

Abstract
In response to a prolonged membrane depolarization, inactivation autoregulates the activity of voltage-gated ion channels. Slow inactivation involving a localized constriction of the selectivity filter (P/C-type mechanism) is prevalent in many voltage-gated K+ channels of the Kv1 subfamily. However, the generalization of this mechanism to other Kv channel subfamilies has remained uncertain and controversial. In agreement with a “foot-in-the-door” mechanism and the presence of ion-ion interactions in the pore, elevated external K+ slows the development of P/C-type inactivation and accelerates its recovery. In sharp contrast and resembling the regulation of the hippocampal A-type K+ current, we found that Kv4.x channels associated with KChIP-1 (an auxiliary subunit) exhibit accelerated inactivation and unaffected recovery from inactivation when exposed to elevated external K+. This regulation depends on the ability of a permeant ion to enter the selectivity filter (K+ = Rb+ = NH4+ > Cs+ > Na+); and the apparent equilibrium dissociation constant of a single regulatory site is 8 mM for K+. By applying a robust quantitative global kinetic modeling approach to all macroscopic properties over a 210-mV range of membrane potentials, we determined that elevated external K+ inhibits unstable closed states outside the main activation pathway and thereby promotes preferential closed-state inactivation. These results suggest the presence of a vestigial and unstable P/C-type mechanism of inactivation in Kv4 channels and strengthen the concept of novel mechanisms of closed-state inactivation. Regulation of Kv4 channel inactivation by hyperkalemia may help to explain the pathophysiology of electrolyte imbalances in excitable tissues.
INTRODUCTION
Inactivation of voltage-gated ion channels is a time-dependent autoregulatory process that is essential to shape and orchestrate active electrical signaling in excitable tissues (1). The C-type mechanism is the most extensively studied mechanism of inactivation in voltage-gated K+ (Kv) channels within the Shaker Kv1 subfamily (2–14). It involves regions at the C-terminal side of the membrane-spanning core of the Kv channel, which includes the selectivity filter and the activation gate (2,4,15), and results from a cooperative local conformational change that constricts the external mouth of the selectivity filter (6–8,16–19). Given the central role of the narrow K+ pore in this mechanism, several studies have referred to it as P/C-type inactivation (10,13,20). Typically, the development of P/C-type inactivation in Kv1 channels is slowed by external pore blockers (TEA) and elevated external K+ (3,5,13,16,21); and the latter also accelerates the recovery from P/C-type inactivation (22,23). These hallmarks indicate that the regulation of P/C-type inactivation in Kv1 channels involves a “foot-in-the-door” mechanism at an external pore site and ion-ion interactions in the selectivity filter (4,23–26). In sharp contrast, inactivation of Kv4 channels is not affected by high concentrations of external TEA, and elevated external K+ accelerates the development of inactivation and has no effect on the recovery from inactivation (27,28), which supports the concept of an alternative mechanism of inactivation in non-Shaker Kv channels (13,29–31). Native Kv4 channels in hippocampal neurons exhibit similar regulation by external K+ (32). Although these observations are physiologically and mechanistically relevant, the underlying mechanism has been difficult to solve because fast N-type-like inactivation of Kv4 channels limits the resolution of the results and confounds their interpretation; and the kinetic analysis of Kv4 channel gating in previous studies has not been sufficiently constrained experimentally to derive a sound biophysical framework.
To gain firmer biophysical insights into the mechanism of slow Kv4 inactivation and its modulation by external K+, we investigated the heterologously expressed Kv4.x:KChIP-1 complex in the presence of normal or elevated external K+. KChIP-1 (K-channel-interacting-protein-1) is an accessory subunit that binds to the channel's N-terminus tightly and thereby remodels Kv4 inactivation gating (29,33,34). This physiologically relevant Kv4.x:KChIP-1 complex lacks a fast N-type-like mechanism of inactivation (35) because KChIP-1 sequesters the N-terminal inactivation gate constitutively (33,34,36); and therefore, the complexity of inactivation in the Kv4.x:KChIP-1 complex is reduced. By examining the macroscopic properties of the Kv4.3:KChIP-1 complex over a broad range of membrane potentials (∼200 mV), we determined that elevated external K+ has multiple effects on voltage-dependent activation and inactivation. These effects appeared to be inconsistent with the P/C-type inactivation mechanism and the “foot-in-the door” paradigm; however, additional evidence showed that the modulation by external K+ involves a single external site in the channel's selectivity filter. Highly constrained global kinetic modeling applied to all macroscopic observations revealed a feasible explanation for this unusual behavior. Essentially, external K+ regulates the presence of unstable closed states outside the main voltage-dependent activation pathway. We discuss this simple outcome in terms of its physiological impact, the presence of vestigial P/C-type inactivation in Kv4 channels, and the concept of an alternative mechanism of closed-state inactivation (CSI), which may involve the channel's activation gate.
MATERIALS AND METHODS
Molecular biology
Kv4.3 complementary DNA (cDNA) was provided by Dr. J. Nerbonne (Washington University, St. Louis, MO) and was maintained in pBK-CMV (Strategene, La Jolla, CA). KChIP-1 cDNAs for oocyte and mammalian cell expression were provided by Dr. Mark Bowlby (Weyth-Ayerst Research, Princeton, NJ) and Dr. Paul J. Pfaffinger (Baylor College of Medicine, Houston, TX), respectively. The KChIP-1 cDNAs were maintained in pBJ/KSM (W. J. Joiner, Yale University) and pCMV (Henry Jerng, Baylor College of Medicine). Kv4.2 cDNA was provided by M. Sheng (Massachusetts Institute of Technology, Cambridge, MA) and maintained in pRc-CMV (Invitrogen, Carlsbad, CA). Capped cRNA for expression in Xenopus laevis oocytes was produced by in vitro transcription using the mMessage mMachine kit driven by T7 RNA polymerase (Ambion, Austin, TX). The transfection of tsA-201 cells (provided by Dr. R. Horn, Thomas Jefferson University, Philadelphia, PA) was accomplished by the calcium-phosphate method. Cells were cotransfected with the Kv4.2 and KChIP-1 plasmid DNAs at a 1:1 mass ratio. A plasmid containing the CD8 gene (5 μg) was included in the cotransfection to allow the identification of individual transfected cells by decorating them with beads bearing anti-CD8 antibody (Dynal Biotech, Brown Deer, WI) (37).
Electrophysiology
The Kv4.3 and KChIP-1 cRNAs were microinjected into defolliculated X. laevis oocytes (∼50 ng/cell) using a Nanoject microinjector (Drummond, Broomall, PA). The currents were recorded 1–3 days postinjection using either the two-electrode voltage-clamp technique or cell-attached configuration of the patch-clamp method. The bath/pipette solution (2 mM K+; same as ND-96) contained (mM): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES (pH 7.4, adjusted with NaOH). For the 98 mM-K+ bath solution, all NaCl was substituted with KCl. Kv4.2:KChIP-1 currents expressed in tsA-201 cells were measured in the tight-seal whole-cell configuration of the patch-clamp method with the following pipette (intracellular) solution: 120 mM KF, 1 mM CaCl2, 2 mM MgCl2, 11 mM EGTA, and 10 mM HEPES, pH 7.2, adjusted with KOH; and the following external bath solution: 132 mM NaCl, 2 mM KCl, 1.5 mM CaCl2, 1 mM MgCl2, 20 mM TEA (tetraethylammonium chloride), and 10 mM HEPES, pH 7.4, adjusted with NaOH; for the 132 mM-K+ bath solution all NaCl was substituted with KCl (pH 7.4, adjusted with KOH). The calculated free [Ca2+] and [Mg2+] in the intracellular solution were ∼40 nM and ∼1.4 mM, respectively (MaxChelator, ‘WEBMAXC v2.10’; http://www.stanford.edu/∼cpatton/maxc.html). In tsA-201 whole-cell recordings, TEA was necessary to eliminate a small but significant endogenous delayed rectifier K+ current. The experiment began once current kinetics and amplitude became stable. In the whole-cell recording configuration, series resistances (Rs = 2–5 MΩ) were compensated to yield a total voltage error of <3 mV. A P/4 leak subtraction protocol consisting of four subpulses from a subsweep holding potential of −110 mV was used to subtract passive components of the total current. Currents were filtered at 2 kHz and sampled at 10 kHz. Analyses and graphical displays were produced with pClamp 9.0 (Axon Instruments, Foster City, CA) and Origin 7.5 (OriginLab, Northhampton, MA) software. All measurements were taken at room temperature (22°C–24°C), and results are expressed as mean ± SE.
Quantitative global kinetic modeling
To test specific Markov models of Kv channel gating quantitatively, we implemented a robust global kinetic modeling approach constrained simultaneously by several electrophysiological and kinetic measurements. The goal of this analysis is to determine a physiologically plausible kinetic model that quantitatively describes the time- and voltage-dependent macroscopic properties of the Kv4.3:KChIP-1 channel in the presence of 2 and 98 mM external K+ over a 210-mV range of membrane potentials. The modeling employed a modified version of the program IChSim (www.ICHMASCOT.org). In this program, the characteristic differential equations of the models are solved by numerical integration and the “Q-Matrix” method (38,39). To conduct the quantitative global kinetic modeling, all macroscopic properties were evaluated simultaneously in terms of the voltage dependencies of peak conductance, steady-state inactivation, and the time constants of activation, deactivation, and inactivation. In general, the experimental time constants were obtained from exponential fits (see below); and the best-fit parameters were obtained from the minimization of the squared differences between the experimental observation and the model according to
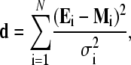 |
where Ei and Mi are the experimental observations and corresponding modeled values, respectively; N is the total number of experimental observations in the data set (N = 63 data points); and σ is the experimental standard deviation of each data point. The division by σ2 corrects for the bias that results from the large numerical differences in the absolute values of the observations (e.g., Gp-V and time constant-V relations; Fig. 1). The quantity d was minimized by using the simplex algorithm. To converge, this algorithm requires a large number of evaluations for different sets of rate constants. Thus, for a set of rate constants, a separate program generated these evaluations automatically by simulating the currents evoked by the experimental pulse protocols; and the minimization proceeded automatically until it converged and returned the best-fit model parameters. For a given model, we started by fitting the 98 K data set and all parameters were allowed to vary. Then, to fit the 2 K data set, the best-fit parameters from the 98 K data set were used as starting parameter values, and all equivalent charges in the voltage-dependent transitions were fixed; all other adjustable parameters were allowed to vary (Table 1). The maximum open probability ratio was constrained to be 1.7 (POmax-98K/POmax-2K = 0.85:0.5), which corresponds to the observed Gpmax ratio (Fig. 1 B). Assuming POmax values ranging between 0.4–0.9 did not affect the outcome of the global fits significantly. These open probabilities are comparable to those estimated from single-channel records. The total gating charge per Kv4 channel (QT) has not been measured. However, the S4 segments of Kv4 and Kv2 channels are similar and the estimated QT of Shab/Kv2 channels ranges 8–12 e0 (40), which is in agreement with the outcome of the global fit (QT = 10.9 e0).
FIGURE 1.

Gating properties of Kv4.3:KChIP-1 channels in normal and elevated external K+. (A) Whole-oocyte K+ currents evoked by step depolarizations between −90 and +70 (ΔV = 20 mV) from a holding potential of −100 mV at 2 and 98 mM KCl as indicated. Bottom panel shows overlay of traces at +70 mV. For direct comparison, the trace at 98 mM (dotted line) was scaled (thin line) and superimposed with the original trace at 2 mM. Note a faster current decay in 98 mM external K+. The arrow marks the level of the peak current in the presence of 98 mM external K+. (B) Normalized peak conductance-voltage relations in the presence of normal and elevated external K+. The peak chord conductance (G) was calculated as G = Ip/(Vm − VR), where Ip, Vm, and VR are the peak current, the test potential, and the reversal potential, respectively. The solid lines across the data points are the best fits assuming a fourth-order Boltzmann function with the parameters indicated in the graphs (V0.5 corresponds to the midpoint potential, and k is the slope factor). Note that elevated external K+ increases Gpmax. (C) Same as in B, scaled and normalized. (D) Voltage dependence of the time constants of inactivation. The combined time constants are derived from the kinetic analysis of the development of inactivation (−60 to +70 mV, circles) and the recovery from inactivation (−120, −100, −90, −80, and −70 mV, triangles) (Supplementary Material, Figs. S1 and S2). The development of inactivation at negative voltages (−60 and −50 mV) was best described by the sum of two exponential terms but was dominated by a slow component (>80%). As an approximation for the quantitative global kinetic modeling (Figs. 6 and 7), we plotted the weighted mean time constants for these voltages. The dashed lines depict the corresponding steady-state inactivation curves (best-fit Boltzmann functions with the following parameters: V0.5 (2 K) = −68 mV, k (2 K) = 4.5 mV; V0.5 (98 K) = −68 mV, k (98 K) = 4.6 mV). Data points are plotted in Figs. 6 B and 7 B. To ensure steady state in these experiments, the prepulse duration was 10 s. (E) Voltage dependence of the combined time constants of deactivation (from −140 to −70 mV, triangles) and activation (from −40 to +50 mV, circles). The corresponding current traces were recorded in the cell-attached configuration (Supplementary Material, Figs. S1 and S3). The time constants are derived from the kinetic analysis of the currents' rising phase and the tail current relaxations (Supplementary Material, Fig. S1). Between −140 and −100 mV, the tail current relaxations from channels exposed to 98 mM K+ were best described by the sum of two exponential terms; and at more depolarized voltages, a fast component was dominant (≥70%) or a simple exponential was sufficient to describe the relaxation. As an approximation for the quantitative global kinetic modeling (Figs. 6 and 7), we plotted the weighted mean time constants when the descriptive kinetic analysis yielded two time constants (Materials and Methods).
TABLE 1.
Rate constant estimates from global kinetic modeling
| Rate constant* s−1 | 98 mM KOUT Scheme IV | 2 mM KOUT Scheme V | 2 mM KOUT Scheme VI | Fold change IV vs. V | Fold change IV vs. VI |
|---|---|---|---|---|---|
| α(V = 0) | 512 | 612 | 493 | −1.2 | 1 |
| β(V = 0) | 37 | 37 | 34 | 1 | 1.1 |
| γ(V = 0) | 3512 | 248 | 271 | 14 | 13 |
| δ(V = 0) | 14.5 | 10 | 13.9 | 1.4 | 1 |
| ɛ(V = 0) | 7567 | 14,622 | 14,783 | −1.9 | −1.9 |
| Φ(V = 0) | 919 | 1356 | 658 | −1.5 | 1.4 |
| kci | 70 | 60 | 127 | 1.2 | −1.8 |
| kic | 0.045 | 0.043 | 0.041 | 1 | 1.1 |
| kxp | - | 2076 | 543 | - | - |
| kpx | - | 3000 | 800 | - | - |
Rate constants that control the voltage-dependent transitions (α, β, γ, δ, ɛ, and φ) are assumed to depend exponentially on membrane potential according to these relationships: α(V) = α(V = 0)exp(zαeV/kT), β(V) = β(V = 0)exp(zβeV/kT), γ (V) = γ (V = 0)exp(zγeV/kT), δ (V) = δ (V = 0)exp(zδeV/kT), ɛ (V) = ɛ (V = 0)exp(zɛeV/kT), φ (V) = φ (V = 0)exp(zφeV/kT) where zα, zβ, zγ, zδ, zɛ, and zφ are the corresponding equivalent electronic charges 0.72, −1.42, 0.5, −1.68, 0.051, and −0.084 e0, respectively, e is the electronic charge, V is the membrane potential, k is the Boltzmann constant, and T is the absolute temperature. All equivalent charges were estimated from modeling the 98 K data set (Materials and Methods). The total gating charge per channel assumed in these models is 10.9 e0. The allosteric factors f and g were 0.17 and 5.1 in 2 mM external K+; and 0.17 and 3.4 in 98 mM external K+, respectively. Negative and positive fold-change values denote a decrease and increase in the corresponding parameter, respectively.
To compare the best fits to different models (Table 2), we estimated the badness of fit, b, according to
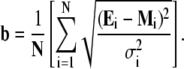 |
For the total number of individual experimental values (N) in the global data sets, this expression yields the average error of the global fit relative to the experimental σ (badness of fit increases as b increases).
TABLE 2.
Best-fit model comparison (badness of fit)
NC = program did not converge. Badness of fit b was calculated as explained in Materials and Methods. Accordingly, the best global fit improves as b decreases. Schemes IV, V, and VI produced equivalent best-global fits for the 98 K data set (b = 0.6) because Schemes V and VI reduce to Scheme IV when the Ci state is virtually eliminated (Results; Figs. 4 and 5).
Limitations of kinetic modeling
The strength of our global kinetic modeling method rests on the simultaneous evaluation of multiple macroscopic measurements, which investigate activation, deactivation, and inactivation over a broad range of physiologically relevant membrane potentials. However, there is a potential limitation because it relies on approximating the kinetics as simple exponential relaxations (inactivation and deactivation) and fourth-order exponential relaxations (activation) to extract the observed time constants. Although for most of the membrane potential range these are excellent approximations (Supplementary Material, Figs. S1 and S2) at hyperpolarized voltages (−140 to −100 mV) tail currents were best described by the sum of two exponentials. Thus, to include this voltage range in the modeling, we plotted the weighted mean time constant as an approximation of deactivation kinetics. Tail current description by assuming the sum of exponential terms suggests complexities that the examined models are not explicitly accounting for. For instance, the modeled open state may be an aggregate of distinct open states, which is consistent with the presence of subconducting states at the single-channel level in Kv4 channels (36). Keeping this limitation in mind, we will show that a reduced set of models can account for the overall effects of external K+ on deactivation and all other macroscopic properties combined.
RESULTS
Modulation of the Kv4.3:KChIP-1 channel gating by external K+
To investigate the biophysical basis of the modulation of Kv4 channels by external K+, we examined the macroscopic voltage- and time-dependent properties of the heterologously expressed Kv4.3:KChIP-1 complex over a broad range of membrane potentials (∼200 mV). Examining this binary complex was advantageous because it is physiologically relevant (29,41) and lacks fast N-type inactivation. The latter was especially important because it allowed us to focus on the slower inactivation process without kinetic interference, which may confound the interpretation of the data. Relative to a normal concentration (2 mM), elevated external K+ (98 mM) modulated activation, inactivation, and deactivation of the Kv4.3:KChIP-1 complex. As expected, bath exposure to 98 mM K+ suppressed the peak current; however, this inhibition appeared to be significantly less than that predicted by the reduction in driving force (Fig. 1 A). Analysis of the peak chord-conductance-voltage relations (Gp-V curves) demonstrated that elevated external K+ increases the maximum Gp (Fig. 1 B), which suggests that Kv4.3 channels under normal ionic conditions operate at a submaximal open probability and that elevated external K+ increases the maximum open probability. In addition, consistent with a relative stabilization of the channel's open state and in agreement with the classical modulation of K+ channel gating by permeant ions (24,42), the Gp-V curve in the presence of 98 mM external K+ was leftward shifted (−16 mV; Fig. 1, B and C). A particularly intriguing feature of the modulation was that elevated external K+ uniformly reduced the time constant of inactivation by approximately two- to threefold over a broad range of membrane potentials (−60 to +70 mV; Fig. 1 D). This effect did not affect the voltage dependence of steady-state inactivation (Fig. 1 D) but is opposite to the classical predictions of a “foot-in-the-door” mechanism acting on the pore gate to regulate P/C-type inactivation, which is typically slowed by elevated external permeant ions (5). It is also significant that, between −120 and −70 mV, the recovery from inactivation of the Kv4.3:KChIP-1 complex was not affected by elevated external K+ (Fig. 1 D; Supplementary Material, Fig. S2). Under similar conditions, recovery from P/C-type inactivation is typically accelerated through interactions in the selectivity filter of Shaker Kv1 channels (22,23). In addition, the Kv4.3:KChIP-1 complex exhibited accelerated deactivation in the presence of 98 mM external K+ at hyperpolarized membrane potentials (−140 to −90 mV; Fig. 1 E). Similarly, activation kinetics between −30 and +50 mV is slightly faster (Fig. 1 E; Supplementary Material, Fig. S1). The acceleration of inactivation and deactivation of the Kv4.3:KChIP-1 channel complex by elevated external K+ is consistent with a previously reported kinetic relationship between these processes (27,43,44). More typically, however, elevated external concentrations of permeant ions slow deactivation of native and recombinant Kv channels at hyperpolarized membrane potentials (24,27,45).
Overall, this unusual modulation and similar observations reported previously (13,29,31) have suggested distinct and poorly understood mechanisms of inactivation that must exist in non-Shaker Kv channels. Ruling out that the observed modulation of inactivation resulted from the recording conditions or an active role of KChIP-1, we found qualitatively identical results in cell-attached macropatches or when the N-terminal-deleted Kv4.3 channel (Δ2–19 Kv4.3) was expressed alone (Supplementary Material, Fig. S3; Fig. 2). Furthermore, other results showed that this modulation was not dependent on the expression system or the Kv4 channel isoform (see below). The observation of similar results in different recording configurations and expression systems that produce a broad range of current magnitudes (pA–μA) also rules out a significant impact of uncompensated series resistance on the reported results.
FIGURE 2.

Gating properties of the Δ2–19 Kv4.3 channel in normal and elevated external K+. (A) Whole-oocyte outward K+ currents evoked by step depolarizations between −90 and +70 (ΔV = 20 mV) from a holding potential of −100 mV. Bottom panel shows overlay of traces at +70 mV. For direct comparison, the traces were scaled and superimposed. Note that elevated external K+ barely decreases the peak current and induces faster current decay. (B) Normalized peak conductance-voltage relations in the presence of normal and elevated external K+. The peak chord conductance (Gp) was calculated as explained in the Fig. 1 legend. The lines across the data points are the best fits assuming a fourth-order Boltzmann function with the parameters indicated in the graphs (V0.5 corresponds to the midpoint potential and k is the slope factor). Note that elevated external increases Gpmax. (C) Same as in B, normalized. (D) Voltage dependence of combined time constants of recovery from inactivation (from −120 to −60 mV, triangles) and inactivation (from −50 to +70 mV, circles). (E) Relation between prepulse potential and the normalized peak current. The relation was described by assuming a Boltzmann distribution with the following best-fit parameters: V0.5 (2 K) = −67 mV, k (2 K) = 5 mV; V0.5 (98 K) = −70 mV, k (98 K) = 5 mV.
The modulation of the Kv4.2:KChIP-1 complex by external K+ involves an external site in the selectivity filter of the channel
To test whether the effects described above are mediated by the binding of external K+ to the selectivity filter of the Kv4 channel, we examined the permeant ion and concentration dependencies of the acceleration of inactivation in the Kv4.2:KChIP-1 channel complex expressed in tsA-201 cells. According to typical permeability sequences of highly selective K+ channels (1), we found that highly permeant ions (K+ and Rb+) are similarly effective in accelerating the development of inactivation. Although 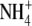 was also an effective modulator, other weakly permeant ions (Cs+ and Na+) have little or no effect on inactivation (Fig. 3, A and B). That is, inactivation kinetics in the presence of Cs+ and Na+ are similar to those observed in 2 mM external K+. Also, we found that the time constant of the development of inactivation depends on the concentration of external K+ in a manner that is consistent with the presence of a single external modulatory binding site (KD = 8 mM, nH = 1; Fig. 3 C). Altogether, these observations suggest strongly that the channel's selectivity filter plays a central role in the modulation of inactivation of the Kv4.x:KChIP-1 complex by external K+.
was also an effective modulator, other weakly permeant ions (Cs+ and Na+) have little or no effect on inactivation (Fig. 3, A and B). That is, inactivation kinetics in the presence of Cs+ and Na+ are similar to those observed in 2 mM external K+. Also, we found that the time constant of the development of inactivation depends on the concentration of external K+ in a manner that is consistent with the presence of a single external modulatory binding site (KD = 8 mM, nH = 1; Fig. 3 C). Altogether, these observations suggest strongly that the channel's selectivity filter plays a central role in the modulation of inactivation of the Kv4.x:KChIP-1 complex by external K+.
FIGURE 3.

Modulation of Kv4.2:KChIP-1 channels by external permanent ions. (A) The overlay of traces at +50 mV in the presence of 132 mM K+ (black trace) or 132 mM of the test monovalent cation as indicated (gray trace). (B) Inactivation time constants versus permeability ratio. Permeability ratios under bionic conditions were calculated by applying the Goldman-Hodgkin-Katz equation (1). (C) Time constants of inactivation at +70 mV against concentration of external K+. Solid line is the best-fit Hill equation with the apparent equilibrium dissociation constant (KD) of 8 mM and a Hill coefficient, nH = 1. Inset shows normalized currents at +70 mV obtained by application of various external K+ concentration (0, 2, 5, 15, 50, and 132 mM). Note the gradual acceleration of the development of inactivation upon raising external [K+].
Modeling gating of the Kv4.x:KChIP-1 complex and its modulation by external K+
Four salient features characterize the modulation of the Kv4:KChIP-1 complex by elevated external K+: 1), accelerated inactivation, 2), increased Gpmax, 3), a leftward-shifted Gp-V relation, and 4), accelerated activation and deactivation. This combination of effects is generally inconsistent with traditional predictions of the “foot-in-the-door” paradigm involving the selectivity filter and the gating machinery. At the same time, however, ion substitution experiments and the concentration-response relation suggest that an external K+ site in the selectivity filter contributes to the modulation. Therefore, to determine a viable solution that explains the unusual modulation of the Kv4:KChIP-1 complex by elevated external K+, the biophysical framework must account for pore interactions that either directly or indirectly may produce broad effects on activation and inactivation gating.
Previous studies have proposed CSI as a significant pathway of inactivation in Kv4 channels (27,36,43,44,46). However, the importance of this pathway in Kv4 channels has remained controversial (47,48). To reassess the mechanistic viability of CSI under more stringent conditions and explain gating modulation by external K+, we applied a quantitative global kinetic modeling approach to test physiologically plausible Markov models (Schemes I–VI; Figs. 4 and 5) constrained simultaneously by complete sets of macroscopic measurements encompassing time- and voltage-dependent gating over a broad range of membrane potentials (210 mV; Fig. 1; Materials and Methods). First, we considered kinetic models proposed previously to explain Kv4 channel inactivation (Schemes I and II) (43,46,47) and applied them to the 98 K data set. Scheme I was readily ruled out because it does not predict the bell-shaped voltage dependence of the time constants of inactivation (Supplementary Material, Fig. S4), a hallmark of our measurements (Fig. 1 D); and attempts to obtain a global fit with Scheme I did not converge (Table 2). Scheme II describes the overall features of the data qualitatively; however, the global best fit is relatively poor because it does not accurately describe the voltage dependencies of the time constants of activation, deactivation, and inactivation (Fig. 6, C and D; Table 2). Thus, to account for the 98 K data set more accurately and globally, we built Scheme IV (Fig. 5), which results from adding an inactivation-permissive preopen closed state (C5) to Scheme II (Fig. 5). Scheme IV encompasses two parallel pathways: 1), a sequential activation pathway (Ci) that includes strongly voltage-dependent activation (corresponding to a total charge movement of 8.6 e0) and two final weakly voltage-dependent transitions (corresponding to a total charge movement of 2.3 e0) leading to the opening of the channel; and 2), a CSI pathway (Ii) coupled to the Ci pathway by the allosteric factor (f). There is no open-state inactivation because KChIP-1 eliminates N-type-like inactivation in Kv4 channels (Introduction). The preopen closed state C5 has been proposed for Kv channels in previous studies (49,50). Scheme IV was sufficient to produce a significantly improved global best fit that accounts for the voltage dependencies of all time-dependent variables accurately (Fig. 6; Tables 1 and 2). Moreover, the highly constrained global fit demonstrates that CSI is sufficient to explain the gating of the Kv4.3:KChIP-1 complex.
FIGURE 4.

Putative kinetic schemes of Kv4:KChIP-1 channel gating: 5T models. These kinetic schemes include five transitions in the activation pathway and are based on the models proposed by Wang et al. (Scheme I), Beck and Covarrubias, and Bähring et al. (Schemes II and III) (43,46,47). Sequential transitions between C0 and C4 correspond to activation steps governed by the strongly voltage-dependent rate constants α and β. The cooperative opening equilibrium is governed by the weakly voltage-dependent rate constants ɛ and φ (or by voltage-independent rate constants k1 and k2 in Scheme I). Transitions to and from inactivated states in all schemes are voltage independent. Transitions to and from P6 are also voltage-independent, and the forward transition (kxp) decreases as external [K+] increases. Thus, the latter predicts virtual elimination of P6 at saturating concentrations of external K+; and therefore, Scheme III reduces to Scheme II.
FIGURE 5.

Putative kinetic schemes of Kv4:KChIP-1 channel gating: 6T models. These kinetic schemes include six transitions in the activation pathway and are based on the models proposed by Ayer and Sigworth, Beck and Covarrubias, Bähring et al., Klemic et al., and Kurata et al. (31,43,46,50,51). Sequential transitions between C0 and C4 correspond to activation steps governed by the strongly voltage-dependent rate constants α and β. Opening occurs in two cooperative and weakly voltage-dependent steps as proposed by Schoppa and Sigworth (49). This is the difference between Schemes II (Fig. 4) and IV. Scheme V assumes an additional closing transition outside the main activation pathway as explained in Fig. 4 legend; and Scheme VI further assumes that this transition may occur equally well from all states in the main activation pathway. The forward rate constant (kxp) of the transitions between the Ci pathway and the Pi pathway decreases as external [K+] increases. Thus, the latter predicts virtual elimination of P6 at saturating concentrations of external K+; and therefore, Schemes V and VI reduce to Scheme IV.
FIGURE 6.

Global kinetic modeling of Kv4.3:KChIP-1 channel gating in 98 mM external K+. The observations and best global fit (assuming Schemes II and IV) are shown in gray, blue, and red, respectively. For direct comparison against the theoretical values, all experimental observations are replotted from Fig. 1. (A) Family of Kv4.3:KChIP-1 current traces. (B) Gp-V relation and steady-state inactivation curve. (C) Voltage dependence of the time constants of recovery from inactivation and the development of inactivation. The inset in this graph shows an expanded view between −50 and +80 mV. (D) Voltage dependence of the time constants of activation and deactivation. The global best-fit parameters are summarized in Table 1, and badness of the fit model comparisons are summarized in Table 2.
Then, we turned to the 2 K data set and determined that Scheme IV was insufficient to account for the results globally because it did not describe deactivation (Fig. 7 D) and inactivation kinetics at positive voltages (Fig. 7 C, inset; Table 2). A better solution hypothesized the presence of an unstable P/C-type inactivated state to explain the permeant ion and dose dependencies (Fig. 3). Scheme V, which results from simply adding a short-lived closed state coupled to the open state in Scheme IV (P6), is the explicit representation of this hypothesis. Assuming Scheme V, the global best fit for the 2 K data set was excellent (Fig. 7; Tables 1 and 2). Therefore, elevated external K+ may simply eliminate the O→P6 transition through a “foot-in-the-door” mechanism that prevents a rapid but unstable pore collapse and thus stabilizes the open state. This explanation implies that the forward rate constant governing the O→P6 transition (kxp) decreases as the concentration of external K+ increases, which is in agreement with the gradual inverse relationship between the time constant of inactivation and the concentration of external K+ (Fig. 3 C). That is, to explain the K+-dependent modulation, Scheme V reduces to Scheme IV at saturating concentrations of external K+. Neither Scheme II nor Scheme II plus the P6 state (Scheme III) produced satisfactory global fits for the 2 K data set (Fig. 4; Supplementary Material, Fig. 5S; Table 2).
FIGURE 7.

Global kinetic modeling of Kv4.3:KChIP-1 channel gating in 2 mM external K+. The observations and best global fit (assuming Schemes IV and VI) are shown in gray, black, and olive, respectively. The best global fit assuming Scheme V was virtually indistinguishable from Scheme VI shown here. For direct comparison against the theoretical values, all experimental observations are replotted from Fig. 1. (A) Family of Kv4.3:KChIP-1 current traces. (B) Gp-V relation and steady-state inactivation curve. (C) Voltage dependence of the time constants of recovery from inactivation and the development of inactivation. The inset in this graph shows an expanded view between −50 and +80 mV. (D) Voltage dependence of the time constants of activation and deactivation. The global best-fit parameters are summarized in Table 1, and badness of the fit model comparisons are summarized in Table 2.
Due to the relatively complex gating and small unitary conductance of Kv4 channels, the demonstration of the short-lived closed state at the single-channel level is still lacking. However, further modeling showed an alternative solution, which explains the 2 K data set without predicting rapid gating at the unitary level. This solution (Scheme VI, Fig. 5) is a variation of a coupled CSI kinetic model proposed by Klemic et al. and Kurata et al. (31,51). Essentially, to build Scheme VI, we added a parallel upper pathway of closed states (Pi) to Scheme IV. This modification assumes that channels may also undergo unstable P/C-type CSI from resting and activated states (52) and that the bottom inactivation pathway is responsible for most of the observable macroscopic inactivation. Importantly, it is critical to note that both Schemes V and VI result from adding the same pair of additional adjustable parameters to Scheme IV. The main difference between Schemes V and VI is that the latter allows the putative closing transition to occur from all states in the activation pathway. Therefore, the total number of states increases, but the number of adjustable parameters remains the same. Scheme VI yielded an excellent global best fit that accounts for the 2 K data set (Fig. 7; Tables 1 and 2). The successful application of Schemes IV, V, and VI to describe the electrophysiological properties of the Kv4.3:KChIP-1 complex and its modulation by K+ explains how the elevation in the external [K+] ultimately accelerates macroscopic inactivation. Basically, the rate constant kxp slows dramatically by saturating an external K+ site in the pore. Therefore, the relatively unstable Pi states are virtually eliminated and the Pomax increases. The latter increases the effective closing rate and the occupancy of the preopen inactivation-permissive state C5 before significant inactivation begins to develop. The increase in the C5 probability is directly responsible for the observed acceleration of inactivation. Note that except for the elimination of the Pi state(s) and a 14-fold increase in the preopening rate constant γ, most adjustable parameters did not change or changed less than twofold when comparing the outcomes of the 2 K and 98 K best global fits (Table 1). Overall, these analyses strongly support the prominent role of CSI in Kv4 channels and suggest a sound mechanistic working hypothesis that explains the modulation of the Kv4.3:KChIP-1 complex by external K+.
DISCUSSION
To our knowledge, this study is the first attempt to apply highly constrained global kinetic analysis to investigate the gating of Kv4 channels and provide a sound biophysical framework that explains inactivation and the modulation of these channels by external K+. The systematic kinetic analysis of the Kv4:KChIP-1 complex lead to a reduced set of plausible gating models (6T Models, Fig. 5) that can describe macroscopic gating quantitatively over a 210 mV range of membrane potentials and under two different ionic conditions (2 and 98 mM external K+). Although the badness of the fit was slightly smaller for Scheme VI relative to Scheme V (Table 2), the available macroscopic data sets did not allow a conclusive discrimination between these models. This outcome is expected because both schemes similarly assume the presence of an unstable closed state or a set of unstable closed states outside the main activation pathway. Furthermore, this presence depends on the concentration of external K+ in both schemes. The unanswered questions concern the determination of the most likely pathway: is it a short-lived closed state linked to the open state only; or is it a parallel pathway of unstable closed states originating from all states in the activation pathway? Kinetic analysis of single-channel recordings outside the scope of this study would help to answer these questions. Regardless of the exact pathway and from a molecular perspective, we hypothesize that these unstable closed states correspond to vestigial P/C-type inactivation in Kv4 channels because elevating the concentration of external K+ suppresses a putative pore collapse as predicted by the “foot-in-the-door” paradigm. Conversely, the pore collapse would become more favorable when the external K+ is nominally reduced to zero, as shown by others (53). The low kinetic stability of the pore collapse may result from overall high structural stability of the Kv4 selectivity filter, which makes it resistant to P/C-type inactivation. Indeed, several residues that may increase the structural stability of the conducting selectivity filter and oppose P/C-type inactivation in Kv1 channels (5,13,18,25) are found naturally at critical highly conserved positions in regions encompassing the pore loop and the S6 segment of Kv4 channels. For instance, Lys, Tyr, Val, and Ser in Kv4 channels occupy positions equivalent to Ala419, Trp435, Thr449, and Ala463 in the Shaker-B Kv channel, respectively (13,14,29). Also, charged and polar residues (Lys, Ser, or Glu) are found in Kv4 channels at a position normally occupied by Val451 in the Shaker-B Kv channel (14).
A putative mechanism of slow inactivation in Kv4 channels
If the pore collapse underlying the P/C-type mechanism is not stable, how do Kv4:KChIP-1 channels ultimately inactivate so effectively? Distinct slow inactivation phenotypes in Kv channels may result from the way the voltage sensors (VS) communicate with two distinct gates: the external pore gate involving the selectivity filter and the internal activation gate involving the S6 “bundle crossing” (P-gate and A-gate, respectively). If the VS interacts with a P-gate that readily adopts a stable collapsed conformation, it promotes and slowly stabilizes P/C-type inactivated states as found in Shaker-B Kv channels (10,14,19). The latter arises from slow gating charge “immobilization” (10,11,13,14,19). Kv4 channels also undergo profound slow gating charge “immobilization” at hyperpolarized membrane potentials (K. Dougherty, J. A. De Santiago-Castillo, and M. Covarrubias, unpublished results); and yet, the results of this study do not support a major contribution of a P/C-type mechanism to Kv4 channel inactivation. Thus, if the Kv4 P-gate is resistant to collapse (see above) and weakly coupled to the VS, the selectivity filter may spontaneously fluctuate between the collapsed and conducting conformations, and slow VS “immobilization” would not stabilize the collapsed selectivity filter and may in fact stabilize its conducting conformation (54). Conceivably, CSI of Kv4 channels may then result from a conformational change at the A-gate (55). Although the presence of such a conformational change and its mechanism remain to be investigated, it may also explain the so-called “U-type” inactivation in other Kv channels (13,30,31) and an apparent mutually exclusive interaction between slow inactivation and fast N-type-like inactivation in Kv4 channels, which is relieved by KChIP-1 or deletion of the N-terminus (36,46). Whether the collapse of the P-gate and the putative closed-inactivated conformation of the A-gate can coexist is uncertain; however, such an assumption is not necessary to explain the observations reported here.
Shedding light on the mechanisms of inactivation and modulation of Kv4 channels by external K+ is physiologically relevant because ischemia and renal dysfunction may induce hyperkalemia that is in the range of the apparent KD of the modulatory site (56); and this hyperkalemia may trigger cardiac arrhythmias (57). Furthermore, external K+ also accelerates inactivation of the hippocampal somatodendritic A-type K+ current with an apparent affinity of ∼10 mM (32); and most likely, this current is mediated by a Kv4 channel complex, which is associated with long-term potentiation (29,58–60).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We are grateful to Drs. Carol Deutsch, Richard Horn, and Toshinori Hoshi for their critical reading of the manuscript. Also, we thank Mr. Aditya Bhattacharji and Mr. Benjamin Kaplan for their skillful technical help with Xenopus oocyte experiments and the rest of the Covarrubias lab for constructive feedback.
This work was supported by National Institutes of Health research grant R01 NS032337.
Yu. A. Kaulin and J. A. De Santiago-Castillo contributed equally to this work.
Editor: Kenton J. Swartz.
References
- 1.Hille, B. 2001. Ionic Channels of Excitable Membranes. Sinauer Associates, Sunderland, MA.
- 2.Hoshi, T., W. N. Zagotta, and R. W. Aldrich. 1991. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 7:547–556. [DOI] [PubMed] [Google Scholar]
- 3.Choi, K. L., R. W. Aldrich, and G. Yellen. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 88:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yellen, G. 1998. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31:239–295. [DOI] [PubMed] [Google Scholar]
- 5.Lopez-Barneo, J., T. Hoshi, S. H. Heinemann, and R. W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- 6.Liu, Y., M. E. Jurman, and G. Yellen. 1996. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 16:859–867. [DOI] [PubMed] [Google Scholar]
- 7.Panyi, G., Z. Sheng, and C. Deutsch. 1995. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys. J. 69:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogielska, E. M., W. N. Zagotta, T. Hoshi, S. H. Heinemann, J. Haab, and R. W. Aldrich. 1995. Cooperative subunit interactions in C-type inactivation of K channels. Biophys. J. 69:2449–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiss, L., J. LoTurco, and S. J. Korn. 1999. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 76:253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loots, E., and E. Y. Isacoff. 1998. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. J. Gen. Physiol. 112:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olcese, R., R. Latorre, L. Toro, F. Bezanilla, and E. Stefani. 1997. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J. Gen. Physiol. 110:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmusson, R. L., M. J. Morales, S. Wang, S. Liu, D. L. Campbell, M. V. Brahmajothi, and H. C. Strauss. 1998. Inactivation of voltage-gated cardiac K+ channels. Circ. Res. 82:739–750. [DOI] [PubMed] [Google Scholar]
- 13.Kurata, H. T., and D. Fedida. 2006. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 92:185–208. [DOI] [PubMed] [Google Scholar]
- 14.Larsson, H. P., and F. Elinder. 2000. A conserved glutamate is important for slow inactivation in K+ channels. Neuron. 27:573–583. [DOI] [PubMed] [Google Scholar]
- 15.Panyi, G., and C. Deutsch. 2006. Cross talk between activation and slow inactivation gates of Shaker potassium channels. J. Gen. Physiol. 128:547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiss, L., and S. J. Korn. 1998. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys. J. 74:1840–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- 18.Cordero-Morales, J. F., L. G. Cuello, Y. Zhao, V. Jogini, D. M. Cortes, B. Roux, and E. Perozo. 2006. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 13:311–318. [DOI] [PubMed] [Google Scholar]
- 19.Gandhi, C. S., E. Loots, and E. Y. Isacoff. 2000. Reconstructing voltage sensor-pore interaction from a fluorescence scan of a voltage-gated K+ channel. Neuron. 27:585–595. [DOI] [PubMed] [Google Scholar]
- 20.De Biasi, M., H. A. Hartmann, J. A. Drewe, M. Taglialatela, A. M. Brown, and G. E. Kirsch. 1993. Inactivation determined by a single site in K+ pores. Pflugers Arch. 422:354–363. [DOI] [PubMed] [Google Scholar]
- 21.Baukrowitz, T., and G. Yellen. 1995. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 15:951–960. [DOI] [PubMed] [Google Scholar]
- 22.Levy, D. I., and C. Deutsch. 1996. A voltage-dependent role for K+ in recovery from C-type inactivation. Biophys. J. 71:3157–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy, D. I., and C. Deutsch. 1996. Recovery from C-type inactivation is modulated by extracellular potassium. Biophys. J. 70:798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swenson, R. P., Jr., and C. M. Armstrong. 1981. K+ channels close more slowly in the presence of external K+ and Rb+. Nature. 291:427–429. [DOI] [PubMed] [Google Scholar]
- 25.Ogielska, E. M., and R. W. Aldrich. 1999. Functional consequences of a decreased potassium affinity in a potassium channel pore. Ion interactions and C-type inactivation. J. Gen. Physiol. 113:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ray, E. C., and C. Deutsch. 2006. A trapped intracellular cation modulates K+ channel recovery from slow inactivation. J. Gen. Physiol. 128:203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shahidullah, M., and M. Covarrubias. 2003. The link between ion permeation and inactivation gating of Kv4 potassium channels. Biophys. J. 84:928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jerng, H. H., and M. Covarrubias. 1997. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys. J. 72:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jerng, H. H., P. J. Pfaffinger, and M. Covarrubias. 2004. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol. Cell. Neurosci. 27:343–369. [DOI] [PubMed] [Google Scholar]
- 30.Klemic, K. G., C. C. Shieh, G. E. Kirsch, and S. W. Jones. 1998. Inactivation of Kv2.1 potassium channels. Biophys. J. 74:1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klemic, K. G., G. E. Kirsch, and S. W. Jones. 2001. U-type inactivation of Kv3.1 and Shaker potassium channels. Biophys. J. 81:814–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirichok, Y. V., A. V. Nikolaev, and O. A. Krishtal. 1998. [K+]out accelerates inactivation of Shal-channels responsible for A-current in rat CA1 neurons. Neuroreport. 9:625–629. [DOI] [PubMed] [Google Scholar]
- 33.Wang, H., Y. Yan, Q. Liu, Y. Huang, Y. Shen, L. Chen, Y. Chen, Q. Yang, Q. Hao, K. Wang, and J. Chai. 2007. Structural basis for modulation of Kv4 K+ channels by auxiliary KChIP subunits. Nat. Neurosci. 10:32–39. [DOI] [PubMed] [Google Scholar]
- 34.Pioletti, M., F. Findeisen, G. L. Hura, and D. L. Minor Jr. 2006. Three-dimensional structure of the KChIP1-Kv4.3 T1 complex reveals a cross-shaped octamer. Nat. Struct. Mol. Biol. 13:987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gebauer, M., D. Isbrandt, K. Sauter, B. Callsen, A. Nolting, O. Pongs, and R. Bähring. 2004. N-type inactivation features of Kv4.2 channel gating. Biophys. J. 86:210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beck, E. J., M. Bowlby, W. F. An, K. J. Rhodes, and M. Covarrubias. 2002. Remodelling inactivation gating of Kv4 channels by KChIP-1, a small-molecular-weight calcium binding protein. J. Physiol. (Lond.). 538:691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jurman, M. E., L. M. Boland, Y. Liu, and G. Yellen. 1994. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 17:876–881. [PubMed] [Google Scholar]
- 38.Radharkrishnan, K. A., and A. C. Hindmarsh. 1993. Description and Use of LSODE, the Livermore Solver of Ordinary Differential Equations. Technical Report UCRL-ID-113855. NASA Reference Publication 1327.
- 39.Colquhoun, D., and A. G. Hawkes. 1995. A Q-matrix cookbook: how to write only one program to calculate the single-channel and macroscopic predictions for any kinetic mechanism. In Single-Channel Recording. Plenum Publishing, New York.
- 40.Islas, L. D., and F. J. Sigworth. 1999. Voltage sensitivity and gating charge in Shaker and Shab family potassium channels. J. Gen. Physiol. 114:723–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jerng, H. H., K. Kunjilwar, and P. J. Pfaffinger. 2005. Multiprotein assembly of Kv4.2, KChIP3 and DPP10 produces ternary channel complexes with ISA-like properties. J. Physiol. 568:767–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matteson, D. R., and R. P. Swenson Jr. 1986. External monovalent cations that impede the closing of K channels. J. Gen. Physiol. 87:795–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beck, E. J., and M. Covarrubias. 2001. Kv4 channels exhibit modulation of closed-state inactivation in inside-out patches. Biophys. J. 81:867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jerng, H. H., M. Shahidullah, and M. Covarrubias. 1999. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J. Gen. Physiol. 113:641–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zagotta, W. N., T. Hoshi, J. Dittman, and R. W. Aldrich. 1994. Shaker potassium channel gating. II: Transitions in the activation pathway. J. Gen. Physiol. 103:279–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bähring, R., L. M. Boland, A. Varghese, M. Gebauer, and O. Pongs. 2001. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J. Physiol. 535:65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang, S., V. E. Bondarenko, Y. J. Qu, G. C. Bett, M. J. Morales, R. L. Rasmusson, and H. C. Strauss. 2005. Time- and voltage-dependent components of Kv4.3 inactivation. Biophys. J. 89:3026–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel, S. P., and D. L. Campbell. 2005. Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J. Physiol. 569:7–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schoppa, N. E., and F. J. Sigworth. 1998. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J. Gen. Physiol. 111:313–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ayer, R. K. Jr., and F. J. Sigworth. 1997. Enhanced closed-state inactivation in a mutant Shaker K+ channel. J. Membr. Biol. 157:215–230. [DOI] [PubMed] [Google Scholar]
- 51.Kurata, H. T., K. W. Doerksen, J. R. Eldstrom, S. Rezazadeh, and D. Fedida. 2005. Separation of P/C- and U-type inactivation pathways in Kv1.5 potassium channels. J. Physiol. 568:31–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Claydon, T. W., M. Vaid, S. Rezazadeh, D. C. Kwan, S. J. Kehl, and D. Fedida. 2007. A direct demonstration of closed-state inactivation of K+ channels at low pH. J. Gen. Physiol. 129:437–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eghbali, M., R. Olcese, M. M. Zarei, L. Toro, and E. Stefani. 2002. External pore collapse as an inactivation mechanism for Kv4.3 K+ channels. J. Membr. Biol. 188:73–86. [DOI] [PubMed] [Google Scholar]
- 54.Olcese, R., D. Sigg, R. Latorre, F. Bezanilla, and E. Stefani. 2001. A conducting state with properties of a slow inactivated state in a shaker K+ channel mutant. J. Gen. Physiol. 117:149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin, K. S., C. Maertens, C. Proenza, B. S. Rothberg, and G. Yellen. 2004. Inactivation in HCN channels results from reclosure of the activation gate: desensitization to voltage. Neuron. 41:737–744. [DOI] [PubMed] [Google Scholar]
- 56.Leis, J. A., L. K. Bekar, and W. Walz. 2005. Potassium homeostasis in the ischemic brain. Glia. 50:407–416. [DOI] [PubMed] [Google Scholar]
- 57.Parham, W. A., A. A. Mehdirad, K. M. Biermann, and C. S. Fredman. 2006. Hyperkalemia revisited. Tex. Heart Inst. J. 33:40–47. [PMC free article] [PubMed] [Google Scholar]
- 58.Kim, J., D. S. Wei, and D. A. Hoffman. 2005. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones 1. J. Physiol. 569:41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lauver, A., L. L. Yuan, A. Jeromin, B. M. Nadin, J. J. Rodriguez, H. A. Davies, M. G. Stewart, G. Y. Wu, and P. J. Pfaffinger. 2006. Manipulating Kv4.2 identifies a specific component of hippocampal pyramidal neuron A-current that depends upon Kv4.2 expression. J. Neurochem. 99:1207–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim, J., S. C. Jung, A. M. Clemens, R. S. Petralia, and D. A. Hoffman. 2007. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 54:933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]