

Abstract
Sclerosteosis, a skeletal disorder characterized by high bone mass due to increased osteoblast activity, is caused by loss of the SOST gene product, sclerostin. The localization in bone and the mechanism of action of sclerostin are not yet known, but it has been hypothesized that it may act as a bone morphogenetic protein (BMP) antagonist. We show here that SOST/sclerostin is expressed exclusively by osteocytes in mouse and human bone and inhibits the differentiation and mineralization of murine preosteoblastic cells (KS483). Although sclerostin shares some of the actions of the BMP antagonist noggin, we show here that it also has actions distinctly different from it. In contrast to noggin, sclerostin did not inhibit basal alkaline phosphatase (ALP) activity in KS483 cells, nor did it antagonize BMP-stimulated ALP activity in mouse C2C12 cells. In addition, sclerostin had no effect on BMP-stimulated Smad phosphorylation and direct transcriptional activation of MSX-2 and BMP response element reporter constructs in KS483 cells. Its unique localization and action on osteoblasts suggest that sclerostin may be the previously proposed osteocyte-derived factor that is transported to osteoblasts at the bone surface and inhibits bone formation.
Keywords: sclerosteosis, SOST, osteoblast, Smad phosphorylation, MSC
Introduction
Sclerosteosis (OMIM 269500) is a rare sclerosing skeletal disorder characterized by generalized progressive bone overgrowth leading to tall stature, facial distortion, entrapment of cranial nerves, and raised intracranial pressure that predisposes to sudden death (1–4). Syndactyly, radial deviation of terminal phalanges, and dysplasia of the nails are variable manifestations. The majority of patients belong to the Afrikaner population of South Africa (5), but several patients have been described in other parts of the world (6–13). In sclerosteosis, bone formation is increased, whereas bone resorption is not affected or mildly diminished (11, 14). Bone biopsies from patients exhibit predominance of cuboidal active-appearing osteoblasts, increased double tetracycline label spacing, and increased osteoid levels that mineralize normally. Serum alkaline phosphatase (ALP) levels are elevated in some, but not all patients.
Recently, sclerosteosis was found to result from loss of the SOST gene product sclerostin, a newly cloned gene localized on chromosome region 17q12-q21 (15, 16). Van Buchem disease (OMIM 239100), a condition resembling sclerosteosis but differentiated by its less severe character and absence of hand malformations (3, 5) was recently found to result from a homozygous 52-kb deletion some 35-kb downstream of the SOST gene (17, 18). Absence of any evidence for a gene within this deleted region suggests that loss of regulatory elements within the 52-kb deletion results in down-regulation of SOST expression. These data suggest that SOST/sclerostin is a regulator of bone mass in humans.
However, the localization of SOST/sclerostin expression in bone and the mechanism by which it affects bone formation are yet unknown. The amino acid sequence of sclerostin shares similarity with the cystine knot–containing factors belonging to the DAN family of secreted glycoproteins (16, 19). The DAN family includes DAN, cerberus, gremlin, protein related to DAN, and cerberus, caronte, and dante that share the ability to antagonize bone morphogenetic protein (BMP) activity (20, 21). BMPs are stimulators of bone formation and their activity is controlled at several levels, including extracellular antagonists that bind BMPs and prevent their binding to signaling receptors (22, 23). Of these BMP antagonists, noggin and gremlin have been shown to regulate osteoblastic differentiation and function. Together, these data suggest that increased bone formation in patients with sclerosteosis may be due to unopposed BMP-stimulated osteoblastic activity (16). BMPs elicit the cellular responses via specific BMP types I and II serine/threonine kinase receptors and their downstream transcription effector proteins called Smads (24).
Therefore, in the present work, we examined the localization and mechanism of action of sclerostin, and we show that it is exclusively expressed in osteocytes and inhibits the bone-forming activity of osteoblasts by a mechanism different from that described for classic BMP antagonists.
Materials and Methods
Compounds.
Human sclerostin and control medium (CoM) were provided by D.G. Winkler (Celltech Inc., Bothell, WA). Sclerostin was purified using a Heparin Hitrap column (Amersham Biosciences) from conditioned medium of insect Spodoptera frugiperda (SF9) cells infected with human SOST sequence. CoM of uninfected SF9 cells was handled similar to the medium of SOST expressing SF9 cells. Recombinant human BMPs were purchased from R&D Systems and parathyroid hormone-related protein (PTHrP) 1–34 was obtained from Bacham AG.
Isolation of Total RNA.
RNA isolation was performed according to the method described by Chomczynski and Sacchi (25). Human bone biopsies from a femur head of an adult male, a mastoid from a patient with sclerosteosis, and mouse tibiae of young adult (8 wk old) female Swiss albino mice were cleaned from connective tissue and snap frozen in liquid nitrogen. The frozen bone biopsies were pulverized in liquid nitrogen cooled in 7-ml stainless steel flasks at 1,500 revolutions/min for 2 min using a Braun Mikro-Dismembrator U (Salm en Kipp N.V.). Pulverized bones were resuspended in 4 M guanidinium isothiocyanate lysis buffer and centrifuged at 6,000 revolutions/min for 10 min to remove bone grit. RNA isolation from cells was performed as described previously (26).
RT-PCR.
Denatured RNA was reverse transcribed into cDNA and standardized by competitive PCR using internal standards (mouse pMUS and human pQB-2) as described previously (26). Subsequently, expression of various genes was examined by semi-quantitative PCR. cDNA was denatured at 94°C for 3 min followed by repeated cycles of 30 s at 94°C, 45 s at 56°C, and 30 s at 72°C. Primer sets used crossed intron/exon boundaries so that eventual contaminations with genomic DNA would not be amplified in the amplification process or would generate amplicons of larger size. Primer sets used were as follows: human β2-microglobulin, sense, 5′-CCAGCAGAGAATGGAAAGTC-3′, antisense, 5′-GATGCTGCTTACATGTCTCG-3′; mouse β2-microglobulin, sense, 5′-TGACCGGCTTGTATGCTATC-3′, antisense, 5′-CAGTGTGAGCCAGGATATAG-3′; human osteocalcin, sense, 5′-GTAGTGAAGAGACCCAGGCG-3′, antisense, 5′-GGGAAGAGGAAAGAAGGGTG-3′; mouse osteocalcin primer set 1, sense, 5′-TCTGACAAAGCCTTCATGTCC-3′, antisense, 5′-CGCATCTACGGTATCACTATTT-3′; mouse osteocalcin primer set 2, sense, 5′-GCAGCTTGGTGCACACCTAG-3′, antisense, 5′-ATGGATGTCACAGCACGCTCC-3′; human SOST, sense, 5′-CCGGAGCTGGAGAACAACAAG-3′, antisense, 5′-GCACTGGCCGGAGCACACC-3′; and mouse SOST, sense, 5′-TCCTCCTGAGAACAACCAGAC-3′, antisense, 5′-TGTCAGGAAGCGGGTGTAGTG-3′.
Radioactive In Situ Hybridization.
8-wk-old female Swiss albino mice were fixed in vivo with 2% paraformaldehyde, 75 mM lysine monohydrochloride, and 10 mM sodium periodate in 0.1 M phosphate buffer under general anesthesia (xylazine, Bayer; ketamine HCl, Eurovet; reference 27). Tibiae and femurs were removed and decalcified for 2 wk in 10% EDTA and 0.5% paraformaldehyde. 17-d-old mouse embryos were fixed in 3.7% phosphate-buffered formaldehyde, pH 7.2, for 5 d. 5-μm-thick sections were prepared from paraffin-embedded blocks of decalcified mouse tibiae and femurs and undecalcified mouse embryos.
In situ hybridization was performed as described previously with some modifications (27). α-[35S]CTP–labeled mouse sense and antisense SOST probes were generated from SOST cDNA containing pCR2.1 vectors provided by K. Staehling-Hampton (Celltech Inc., Bothell, WA). Sections were hybridized overnight with α-[35S]CTP labeled probes at 60°C. Sections were coated with K5 emulsion (Ilford Limited), developed with Kodak D19, and fixed with Kodak Unifix.
Immunohistochemistry.
Human bone biopsies from 6 patients with sclerosteosis and 16 control subjects were obtained after surgical treatment and fixed in 4% phosphate-buffered formaldehyde and 70% ethanol before decalcification in 17% formic acid and 2.6% sodium formate. 5-μm-thick paraffin sections were stained as described previously (27). Primary mouse monoclonal IgG-1κ antibodies generated against full-length human sclerostin were provided by D.G. Winkler and used at a 1:1,000 dilution. Osteoclasts were stained using TRAcP staining as a marker. For this, naphthol ASBI phosphate was used as substrate, pararosaniline was used as a coupler, and 30 mM L+-tartaric acid was used as an inhibitor (28).
Cell Line Cultures.
Mouse preosteoblastic KS483 cells form mineralized bone nodules when cultured under osteogenic cultures conditions; i.e., in αMEM without phenol red (GIBCO BRL) supplemented with 10% FCS (Integro), 50 μg/ml ascorbic acid from day 3 or 4 onwards (Merck), and 10 mM β-glycerophosphate from day 10 onwards (Sigma-Aldrich; references 29–32). KS483 cells were seeded at a density of 15,000 cells/cm2, and agents were added when cells reached confluence after 3 or 4 d of culture. In short-term experiments, ALP activity was measured kinetically in the cell layer after another 4 d of culture (31). In long-term differentiation experiments, ALP activity and mineralization were analyzed by ALP and alizarin red staining, respectively (31). Medium was renewed every 3 to 4 d.
Mouse myoblastic C2C12 cells were seeded at a density of 20,000 cells/cm2 and cultured until confluence (3 or 4 d of culture) in DMEM and 20% FCS. At confluence, FCS serum concentration was reduced to 5%, and agents were added. ALP activity was measured kinetically in the cell layer after another 4 d of culture.
Mesenchymal Stromal Cell (MSC) Cultures.
Mouse bone marrow–derived MSCs were obtained from young adult (8 wk old) female Swiss albino mice by removing both ends of the femora and tibia and subsequent flushing with PBS. Cells were resuspended to obtain a single cell suspension and seeded at a density of 1.67 × 106 cells/cm2 in αMEM, 10% heat-inactivated FCS, 50 μg/ml ascorbic acid, and 10 mM β-glycerophosphate from day 13 onwards. Two-thirds medium was renewed every 3 to 4 d.
Liquid nitrogen stored Ficoll-separated human bone marrow cells from healthy donors obtained by aspiration from the posterior iliac crest were provided by W.A. Noort (Leiden University Medical Center, Leiden, Netherlands; reference 33). Thawed human bone marrow cells were seeded at a density of ± 107 cells/25 cm2 culture flask in 8 ml αMEM and 10% heat-inactivated FCS and allowed to attach without disturbance for 7 d. After the attachment period, half of the culture medium was renewed at 3–4 d intervals until cultures reached subconfluence (after ± 2 wk). The enriched human bone marrow–derived MSCs were reseeded at a density of 2.5 × 104 cells/cm2 in αMEM, 10% heat-inactivated FCS, 50 μg/ml ascorbic acid, 10−7 M dexamethasone (Organon N.V.), and 5 mM β-glycerophosphate from day 7 onwards. Medium was renewed every 3–4 d. In short-term experiments, cells were stimulated after 4 d of culture (i.e., at confluence), and ALP activity was measured kinetically in the cell layer after another 4 d of culture.
Transfections and Reporter Assays.
KS483 cells were seeded at a density of 10,000/cm2 in 24-well plates and transiently transfected with 1 μg of the BMP-responsive luciferase reporter construct MSX-2 (34) or BMP response element (BRE; reference 35) using Fugene™6 transfection reagent (Roche) according to the manufacturer's protocol. To correct for transfection efficiency, 100 ng of Renilla luciferase vector (pRL-SV40; Promega) was cotransfected. 12 h after transfection, the medium was changed for medium containing 0.2% FCS. The cells were kept in 0.2% FCS medium for 1 d and left either nonstimulated or stimulated for an additional 24 h. Luciferase assays were performed with the Dual-Luciferase Reporter assay system (Promega) according to the manufacturer's instructions. 10 μl of cell lysate was first assayed for firefly luciferase and then for Renilla luciferase activity, using the Wallac 1450 Microbeta Trilux luminescence counter (PerkinElmer). Firefly luciferase activity was corrected for Renilla luciferase activity to control for differences in transfection efficiency.
Smad Phosphorylation.
KS483 cells were seeded at a density of 15,000 cells/cm2 and cultured until confluence when FCS concentration was reduced to 0.2%. The next day, cultures were stimulated for 45 min with the agents indicated. Phosphorylated Smad-1, -5, and/or -8 were detected specifically by phospho–Smad-1 antibodies as described previously (36).
Statistics.
Values are expressed as mean ± SEM. Statistical differences between values were examined by one-way analysis of variance followed by Tukey's multiple comparison test and considered to be significantly different at P < 0.05.
Results
SOST mRNA Expression in Human and Mouse Bone.
Total RNA was isolated from mouse and human bone and from bone of a patient with sclerosteosis. Using RT-PCR, SOST mRNA expression was found in tibia of young adult mice and a human femur head obtained during hip replacement (Fig. 1 A). SOST mRNA expression was also found in mastoid of a patient with sclerosteosis from South Africa, suggesting that the mutation found in these patients does not lead to an increased degradation of SOST mRNA. Sequence analysis confirmed that the amplified mouse and human sclerostin amplicons were from sclerostin mRNA origin.
Figure 1.
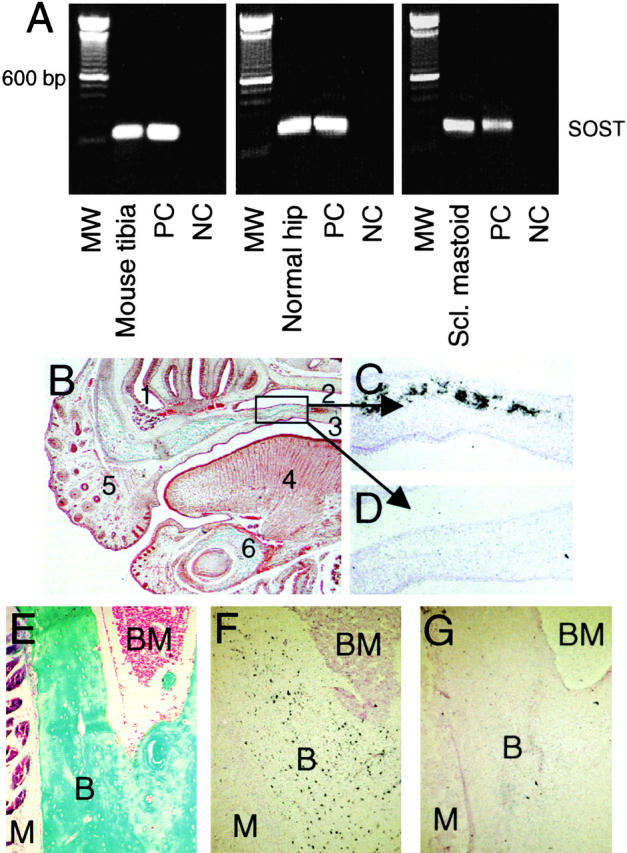
Localization of SOST mRNA expression in vivo. RT-PCR analysis of SOST mRNA expression in tibia of a young adult mouse, human hipbone, and mastoid of a patient with sclerosteosis (A). Amplicons were amplified over 35 cycles and had the predicted size of 185 bp. In situ hybridization analysis of SOST mRNA expression in 17.5-d-old fetal mouse embryos (B–D) and adult mouse tibia (E–G). Goldner staining of the upper and lower jaw to visualize mineralized bone (B, green/blue). Details of the palatal shelf in the upper jaw showing specific SOST mRNA expression in mineralized bone using antisense probe against SOST (C) and sense probe as control (D). Goldner staining of adult mouse tibia (E). Specific SOST mRNA expression in osteocytes using antisense probe against SOST (F) and sense probe as control (G). Counterstaining was hematoxylin and eosin (C, D, F, and G). Magnification is (B) 40, (C and D) 200, and (E–G) 100. (1) Nasal cavity. (2) Nasopharynx. (3) Oropharynx. (4) Tongue. (5) Nose. (6) Mandible. B, Bone. BM, bone marrow. M, muscle. MW, 100-bp molecular weight marker. NC, negative control. PC, positive control.
To locate expression in bone, we further studied SOST mRNA expression using radioactive in situ hybridization on sections. In 17.5-d-old mouse embryos, SOST mRNA was expressed in all mineralized bones (i.e., the palatal shelf; Fig. 1, B–D). The expression pattern of SOST mRNA was confirmed with a second RNA probe directed to an untranslated region of SOST mRNA. In 5-d-old neonatal ulnae, radii, and tail vertebrae and in tibia of young adult mice, SOST mRNA expression was restricted to osteocytes with the highest expression in the center of cortical bone (Fig. 1, E–G). SOST mRNA expression was not found in articular cartilage at any of the examined stages.
Sclerostin Protein Expression in Human Bone.
To study sclerostin expression at the protein level, bone biopsies of 18 control subjects were analyzed with a panel of 30 monoclonal antibodies against human sclerostin. Eight of these clones showed exclusive sclerostin staining in osteocytes in both cortical and trabecular bone, whereas the other clones were negative for specific sclerostin staining (Fig. 2, A and B). Clones H7 and H11 provided the strongest signals. High power magnification showed that the osteocyte canaliculi and/or lacunae were positive for sclerostin expression (Fig. 2 C). Sclerostin expression was found in osteocytes in bone biopsies from adults (range 28–69 yr; Fig. 2, A–C) as well as children (range 6 mo–12 yr; Fig. 2 D). Importantly, no sclerostin staining was observed in lining cells or active bone-forming osteoblasts. Furthermore, articular (Fig. 2, E and F) cartilage was negative for sclerostin staining.
Figure 2.
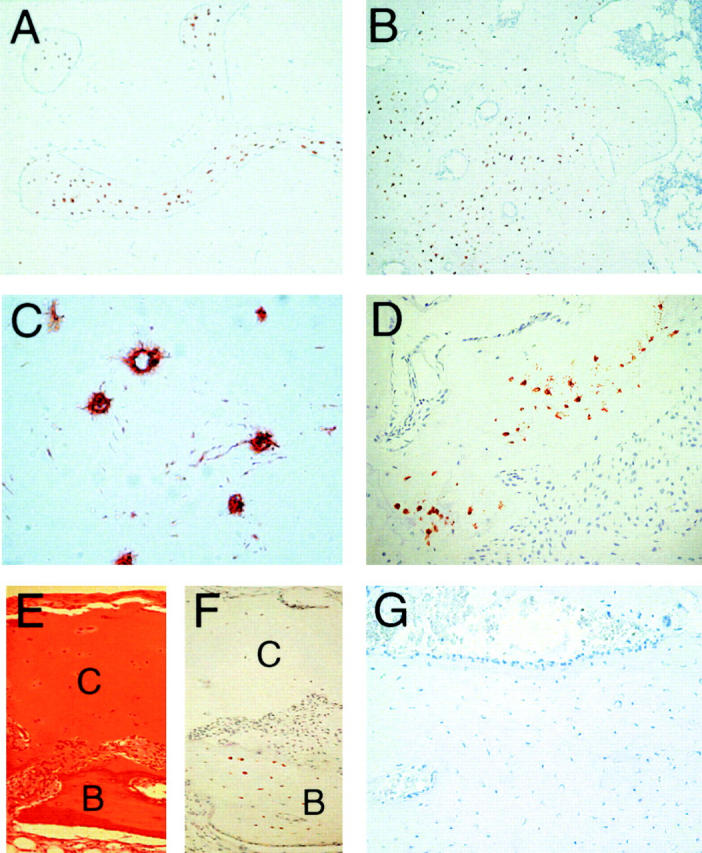
Sclerostin protein is expressed by osteocytes in human bone biopsies. Sclerostin protein expression was analyzed in control and sclerosteosis human bone biopsies using mouse monoclonal antibodies generated against human sclerostin. Staining with clone H11 showed sclerostin expression in osteocytes of both trabecular (A) and cortical (B) normal femur. Canaliculi and/or lacunae of osteocytes were positive for sclerostin (C). Sclerostin staining of a toe bone biopsy of a 1-yr-old boy showed clear restricted expression of sclerostin to osteocytes (D). No sclerostin expression was found in articular cartilage (E and F) of a 12-yr-old girl. No sclerostin staining was found in mastoid of a patient with sclerosteosis (G). Note the presence of active bone-forming osteoblasts on the bone surface that is characteristic for a site of active bone formation. Counterstaining was Mayers Hematoxylin. C, cartilage. B, bone. Magnification is (A and B) 40, (C) 400, and (D–G) 100.
Specificity of sclerostin signal was demonstrated by the absence of any binding with clones H7 and H11 in bone biopsies of six patients with sclerosteosis from South Africa (Fig. 2 G). These patients should not express sclerostin due to a stop codon in the first exon of the SOST gene caused by a C69T transition (16). Note the predominance of cuboidal active-appearing osteoblasts that are characteristic for sclerosteosis bone. Staining with a mouse monoclonal antibody of the same isoform IgG-1κ against human PECAM-1/CD31 showed no signal in osteocytes, whereas blood vessels in the same bone biopsy stained positive (unpublished data).
SOST/Sclerostin Is Not Expressed in Osteoclasts.
To investigate whether or not SOST mRNA and sclerostin are expressed in osteoclasts, we studied SOST/sclerostin expression in combination with TRAcP staining to identify osteoclasts in mouse and human bone. No SOST mRNA expression was found in TRAcP+ osteoclasts in 17-d-old mouse embryos, whereas it was found in the bone collars of these ribs that were devoid of osteoclasts (Fig. 3, A–C). In postnatal mouse bones, SOST mRNA expression was also not found in TRAcP+ osteoclasts (unpublished data). Similarly, we have never seen any TRAcP+ osteoclasts being positive for sclerostin expression in any of the postnatal or adult human bone biopsies studied, whereas osteocytes were positive (Fig. 3, D and E).
Figure 3.
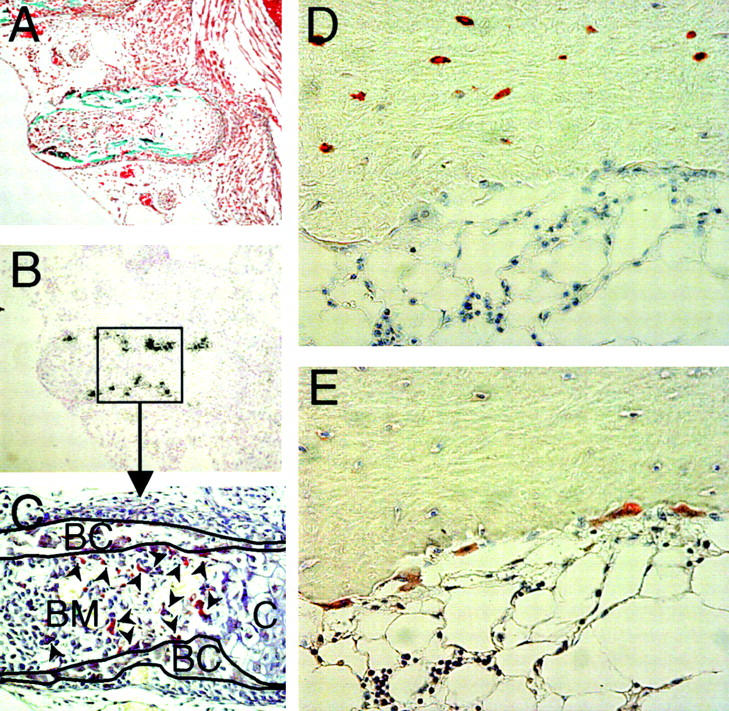
Sclerostin protein is not expressed by osteoclasts in mouse and human bone. Goldner staining of a rib of a 17.5-d-old fetal mouse embryo (A). Specific SOST mRNA expression in mineralized bone of the rib using antisense probe against SOST (B). TRAcP staining of the rib showing that osteoclasts do not colocalize with SOST mRNA expression. TRAcP+ osteoclasts (arrowheads) are present in the bone marrow and on the surface of the bone collars, whereas SOST mRNA is located within the bone collar (C). Sclerostin staining of human bone biopsy of a 12-yr-old girl shows that sclerostin is expressed by osteocytes, but not by multinuclear osteoclasts on the bone surface (D). TRAcP staining to identify multinuclear osteoclasts on the bone surface (E). Magnification is (A and B) 100 and (C–E) 200.
SOST mRNA Expression during Osteoblast Differentiation In Vitro.
The presence of SOST mRNA expression in mineralized bone during embryogenesis and the restricted expression of SOST mRNA and sclerostin protein in osteocytes postnatally suggested a differentiation dependent expression of the gene. To examine this, we studied the onset of SOST mRNA expression during osteoblastic differentiation of mouse and human bone marrow–derived MSCs and of mouse preosteoblastic KS483 cells. All these cells differentiate into mature osteoblasts that form a mineralized matrix when cultured under osteogenic conditions, including ascorbic acid and β-glycerophosphate and, in the case of human MSCs, also dexamethasone. Mouse MSCs formed ALP positive bone nodules after ± 7 d of culture and these nodules mineralized from ± 12 d onwards. SOST mRNA expression was analyzed in relation to that of the known osteoblast differentiation marker osteocalcin. In noncultured mouse bone marrow, day 0, both osteocalcin and SOST mRNA were not expressed (Fig. 4 A). Induction of osteocalcin mRNA expression was seen at day 11 and was further elevated from day 14 onwards. SOST mRNA expression was induced after the onset of osteocalcin mRNA expression and was found from day 14 onwards.
Figure 4.

SOST mRNA is expressed during the mineralization phase of osteoblastic cultures. Mouse MSCs (A), KS483 cells (B), and human MSCs (C) were cultured under osteogenic conditions. SOST mRNA expression during osteoblastic differentiation is shown in relation to the late osteoblast differentiation marker osteocalcin. Osteocalcin was amplified over 30, 30, and 35 cycles for mouse MSCs, KS483 cells, and human MSCs, respectively. The amplicons had the predicted size of 199 bp (mouse primer set 1), 430 bp (mouse primer set 2), and 242 bp (human primer set). SOST was amplified over 35, 35, and 40 cycles, respectively, and the amplicons had the predicted size of 137 bp (mouse primer set) and 185 bp (human primer set). KS483 cells were cultured in the absence or presence of 10−8 M PTHrP with continuous treatment from day 4 onwards. MW, 100-bp molecular weight marker. PC, positive control.
However, mouse MSC cultures consist of a heterogeneous cell population. Therefore, we also studied expression of SOST mRNA in the preosteoblastic KS483 cell line. Similar to mouse bone marrow cells, these cells formed ALP positive bone nodules during the first week of culture that mineralized from ± 11 d onwards. Osteocalcin mRNA expression was induced at day 10 of culture and was further elevated from day 17 onwards (Fig. 4 B). Similar to mouse MSCs, SOST mRNA expression was induced only after the onset of osteocalcin mRNA expression and was found from day 17 onwards. Differentiation of KS483 cells can be inhibited by treatment with a continuous high dose of 10−8 M PTHrP (1–34), resulting in the absence of ALP positive and mineralized bone nodules (32). Addition of PTHrP delayed and reduced osteocalcin mRNA expression and completely prevented induction of SOST mRNA.
In vitro, human MSCs do not form bone nodules, but a rather homogeneous cell layer of ALP positive cells and areas of more and less condense mineralized matrix. Osteocalcin mRNA was already expressed in undifferentiated cells, and the expression was elevated from day 14 onwards, which coincides with onset of mineralization (Fig. 4 C). SOST mRNA expression was not detected in undifferentiated cells and was always detected in late differentiated cells (i.e., 21 d of culture). However, the exact moment of induction of SOST mRNA expression could not be determined and varied between 10 and 21 d of culture. In addition, SOST mRNA was expressed at low levels and was only detected after 40 PCR cycles. In summary, we show that SOST mRNA was expressed only after ALP and osteocalcin expression in all three culture systems, which is consistent with a differentiation-dependent expression. This was further supported by the observation that inhibition of differentiation of KS384 cells by PTHrP also suppressed SOST mRNA induction.
Sclerostin Inhibits Bone-forming Activity of Osteoblasts In Vitro.
Protein staining and Western blot analysis showed that the sclerostin preparation contained three bands of ∼18, 23, and 30 kD that may correlate to the two possible N-glycosylation sites of human sclerostin (Fig. 5 A; reference 16). No other protein bands were found using the sensitive silver protein staining method, demonstrating its high degree of purification. The sclerostin preparation ran similar to sclerostin containing medium of KS483 and CHO cells with transient and stable expression of human SOST cDNA, respectively. Using an ELISA-based binding assay with 200 ng of human sclerostin–coated on the surface or an in vitro binding and covalent cross-linking assay with iodinated sclerostin, we found that sclerostin bound BMP-6 dose dependently over a concentration range of 5–400 ng/ml (unpublished data).
Figure 5.

Sclerostin inhibits unstimulated and BMP-stimulated osteoblastic differentiation of KS483 and primary human MSCs, but not of C2C12 cells. Silver staining (lanes 1 and 2) and Western blotting with a rabbit anti–human sclerostin antibody (lanes 3–8) of human sclerostin (lanes 2 [100 ng/ml], 3 [500 ng/ml], 5 [100 ng/ml], and 7 [100 ng/ml]), sclerostin containing medium of KS483 (lane 4), CHO cells (lane 6), and immunodepleted sclerostin preparation (lane 8, equal volume as 100 ng/ml sclerostin preparation) run under reducing conditions (A). Long-term (18 d) osteogenic KS483 cell cultures were treated with sclerostin (2.5 μg/ml or concentrations indicated) or CoM (equal volume) from day 4 onwards and analyzed for ALP activity (B) and mineralization (C). Confluent KS483 were stimulated with BMPs (50 ng/ml BMP-2, 50 ng/ml BMP-4, 300 ng/ml BMP-5, 100 ng/ml BMP-6, and 300 ng/ml BMP-7) in the absence or presence of the dose range of sclerostin or CoM. ALP activity was measured kinetically 4 d after stimulation (D). Confluent C2C12 cells were stimulated with 50 ng/ml BMP-4 in the absence or presence of the dose range of sclerostin or CoM. ALP activity was measured kinetically 4 d after stimulation (E). Confluent hMSCs were stimulated with 100 ng/ml BMP-4 in the absence or presence of the dose range of sclerostin. ALP activity was measured kinetically 4 d after stimulation (F). CoM, Control medium. hMSCs, human mesenchymal stromal cells. MW, molecular weight marker. Scl, sclerostin. Scl CHO, sclerostin containing medium of CHO cells. Scl ID, Immunodepleted sclerostin preparation. Scl KS483, sclerostin containing medium of KS483 cells. Results are expressed as mean ± SEM of triplicate (B and C) or octagonal (D–F) experiments relative to control (B and C) and BMP stimulation (D–F) that was set at 100%. *, Significant versus CoM (P < 0.05). Statistical differences were omitted from C for clarity.
To study the effect of sclerostin on the formation of bone matrix, long-term osteoblastic KS483 cell cultures were treated with sclerostin at concentrations of 0.025–2.5 μg/ml. As shown in Fig. 5 B, sclerostin slightly but significantly inhibited ALP activity only at the highest concentration used. In contrast, sclerostin had a dramatic inhibitory effect on calcium deposition (Fig. 5 C). This effect of sclerostin is different from that of the BMP antagonist noggin that inhibited both ALP activity and mineralization completely at equal concentrations in these cells (31).
We examined the effect of sclerostin in cultures stimulated with BMPs. In KS483 cells, sclerostin dose dependently antagonized the stimulation of ALP activity by two different subclasses of the BMP family that have the most pronounced effects on bone formation (i.e., BMP-2 and BMP-4 vs. BMP-5, BMP-6, and BMP-7; Fig. 5 D). Sclerostin did not show any preference for the different BMPs, nor did it suppress ALP activity below basal level. This again contrasts the action of noggin that inhibited BMP-stimulated ALP activity below the basal level (31) and showed a preference for antagonizing BMP-2, -4, and -7 in these cells (unpublished data). Specificity of the BMP antagonistic effect of the human sclerostin preparation was proven by loss of activity after immunodepletion with a rabbit anti–human sclerostin antibody. Depletion of sclerostin was checked by Western blotting (Fig. 5 A, lanes 7 and 8).
The aforementioned experiments revealed differences in the effects of sclerostin and noggin on basal and BMP-stimulated differentiation of preosteoblastic cells. To further examine this, we used another cell line (i.e., mouse C2C12 cells that acquire osteoblastic features after BMP stimulation). In this cell line, sclerostin did not antagonize ALP activity induced by BMP-4 (Fig. 5 E), BMP-2, nor BMP-6 (not depicted). Similarly, BMP-induced outgrowth of distal bone ends and inhibition of chondrocyte differentiation of cultured fetal mouse metatarsals (37) was not antagonized by sclerostin.
To study whether the observed BMP antagonistic effect of sclerostin was not restricted to the KS483 cell line, we tested sclerostin on primary human MSCs. Similar to the effect on KS483 cells, sclerostin antagonized BMP-4–stimulated ALP activity in primary human MSCs dose dependently (Fig. 5 F).
Although sclerostin's action was different from that of noggin, it antagonized BMP activity under some conditions. To examine the BMP antagonistic activity of sclerostin in more detail, we studied the effect of sclerostin on early BMP responses as Smad phosphorylation and direct target gene transcription. BMP-4–stimulated phosphorylation of Smad-1, -5, and/or -8 was not antagonized by sclerostin, whereas it was antagonized by noggin (Fig. 6 A). Similarly, BMP stimulation of two BMP/Smad-dependent promoter constructs (i.e., a luciferase reporter construct containing the MSX-2 promoter and a synthetic BRE luciferase reporter construct containing multimerized Smad-binding elements from the Id-1 promoter) was not antagonized by sclerostin (Fig. 6, B and C, respectively). However, noggin completely inhibited BMP-stimulated luciferase activity. These data were confirmed by examining mRNA expression levels as sclerostin treatment did not prevent the elevation in MSX-2 and Id-1 mRNA levels 4 h after BMP stimulation, whereas noggin treatment did (unpublished data).
Figure 6.

Sclerostin does not antagonize early BMP responses in KS483 cells. Western analysis of Smad-1, -5, and/or -8 phosphorylation in confluent KS483 cells that were stimulated for 45 min with 50 ng/ml BMP-4 in the absence or presence of 2.5 μg/ml sclerostin or 500 ng/ml noggin (A). Subconfluent KS483 cells were transfected with MSX-2 luciferase reporter construct (B) or BRE luciferase reporter construct (C) and stimulated for 24 h with 100 ng/ml BMP-6 or 50 ng/ml BMP-4 in the absence or presence of the dose range of sclerostin, CoM, or noggin (500 ng/ml), respectively. CoM, Control medium. Scl, sclerostin. Smad-P, phosphorylated Smad-1, -5, and/or -8. Results are expressed as mean ± SEM of triplicate experiments relative to BMP stimulation that was set at 100%. *, Significant versus CoM. #, Significant versus BMP-4. P < 0.05.
The observation that sclerostin antagonized the late BMP response of ALP activity in KS483 cells but not direct BMP responses suggests that sclerostin may act on or in combination with a BMP-inducible factor. To study this, sclerostin was added at several time points after BMP-6 stimulation; it was found to inhibit ALP activity even when it was added as long as 24 h after initial BMP stimulation (Fig. 7 A). The BMP-induced factor may act in synergism with BMPs to stimulate ALP activity. If this is true, BMP presence is not only required at the initial time point of stimulation but also during the culture period after stimulation. To study this, we removed BMP-4 from the medium or antagonized its activity at several time points after BMP stimulation by changing the medium or adding soluble truncated BMP receptor-1A, respectively. Using both methods, it was found that BMP-4 needed to be present for >24 h to stimulate ALP activity significantly in KS483 cells (Fig. 7 B). Thereafter, ALP activity increased gradually with 72 h of BMP-4 presence being similar to continuous BMP-4 presence. Similar data were obtained with BMP-6 (unpublished data).
Figure 7.

Time-dependent effects of sclerostin and BMPs on ALP activity. Sclerostin antagonizes BMP responses in KS483 cells when added as long as 24 h after BMP stimulation (A). Confluent KS483 cells were stimulated with 100 ng/ml BMP-6 in the absence or presence of 1.0 μg/ml sclerostin that was added at the indicated time points after BMP stimulation. ALP activity was measured kinetically 3 d after stimulation. BMPs need to be present for >24 h to stimulate ALP activity (B). Confluent KS483 cells were stimulated with 50 ng/ml BMP-4 for indicated time periods. BMP-4 was removed from the medium or its activity antagonized by changing the medium (light gray bars) or adding soluble truncated BMP receptor-1A (250 ng/ml; dark gray bars), respectively. ALP activity was measured kinetically 4 d after stimulation. Results are expressed as mean ± SEM of octagonal experiments. *, Significant versus BMP-6. #, Significant versus control (P < 0.05). strBMPR-1A, soluble truncated BMP receptor-1A.
Discussion
In the present work, we show that sclerostin is a negative regulator of bone formation that is expressed exclusively in osteocytes of mouse and human bone and suppresses bone formation by inhibiting the differentiation of osteoblasts. Recently, sclerostin was hypothesized to act as a BMP antagonist (16). However, its unique localization and its inability to antagonize several BMP responses in osteoblastic cells differentiate sclerostin from the classical BMP antagonists noggin and gremlin that have also been reported to play an important role in bone formation (22).
Sclerostin deficiency due to SOST mutations causes sclerosteosis (15, 16), a generalized progressive skeletal disorder characterized by high bone mass due to increased osteoblast activity (Fig. 8, A and B; references 5, 11, 14). However, the mechanism by which sclerostin regulates bone formation is unknown. We show here that SOST mRNA was expressed in all mineralized bones of both intramembraneous and endochondral origin during mouse embryogenesis. At this early stage of bone development, it is difficult to determine whether SOST mRNA expression was restricted to osteocytes or whether osteoblasts expressed SOST mRNA as well. However, postnatally, it was obvious that SOST mRNA as well as sclerostin protein expression were restricted to osteocytes in trabecular and cortical bones of both endochondral and intramembraneous origin in mice and humans. These results are in accordance with RT-PCR findings showing SOST mRNA expression in mouse and human bone biopsies in the present work as well as in a human bone biopsy reported previously by Brunkow et al. (16). Recently, Kusu et al. (38) reported that SOST mRNA is expressed by osteoclasts based on similarities between patterns of MMP-9 and SOST mRNA expression in bone samples of newborn mice. However, here we provide unequivocal evidence that SOST/sclerostin is not expression in osteoclasts in embryonic, neonatal, and adult mouse and human bone using in situ hybridization and immunohistochemistry to detect SOST/sclerostin expression and the classical specific TRAcP staining method to identify osteoclasts.
Figure 8.

Histological pictures and a schematic model of increased bone formation in sclerosteosis due to sclerostin deficiency. Representative Goldner staining of calvarial bones of a control (A) and a patient with sclerosteosis (B). Schematic model of sclerostin's mechanism of action in bone remodeling (C). Sclerostin produced by osteocytes may be transported through lacunae to the bone surface, where it inhibits osteoblastic bone formation. In sclerosteosis, loss of sclerostin may prolong the active bone-forming phase of osteoblasts and thereby increase the amount of bone formed by osteoblasts. The increased osteoid levels in sclerosteosis reflect this. In the absence of increased osteoclastic bone resorption, the increased bone formation results in a positive bone balance and, subsequently, in the excess bone mass of normal architecture and increased strength found in sclerosteosis. LC, lining cell. OB, osteoblast. OC, osteoclast. OCYT, osteocyte.
Specificity of sclerostin protein expression in human bone biopsies was demonstrated by the absence of sclerostin staining in bone biopsies of six patients with sclerosteosis. Positive sclerostin staining of osteocyte canaliculi and/or lacunae, with which osteocytes are connected to neighboring osteocytes in an osteon or to osteoblasts and lining cells on the bone surface, suggests that sclerostin is transported to neighboring cells (possibly to active bone-forming osteoblasts). This transport might be intracellular from cell to cell because osteocyte canaliculi are connected at their tips by gap junctions (39). Alternatively, and perhaps more likely because of the presence of a putative secretion signal in sclerostin (16), the transport may occur extracellularly from lacuna to lacuna. The notion that sclerostin is a secreted protein is supported further by the presence of sclerostin in culture medium of human SOST cDNA expressing osteoblastic KS483 cells.
In vitro, SOST mRNA expression was restricted to the mineralization phase of osteoblastic cultures. SOST mRNA levels appeared to be higher in mouse MSCs than in KS483 cell cultures and were low in human MSC cultures. Low levels of SOST mRNA expression have been detected previously in late-stage (i.e., 21 d old) primary human osteoblastic cultures of a male Caucasian donor (15). Differences in expression levels between the culture systems used may be related to the absence or presence as well as the size of bone nodules. Mouse MSC cultures contained bone nodules that were much larger than those formed in KS483 cultures. In human MSC cultures, bone nodules did not develop. The relative low levels of SOST mRNA expression in all three culture systems suggests that SOST mRNA is restricted to only a few cells, possibly osteocytes in the bone nodules. The presence of osteocyte-like cells in bone nodules has been demonstrated previously in late mineralized fetal mouse calvaria-derived osteoblastic cultures (40), and we have indication for the presence of osteocytes in the bone nodules of mouse MSC cultures as well (unpublished data). Whether SOST mRNA expression is more pronounced or restricted to osteocytes in bone nodules remains to be established.
Because increased bone formation in sclerosteosis results from sclerostin deficiency, sclerostin is expected to have an inhibitory effect on bone formation. In the present work, we show that, in contrast to the BMP antagonist noggin (31), sclerostin only slightly inhibited the early osteoblast differentiation marker ALP activity. However, similar to noggin, sclerostin strongly inhibited the formation of mineralized bone nodules, the end-stage marker of osteoblast activity. Together, these data suggest that sclerostin produced by osteocytes is transported to the bone surface, where it inhibits the later stages of bone formation (Fig. 8 C). This makes sclerostin a perfect candidate for the osteocyte-derived factor that inhibits active bone-forming osteoblasts on the bone surface as proposed by Marotti (41) and Martin (42). These investigators suggested that an osteocyte sufficiently covered by new bone and osteoid sends an inhibitory signal through their dendritic processes to reduce the bone deposition rate of osteoblasts. In sclerosteosis, the absence of the inhibitory signal sclerostin would keep osteoblasts in an active deposition state and, as a consequence, would increase the deposition of bone by osteoblasts. This model can thereby explain the phenotype of sclerosteosis in which loss of sclerostin results in increased bone formation that is not associated with increased osteoclastic bone resorption. Alternatively, sclerostin may act on the osteocyte itself and inhibit the production of a signal that inhibits osteoblast activity.
However, the mechanism by which sclerostin produced by osteocytes inhibits bone formation is yet unknown. Brunkow et al. (16) suggested that sclerostin may inhibit bone formation by antagonizing BMP-stimulated osteoblast activity. In line with this, sclerostin inhibited BMP-stimulated ALP activity in mouse osteoblastic KS483 cells. However, in contrast to the restricted antagonistic activity of sclerostin for BMP-6 and BMP-7 recently reported by Kusu et al. (38), we found no obvious differences in the capacity of sclerostin to inhibit stimulation of ALP activity by BMP-2, -4, -5, -6, and -7. In addition, and in contrast to sclerostin's low binding affinity (10−8 M) for BMPs, Kusu et al. reported that stimulation of ALP activity by 10 ng/ml BMP-6 was antagonized by 0.1 ng/ml sclerostin. This is at least a 1,000-fold lower than in our experience in which ALP activity stimulated by 100 ng/ml BMP-6 was antagonized by 2.5 μg/ml sclerostin. Importantly, sclerostin did not antagonize BMP-stimulated ALP activity in mouse C2C12 cells and BMP-induced morphological changes in cultured fetal mouse metatarsals. These data differentiate sclerostin from the classical BMP antagonists that all inhibit BMP bioactivity independently of the bioassay used. Confirmation of sclerostin being different from the classical BMP antagonists was provided by its inability to inhibit BMP stimulation of Smad phosphorylation and BMP/Smad-dependent reporter constructs MSX-2 and BRE in KS483 cells. Furthermore, addition of sclerostin 24 h after BMP stimulation still inhibited ALP activity. The trend toward a better inhibition of BMP-stimulated ALP activity when sclerostin was added later may be explained by increased instability or loss of bioactivity of sclerostin in medium supplemented with 10% FCS at 37°C.
The observation that sclerostin antagonized the late BMP response of ALP activity in KS483 cells, but not direct BMP responses may be explained by different mechanisms of action of sclerostin.
First, sclerostin may function as a BMP antagonist only when a BMP-inducible factor is coexpressed (Fig. 9, Option I). This cofactor is necessary for sclerostin to prevent BMP binding to its receptor in such a way that activation of the receptor is prevented. Such a cofactor may be similar to twisted gastrulation for the BMP antagonist chordin (43). This option is supported by sclerostin binding to BMPs as recently reported by Kusu et al. (38) and also found by us using ELISA-based binding studies, in vitro binding, and covalent cross-linking of iodinated sclerostin to BMP-6 (unpublished data). However, there is a large difference in the binding affinities of noggin and sclerostin for BMPs, 10−12 M versus 10−8 M, respectively, that is not reflected in the small difference in BMP-antagonizing activities, 10−5 M versus 10−4 M, respectively. The proposed BMP-inducible cofactor may increase sclerostin's binding affinity for BMPs similar to that of noggin. If true, sclerostin may antagonize early BMP responses in cells already expressing the cofactor, whereas it will not antagonize any BMP response in cells in which the cofactor cannot be induced, possibly C2C12 cells.
Figure 9.

Two schematic models that may explain the mechanism of action of sclerostin. Both models provide a mechanism by which sclerostin may antagonize the late BMP response ALP activity, but not the early BMP responses of Smad phosphorylation and direct gene transcription (BRE and MSX-2 reporter construct activation). (Option I) Sclerostin functions as a BMP antagonist when a BMP-inducible factor is coexpressed. The cofactor is necessary for sclerostin to prevent BMP binding to its receptor. The BMP-inducible cofactor may, for example, increase sclerostin's binding affinity for BMPs. (Option II) Sclerostin prevents receptor binding of an yet unknown BMP-inducible factor that stimulates ALP activity. BMPR, BMP receptor. OB, osteoblast. Smad-P, phosphorylated Smad.
Second, sclerostin may antagonize another yet unknown BMP-inducible factor that stimulates bone formation (Fig. 9, Option II). This option is supported by the observation that sclerostin does not have any preference for the different types of BMPs. In contrast, noggin preferentially antagonizes BMP-2, -4, and -7. In addition, the differences between the binding affinities and antagonizing activities of noggin and sclerostin may also favor the existence of another factor antagonized by sclerostin. This unknown factor may act in synergism with BMPs because BMPs needed to be present for >24 h to stimulate ALP activity in KS483 cells.
Alternatively, sclerostin may bind to its own, yet unknown BMP-inducible receptor and inhibit bone formation. However, in preliminary experiments, we did not find any binding of iodinated sclerostin to BMP-stimulated KS483 cells.
In summary, sclerostin is expressed in osteocytes and inhibits bone formation by a mechanism different from that of known BMP antagonists. Its localization and characteristic of being a secreted protein suggest that sclerostin is transported to the bone surface, where it inhibits osteoblast differentiation and function. These observations suggest that inactivation of sclerostin by small molecules or humanized neutralizing antibodies may induce a positive bone balance, an effect that may have therapeutic implications for patients with osteoporosis.
Acknowledgments
The authors thank their colleagues from Celltech Inc., especially J.A. Latham, D.G. Winkler, and M.K. Sutherland, who supported this work both practically and financially.
B.A.J. Roelen was supported by The Netherlands Organization for Scientific Research (ALW 809.67.024).
Portions of this work were presented at the 24th Annual Meeting of the American Society for Bone and Mineral Research in San Antonio, TX on 20–24 September 2002.
Abbreviations used in this paper: ALP, alkaline phosphatase; BMP, bone morphogenetic protein; BRE, BMP response element; CoM, control medium; MSC, mesenchymal stromal cell; PTHrP, parathyroid hormone-related protein; SF9, Spodoptera frugiperda.
References
- 1.Truswell, A.S. 1958. Osteopetrosis with syndactyly, a morphologic variant of Albers-Schonberg disease. J. Bone Joint Surg. Br. 40:208–218. [PubMed] [Google Scholar]
- 2.Hansen, H. 1967. Sklerosteose. Handbuch der Kinderheilkunde. H. Opitz and F. Schmid, editors. Springer-Verlag, Berlin. 351–355.
- 3.Beighton, P., A. Barnard, H. Hamersma, and A. Van Der Wouden. 1984. The syndromic status of sclerosteosis and van Buchem disease. Clin. Genet. 25:175–181. [DOI] [PubMed] [Google Scholar]
- 4.Beighton, P. 1988. Sclerosteosis. J. Med. Genet. 25:200–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamersma, H., J. Gardner, and P. Beighton. 2003. The natural history of sclerosteosis. Clin. Genet. 63:192–197. [DOI] [PubMed] [Google Scholar]
- 6.Higinbotham, N.L., and S.F. Alexander. 1941. Osteopetrosis: four cases of one family. Am. J. Surg. 53:444–454. [Google Scholar]
- 7.Kelley, C.H., and J.W. Lawlah. 1946. Albers-Schonberg disease: a family survey. Radiology. 47:507–513. [DOI] [PubMed] [Google Scholar]
- 8.Pietruschka, G. 1958. Weitere Mitteilungen uber die Marmorknochenkrankheit (Albers-Schonbergsche Krankheit) nebst Bemerkungen zur Differentialdiagnose. [Further information on marble bones (Albers-Schonberg disease) with remarks on differential diagnosis.] [In German.] Klin Monatsbl Augenheilkd. 132:509–525. [PubMed] [Google Scholar]
- 9.Sugiura, Y., and T. Yasuhara. 1975. Sclerosteosis: A case report. J. Bone Joint Surg. Am. 57:273–276. [PubMed] [Google Scholar]
- 10.Freire de Paes Alves, A., J. Rubim, L. Cardoso, and M. Rabelo. 1982. Sclerosteosis: A marker of Dutch ancestry? Rev. Brasil Genet. 5:825–834. [Google Scholar]
- 11.Stein, S., C. Witkop, S. Hill, M. Fallon, L. Viernstein, G. Gucer, P. McKeever, D. Long, J. Altman, N.R. Miller, et al. 1983. Sclerosteosis: neurogenetic and pathophysiologic analysis of an American kinship. Neurology. 33:267–277. [DOI] [PubMed] [Google Scholar]
- 12.Bueno, M., G. Olivan, A. Jimenez, J. Garragori, A. Sarria, A. Bueno, M.J. Bueno, and F. Ramos. 1994. Sclerosteosis in a Spanish male: first report in a person of Mediterranean origin. J. Med. Genet. 31:976–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tacconi, P., P. Ferrigno, L. Cocco, A. Cannas, G. Tamburini, P. Bergonzi, and M. Giagheddu. 1998. Sclerosteosis: report of a case in a black African man. Clin. Genet. 53:497–501. [DOI] [PubMed] [Google Scholar]
- 14.Hill, S.C., S.A. Stein, A. Dwyer, J. Altman, R. Dorwart, and J. Doppman. 1986. Cranial CT findings in sclerosteosis. Am. J. Neuroradiol. 7:505–511. [PMC free article] [PubMed] [Google Scholar]
- 15.Balemans, W., M. Ebeling, N. Patel, E. Van Hul, P. Olson, M. Dioszegi, C. Lacza, W. Wuyts, J. Van Den Ende, P. Willems, et al. 2001. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 10:537–543. [DOI] [PubMed] [Google Scholar]
- 16.Brunkow, M.E., J.C. Gardner, J. Van Ness, B.W. Paeper, B.R. Kovacevich, S. Proll, J.E. Skonier, L. Zhao, P.J. Sabo, Y. Fu, et al. 2001. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 68:577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balemans, W., N. Patel, M. Ebeling, E. Van Hul, W. Wuyts, C. Lacza, M. Dioszegi, F.G. Dikkers, P. Hildering, P.J. Willems, et al. 2002. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 39:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Staehling-Hampton, K., S. Proll, B.W. Paeper, L. Zhao, P. Charmley, A. Brown, J.C. Gardner, D. Galas, R.C. Schatzman, P. Beighton, et al. 2002. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am. J. Med. Genet. 110:144–152. [DOI] [PubMed] [Google Scholar]
- 19.Avsian-Kretchmer, O., and Hsueh, A.J.W. 2004. Comparative genomics analysis of the eight-membered-ring cystine-knot-containing bone morphogenetic protein (BMP) antagonists. Mol. Endocrinol. 18:1–12; First published on October 2, 2003; 10.1210/me.2003-0227 [DOI] [PubMed] [Google Scholar]
- 20.Hsu, D.R., A.N. Economides, X. Wang, P.M. Eimon, and R.M. Harland. 1998. The Xenopus dorsalizing factor Gremlin identifies a novel family of secreted proteins that antagonize BMP activities. Mol. Cell. 1:673–683. [DOI] [PubMed] [Google Scholar]
- 21.Pearce, J.J., G. Penny, and J.A. Rossant. 1999. Mouse cerberus/Dan-related gene family. Dev. Biol. 209:98–110. [DOI] [PubMed] [Google Scholar]
- 22.Reddi, A.H. 2001. Interplay between bone morphogenetic proteins and cognate binding proteins in bone and cartilage development: noggin, chordin and DAN. Arthritis Res. 3:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canalis, E., A.N. Economides, and E. Gazzerro. 2003. Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr. Rev. 24:218–235. [DOI] [PubMed] [Google Scholar]
- 24.Heldin, C.H., K. Miyazono, and P. Ten Dijke. 1997. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 390:465–471. [DOI] [PubMed] [Google Scholar]
- 25.Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162:156–159. [DOI] [PubMed] [Google Scholar]
- 26.Van Bezooijen, R.L., H.C. Farih-Sips, S.E. Papapoulos, and C.W. Löwik. 1998. IL-1alpha, IL-1beta, IL-6, and TNF-alpha steady-state mRNA levels analyzed by reverse transcription-competitive PCR in bone marrow of gonadectomized mice. J. Bone Miner. Res. 13:185–194. [DOI] [PubMed] [Google Scholar]
- 27.Van Der Eerden, B.C., M. Karperien, E.F. Gevers, C.W. Löwik, and J.M. Wit. 2000. Expression of Indian hedgehog, parathyroid hormone-related protein, and their receptors in the postnatal growth plate of the rat: evidence for a locally acting growth restraining feedback loop after birth. J. Bone Miner. Res. 15:1045–1055. [DOI] [PubMed] [Google Scholar]
- 28.Barka, T., and P.J. Anderson. 1962. Histochemical method for acid phosphatase using hexazonium pararosanilin as coupler. J. Histochem. Cytochem. 10:741–753. [Google Scholar]
- 29.Yamashita, T., H. Ishii, K. Shimoda, T.K. Sampath, T. Katagiri, M. Wada, T. Osawa, and T. Suda. 1996. Subcloning of three osteoblastic cell lines with distinct differentiation phenotypes from the mouse osteoblastic cell line KS-4. Bone. 19:429–436. [DOI] [PubMed] [Google Scholar]
- 30.Dang, Z.C., R.L. Van Bezooijen, M. Karperien, S.E. Papapoulos, and C.W. Löwik. 2002. Exposure of KS483 cells to estrogen enhances osteogenesis and inhibits adipogenesis. J. Bone Miner. Res. 17:394–405. [DOI] [PubMed] [Google Scholar]
- 31.Van Der Horst, G., R.L. Van Bezooijen, M.M. Deckers, J. Hoogendam, A. Visser, C.W. Löwik, and M. Karperien. 2002. Differentiation of murine preosteoblastic KS483 cells depends on autocrine bone morphogenetic protein signaling during all phases of osteoblast formation. Bone. 31:661–669. [DOI] [PubMed] [Google Scholar]
- 32.Deckers, M.M., M. Karperien, C. Van Der Bent, T. Yamashita, S.E. Papapoulos, and C.W. Löwik. 2000. Expression of vascular endothelial growth factors and their receptors during osteoblast differentiation. Endocrinology. 141:1667–1674. [DOI] [PubMed] [Google Scholar]
- 33.Fibbe, W.E., H.M. Goselink, G. Van Eeden, J. Van Damme, A. Billiau, P.J. Voogt, R. Willemze, and J.H. Falkenburg. 1988. Proliferation of myeloid progenitor cells in human long-term bone marrow cultures is stimulated by interleukin-1 beta. Blood. 72:1242–1247. [PubMed] [Google Scholar]
- 34.Sirard, C., S. Kim, C. Mirtsos, P. Tadich, P.A. Hoodless, A. Itie, R. Maxson, J.L. Wrana, and T.W. Mak. 2000. Targeted disruption in murine cells reveals variable requirement for Smad4 in transforming growth factor beta-related signaling. J. Biol. Chem. 275:2063–2070. [DOI] [PubMed] [Google Scholar]
- 35.Korchynskyi, O., and P. Ten Dijke. 2002. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 277:4883–4891. [DOI] [PubMed] [Google Scholar]
- 36.Goumans, M.J., G. Valdimarsdottir, S. Itoh, A. Rosendahl, P. Sideras, and P. Ten Dijke. 2002. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 21:1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haaijman, A., M. Karperien, B. Lanske, J. Hendriks, C.W. Löwik, A.L. Bronckers, and E.H. Burger. 1999. Inhibition of terminal chondrocyte differentiation by bone morphogenetic protein 7 (OP-1) in vitro depends on the periarticular region but is independent of parathyroid hormone-related peptide. Bone. 25:397–404. [DOI] [PubMed] [Google Scholar]
- 38.Kusu, N., J. Laurikkala, M. Imanishi, H. Usui, M. Konishi, A. Miyake, I. Thesleff, and N. Itoh. 2003. Sclerostin is a novel secreted osteoclast-derived bone morphogenetic protein antagonist with unique ligand specificity. J. Biol. Chem. 278:24113–24117. [DOI] [PubMed] [Google Scholar]
- 39.Doty, S.B. 1981. Morphological evidence of gap junctions between bone cells. Calcif. Tissue Int. 33:509–512. [DOI] [PubMed] [Google Scholar]
- 40.Pockwinse, S.M., L.G. Wilming, D.M. Conlon, G.S. Stein, and J.B. Lian. 1992. Expression of cell growth and bone specific genes at single cell resolution during development of bone tissue-like organization in primary osteoblast cultures. J. Cell. Biochem. 49:310–323. [DOI] [PubMed] [Google Scholar]
- 41.Marotti, G. 1996. The structure of bone tissues and the cellular control of their deposition. Ital. J. Anat. Embryol. 101:25–79. [PubMed] [Google Scholar]
- 42.Martin, R.B. 2000. Does osteocyte formation cause the nonlinear refilling of osteons? Bone. 26:71–78. [DOI] [PubMed] [Google Scholar]
- 43.Larrain, J., M. Oelgeschlager, N.I. Ketpura, B. Reversade, L. Zakin, and M.E. De Robertis. 2001. Proteolytic cleavage of Chordin as a switch for the dual activities of Twisted gastrulation in BMP signaling. Development. 128:4439–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]