

Abstract
Vascular endothelial growth factor (VEGF) is a protein factor which has been found to play a significant role in both normal and pathological states. Its role as an angiogenic factor is well-established. More recently, VEGF has been shown to protect neurons from cell death both in vivo and in vitro. While VEGF’s potential as a protective factor has been demonstrated in hypoxia–ischemia, in vitro excitotoxicity, and motor neuron degeneration, its role in seizure-induced cell loss has received little attention. A potential role in seizures is suggested by Newton et al.’s [Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, Duman RS (2003) Gene profile of electroconvulsive seizures: Induction of neurotrophic and angiogenic factors. J Neurosci 23:10841–10851] finding that VEGF mRNA increases in areas of the brain that are susceptible to cell loss after electroconvulsive-shock induced seizures. Because a linear relationship does not always exist between expression of mRNA and protein, we investigated whether VEGF protein expression increased after pilocarpine-induced status epilepticus. In addition, we administered exogenous VEGF in one experiment and blocked endogenous VEGF in another to determine whether VEGF exerts a neuroprotective effect against status epilepticus-induced cell loss in one vulnerable brain region, the rat hippocampus. Our data revealed that VEGF is dramatically up-regulated in neurons and glia in hippocampus, thalamus, amygdala, and neocortex 24 h after status epilepticus. VEGF induced significant preservation of hippocampal neurons, suggesting that VEGF may play a neuroprotective role following status epilepticus.
Keywords: neuroprotection, pilocarpine, thalamus, amygdala, neocortex, neurotrophic
Vascular endothelial growth factor (VEGF) is a secreted protein mitogen initially studied for its potent effects as a vascular growth factor (for review see Yancopoulos et al., 2000). In addition to its well-established effects on vasculature, recent findings illustrate that VEGF has direct neurotrophic effects (for reviews, see Carmeliet and Storkebaum, 2002; Krum and Rosenstein, 2004). During development, VEGF stimulates axonal outgrowth and increases the survival of dorsal root ganglion neurons and developing retinal cells (Sondell et al., 1999; Yourey et al., 2000). In cell culture, it protects neurons from a variety of insults, including hypoxia, ischemia, and excitotoxicity (Jin et al., 2000, 2001; Matsuzaki et al., 2001). VEGF has also been shown to protect against ischemic neuronal damage in vivo (Wang et al., 2005; Bellomo et al., 2003; Sun et al., 2003; Hayashi et al., 1998), though this protection is sometimes accompanied by undesirable vascular effects (Wang et al., 2005). Indeed, VEGF is up-regulated after stroke in brain, perhaps in an adaptive effort to increase blood supply while simultaneously protecting cells (for review see Croll and Wiegand, 2001). VEGF may also work to protect neurons against neurodegenerative processes as it protects motoneurons in animal models of amyotrophic lateral sclerosis (Storkebaum et al., 2005; Zheng et al., 2004).
In addition to its up-regulation after acute ischemic insults, VEGF mRNA is increased after electroconvulsive shock-induced seizures in brain regions susceptible to seizure-induced cell damage such as hippocampus (Newton et al., 2003). The current study was conducted to determine if VEGF protein, like its mRNA, would be increased after seizures using pilocarpine to induce status epilepticus. In addition, a VEGF receptor immunoadhesin was used to sequester endogenous VEGF to determine if increases in VEGF protein after status epilepticus help protect neurons from cell death. Finally, because exogenous VEGF has been found to be neuroprotective in many models, we chronically infused exogenous VEGF and studied its effect on status epilepticus-induced cell loss.
EXPERIMENTAL PROCEDURES
Subjects
All subjects were adult male Sprague–Dawley rats (Charles River Laboratories, Kingston, NY, USA) weighing 250–350 g. Animals were housed three per cage within a temperature-stabilized animal facility with food and water available ad libitum. Animals were maintained on a 12-h light/dark cycle (lights on 07:00 h). All experiments were approved by the Queens College Institutional Animal Care and Use Committee which operates under federal and state animal care guidelines. All experiments conformed to international guidelines on the ethical use of animals, and every effort was made to minimize both the number of animals used and their suffering.
Proteins
The VEGF used for protein infusions was human recombinantVEGFA165 (a generous gift of Regeneron Pharmaceuticals, Tarrytown, NY, USA). VEGF was stored frozen until use and then diluted in sterile phosphate-buffered saline (PBS) (Sigma-Aldrich, St. Louis, MO, USA) to attain doses of 15 ng/day, 30 ng/day, 45 ng/day, and 60 ng/day when delivered at 0.5 μl/h via osmotic minipump. These doses were chosen based on pilot data (not shown) demonstrating no significant effect of human VEGF when infused at a dose of 15 ng/day or lower, as well as data demonstrating that infusion of more than 30 ng/day of mouse VEGF 164 (Croll et al., 2004b) or 60 ng/day of human VEGF 165 (S. K. Shah and S. D. Croll, unpublished observations) resulted in overt angiogenesis in brain. Because we used human VEGF in the current experiments, no animal was treated with a dose over 60 ng/day in order to avoid the induction of overt angiogenesis. Flt-Fc, an immunoadhesin designed to sequester endogenous VEGF, was used at a dose of 12 μg/day to interfere with endogenous VEGF receptor binding (a generous gift of Regeneron Pharmaceuticals). This reagent is a forced dimer of regions 1–3 of fetal liver kinase (receptor) (Flt; VEGF receptor 1) fused to the Fc domain of human IgG (hFc) and dissolved in sterile PBS. It has been previously shown to interfere with VEGF’s effects in vivo (Holash et al., 2002). Equimolar hFc was used as a control protein for Flt-Fc. PBS was purchased in powder form, mixed with distilled water, sterilized, and used as a control. All protein reagents were continuously infused starting 5 days before seizure induction, with infusion continuing until the animals were killed.
Pump implantation and protein infusion
Animals receiving protein infusions were anesthetized using 6 mg/kg chlorpromazine injected intraperitoneally followed by 210 mg/kg ketamine (Sigma-Aldrich) administered intramuscularly. The scalp was shaved, cleaned with alcohol, and treated with iodine. Animals were placed into a stereotaxic apparatus and a longitudinal incision was made along the scalp. Two burr holes were drilled and anchor screws (Plastics One, Roanoke, VA, USA) were inserted. A sterile 4 mm cannula (Plastics One), with an attached heat-sealed polyvinyl catheter (Plastics One) containing sterile PBS, was implanted unilaterally into the dorsal hippocampus (3.8 mm posterior and 2.7 mm lateral as measured from bregma, so that the tip would be positioned in the lateral portion of the dentate hilus) of each animal. This location was chosen based on data demonstrating that VEGF diffuses over a 1.5 mm radius (Croll et al., 2004b). Dental acrylic was then applied to secure the cannula and anchor screws in place. Nylon sutures were used to close the incision, topical antimicrobial ointment was applied, and animals were placed under a heat lamp to recover.
One week following cannula implantations, animals were re-anesthetized following the same procedure and an incision was made at the nape of the neck. The heat-sealed tip of the catheter was snipped and an Alzet osmotic minipump (Durect Corporation, Palo Alto, CA, USA), containing either rhVEGF165 (15, 30, 45, or 60 ng/day) or sterile PBS (Sigma-Aldrich), infusing 0.5 μl per hour was attached to the catheter and glued. Additional controls were used in some cohorts, which included the protein controls BSA (bovine serum albumin, to control for protein load), hFc (a recombinant human control protein), and inactivated VEGF. VEGF was inactivated by repeated freeze–thaw cycles, which has previously been shown to eliminate VEGF’s bioactivity (N. Papadopoulos, unpublished observations using human umbilical vein endothelial cell survival assays), rather than by heat, which results in a precipitate. As these control groups produced comparable results to PBS, the data were collapsed into a single category termed “controls.” The pump was inserted into the s.c. space at the nape of the neck and the incision was closed with wound clips. Animals were placed under a heat lamp to recover. Fig. 1 illustrates the timeline for these experiments.
Fig. 1.

Experimental timeline showing timing of VEGF infusion and seizure induction.
Some animals received infusions of BowAng1, a fusion of four molecules of the vascular growth factor angiopoietin-1 with two molecules of hFc (Davis et al., 2003), along with VEGF. Angiopoietin-1 has been shown to block VEGF’s vascular permeabilizing effects (Thurston et al., 2000) but not its angiogenic effects in brain (Croll et al., unpublished observations), and was used to determine the role of vascular leak in VEGF’s effects after seizures.
Acute seizure induction
Five days following pump implantations for protein infusions, animals were pre-treated with 1 mg/kg atropine methylbromide (Sigma-Aldrich) injected s.c. 30 min prior to receiving 350 mg/kg pilocarpine hydrochloride (Sigma-Aldrich) intraperitoneally. Seizures were scored from stages 1–5 based on a previously published (Rudge et al., 1998) modification of Racine’s (1972) scale. Briefly, stages 1–4 were identical to Racine’s stages. However, the scale was expanded to define stage 5 as sudden, but transient, whole-body tonus. Stages 6 and 7 were status epilepticus, which was defined as over 5 min of continuous seizures without intervening return to normal behavior. Stage 6 involved continuous rearing and falling whereas stage 7 included the episodes of stage 5 seizures. Stage 8 was defined as death occurring during status epilepticus (Rudge et al., 1998).
Status epilepticus was truncated with 10 mg/kg diazepam (Henry Schein, Melville, NY, USA) administered intraperitoneally after 60 min. Animals not reaching status epilepticus were excluded from all remaining analyses. Each control animal received an injection of atropine and subsequently diazepam concurrent with animals that had seizures, but 0.9% NaCl (saline) was injected instead of pilocarpine, using an equivalent volume.
VEGF enzyme-linked immunosorbent assays (ELISA)s
Rat VEGF ELISAs
Animals were killed with Euthasol veterinary euthanasia solution (Henry Schein, Inc.) 24 h following injection of saline or pilocarpine for analysis of VEGF protein levels by ELISA. Brains were removed and placed on ice for 3 min until firm. They were then placed in an acrylic brain matrix (MyNeuroLab, Inc., www.myneurolab.com) and cut into 1.5 mm slabs with a thin razor. Two slabs containing the full medial–lateral extent of dorsal hippocampus were selected, and a 3 mm wide sample was cut from the center of dorsal hippocampus and the overlying cortex (selected to match the region of VEGF infusion in later studies). Tissue samples were frozen in Eppendorf tubes on dry ice. An Immuno Maxisorp plate (VWR Scientific, West Chester, PA, USA) was coated with 100 μl per well rhVEGF165 (Regeneron Pharmaceuticals) at 2 μg/ml in carbonate/bicarbonate buffer (Sigma-Aldrich) and incubated overnight at 4 °C. The plate was then washed with KPL (Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA) buffer followed by 300 μl 0.2% I-blocking buffer (Tropix, Foster City, CA, USA) at room temperature for 1 h. The standard was diluted to 100 ng/ml and serially diluted in diluent with normal mouse serum. Samples were diluted and 100 μl was placed in each well, in duplicate, and incubated with VEGF antibody (R & D Systems, Minneapolis, MN, USA) for 2 h at room temperature. The plate was then washed four times with 300 μl wash buffer. Goat anti-human IgG Fc conjugated to horseradish peroxidase (Sigma-Aldrich) at 1:20,000 was added in diluent and incubated for 1 h at room temperature. The plate was washed again four times, followed by 100 μl per well of Tris–mercapto-ethanol buffer substrate (Sigma-Aldrich), and developed at room temperature for 30 min. Development was stopped by adding 100 μl per well 2 N H2SO4. The plate was read at 450–570 nm, and samples were normalized to standards where the range of the standard curve was 0.14–100 ng/ml.
Human VEGF ELISAs
After 5 days of continuous infusion of human recombinant VEGF or control protein (hFc) at 30 ng/day, rats were killed and tissue was collected and homogenized as described for the rat VEGF ELISAs. Human VEGF ELISAs were performed using the Quantikine Human VEGF ELISA kit from R & D Systems (RNDsystems.com) in accordance with manufacturer’s instructions.
Immunocytochemistry
Animals were deeply anesthetized with an overdose of a pentobarbital-based killing solution, and were subsequently exsanguinated with heparinized isotonic (0.9%) saline perfused through the heart. Following saline perfusion, animals were perfusion-fixed with 4% paraformaldehyde in first acetate and then borate buffer, as previously described (Croll et al., 1999). The brains were removed and placed in 30% sucrose borate buffer at 4 °C until they were sectioned. After 3–7 days, brains were sectioned coronally at 40 μm and stored in cryoprotectant (Watson et al., 1986) at −20 °C until they were stained. Sections were immunostained as previously described (Scharfman et al., 2000) using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA) and an anti-VEGF (goat polyclonal, 1:1000, R & D Systems) antibody; other sections were additionally double-immunostained with VEGF and a second antibody, glial fibrillary acidic protein (GFAP rabbit polyclonal, 1:60,000, Dako, Carpinteria, CA, USA), an astrocyte marker, to detect co-localization of VEGF protein with GFAP. Brains were also stained with an anti-RECA (rat endothelial cell antigen, 1:250, Serotec, Raleigh, NC, USA) antibody to visualize vasculature. Before staining brain tissue, we verified the specificity of the VEGF antibody by immunostaining sections adjacent to sections used for VEGF in situ hybridization in developing embryos and adult rat ovaries. Expression patterns for immunostaining matched those for in situ hybridization (data not shown). In addition, for all stains, the primary antibody was not added to some sections, and these sections showed no specific staining pattern. Sections were stained with Cresyl Violet for evaluation of cell damage using a previously described rating scale (Rudge et al., 1998). Additional sections were stained with Methylene Blue for stereological quantification of cell damage. Sections were hydrated through graded ethanols and then stained with a 1.6%/1% Methylene Blue/Azure II solution following exposure to 1% periodic acid.
Quantification of vascular parameters
Vascular density and vascular diameters were measured in RECA-immunostained tissue sections as previously described (Croll et al., 2004b). Briefly, images were viewed under a Nikon Eclipse E400 microscope (Morrell Instruments, Melville, NY, USA) captured with a digital video camera into SPOT software and imported into the public access image analysis program, NIH Image (National Institutes of Health, Bethesda, MD, USA). Vascular density was measured as proportion of area occupied by RECA-positive lumens in equatorial sections by point-count stereology using a randomly-oriented acetate grid overlay. Vascular diameters were measured by taking the smallest diameter across cross-sectional vascular profiles, and the perpendicular distance across longitudinally-oriented vessels. Both measures were taken using the NIH Image length function on those vessels randomly selected by the grid used in point-count stereology. All measurements were conducted by experimenters blind to the treatment groups of the animals.
Quantification of neuronal density
Precise estimates of cornus ammonis (CA) 1 density were determined using the optical fractionator method (West et al., 1991). The total number of neurons was determined within a region of interest in area CA1 that was within the diffusion range, 1.5 mm radius, of the cannula tip (verified by VEGF immunostaining, data not shown). The region of interest was defined as the area of the CA1 pyramidal cell layer between area CA2 and the subiculum in the medial–lateral axis, and from the initial appearance of the CA1 pyramidal cell layer to the portion of the hippocampus where the dorsal and ventral portions of area CA3 united in the rostral caudal axis. Forty micron coronal sections in a one-in-six series, with a random starting point, were mounted on slides and stained with a 1.6%/1% Methylene Blue/Azure II solution following exposure to 1% periodic acid.
Sections were viewed with an Olympus BX-51 m icroscope and Optronics video camera. Using a stereological software package (Stereo Investigator, Microbrightfield Inc., Williston, VT, USA), a pre-determined counting frame (25 μm2) was systematically moved along a randomly placed grid (125 μm2), and the number of cell nucleoli that came into focus within a portion of the section (excluding 4 μm upper and lower guard zones) was counted. Only cells that had a darkly stained nucleolus surrounded by a lightly stained nucleus and cytoplasm were counted. In the event that two nucleoli could not be distinguished as belonging to two separate neurons, only one neuron was counted. Neurons that were pyknotic, i.e. had a dense, collapsed, profile, were not considered viable and were excluded from the analysis. The total number of neurons within the region of interest was estimated with the formula: N=sum Q−×1/tsf×1/asf/1/ssf where the number of neurons counted (sum Q−) was multiplied by the reciprocal value of the sampling probabilities based on the proportion of section thickness (tsf), cell layer area (asf), and total number of sections (ssf). All histological and stereological analyses were conducted by an independent evaluator blind to the treatment conditions for each animal.
Statistical analyses
A two-way factorial analysis of variance (ANOVA) was conducted on rat VEGF ELISA values to examine the effect of treatment (saline vs. pilocarpine) by region (hippocampus vs. cortex). A one-way independent groups ANOVA was conducted to compare CA1 neuronal densities for various VEGF doses versus vehicle-infused brains. Post hoc analyses were conducted to determine significant effective doses of VEGF. Student’s independent groups t-tests were used to compare vascular densities and cell loss scores for controls versus VEGF, hFc versus Flt-Fc, and/or VEGF versus VEGF+BowAng1. All analyses were conducted using an α value of 0.05.
RESULTS
VEGF ELISA
VEGF ELISAs were used to quantify changes in VEGF protein in tissue of animals that had status epilepticus vs. saline controls. None of these animals received protein infusions or cannula implantations. ELISA data revealed a statistically significant doubling of VEGF protein 24 h after pilocarpine-induced status epilepticus in both cortex and hippocampus (treatment effect F(1,8)=50.344, P=0.0001; Fig. 2).
Fig. 2.
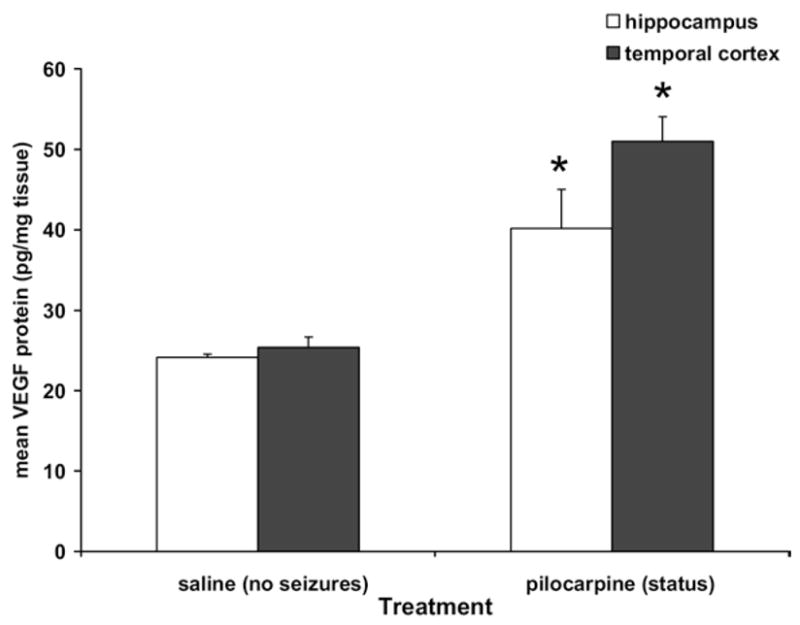
VEGF protein as measured by ELISA 24 h after pilocarpine-induced status epilepticus. VEGF protein was doubled in both hippocampus and temporal cortex 24 h after pilocarpine-induced seizures (n=3), * significantly different than saline (n=3), P<0.05
VEGF immunostaining
Because ELISA does not provide information about cellular localization of increased VEGF protein, immunostaining for VEGF was conducted (n=7–8 per group). Preliminary findings from VEGF immunostains have been previously reported (Croll et al., 2004a, 2005). In saline controls, VEGF immunostaining was very light, and was observed at negligibly low levels in neurons. In contrast, staining revealed a marked expression of VEGF protein in neurons of hippocampal CA1 and CA3 (Fig. 3), as well as the temporal neocortex (Fig. 4) 24 h after status epilepticus, which resolved by 7 days after status (data not shown). This immunostaining appeared to be cytosolic, particularly in CA3 (Fig. 3D), given that the darkest staining was observed between the outer membrane and the nucleus (which was unstained).
Fig. 3.
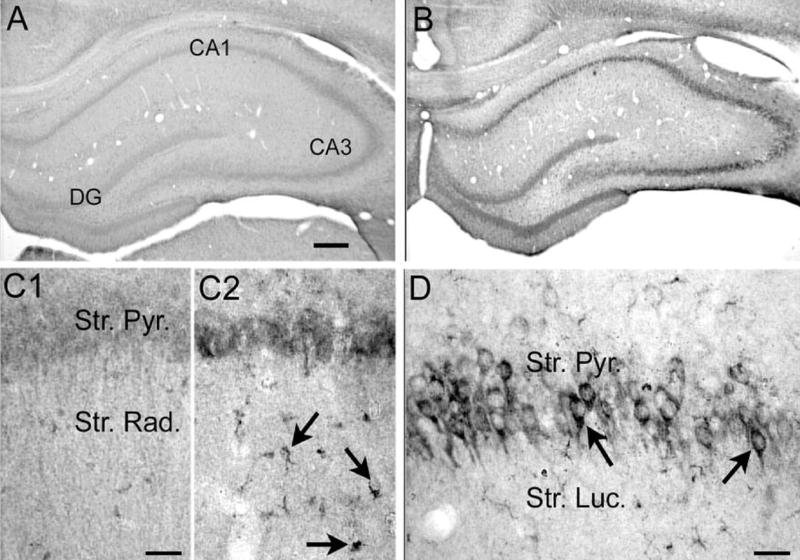
Increased VEGF protein expression in hippocampus 24 h after pilocarpine-induced status epilepticus. (A, B) Sections from an (A) saline- and (B) pilocarpine-treated rat that had 60 min of status epilepticus were processed together using an antibody to VEGF (see Experimental Procedures). Increased VEGF protein was evident in the cell layers. DG=dentate gyrus. Scale bar=200 μm. (C) Increased magnification of area CA1 from (C1) and (C2) shows that glial-like structures were associated with increased VEGF immunoreactivity after pilocarpine-induced status compared with controls. Scale bar=50 μm. (D) A different section from a pilocarpine-treated rat that had status epilepticus, showing increased VEGF immunore-activity in area CA3 pyramidal cell somata. Scale bar=100 μm.
Fig. 4.
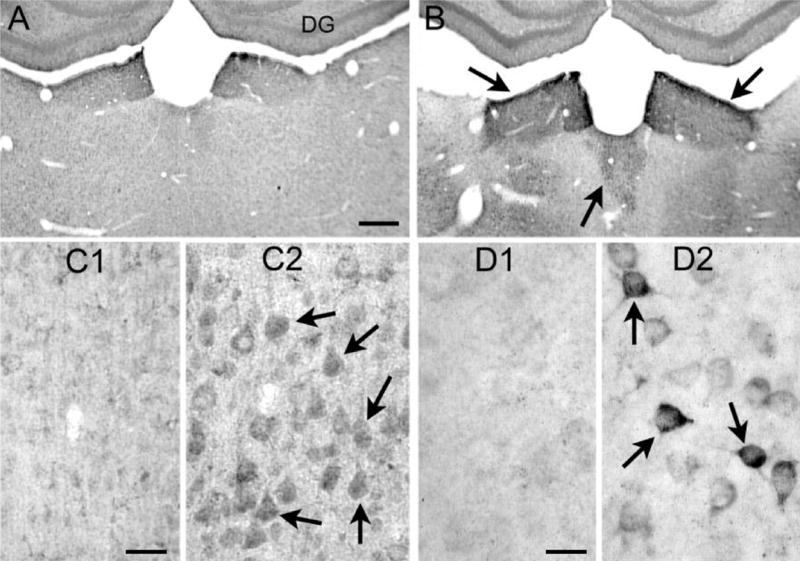
Increased VEGF protein expression in thalamus, neocortex, and amygdala 24 h after pilocarpine-induced status epilepticus. (A, B) Sections from an (A) saline- and (B) pilocarpine-treated rat that had 60 min of status epilepticus (B) were processed together using an antibody to VEGF (see Experimental Procedures). Increased VEGF protein was particularly evident in the dorsal midline thalamic nuclei, indicated by the arrows. DG=dentate gyrus. Scale bar=200 μm. (C) Sections from a (C1) saline- and (C2) pilocarpine-treated rat that had 60 min of status epilepticus (C2). Increased VEGF protein was evident in pyramidal cell somata (arrows) in temporal neocortex. Scale bar=50 μm. (D) Sections from a (D1) saline- and (D2) pilocarpine-treated rat that had 60 min of status epilepticus (D2). Increased VEGF protein was evident in somata of amygdala neurons (arrows). Scale bar=25 μm.
In addition to VEGF expression in neurons, VEGF protein was consistently evident in cells throughout the hippocampus and cortex that appeared to have a glial morphology. While both saline- and pilocarpine-treated animals showed this staining pattern, it was much more pronounced in tissue from the pilocarpine-treated rats (Fig. 3C2 versus 3C1). On these cells, staining was punctate and marginal, suggesting the possibility of cell-surface staining. To verify that these cells were, indeed, astrocytes, tissue sections treated with the VEGF antibody were also processed using an antibody to GFAP, a marker of mature astrocytes. The glia-like cells that expressed VEGF also expressed GFAP, confirming that the cells were in fact astroglia (Fig. 5). Astroglial profiles appeared larger in post-status animals than in controls, most likely due either to post-seizure reactivity or due to the effects of VEGF, which has been previously shown to induce astroglial hypertrophy (Ackerman et al., 2003).
Fig. 5.
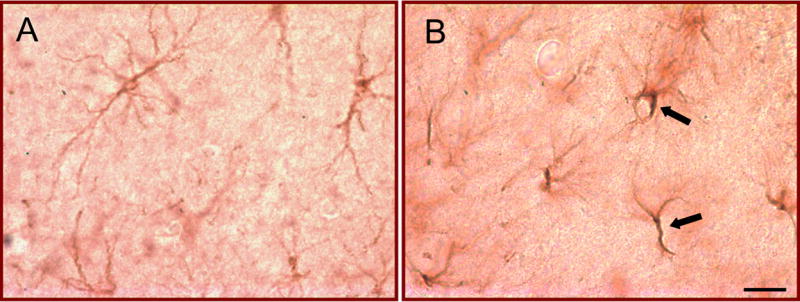
Increased VEGF protein expression in astrocytes 24 h after pilocarpine-induced status epilepticus. (A, B) Sections from a (A) saline- and (B) pilocarpine-treated rat that had 60 min of status epilepticus (B) were processed together using antibodies to both VEGF and GFAP (see Experimental Procedures). Increased VEGF protein was evident in glial profiles after status epilepticus, as indicated by the arrows. Scale bar=25 μm.
Similar patterns of VEGF protein up-regulation after status epilepticus were observed in other regions of the brain commonly implicated in seizures, including the dorsal midline thalamus and amygdala (Fig. 4), with the neuronal up-regulation particularly striking in amygdala (Fig. 4D).
Flt-Fc infusion
To determine whether endogenous VEGF serves a protective role in cell loss following status epilepticus, the VEGF blocker Flt-Fc was continuously infused by an osmotic pump for 5 days before and during status epilepticus; the pump continued to infuse for 1 day thereafter (n=5; see Experimental Procedures). Other animals were infused with hFc as a control protein (n=4). Stereological estimates of neuronal density revealed a statistically significant decrease in neuronal density in animals treated with Flt-Fc relative to their hFc controls (t(7)=3.482, P<0.01, Fig. 6). Seizure scores in the Flt-Fc-infused animals were not different than those in the hFc controls (t(7)=.412, P=0.693, data not shown), suggesting that there was no overt effect on the severity of seizures that could explain the result.
Fig. 6.
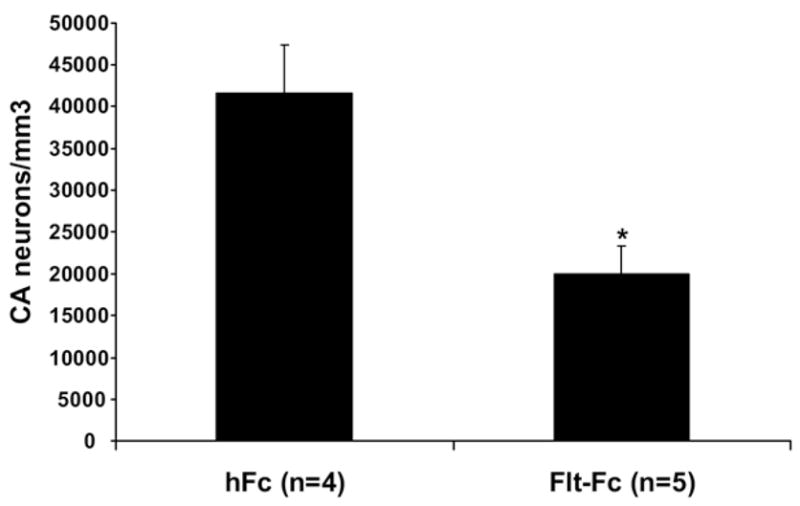
Neuronal density estimates after treatment with Flt-Fc, which sequesters endogenous VEGF, during status epilepticus. Bar graph shows significantly worse cell loss in animals treated with Flt-Fc relative to those treated with the hFc control protein. * Significantly different than hFc, P<0.01.
CA1 cell loss
Because infusions with Flt-Fc protein caused significantly more cell loss after seizures, we hypothesized that VEGF plays a neuroprotective role after status epilepticus. To further investigate this hypothesis, as well as to determine whether exogenous VEGF would provide additional protection to neurons, animals were infused with rhVEGF protein during status epilepticus. Human VEGF ELISAs used to estimate the tissue concentration of VEGF during infusions of 30 ng/day VEGF revealed an average of 7.2 ng hVEGF per mg tissue, in contrast to the 40 pg rat VEGF per mg tissue measured in post-status hippocampus (refer to Fig. 1).
Nissl staining revealed a loss of neurons 24 h after status epilepticus in CA1 of the hippocampus (Fig. 7A). Animals infused with VEGF appeared to have less neuronal loss and fewer neurons with pyknotic profiles (Fig. 7B). Neuronal density estimates, quantified stereologically, revealed that animals infused with 30 ng/day (n=10) or 60 ng/day (n=11) VEGF had significantly higher neuronal densities than animals given pilocarpine and infused with PBS or control proteins (n=14) (Fig. 7C, F(4,47)=2.577; P<0.05). It should be noted that although only CA1 was quantified, subjective evaluation of cell loss by reviewers blind to the treatment groups of the animals suggested that CA3 neurons were also present in greater density in VEGF-treated animals than in controls. Images of post-status CA3 neurons with or without VEGF treatment have already been published (Croll et al., 2004a, 2005).
Fig. 7.

VEGF-treated animals show less hippocampal damage than controls 24 h after seizures. (A, B) Representative photomicrographs of the CA1region 24 h following status epilepticus show more pyramidal cell loss in (A) PBS-infused hippocampus than in (B) rhVEGF165-infused hippocampus. Scale bars=50 μm. (C) Neuronal density estimates calculated using the optical disector method revealed that infusion of rhVEGF165 (15 ng/day (n=6), 30 ng/day (n=10), 45 ng/day (n=11), and 60 ng/day (n=11)) 5 days prior to and 1 day following pilocarpine-induced status epilepticus significantly attenuated pyramidal cell loss compared with infusion of PBS vehicle (n=14), * significantly different than PBS, P<0.05. (D) VEGF infusion at 30 ng/day does not increase vascular density in animals exposed to status epilepticus, NS. (E, F) Representative photomicrographs of the CA1 region stained with anti-RECA for detection of vascular endothelial cells 24 h following status epilepticus. No differences were detected between(A) PBS-infused hippocampus and (B) rhVEGF165-infused hippocampus. Scale bars=50μm
Comparison of seizure severity in VEGF- versus PBS-infused animals across studies revealed no significant difference in behavioral rating of status (i.e. seizure scores of 1–8; see Experimental Procedures; t(147)=1.636; P>0.104).
Vascular changes after VEGF infusions
Previous studies suggest that VEGF protects neurons via direct, receptor-mediated neuroprotection (Matsuzaki et al., 2001; Jin et al., 2001; Wick et al., 2002; Sun and Guo, 2005), but there are other possible mechanisms for its neuroprotective effect. One mechanism might be related to the fact that VEGF has vascular effects, the most pronounced of which are angiogenesis and vascular permeability.
First, VEGF infusion could have increased vascular density, thereby providing greater blood supply to the cells that need it the most, those which are metabolically overactive during status epilepticus. To rule out this possibility, we measured vascular density. There was no significant difference in vascular density (Fig. 7D, t(23)=0.654, P>0.52) or vascular diameter (data not shown, t(23)=1.03, P>0.32) between animals infused with VEGF and control animals.
Second, increased vascular permeability could have increased distance between cells in the region of VEGF diffusion, hence reducing excitotoxic transmission. To address this possibility, some animals were co-infused with both VEGF and BowAng1, a potent form of angiopoietin-1, an inhibitor of VEGF-induced vascular permeability. Animals co-treated with BowAng1 showed no reduction in neuroprotection (data not shown, F(1,20)=0.528, P=0.525).
DISCUSSION
Summary
Our results revealed that VEGF protein was dramatically up-regulated in both neurons and glia in hippocampus, thalamus, amygdala, and neocortex 24 h after pilocarpine-induced seizures. The function of this up-regulation of endogenous VEGF following seizure activity remains unknown. However, our data showed that local infusion of Flt-Fc, a VEGF trap, increased neuronal loss after status epilepticus, while infusion of exogenous VEGF into the hippocampus protected against neuronal loss. These findings suggest the possibility that the up-regulation of endogenous VEGF after seizures may serve a neuroprotective role, and that the VEGF receptor system has potential as a novel therapeutic pathway for the development of exogenous ligands to prevent cell loss after severe seizures. Vascular density was not significantly increased by the doses of VEGF used in this paradigm, and hence was not likely to contribute to the protective effect of VEGF.
VEGF immunostaining
Studies in rodents (for example, see Borges et al., 2003) and in humans (Bernasconi et al., 2003; Jutila et al., 2001) have shown that structures including hippocampus, amygdala, entorhinal, and perirhinal cortices are compromised as a result of severe seizures. Accordingly, our data revealed that VEGF is up-regulated in these same areas after pilocarpine-induced status epilepticus, suggesting a preferential induction of VEGF in vulnerable regions of brain. The up-regulation of VEGF in this model suggests that VEGF may play a role in seizures or their sequelae, although it could also be an epiphenomenon given that the correspondence between affected brain regions and VEGF up-regulation is correlational in nature. Given that VEGF has been shown to have neuroprotective effects across a wide variety of manipulations (for review see Carmeliet and Storkebaum, 2002; Storkebaum et al., 2004; Krum and Rosenstein, 2004), we first suspected that VEGF up-regulation reflected compensatory mechanisms of neurons to protect themselves from death. However, whether the levels of up-regulated VEGF would be sufficient to provide protection remained to be determined.
Neuroprotection
Our finding of decreased CA1 neuronal loss at the site of exogenous VEGF infusion suggests that VEGF has a neuroprotective effect after status epilepticus. Whether up-regulation of endogenous VEGF protein represents an adaptive response by the brain to try to protect cells is currently unproven. However, given that significant neuroprotection only occurred when exogenous VEGF was administered at doses of 30 ng/day or higher, and our ELISAs suggest that these levels may not be reached even after up-regulation, it seems unlikely that endogenous levels of VEGF are sufficient to mediate neuroprotection. However, there is an argument for a role of endogenous VEGF in neuroprotection based on the finding that Flt-Fc, which sequesters endogenous VEGF, significantly increased neuronal loss. This result strongly suggests that endogenous VEGF plays at least some role in neuronal preservation. It is possible, for instance, that endogenous VEGF has better access to its receptors, thereby providing auto-crine and paracrine trophic support in the local microenvironment.
Vascular density
VEGF protein increases vascular density in most systems in which it has been administered (for review see Carmeliet, 2000). This effect is important to consider when interpreting VEGF’s beneficial effects on neuronal survival after status epilepticus. Neurons require more glucose and oxygen, and hence more blood supply, when they increase their metabolic activity. However, the current experiments were designed to use a dose of VEGF below the doses that are optimal for angiogenesis (Croll et al., 2004b). Indeed, VEGF protein induced no increase in vascular density in the present experiments. Therefore, it seems unlikely that VEGF is mediating its protective effect by increasing the availability of vasculature.
Mechanisms of neuroprotection
The exact mechanism of VEGF’s neuroprotection after status epilepticus is currently unknown. However, elegant studies have shown that the neuroprotective effects of VEGF can be mediated via one of its receptors, VEGFR2 (Sun and Guo, 2005; Jin et al., 2001; Matsuzaki et al., 2001; Wick et al., 2002). VEGFR2 signals through the Akt cell survival pathway, and hence could be causing its protective effects through this activation (Sun and Guo, 2005; Mazure et al., 1997; Gerber et al., 1998).
While VEGFR2 is not localized to neurons constitutively, its mRNA or protein has been consistently observed on neurons in culture (Sondell et al., 2000; Matsuzaki et al., 2001) as well as in adult brain after insults such as cerebral ischemia (Croll and Wiegand, 2001) and status epilepticus (Nicoletti et al., 2005). Therefore, VEGF receptors appear to be inducible in neurons upon exposure to pathological states, and could then transduce pro-survival signals upon exposure to VEGF protein.
Recent data from our laboratories showed that VEGF decreased epileptiform activity when administered to hippocampal slices from epileptic rats (McCloskey et al., 2005). VEGF also decreased synaptic transmission in slices from normal rats. While a “quieting” effect of VEGF may be expected to result in lower seizure scores and hence a decrease in cell loss, we did not observe any significant decrease in seizure behaviors in our VEGF-infused animals. However, we still cannot rule out the possibility that subtle “quieting” effects of VEGF led to decreased excitotoxicity in the absence of observable decreases in seizure behaviors, particularly because the current experiment evaluated seizure severity based on seizure behavior rather than on electroencephalogram recordings. Additional research will be necessary to fully elucidate the mechanism of VEGF-induced neuroprotection after status epilepticus.
CONCLUSIONS
It is unlikely that exogenous VEGF will be useful as a therapeutic agent to protect neurons during severe seizures. As a large protein with multiple effects, issues of delivery and specificity of effect will be significant barricades to its use as a drug. However, if the receptor systems underlying these effects could be elucidated, small molecule neuroprotective reagents could be developed with specificity for the relevant receptors. Therefore, the finding that exogenous VEGF protein significantly protects hippocampal neurons from cell death after status epilepticus could lead the way to novel approaches to cell protection in epilepsy.
Acknowledgments
The work described was funded by Queens College Start-up Funds to S.D.C. and NS 37562 to H.E.S. The authors are grateful to Adam McLeod, Needa Waseem, Sidra Khalid, Frank Rotella, Chantal Bruno, and Dean Quinteros for technical contributions.
Abbreviations
- ANOVA
analysis of variance
- asf
area sampling fraction
- CA
cornus ammonis
- ELISA
enzyme-linked immunosorbent assay
- Flt
fetal liver kinase (receptor)
- GFAP
glial fibrillary acidic protein
- hFc
human Fc domain of IgG
- NIH
National Institutes of Health
- PBS
phosphate-buffered saline
- RECA
rat endothelial cell antigen
- ssf
section sampling fraction
- tsf
thickness sampling fraction
- VEGF
vascular endothelial growth factor
References
- Ackerman TF, Krellman JW, Fox L, Elkady A, Fuzailov E, Sideris A, Kasselman LJ, Croll SD. 2003 Abstract Viewer/Itinerary Planner, Program No. 785.5. Washington, DC: Society for Neuroscience; 2003. VEGF induces neuronal and astroglial hypertrophy in adult rat cortex independent of vascular leak or inflammation. http://sfn.scholarone.com/itin2003/index.html. [Google Scholar]
- Bellomo M, Adamo EB, Deodato B, Catania MA, Mannucci C, Marini H, Marciano MC, Marini R, Sapienza S, Giacca M. Enhancement of expression of vascular endothelial growth factor after adeno-associated virus gene transfer is associated with improvement of brain ischemia injury in the gerbil. Pharmacol Res. 2003;48:309–317. doi: 10.1016/s1043-6618(03)00128-2. [DOI] [PubMed] [Google Scholar]
- Bernasconi N, Bernasconi A, Caramanos Z, Antel SB, Andermann F, Arnold DL. Mesial temporal damage in temporal lobe epilepsy: a volumetric MRI study of the hippocampus, amygdala, and parahippocampal region. Brain. 2003;126:462–469. doi: 10.1093/brain/awg034. [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182(1):21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. VEGF gene therapy: stimulating angiogenesis or angiomagenesis? Nat Med. 2000;6:1102–1103. doi: 10.1038/80430. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Storkebaum E. Vascular and neuronal effects of VEGF in the nervous system: implications for neurological disorders. Semin Cell Dev Biol. 2002;13:39–53. doi: 10.1006/scdb.2001.0290. [DOI] [PubMed] [Google Scholar]
- Croll SD, Chesnutt CR, Greene NA, Lindsay RM, Wiegand SJ. Peptide immunoreactivity in aged rat cortex and hippocampus as a function of memory and BDNF infusion. Pharmacol Biochem Behav. 1999;64:625–635. doi: 10.1016/s0091-3057(99)00122-7. [DOI] [PubMed] [Google Scholar]
- Croll SD, Goodman JH, Scharfman HE. VEGF in epilepsy: a double-edged sword? In: Scharfman HE, Binder DK, editors. Molecular mechanisms of epileptogenesis. Georgetown, TX: Landes Bioscience; 2004a. pp. 57–68. [Google Scholar]
- Croll SD, Ransohoff RM, Cai N, Zhang Q, Martin FJ, Wei T, Kasselman LJ, Kintner J, Murphy AJ, Yancopoulos GD, Wiegand SJ. VEGF-mediated inflammation precedes angiogenesis in adult brain. Exp Neurol. 2004b;187:388–402. doi: 10.1016/j.expneurol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Croll SD, McCloskey DP, Nicoletti JN, Scharfman HE. VEGF as a seizure therapeutic: killing two birds with one stone. In: Binder DK, Scharfman HE, editors. Growth factors and epilepsy. New York: Nova Sciences; 2005. pp. 141–157. [Google Scholar]
- Croll SD, Wiegand SJ. Vascular growth factors and cerebral ischemia. Mol Neurobiol. 2001;23:121–135. doi: 10.1385/MN:23:2-3:121. [DOI] [PubMed] [Google Scholar]
- Davis S, Papadopoulos N, Aldrich TH, Maisonpierre PC, Huang T, Kovac L, Xu A, Leidich R, Radziejewska E, Rafique A, Goldberg J, Jain V, Bailey K, Karow M, Fandl J, Samuelsson SJ, Ioffe E, Rudge JS, Daly TJ, Radziejewski C, Yancopoulos GD. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nat Struct Biol. 2003;10(1):38–44. doi: 10.1038/nsb880. [DOI] [PubMed] [Google Scholar]
- Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Abe K, Itoyama Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J Cereb Blood Flow Metab. 1998;18(8):887–895. doi: 10.1097/00004647-199808000-00009. [DOI] [PubMed] [Google Scholar]
- Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Raziejewski C, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS. VEGF trap: A novel VEGF blocker with potent anti-tumor effects. Proc Natl Acad Sci U S A. 2002;99(17):7453–7461. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vivo ischemia. Proc Natl Acad Sci U S A. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Mao XO, Batteur SP, McEachron E, Leahy A, Greenberg DA. Caspase-3 and the regulation of hypoxic neuronal death by vascular endothelial growth factor. Neuroscience. 2001;108:351–358. doi: 10.1016/s0306-4522(01)00154-3. [DOI] [PubMed] [Google Scholar]
- Jutila L, Ylinen A, Partanen K, Alafuzoff I, Mervaala E, Partanen J, Vapalahti M, Vainio P, Pitkanen A. MR volumetry of the entorhinal, perirhinal, and temporal cortices in drug-refractory temporal lobe epilepsy. AJNR Am J Neuroradiol. 2001;22:1490–1501. [PMC free article] [PubMed] [Google Scholar]
- Krum JM, Rosenstein JM. New roles for VEGF in nervous tissue: beyond blood vessels. Exp Neurol. 2004;187:246–253. doi: 10.1016/j.expneurol.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. FASEB J. 2001;15:1218–1220. [PubMed] [Google Scholar]
- Mazure NM, Chen EY, Laderoute KR, Giaccia AJ. Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signlaing pathway in Haras-transformed cells through a hypoxia-inducible factor-1 transcriptional element. Blood. 1997;9:3322–3331. [PubMed] [Google Scholar]
- McCloskey DP, Croll SD, Scharfman HE. Depression of synaptic transmission by vascular endothelial growth factor in adult rat hippocampus and evidence for increased efficacy after chronic seizures. J Neurosci. 2005;28(39):8889–8897. doi: 10.1523/JNEUROSCI.2577-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, Duman RS. Gene profile of electroconvulsive seizures: Induction of neurotrophic and angiogenic factors. J Neurosci. 2003;23:10841–10851. doi: 10.1523/JNEUROSCI.23-34-10841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti JN, Shah SK, Khalid S, Atassi H, Croll SD. 2005 Abstract Viewer/Itinerary Planner, Program No. 668.12. Washington, DC: Society for Neuroscience; 2005. VEGFR2 upregulation following pilocarpine-induced status epilepticus in rat. http://sfn.scholarone.com/itin2005/index.html. [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Mather PE, Pasnikowski EM, Cai N, Corcoran T, Acheson A, Anderson K, Lindsay RM, Wiegand SJ. Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not neuroprotective. Exp Neurol. 1998;149:398–410. doi: 10.1006/exnr.1997.6737. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL. Granule-like neurons at the hilar/CA3 border after SE and their synchrony with area CA3 pyramidal cells: Functional implications of seizure-induced neurogenesis. J Neurosci. 2000;20:6144–6158. doi: 10.1523/JNEUROSCI.20-16-06144.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci. 1999;19:5731–5740. doi: 10.1523/JNEUROSCI.19-14-05731.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondell M, Sundler F, Kanje M. Vascular endothelial growth factor is a neuroprotective factor which stimulates axonal outgrowth through the flk-1 receptor. Eur J Neurosci. 2000;12:4243–4254. doi: 10.1046/j.0953-816x.2000.01326.x. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays. 2004;26(9):943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8(1):85–92. doi: 10.1038/nn1360. [DOI] [PubMed] [Google Scholar]
- Sun FY, Guo X. Molecular and cellular mechanisms of neuroprotection by vascular endothelial growth factor. J Neurosci Res. 2005;79(1–2):180–184. doi: 10.1002/jnr.20321. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest. 2003;111(12):1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6(4):460–463. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kilic E, Kilic U, Weber B, Bassetti CL, Marti HH, Hermann DM. VEGF overexpression induces post-ischaemic neuroprotection, but facilitates haemodynamic steal phenomena. Brain. 2005;128(1):52–63. doi: 10.1093/brain/awh325. [DOI] [PubMed] [Google Scholar]
- Watson RE, Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–159. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231(4):482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci. 2002;22:6401–6407. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- Yourey PA, Gohari S, Su JL, Alderson RF. Vascular endothelial cell growth factors promote the in vitro development of rat photo-receptor cells. J Neurosci. 2000;20:6781–6788. doi: 10.1523/JNEUROSCI.20-18-06781.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Nennesmo I, Fadeel B, Henter JI. Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2004;56(4):564–567. doi: 10.1002/ana.20223. [DOI] [PubMed] [Google Scholar]