

Abstract
Stress of the endoplasmic reticulum (ER stress) is caused by the accumulation of misfolded proteins, which occurs in many neurodegenerative diseases. ER stress can lead to adaptive responses or apoptosis, both of which follow activation of the unfolded protein response (UPR). Heat shock proteins (HSP) support the folding and function of many proteins, and are important components of the ER stress response, but little is known about the role of one of the major large HSPs, HSP105. We identified several new partners of HSP105, including glycogen synthase-3 (GSK3), a promoter of ER stress-induced apoptosis, and GRP78, a key component of the UPR. Knockdown of HSP105 did not alter UPR signaling after ER stress, but blocked caspase-3 activation after ER stress. In contrast, caspase-3 activation by genotoxic stress was unaffected by knockdown of HSP105, suggesting ER stress-specificity in the apoptotic action of HSP105. However, knockdown of HSP105 did not alter cell survival after ER stress, but instead diverted signaling to a caspase-3-independent cell death pathway, indicating that HSP105 is necessary for apoptotic signaling after UPR activation by ER stress. Thus, HSP105 appears to chaperone the responses to ER stress through its interactions with GRP78 and GSK3, and without HSP105 cell death following ER stress proceeds by a non-caspase-3-dependent process.
Keywords: HSP105, GRP78, UPR, apoptosis, GSK3, ER stress
1. Introduction
The endoplasmic reticulum (ER) is a membranous organelle that provides a unique environment for the proper folding and maturation of secreted and membrane-bound proteins. Diseases, environmental factors, and pharmacological agents can perturb ER function and lead to the accumulation of misfolded proteins and ER stress [1,2]. When the ER stress is severe enough to preclude recovery from excessive accumulation of misfolded proteins, cells undergo controlled death by apoptosis [3,4]. This apoptotic method of cell death is important because it evolves internally, enabling the intracellular inactivation of potentially toxic molecules that could be released in necrotic cell death.
Eukaryotic cells have evolved an elegant but complex system to detect and react to misfolded proteins within the ER, encompassing both adaptive and apoptotic signals [1,2,5]. Thus, cells attempt to rectify the misfolded proteins while at the same time preparing for apoptosis should the ER stress be overwhelming [3,4]. A major response to ER stress involves activation of the highly conserved unfolded protein response (UPR). The UPR includes activation of the trans-ER membrane molecules PKR-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). Each of these proteins is held inactive by GRP78 in the ER under basal conditions, and is activated following release when GRP78 is recruited away to misfolded proteins [6,7]. Activation of these UPR mediators leads to attenuation of translation and increased expression of a subset of stress-responsive genes [4,8].
The survival of cells following activation of the UPR depends on the severity and duration of the stress and is largely determined by the proapoptotic molecule C/EBP homologous transcription factor protein (CHOP, also called GADD153) [9,10]. The expression of CHOP, a member of the C/EBP family of transcription factors, is upregulated following ER stress by the ATF4 and ATF6 transcription factors [11]. With mild ER stress, CHOP is rapidly degraded and its expression is reduced, allowing cells to survive. However, prolonged or severe ER stress maintains high CHOP levels to promote apoptotic signaling that induces activation of the executioner caspase, caspase-3, and cell death by apoptosis [10].
In addition to CHOP, many other proteins have been identified that regulate the ultimate decision between adaptation and death following ER stress [12]. One of these is glycogen synthase-3 (GSK3), a constitutively active Ser/Thr kinase composed of two similar, but not identical, isoforms, GSK3α and GSK3β [13]. We previously found [14] that GSK3 inhibitors reduced apoptosis induced by several agents that cause ER stress, including thapsigargin (an ER Ca++-ATPase inhibitor) and tunicamycin (an N-linked glycosylation inhibitor), which has been confirmed in numerous studies [15-20]. Despite this considerable evidence that GSK3 promotes intrinsic apoptotic signaling following ER stress, the underlying mechanisms remain uncertain [21].
Heat shock proteins (HSPs) also have been identified as regulators of the ER stress response [22,23]. HSPs are molecular chaperones that escort and regulate the actions of many intracellular proteins. HSPs bind newly synthesized or misfolded proteins to optimize folding and function, whereas irreparably damaged proteins are directed by HSPs to sites of degradation [24]. Additionally, HSPs regulate multiple steps in the apoptotic signaling cascade [25]. Together these actions likely underlie the widely reported antiapoptotic, or survival-promoting, capabilities of HSPs. Additionally, HSPs play an important role in cellular signaling by ensuring that many key signaling proteins maintain optimally active conformations [26]. Thus, HSPs generally are considered as immensely helpful proteins that ensure proteins fold and function properly and damage is controlled.
Although HSP105 is one of the major mammalian heat shock proteins, little is known about its actions and client proteins compared with the well-known HSP70 and HSP90. HSP105 is a nucleotide exchange factor for HSP70 [27,28] and prevents aggregation of mutant androgen receptors [29] and Zn/Cu superoxide dismutase [30]. HSP105 attenuates staurosporine-induced apoptosis [31], but overexpressed HSP105α in mouse embryonal F9 cells enhanced apoptosis in response to oxidative stress [32]. However, the role of HSP105 in response to ER stress has not previously been examined.
If a cell is lethally damaged, especially during ER stress when many proteins are misfolded, we hypothesized that there must be a mechanism to ensure that mediators of the apoptotic cascade remain functional in a destructive environment. A logical candidate for this role is a molecular chaperone, a protein that serves to optimize protein and cellular functions even though in this case the outcome is cell death. In this report we present evidence that one function of HSP105 is to chaperone the process of apoptosis following ER stress. Therefore, the present study tested the effects of HSP105 on ER stress-induced apoptosis and demonstrates that HSP105 directs signaling to caspase-3 mediated apoptosis.
2. Material and methods
2.1. Cell Culture
SH-SY5Y human neuroblastoma cells where grown in RPMI 1640 medium supplemented with 10% horse serum and 5% fetal clone II (HyClone, Logan, UT). Cells were placed in serum-free media approximately 16 h prior to experimental treatments. HEK293 cells were grown in Dulbecco’s modified eagle medium supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and 15 mM HEPES (Cellgro, Herndon, VA). All media were supplemented with 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (Cellgro), in humidified, 37°C chambers with 5% CO2. Cells were treated for the indicated times with thapsigargin, tunicamycin (Alexis, San Diego, CA), or camptothecin (Sigma Chemical Co., St. Louis, MO).
2.2. Tissue preparation
Adult, male C57BL/6 mice (Frederick Cancer Research, Frederick, MD), were decapitated, and brains were rapidly dissected in ice-cold saline. The cerebral cortex was homogenized in ice-cold lysis buffer containing 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.5% NP-40, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin, 1 mM phenylmethanesulfonyl fluoride, 1 mM sodium vanadate, 50 mM sodium fluoride, and 100 nM okadaic acid. The homogenates were centrifuged at 20,800×g for 10 min to remove insoluble debris.
2.3. Immunoblotting
Cells were washed twice with phosphate-buffered saline (PBS) and were lysed with IP lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.5% NP-40, 1 mM sodium orthovanadate, 100 μM phenylmethanesulfonyl fluoride, 0.1 μM okadaic acid, 50 mM sodium fluoride, and 10 μg/ml each of leupeptin, aprotinin, and pepstatin). The lysates were sonicated and centrifuged at 20,800×g for 15 min. Protein concentrations were determined by the bicinchoninic acid method (Pierce, Rockford, IL). Samples were mixed with Laemmli sample buffer (2% SDS) and placed in a boiling water bath for 5 min. Proteins were resolved in SDS-polyacrylamide gels, transferred to nitrocellulose, and the membranes were incubated with antibodies to phospho-Ser9-GSK3β, phospho-Ser473-Akt, phospho-Thr308-Akt, phospho-Ser51-eIF2α, total eIF2α, cleaved caspase-3, cleaved poly-(ADP-ribose) polymerase (PARP), Bad (Cell Signaling, Danvers, MA), total GSK3β, active Bax (6A7), caspase-9 (BD-PharMingen/Transduction Laboratories, San Diego, CA), HSP90, HSP70, (Stressgen, Victoria, BC, Canada), CHOP/GADD153, GRP78 (Santa Cruz Biotechnology, Santa Cruz, CA), total GSK3 (Upstate Biotechnology Inc., Lake Placid, NY), HSP105 (Novocastra Laboratories, Newcastle upon Tyne, UK), MCL1 (Biovision, Mountain View, CA), GSK3α (Southern Biotech, Birmingham, AL), total Bax, caspase-3, Bcl-XL, IAPs (kind gifts from Dr. Tong Zhou, University of Alabama at Birmingham), β-actin, or V5-tag (Sigma). Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse, goat anti-rabbit, or donkey anti-goat IgG, followed by detection with enhanced chemiluminescence.
2.4. Subcellular Fractionation
Nuclear and cytosolic fractions were prepared as previously described [33] with minor modifications. Cells were washed twice with PBS and then harvested in 200 μl lysis buffer (10 mM Tris, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.05% NP-40, 1 mM EGTA, 1 mM sodium orthovanadate, 100 μM phenylmethanesulfonyl fluoride, 0.1 μM okadaic acid, 50 mM sodium fluoride, and 10 μg/ml each of leupeptin, aprotinin, and pepstatin). Cells were centrifuged at 2700×g for 10 min at 4°C. The supernatant was centrifuged at 20,800×g for 15 min at 4°C to obtain the cytosolic fraction. The pellet, containing nuclei, was washed twice in 200 μl wash buffer (5 mM HEPES, pH 7.4, 3 mM MgCl2, 1 mM EGTA, 250 mM sucrose, 0.1% BSA, with protease and phosphatase inhibitors). The pellet was then resuspended in wash buffer and layered on top of 1 ml 1 M sucrose (with protease and phosphatase inhibitors), and centrifuged at 2700×g for 10 min at 4°C. The nuclear pellet was washed in lysis buffer containing 0.05% NP-40. The nuclear proteins were extracted by resuspending the pellet in nuclear extraction buffer (20 mM HEPES, pH 7.9, 300 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1 mM β-glycerophosphate, 1mM sodium orthovanadate, 100 μM phenylmethanesulfonyl fluoride, 0.1 μM okadaic acid, 50 mM sodium fluoride, and 10 μg/ml each of leupeptin, aprotinin, and pepstatin) and incubating on ice for 30 min. After extraction, the nuclear samples were centrifuged at 20,800×g for 15 min at 4°C, and the supernatant was retained as the nuclear extract.
2.5. Immunoprecipitation
Immunoprecipitations were performed using sheep anti-mouse IgG Dynabeads (Invitrogen, Carlsbad, CA). The Dynabeads, 10 μl of slurry per sample, were washed twice in PBS/0.1% BSA, and all washes were performed using a magnetic particle separator. The beads were incubated with 1 μg anti-GSK3β antibody, 1 μg anti-KDEL (Stressgen, Victoria, BC, Canada), or 1 μl anti-V5 antibody in PBS/BSA at 4°C with end-over-end mixing overnight. The beads were then washed twice in PBS/BSA, followed by the addition of cell lysate (200 μg protein) or cerebral cortex lysate (1 mg protein). The beads plus lysate were incubated at 4°C for 2 h with end-over-end mixing. The beads were then washed three times with PBS/BSA. Alternatively, some immunoprecipitations of GSK3α or GSK3β were performed using anti-GSK3α- or anti-GSK3β-conjugated sepharose beads (Southern Biotech), by incubating 10-20 μl of beads with cell lysate for 2 h at 4°C with end-over-end mixing. The beads were then washed 3-4 times in lysis buffer. Active Bax was examined by immunoprecipitation using the active Bax specific antibody 6A7 and CHAPS lysis buffer (1% CHAPS, 20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, plus inhibitors) was used in place of IP lysis buffer.
2.6. Lentivirus and shRNAi
Lentiviral mediated RNAi was performed using the Lentilox3.7 vector, which co-expresses eGFP, generously provided by Dr. Luk Van Parijs (Massachusetts Institute of Technology). The oligonucleotides containing the HSP105 target sequence (underlined) that were used are: sequence #2, 5’TGAAGGAGAGGACCAAGCTATTCAAGAGATAGCTTGGTCCTCTCCTTCTTTTTTC and sequence #4, 5’TGCAGTAGCCAGAGGATGTGTTCAAGAGACACATCCTCTGGCTACTGCTTTTTTC, and the reverse compliment of each oligonucleotide with an additional AGCT at the 5’ end. Sense and anti-sense oligonucleotides were annealed and ligated into the XhoI, HPA1 cut Lentilox3.7 vector. Lentiviruses were produced using protocols generously provided by Dr. James Lah (Emory University). Protocols were adapted for a four vector system. Twelve 150 mm dishes of 293FT cells (Invitrogen) were co-transfected with 300 μg of the Lentilox3.7 vector, with or without the RNAi hairpin insert, plus 150 μg each of the packaging vectors pLP1, pLP2, and pLP/VSVG (Invitrogen) by the calcium phosphate method. The media was changed approximately 16 h after transfection, and the cells were cultured an additional 48-72 h. The media was then collected, centrifuged at 500×g for 5 min, and filtered through a 0.45 μm filter. The virus-containing media was then centrifuged at 50,000×g for 2 h at 16°C, the supernatant was discarded, and the pellet was resuspended in 500 μl PBS/tube and incubated on ice for 1 h. The viral suspension was pooled and centrifuged again at 50000×g for 2 h at 16°C. The supernatant was discarded and the pellet was resuspended in 150 μl of PBS, aliquoted, and stored at -80°C. The virus titers were determined by transducing HEK293 cells with 10-fold serial dilutions. 72 h post transduction the percentage of GFP positive cells was determined by flow cytometry and the titer was calculated based on the original number of cells. Titers were approximately 1×108 – 1×109 transducing units/ml. For HSP105 overexpression, the same procedure was used for virus production using the DTOPO-V5 vector carrying the HSP105α coding region, and titered using blasticidine selection. The HSP105α was cloned by RT-PCR from the mRNA of HEK293 cells.
2.7. Immunocytochemistry
For cleaved caspase-3 staining, cells were plated in 24 or 48 well plates and treated as indicated. The cells were fixed in 4% paraformaldehyde for 30 min at 4°C, then washed with PBS. The cells were incubated in blocking/permeabilization buffer (PBS, pH 7.2, with 1% BSA, 0.2% non-fat milk, and 0.3% Triton-X 100) for 30 min at room temperature. The cells were then incubated overnight with the cleaved caspase-3 antibody diluted 1:50 in blocking buffer. The cells were washed several times with PBS, then incubated with Texas red-conjugated secondary and 1 μg/ml DAPI for 1 h at room temperature, following by thorough washing with PBS. Three random fields in each well from at least three independent experiments were imaged using a Nikon fluorescence microscope and quantified using Image ProPlus software. To examine cell morphology, cells were plated on poly-D-lysine-coated coverslips and treated as indicted followed by fixation. The coverslips were washed with PBS and then incubated with 100 ng/ml Hoechst 33342 for 15 min at room temperature.
2.8. XBP-1 splicing
The method to examine XBP-1 splicing was adapted from Calfon et al [34]. Cellular mRNA was isolated using an RNeasy mini kit (Qiagen, Germantown, MD) following the manufacture’s protocol. A fragment of XBP-1 was amplified by RT-PCR using the Superscript III one-step RT-PCR system (Invitrogen) with primers flanking the IRE1 cleavage site. The primers used were 5’AGAAGGCGCTGAGGAGGA and 5’CTGAGAGGTGCTTCCTCG. Following 40 cycles of PCR, the product was digested by PstI to remove the unspliced XBP-1 products; IRE1-mediated splicing removes the PstI site. The DNA was then resolved on a 2% agarose gel containing ethidium bromide and imaged using a molecular imager system and Quantity One software (Bio-Rad, Hercules, CA).
2.9. Cell viability
Cells were grown in 24 well plates and treated as indicated. Following treatments, the media was collected and centrifuged at 1000×g for 5 min. An equal volume of media containing 1% triton X-100 was added to the wells and incubated at 37°C for 30 min to lyse the remaining cells, followed by centrifugation at 1000×g for 5 min. The lactate dehydrogenase (LDH) assay (Roche) was then performed in duplicate on both samples from all wells. The two values obtained for each well were added together and used as the total available LDH and the percent release was calculated based on the amount of LDH in the non-detergent media divided by the total LDH obtained by lysing the cells with Triton X-100.
3. Results
3.1. HSP105 client proteins
HSPs are important modulators of cellular responses to misfolded protein accumulation, as occurs with ER stress, but few client proteins specific to HSP105 are known. Two approaches were used to identify HSP105-associated proteins, co-immunoprecipitation methods and the use of mass spectrometry.
We tested if HSP105 associates with GSK3 because much recent evidence demonstrated that GSK3β activation contributes to ER stress-induced apoptotic signaling [21]. To test if these proteins interact in vivo, GSK3β was immunoprecipitated from mouse cerebral cortex homogenates and immunoblotted for HSP105, which demonstrated that HSP105 co-precipitated with GSK3β (Fig. 1A). To verify this interaction, since the commercial antibodies tested could not immunoprecipitate HSP105 we used SH-SY5Y human neuroblastoma cells stably expressing a low level (approximately 1.7-fold that of wild-type) of HSP105 with a V5 tag (HSP105-V5) to allow immunoprecipitation of HSP105 with an antibody to V5. Consistent with the in vivo data, GSK3β co-precipitated with HSP105-V5 from SH-SY5Y cells (Fig. 1B).
Figure 1. HSP105 interacting proteins.
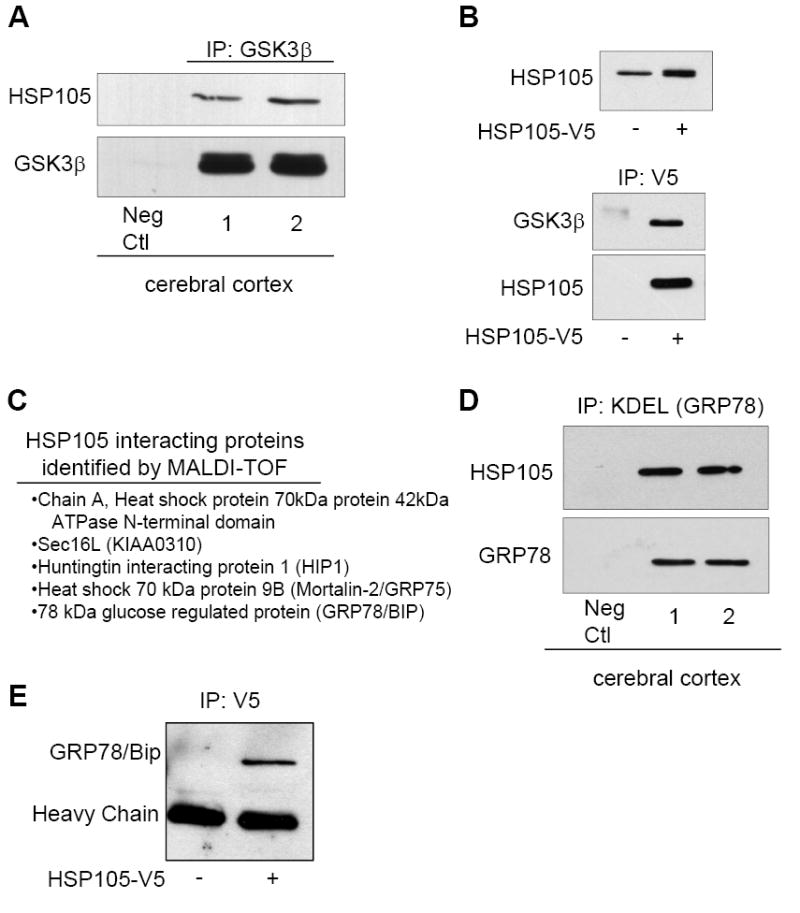
A. GSK3β was immunoprecipitated (IP) from cerebral cortical homogenates of two C57BL/6 mice followed by immunoblotting for HSP105 and GSK3β. B. SH-SY5Y cells stably expressing HSP105-V5 were generated by lentiviral transduction, and the modestly increased expression level of HSP105 was demonstrated by immunoblotting for HSP105. HSP105-V5 was immunoprecipitated using an anti-V5 antibody from non-transduced (-) or HSP105-V5 transduced (+) cells, followed by immunoblotting for GSK3β and HSP105. C. HEK293 cells were transiently transfected with HSP105-V5. HSP105-V5 was then immunoprecipitated from 3 mg of cell lysate, followed by SDS-PAGE and coomassie staining. Several bands were extracted from the gel and the proteins listed were identified by MALDI-TOF. D. GRP78 was immunoprecipitated (IP) from cerebral cortical homogenates of two C57BL/6 mice using an anti-KDEL antibody followed by immunoblotting for HSP105 and GRP78. E. HSP105-V5 was immunoprecipitated with a V5 antibody from HEK293 cells transiently transfected with HSP105-V5 (+) or nontransfected cells (-), followed by immunoblotting for GRP78.
To identify additional HSP105-interacting proteins, HEK293 cells were transiently transfected with HSP105-V5 followed by immunoprecipitation with an anti-V5 antibody, resolution of proteins by electrophoresis, and identification of co-immunoprecipitated proteins by mass spectrometry. The proteins identified by MALDI-TOF included HSP70, which was previously reported to bind HSP105 [35], and four novel HSP105-interacting proteins (Fig. 1C). Of the new HSP105-interacting proteins that were identified, two, Sec16L and GRP78, are associated with the ER. Sec16L (KIAA0310) functions in the export of proteins from the ER [36]. GRP78 is particularly interesting because it is a key regulator of the cellular response to ER stress, as discussed in the introduction. To verify this interaction in vivo, GRP78 was immunoprecipitated, using an anti-KDEL antibody, from mouse cerebral cortex homogenates and immunoblotted for HSP105 and GRP78 (Fig, 1D). To further confirm this interaction, HSP105-V5 was immunoprecipitated from HEK293 cells and immunoblotted for GRP78. Together, this data demonstrates that the two proteins are associated (Fig. 1D). Thus, several new partners of HSP105 were identified, and three of them, GSK3, Sec16L, and GRP78, have important roles in ER function and cellular signaling in response to ER stress.
To assess if stress influences the association of HSP105 with GSK3β, SH-SY5Y cells were subjected to heat shock by incubation at 45°C for 0 to 120 min, followed by immunoprecipitation of GSK3β and immunoblotting HSP105. The amount of HSP105 that co-immunoprecipitated with GSK3β was increased within 7 min of heat shock and the increased association remained evident for approximately 60 min of heat shock (Fig. 2A), suggesting that HSP105 might protect GSK3β from heat-induced damage. During apoptosis induced by heat shock or other stimuli, GSK3β can rapidly translocate to the nucleus [33]. Therefore, we tested if cytosolic or nuclear GSK3 associates with HSP105 and if association in each compartment was altered following heat shock. SH-SY5Y cells were subjected to heat shock for 15 or 30 min, followed by fractionation to isolate nuclei and cytoplasm. HSP105 was detected in both the cytosol and nucleus, and its subcellular distribution was not affected by heat shock (Fig. 2B), whereas heat shock increased nuclear levels of GSK3β (Fig. 2C) and GSK3α (Fig. 2D). Cytosolic GSK3β and GSK3α co-immunoprecipitated HSP105, and the associations were not significantly increased by heat shock. Additionally, significantly increased amounts of HSP105 co-immunoprecipitated with nuclear GSK3α/β in proportion to the increased levels of GSK3α/β in the nucleus. Thus, HSP105 associates with both isoforms of GSK3 in both the cytosol and the nucleus, and heat shock-induced cell stress increases their association in the nucleus. To test if changes in GSK3β phosphorylation or activity influenced its interaction with HSP105, cells were treated with the PI3-kinase inhibitor wortmannin, which induced dephosphorylation of GSK3β at serine-9, or treated with the GSK3 inhibitor lithium (Fig. 2E). Neither wortmannin nor lithium had a significant impact on the association of GSK3β with HSP105 (Fig. 2E), indicating that the interaction is phosphorylation and activation independent.
Figure 2. Characterization of the interaction of HSP105 with GSK3 and shRNAi knockdown of HSP105.
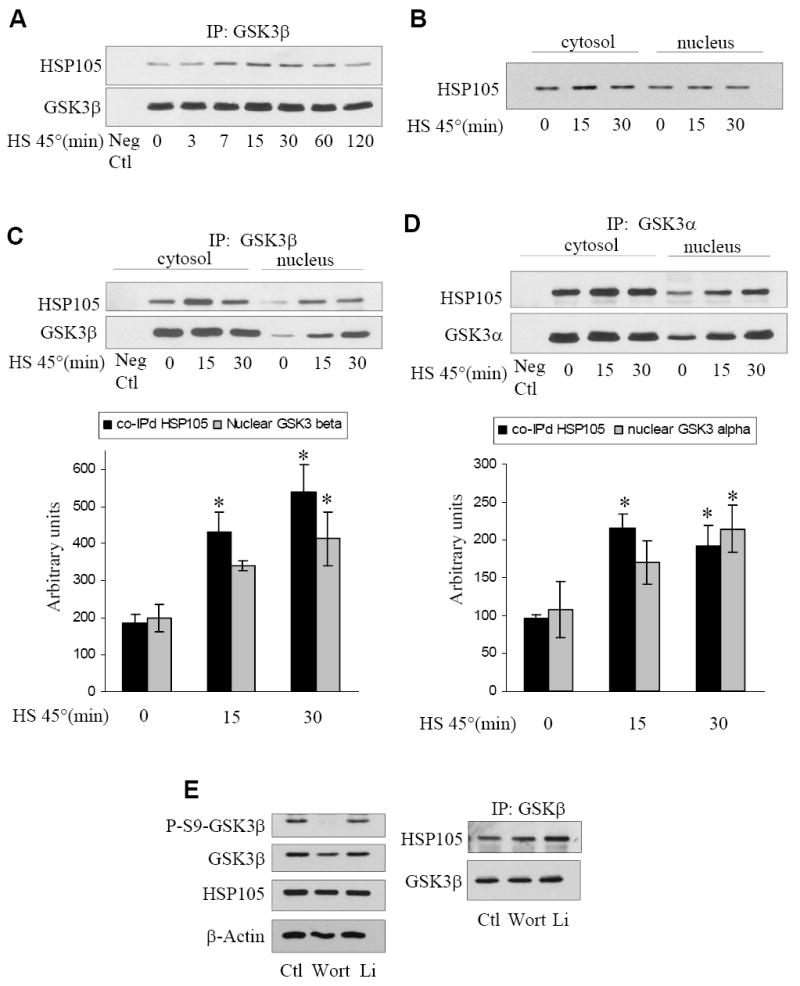
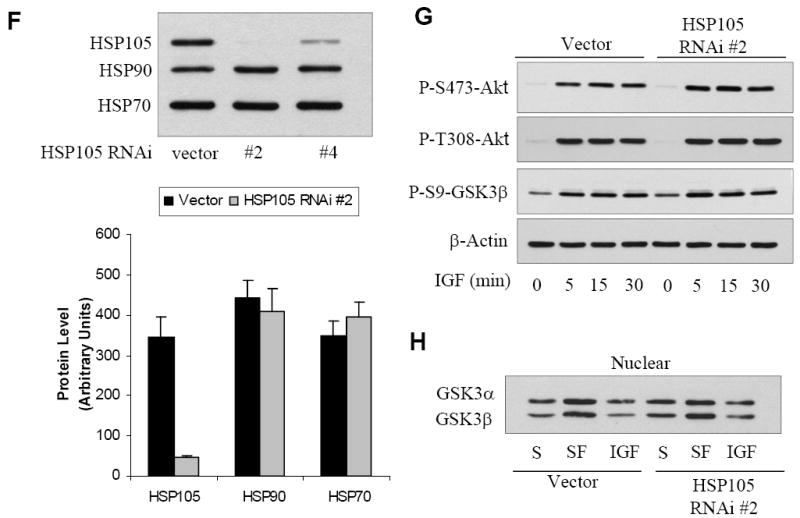
A. SH-SY5Y cells were subjected to heat shock (HS) by incubation in a 45°C water bath for 0-120 min, followed by immunoprecipitation (IP) of GSK3β and immunoblotting for HSP105 and GSK3β. B. SH-SY5Y cells were placed in 45°C water bath for 0-30 min, cytosolic and nuclear fractions were prepared and immunoblotted for HSP105. C. SH-SY5Y cells were placed in a 45°C water bath for 0-30 min, cytosolic and nuclear fractions were prepared, followed by immunoprecipitation of GSK3β and immunoblotting for HSP105 and GSK3β. The levels of nuclear co-immunoprecipitated HSP105 and immunoprecipitated GSK3β were quantified; Means ± SEM; n = 3; *p<0.05 compared with cells not subjected to heat shock. D. SH-SY5Y cells were placed in a 45°C water bath for 0-30 min, cytosolic and nuclear fractions were prepared, followed by immunoprecipitation of GSK3α and immunoblotting for HSP105 and GSK3α. The levels of nuclear co-immunoprecipitated HSP105 and immunoprecipitated GSK3α were quantified; Means ± SEM; n = 3; *p<0.05 compared with cells not subjected to heat shock. E. SH-SY5Y cells were treated with 100 nM wortmannin (Wort) for 1 hr or 20 mM lithium (Li) for 2 hr followed by immunoprecipitation of GSK3β and immunoblotting for HSP105 and GSK3β (right panel). Cell lysates were immunoblotted for phospho-ser9-GSK3β and total levels of GSK3β, HSP105 and β-actin (left panel). F. Stable SH-SY5Y cells were generated by lentiviral transduction using the empty shRNAi vector that still expresses GFP, or with one of two shRNAi directed to HSP105. The RNAi were designated HSP105 RNAi #2 and HSP105 RNAi #4. The efficiency of knockdown of HSP105 and the lack of effects on HSP90 and HSP70 levels were measured in immunoblots, and quantitative values (means ± SEM) were obtained from three independent samples. G. Vector or HSP105 RNAi #2 expressing cells were treated with 50 ng/ml IGF-1 for 0-30 min followed by immunoblotting for phospho-Ser473-Akt, phospho-Thr308-Akt, phospho-Ser9-GSK3β, and β-actin. H. Vector or HSP105 RNAi #2 expressing cells were maintained in serum (S) or placed in serum-free (SF) media for 2 h, then incubated with or without 50 ng/ml IGF-1 for 30 min. The nuclei were isolated and the level of GSK3α/β measured by immunoblot.
3.2. HSP105 regulates caspase-3 activation independently of the UPR
Based on our findings that HSP105 is associated with proteins involved in the response to ER stress, GSK3 and GRP78, we hypothesized that HSP105 might play a role in this signaling cascade. To examine the functional effects of HSP105, shRNAi was used to knock down its levels in cells using an RNAi vector that co-expresses GFP to allow identification of transduced cells. SH-SY5Y cells were stably transduced with lentivirus carrying either the empty RNAi vector or a vector with either of two RNAi sequences specific to HSP105. The level of HSP105 was effectively knocked down by lentivirus-mediated RNAi, with a reduction of greater than 85% with RNAi sequence #2. Specificity was confirmed by the lack of any effect on the levels of two other hsps, HSP90 and HSP70 (Fig. 2F). The efficiency of transduction with vector HSP105 RNAi #2 was near 100%, while the efficiency of HSP105 RNAi #4 was slightly lower, as determined by fluorescence microscopy and flow cytometry (not shown).
Since GSK3 is associated with HSP105, we used cells with knocked down levels of HSP105 to test if HSP105 regulated the serine-phosphorylation of GSK3 after Akt activation or the dynamic flux of nuclear levels of GSK3. Vector cells and cells with knockdown of HSP105 were treated with insulin-like growth factor-1 (IGF-1) and the serine-phosphorylation of Akt and GSK3β were examined by immunoblot analyses using specific phospho-dependent antibodies (Fig. 2G). Stimulation with IGF-1 induced rapid increases in the phosphorylation of both Akt and GSK3β, and these responses were equivalent in cells with or without HSP105. We previously identified mechanisms regulating the nuclear localization of GSK3, which include cytoplasmic sequestration by GSK3 binding partners [37]. Therefore, we tested if reduction of HSP105 influenced the influx or efflux of nuclear GSK3. Cells were placed in serum-free media to promote GSK3 nuclear import, and subsequently stimulated with IGF-1 to promote GSK3 export, followed by fractionation and immunoblotting of nuclear GSK3α/β (Fig 2H). Reduction of HSP105 had no affect on the nuclear import or export of GSK3. Thus, although HSP105 binds GSK3, it is not required for growth factor signaling to serine-phosphorylate GSK3 or for the nuclear flux of GSK3.
To investigate the role of HSP105 in responses to ER stress, we first verified that SH-SY5Y cells respond appropriately to ER stress. Cells were treated with tunicamycin (Fig. 3A) or thapsigargin (Fig. 3B) for 3 to 6 h, followed by immunoblotting for ER stress and apoptotic markers. Treatment with either ER stress-inducing agent resulted in phosphorylation of eIF2α and up-regulation of CHOP, confirming that the UPR was activated. ER stress also led to cleavage (activation) of caspase-3 and cleavage of the caspase-3 substrate PARP. Additionally, ER stress did not affect the association of HSP105 with either GSK3 or GRP78 (not shown). Thus, ER stress-inducing agents activate the UPR and the apoptotic pathway in SH-SY5Y cells.
Figure 3. HSP105 and UPR-induced signaling.
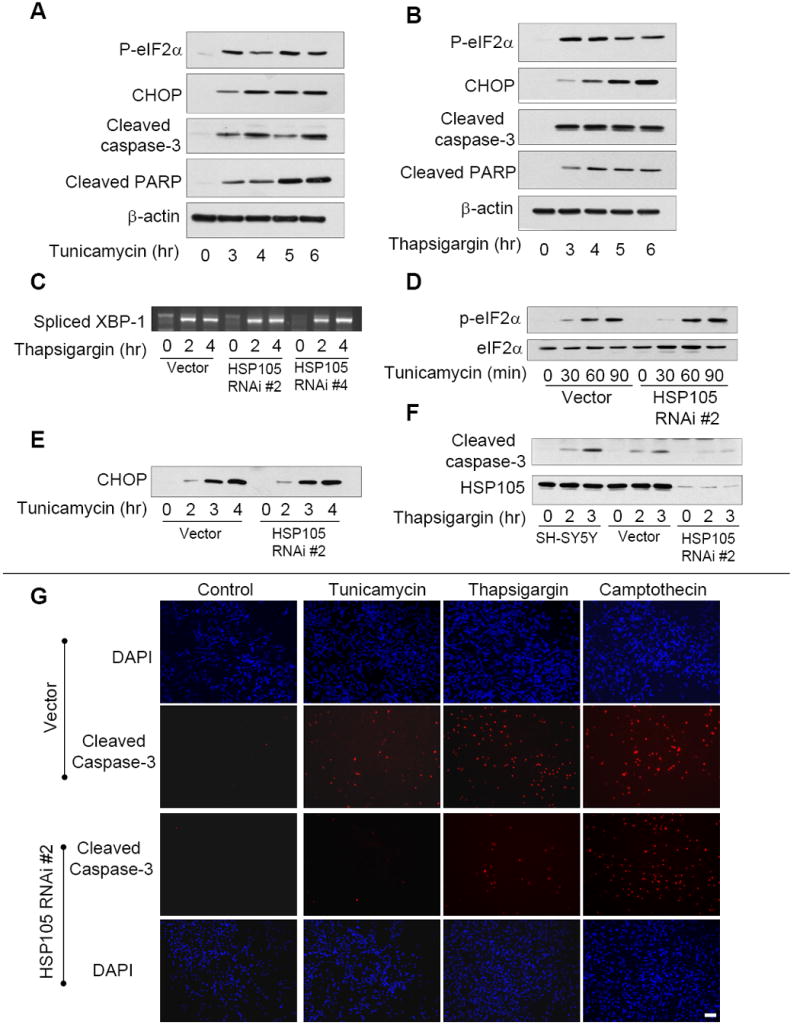
A. SH-SY5Y cells were treated with 4 μg/ml tunicamycin for 0 to 6 h followed by immunoblotting for phospho-eIF2α, CHOP, cleaved caspase-3, cleaved PARP, and β-actin. B. SH-SY5Y cells were treated with 2 μM thapsigargin for 0 to 6 h followed by immunoblotting for phospho-eIF2α, CHOP, cleaved caspase-3, cleaved PARP, and β-actin. C. To examine XBP-1 splicing, vector or HSP105 RNAi expressing cells were treated with 2 μM thapsigargin for 2 or 4 h. The cellular RNA was isolated and spliced XBP-1 examined by RT-PCR. D. Vector or HSP105 RNAi #2 expressing cells were treated with 4 μg/ml tunicamycin for 0 to 90 min, then immunoblotted for phospho-eIF2α or total eIF2α. E. Vector or HSP105 RNAi #2 cells were treated with 4 μg/ml tunicamycin for 0 to 4 h, then immunoblotted for CHOP. F. Untransduced SH-SY5Y cells, vector cells, or HSP105 RNAi #2 expressing cells were treated with 2 μM thapsigargin for 2 or 3 h, then immunoblotted for cleaved caspase-3 and HSP105. G. Vector or HSP105 RNAi#2 expressing cells were treated with 4 μg/ml tunicamycin, 2 μM thapsigargin, or 1 μM camptothecin, for 3 h, then stained for active caspase-3 or DAPI and examined by fluorescence microscopy. Scale bar: 100 μm
To test if HSP105 regulates the UPR or the subsequent apoptotic signaling cascade, ER stress was induced using thapsigargin or tunicamycin in control and HSP105 knockdown cells. RT-PCR analysis following thapsigargin treatment revealed that XBP-1 mRNA splicing induced by ER stress was not overtly affected by HSP105 knockdown (Fig. 3C). Consistent with this, reduction of HSP105 had no impact on the phosphorylation of eIF2α (Fig. 3D) or CHOP expression (Fig. 3E) following ER stress induced by tunicamycin treatment.
In contrast to the unaltered ER stress-induced UPR signaling and CHOP expression after HSP105 knockdown, the subsequent apoptotic signaling was highly dependent on HSP105. Treatment with thapsigargin induced caspase-3 activation in wild-type and vector control cells, but caspase-3 activation was greatly attenuated in HSP105 knockdown cells (Fig. 3F). These results were verified by immunostaining for active caspase-3, which demonstrated that in HSP105 knockdown cells treated with tunicamycin or thapsigargin, active caspase-3 was greatly reduced compared to vector cells (Fig. 3G). In contrast to the profound block by HSP105 knockdown on ER stress-induced caspase-3 activation, HSP105 knockdown did not alter caspase-3 activation induced by the DNA damaging agent camptothecin. Quantification of the immunocytochemical measurements demonstrated that active caspase-3 was significantly reduced in HSP105 knockdown cells compared with vector cells treated with tunicamycin or thapsigargin, but not with camptothecin (Fig. 4). These results were further substantiated by immunoblotting for cleavage of the well-characterized caspase-3 substrate PARP. As with cleaved caspase-3, knockdown of HSP105 greatly reduced PARP cleavage induced by tunicamycin or thapsigargin, but not by camptothecin (Fig. 4). Additionally, when HSP105 was reduced by an alternative HSP105-specific shRNAi, a similar reduction in tunicamycin-induced PARP cleavage was observed (Fig. 4A), thus confirming that the observed affects are due to HSP105 knockdown. Thus, HSP105 knockdown selectively attenuated ER stress-induced apoptotic signaling, and but did not cause a generalized inhibition of caspase-3 activation. Furthermore, knockdown of HSP105 did not affect the level of total caspase-3, caspase-9 or several other modulators of apoptosis including the inhibitors of apoptosis (IAPs), Bad, Bcl-2 and Bcl-XL (Fig. 4D). This demonstrates that HSP105 is an important and specific mediator of ER stress-induced caspase-3 activation.
Figure 4. HSP105 knockdown attenuates ER stress-induced caspase-3 activation.
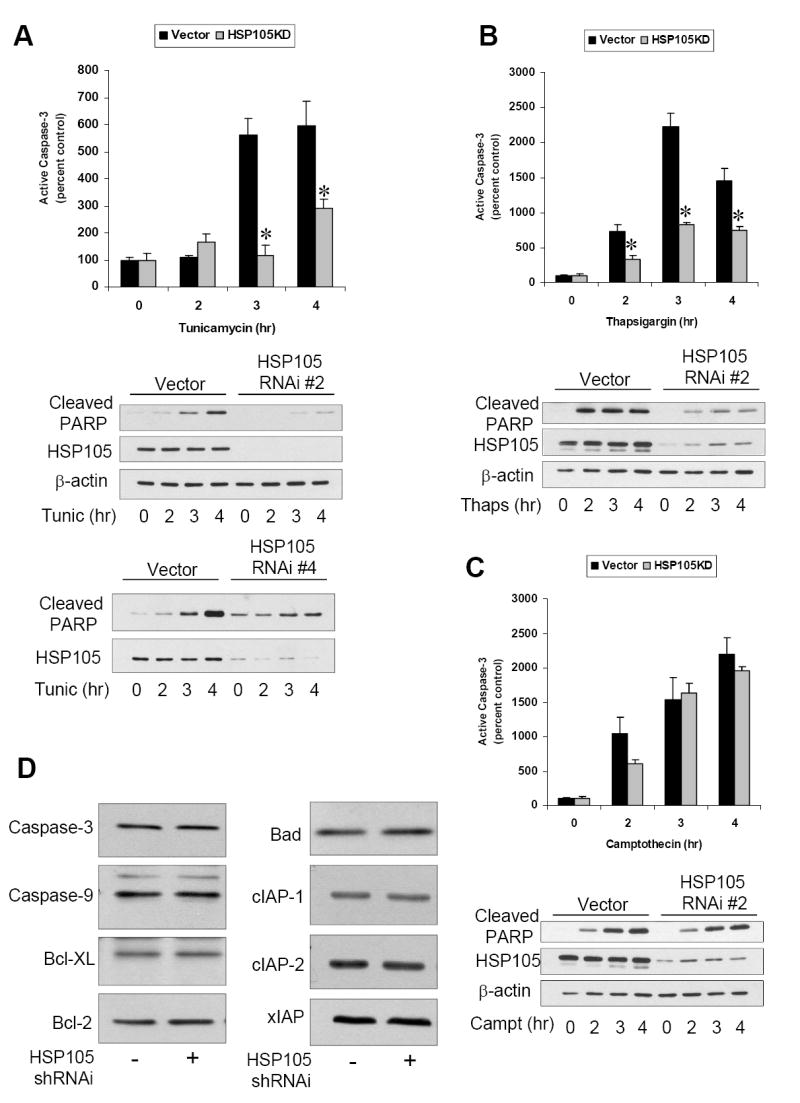
A. Vector cells or HSP105 RNAi #2 expressing cells were treated with 4 μg/ml tunicamycin for 0 to 4 h and active caspase-3 examined by immunocytochemistry. Quantitative values are means ± SEM, n=3, * p<0.05. Cleaved PARP, HSP105, and β-actin were examined by immunoblot. Vector cells and HSP105 RNAi #4 expressing cells were treated with 4 μg/ml tunicamycin (Tunic) for 0 to 4 h and immunoblotted for cleaved PARP and HSP105. B. Vector cells or HSP105 RNAi #2 expressing cells were treated with 2 μM thapsigargin for 0 to 4 h and active caspase-3 examined by immunocytochemistry. Quantitative values are means ± SEM, n=3, * p<0.05. Cleaved PARP, HSP105, and β-actin were examined by immunoblot. C. Vector cells or HSP105 RNAi #2 expressing cells were treated with 1 μM camptothecin for 0 to 4 h and active caspase-3 examined by immunocytochemistry. Quantitative values are means ± SEM, n=3, * p<0.05. Cleaved PARP, HSP105, and β-actin were examined by immunoblot. D. Total levels of caspase-3, caspase-9, Bcl-XL, Bcl-2, Bad cIAP-1, cIAP22, and xIAP were measured by immunoblot in vector control cells and HSP105 RNAi #2 expressing cells.
To further assess the role of HSP105 in ER stress-induced apoptotic signaling, events up-and down-stream of caspase-3 activation were investigated. Following apoptotic stimuli the anti-apoptotic protein MCL1 is rapidly degraded and the pro-apoptotic protein Bax becomes active [38]. Surprisingly, knockdown of HSP105 had no effect on either the tunicamycin-induced reduction in MCL1 level (Fig. 5A) or activation of Bax (Fig. 5B). However, consistent with the reduction in active caspase-3, tunicamycin-induced chromatin condensation was blocked in cells with reduced HSP105 (Fig. 5C). The lack of caspase-3 activation and nuclear condensation after ER stress in cells with the level of HSP105 knocked down raised two possibilities: the cells were better able to adapt to ER stress and survive, or the cells were driven to an alternative pathway of cell death. Measurements of cell viability clearly supported the latter conclusion. Cell death assessed by measurements of LDH release following tunicamycin or thapsigargin treatment demonstrated that the HSP105 knockdown cells died at the same rate as the control cells in response to ER stress (Fig 5D). Thus, HSP105 is required for caspase-3-mediated apoptosis following ER stress-induced activation of the UPR, and in cells with decreased HSP105 an alternative cell death pathway is utilized.
Figure 5. Knockdown of HSP105 does not promote cell survival.
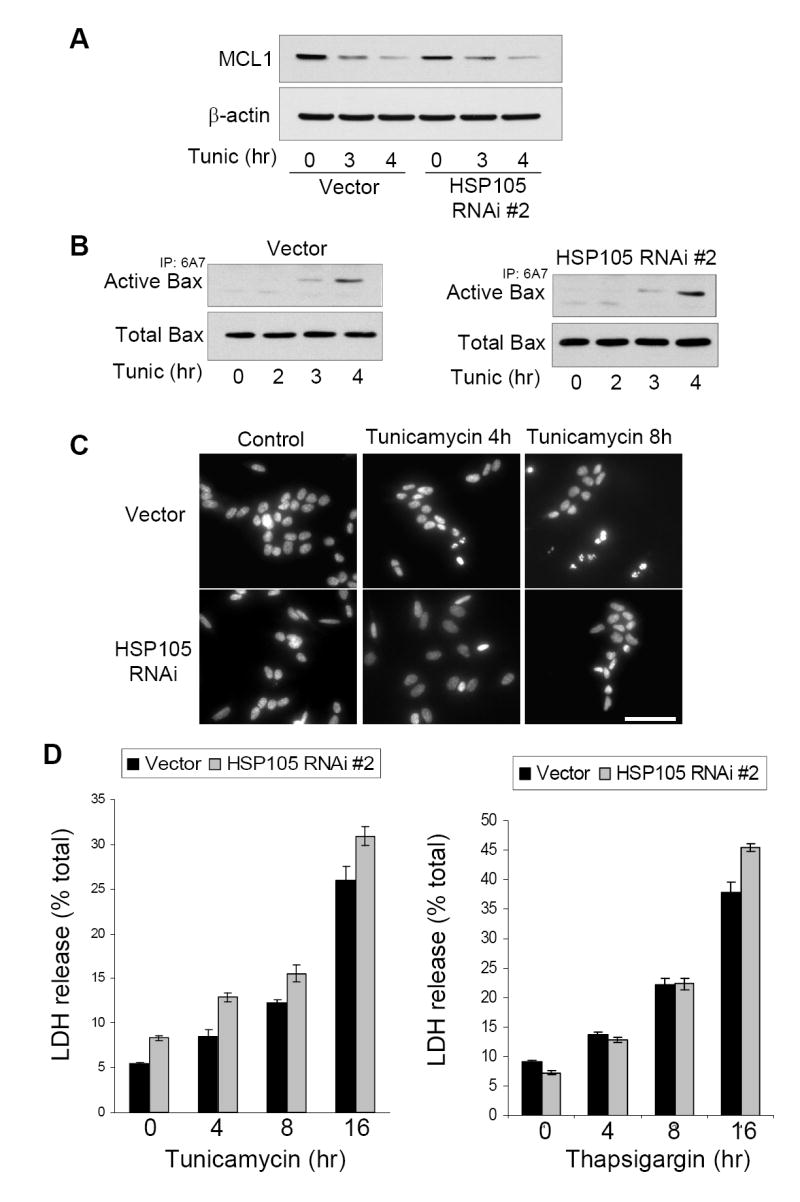
A. Vector or HSP105 RNAi #2 expressing cells were treated with 4 μg/ml tunicamycin for 3 or 4 h, and immunoblotted for MCL1 and β-actin. B. To examine Bax activation, vector or HSP105 RNAi #2 expressing cells were treated with 4 μg/ml tunicamycin for 0 to 4 h. The active conformation specific Bax antibody 6A7 was used to immunoprecipitate active Bax followed by immunoblotting for total Bax in the immunoprecipitate and the cell lysate. C. To examine nuclear morphology, vector or HSP105 RNAi #2 expressing cells were treated with 4 μg/ml tunicamycin for 4 or 8 h, then fixed and stained with Hoechst 33342 and examined by fluorescence microscopy. Scale bar: 100 μm D. Vector or HSP105 RNAi #2 expressing cells were treated with 4 μg/ml tunicamycin or 2 μM thapsigargin for 0 to 16 h, and cell death was assessed by measuring the release of LDH. Quantitative values are means ± SEM, n=3.
4. Discussion
ER stress-specific responses are critical for cellular adaptation and survival and, when these fail, for the proper execution of apoptosis [3,4]. In the present study we identified HSP105 as a crucial component of the apoptotic program following ER stress. HSP105 was found to act downstream of CHOP activation to ensure the apoptotic cascade proceeds to activation of the executioner caspase, caspase-3.
HSPs constitute a large family of proteins involved in many cell functions, but relatively few protein partners, or specific functions, of HSP105 have been identified despite its being one of the major cellular HSPs. HSP105 previously was shown to associate with HSP70 and HSP40 and to suppresses the ability of HSP70/HSP40 to promote protein refolding [29,39]. Although these studies indicated that HSP105 reduces the function HSP70, in opposition to this HSP105 also was reported to be a nucleotide exchange factor for HSP70, thus promoting its activity [27,28]. In addition to interacting with HSP70, bacterial and yeast two-hybrid screenings identified several HSP105-interacting proteins, including α-tubulin, cofilin, dynein light chain 2A, α-adducin, platelet-activating factor, acetylhydrolase α-subunit, and phosphoglycerate kinase 1 [40,41]. Of these, only the interaction with α-tubulin was functionally characterized, as HSP105 was reported to suppress disorganization of microtubules during heat shock via its interaction with α-tubulin [40]. Additionally, HSP105 interacts with the truncated androgen receptor with an extended polyglutamine tract [29], and with mutant Cu/Zn superoxide dismutase [30]. In the current study, we identified several new binding partners of HSP105, including Huntingtin-interacting protein 1, Mortalin, Sec16L, GSK3, and GRP78. Since each of the latter three proteins is involved in some aspect of ER function, HSP105 may be particularly involved in chaperoning events in the ER, especially responses to ER stress since GRP78 plays a central role in the UPR. However, despite the clear association with GRP78, knockdown of HSP105 had no detectable affect on the UPR signaling. Since HSP105 is an abundant protein, it is possible that low levels of HSP105 remaining after shRNAi-mediated knockdown are sufficient for chaperoning GRP78-mediated functions. Alternatively, HSP105 may regulate other GRP78-linked actions in the ER, perhaps acting as a nucleotide exchange factor for GRP78 since HSP105 is a nucleotide exchange factor for HSP70 [27,28], which is highly homologous with GRP78.
GSK3 also was identified as a HSP105-binding protein. These proteins associated in mouse brain, and in both the nucleus and cytosol of SH-SY5Y cells. The two isoforms of GSK3, GSK3α and GSK3β, share a high degree of homology in their large central kinase domains, but less so in their N-terminal and C-terminal regions [42]. Since HSP105 binds both isoforms, it is likely that HSP105 binds within their homologous region. Although many kinases are associated with members of the heat shock family of chaperones [26], HSP105 is the first molecular chaperone found to associate with GSK3 except during translation [43]. The association of GSK3 with HSP105 increased during heat shock, raising the possibility that HSP105 may have a protective action towards GSK3 during cell stresses that have the potential to destabilize GSK3. However, the effect of the HSP105-GSK3 interaction on ER stress signaling remains to be identified. While it is possible that the influence of HSP105 on caspase-3 is through the well characterized GSK3-dependent promotion of apoptotic signaling, it is as likely that HSP105 affects a relatively small pool of GSK3 whose function is still unknown.
The most surprising function of HSP105 identified in this study is that it is required for caspase-3 activation following ER stress. HSPs are usually considered to be anti-apoptotic proteins that assist in adaptation and recovery from potentially lethal insults. However, HSP105 has another role, as it was essential for caspase-3 activation after ER stress, and some specificity for ER stress-induced apoptotic signaling was evident by the lack of a regulatory role of HSP105 in apoptotic signaling induced by the DNA damage pathway caused by camptothecin treatment. ER stress causes accumulation of misfolded proteins, which may threaten the ability of proteins in the apoptotic signaling cascade to function properly in the absence of a chaperone. Therefore, it is possible that HSP105 is not directly a pro-apoptotic protein, but simply performs its normal function, even during apoptosis, as a chaperone to maintain enzymes in active conformations, some of which may be pro-apoptotic. This type of regulation has been demonstrated between HSP60 and pro-caspase-3 (although not in the context of ER stress) in which HSP60 is thought to maintain pro-caspase-3 in an activation or cleavage-competent state [44,45]. HSP105 might have a similar, but stress-specific, role acting on proteins necessary for activation of caspase-3, such as through upstream molecules such as GSK3. It is interesting to note that elevated HSP105 previously was found to be associated with activated caspase-3 during development [46-48]. Alternatively, the finding that knockdown of HSP105 reduced caspase-3 activation could be interpreted as indicating that HSP105 does not facilitate caspase-3 activation but instead suppresses an alternative death pathway. Thus, in the absence of HSP105 this alternative pathway may be de-repressed and activated, so cell death ensues independently of caspase-3. This would be consistent with the report that in other apoptotic conditions overexpression of HSP105 blocked PARP cleavage, a caspase-3 substrate, indicating that HSP105 can reduce signaling to caspase-3 activation [49]. However, others found overexpression of HSP105 enhanced PARP cleavage [32]. These findings could indicate that the role of HSP105 in regulating caspase-3 activation is stimulus-specific, with HSP105 being necessary for caspase-3 activation after ER stress but not with all other apoptotic insults, or overexpressed HSP105 may engage in actions not performed by endogenous HSP105. Our data is consistent with either model, as we observed with knocked down HSP105 reduced capase-3 activation after ER stress but no appreciable change in cell death.
In summary, we identified several new HSP105-interacting proteins, including GSK3 and GRP78. HSP105 was found to be necessary for the proper activation of caspase-3 following ER stress. Thus, HSP105 is an important modulator of the ER stress response by facilitating apoptosis through the appropriate pathways.
Conclusions
The functional role of HSP105 in signaling networks and cell survival is just beginning to be elucidated. In the present study several novel protein-proteins interactions with HSP105 were identified; most notably with GSK3 and GRP78. The identification of these interactions led to the examination of ER stress-induced apoptotic signaling in which HSP105 was found to have a novel role. Knockdown experiments demonstrated that HSP105 is critical for ER stress-induced caspase-3 activation. However knockdown of HSP105 was not sufficient to block ER stress-induced cell death, indicating that in the absence of HSP105 cell death proceeds through an alternative pathway. Overall, the data presented here is consistent with two potential models: 1) HSP105 specifically directs ER stress-induced signals to caspase-3 activation, or 2) HSP105 normally suppresses an ER stress-induced caspase-3-independent cell death pathway.
Acknowledgments
We thank Dr. Van Parijs for the Lentilox3.7 vector, Dr. Tong Zhou for antibodies, Dr. B. Clodfelder-Miller and Chris Yuskaitis for preparation of cerebral cortical lysates, and Dr. James Lah (Emory University) for providing virus production protocols. This research was supported by MH38752 and AG021045 grants from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Boyce M, Yuan J. Cell Death Differ. 2006;13:363. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 2.Lindholm D, Wootz H, Korhonen L. Cell Death Differ. 2006;13:385. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- 3.Szegezdi E, Logue SE, Gorman AM, Samali A. EMBO Rep. 2006;7:880. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 5.Rutkowski DT, Kaufman RJ. Trends Cell Biol. 2004;14:20. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Nat Cell Biol. 2000;2:326. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 7.Shen J, Chen X, Hendershot L, Prywes R. Dev Cell. 2002;3:99. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 8.Schroder M, Kaufman RJ. Annu Rev Biochem. 2005;74:739. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 9.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. Genes Dev. 1998;12:982. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oyadomari S, Mori M. Cell Death Differ. 2004;11:381. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 12.Xu C, Bailly-Maitre B, Reed JC. J Clin Invest. 2005;115:2656. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jope RS, Johnson GVW. Trends Biochem Sci. 2004;29:95. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Song L, De Sarno P, Jope RS. J Biol Chem. 2002;277:44701. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- 15.Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. FASEB J. 2004;18:1162. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- 16.Hiroi T, Wei H, Hough C, Leeds P, Chuang DM. Pharmacogenomics J. 2005;5:102. doi: 10.1038/sj.tpj.6500296. [DOI] [PubMed] [Google Scholar]
- 17.Kim AJ, Shi Y, Austin RC, Werstuck GH. J Cell Sci. 2005;118:89. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan S, Ohsugi M, Liu Z, Fatrai S, Bernal-Mizrachi E, Permutt MA. Diabetes. 2005;54:968. doi: 10.2337/diabetes.54.4.968. [DOI] [PubMed] [Google Scholar]
- 19.Brewster JL, Linseman DA, Bouchard RJ, Loucks FA, Precht TA, Esch EA, Heidenreich KA. Mol Cell Neurosci. 2006;32:242. doi: 10.1016/j.mcn.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Takadera T, Yoshikawa R, Ohyashiki T. Neurosci Lett. 2006;408:124. doi: 10.1016/j.neulet.2006.08.066. [DOI] [PubMed] [Google Scholar]
- 21.Beurel E, Jope RS. Prog Neurobiol. 2006;79:173. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcu MG, Doyle M, Bertolotti A, Ron D, Hendershot L, Neckers L. Mol Cell Biol. 2002;22:8506. doi: 10.1128/MCB.22.24.8506-8513.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oyadomari S, Yun C, Fisher EA, Kreglinger N, Kreibich G, Oyadomari M, Harding HP, Goodman AG, Harant H, Garrison JL, Taunton J, Katze MG, Ron D. Cell. 2006;126:727. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 24.Young JC, Agashe VR, Siegers K, Hartl FU. Nat Rev Mol Cell Biol. 2004;5:781. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 25.Beere HM. J Clin Invest. 2005;115:2633. doi: 10.1172/JCI26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caplan AJ, Mandal AK, Theodoraki MA. Trends Cell Biol. 2007;17:87. doi: 10.1016/j.tcb.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Dragovic Z, Broadley SA, Shomura Y, Bracher A, Hartl FU. EMBO J. 2006;25:2519. doi: 10.1038/sj.emboj.7601138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. EMBO J. 2006;25:2510. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishihara K, Yamagishi N, Saito Y, Adachi H, Kobayashi Y, Sobue G, Ohtsuka K, Hatayama T. J Biol Chem. 2003;278:25143. doi: 10.1074/jbc.M302975200. [DOI] [PubMed] [Google Scholar]
- 30.Yamashita H, Kawamata J, Okawa K, Kanki R, Nakamizo T, Hatayama T, Yamanaka K, Takahashi R, Shimohama S. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04534.x. in press. [DOI] [PubMed] [Google Scholar]
- 31.Yamagishi N, Ishihara K, Saito Y, Hatayama T. Exp Cell Res. 2006;312:3215. doi: 10.1016/j.yexcr.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Yamagishi N, Saito Y, Ishihara K, Hatayama T. Eur J Biochem. 2002;269:4143. doi: 10.1046/j.1432-1033.2002.03109.x. [DOI] [PubMed] [Google Scholar]
- 33.Bijur GN, Jope RS. J Biol Chem. 2001;276:37436. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. Nature. 2002;415:92. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 35.Hatayama T, Yasuda K, Yasuda K. Biochem Biophys Res Commun. 1998;248:395. doi: 10.1006/bbrc.1998.8979. [DOI] [PubMed] [Google Scholar]
- 36.Bhattacharyya D, Glick BS. Mol Biol Cell. 2007;18:839. doi: 10.1091/mbc.E06-08-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meares GP, Jope RS. J Biol Chem. 2007;282:16989. doi: 10.1074/jbc.M700610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gelinas C, White E. Genes Dev. 2005;19:1263. doi: 10.1101/gad.1326205. [DOI] [PubMed] [Google Scholar]
- 39.Yamagishi N, Nishihori H, Ishihara K, Ohtsuka K, Hatayama T. Biochem Biophys Res Commun. 2000;272:850. doi: 10.1006/bbrc.2000.2864. [DOI] [PubMed] [Google Scholar]
- 40.Saito Y, Yamagishi N, Ishihara K, Hatayama T. Exp Cell Res. 2003;286:233. doi: 10.1016/s0014-4827(03)00054-5. [DOI] [PubMed] [Google Scholar]
- 41.Saito Y, Doi K, Yamagishi N, Ishihara K, Hatayama T. Biochem Biophys Res Commun. 2004;314:396. doi: 10.1016/j.bbrc.2003.12.108. [DOI] [PubMed] [Google Scholar]
- 42.Woodgett JR. EMBO J. 1990;9:2431. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V. Mol Cell. 2006;24:627. doi: 10.1016/j.molcel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 44.Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. EMBO J. 1999;18:2040. doi: 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xanthoudakis S, Roy S, Rasper D, Hennessey T, Aubin Y, Cassady R, Tawa P, Ruel R, Rosen A, Nicholson DW. EMBO J. 1999;18:2049. doi: 10.1093/emboj/18.8.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evrard L, Vanmuylder N, Dourov N, Glineur R, Louryan S, Craniofac J. Genet Dev Biol. 1999;19:24. [PubMed] [Google Scholar]
- 47.Evrard L, Vanmuylder N, Dourov N, Hermans C, Biermans J, Werry-Huet A, Rooze M, Louryan S, Craniofac J. Genet Dev Biol. 2000;20:183. [PubMed] [Google Scholar]
- 48.Gashegu J, Ladha R, Vanmuylder N, Philippson C, Bremer F, Rooze M, Louryan S. J Anat. 2007;210:532. doi: 10.1111/j.1469-7580.2007.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hatayama T, Yamagishi N, Minobe E, Sakai K. Biochem Biophys Res Commun. 2001;288:528. doi: 10.1006/bbrc.2001.5802. [DOI] [PubMed] [Google Scholar]