. 1985;84:269–83. doi: 10.1007/BF01871390. [DOI] [PubMed] [Google Scholar]