

Abstract
Neointimal lesions are characterized by accumulation of cells within the arterial wall and are a prelude to atherosclerotic disease. Here we report that a brief exposure to either alkyl ether analogs of the growth factor–like phospholipid lysophosphatidic acid (LPA), products generated during the oxidative modification of low density lipoprotein, or to unsaturated acyl forms of LPA induce progressive formation of neointima in vivo in a rat carotid artery model. This effect is completely inhibited by the peroxisome proliferator-activated receptor (PPAR)γ antagonist GW9662 and mimicked by PPARγ agonists Rosiglitazone and 1-O-hexadecyl-2-azeleoyl-phosphatidylcholine. In contrast, stearoyl-oxovaleryl phosphatidylcholine, a PPARα agonist and polypeptide epidermal growth factor, platelet-derived growth factor, and vascular endothelial growth factor failed to elicit neointima. The structure-activity relationship for neointima induction by LPA analogs in vivo is identical to that of PPARγ activation in vitro and disparate from that of LPA G protein–coupled receptor activation. Neointima-inducing LPA analogs up-regulated the CD36 scavenger receptor in vitro and in vivo and elicited dedifferentiation of cultured vascular smooth muscle cells that was prevented by GW9662. These results suggest that selected LPA analogs are important novel endogenous PPARγ ligands capable of mediating vascular remodeling and that activation of the nuclear transcription factor PPARγ is both necessary and sufficient for neointima formation by components of oxidized low density lipoprotein.
Keywords: neointima, LPA, PPAR, atherogenesis, lipid mediator
Introduction
Atherosclerotic disease is responsible for more than half of all mortality in developed countries (1–3). Neointima formation is an early step in the development of atherosclerotic plaques (2, 3). Atherogenic lesions progress through a prolonged process of lipid accumulation, calcification, and inflammation; subsequent rupture of the atherosclerotic plaque can trigger heart attack and stroke (4, 5). Native low density lipoprotein (nLDL) is a transporter of phospholipids and cholesterol in blood. Oxidative modification of nLDL by stresses such as cigarette smoke exposure (6) results in the generation of novel lipid mediators. This process renders the resulting minimally oxidized LDL (moxLDL) highly atherogenic (6, 7). We have reported previously that oxidative modification of LDL leads to the generation of new, active components with biological and pharmacological properties similar to lysophosphatidic acid (LPA) (8). LPA elicits pleiotropic growth factor–like effects on most cell types through the activation of four specific G protein–coupled receptors (GPCRs) (9, 10). Recent evidence supports a role for LPA as a direct agonist of nuclear transcription factor peroxisome proliferator-activated receptor (PPAR)γ (11).
LPA is produced in serum after the activation of multiple biochemical pathways linked to platelet activation and reaches concentrations at the 10-micromolar range (9, 10). LPA accumulates in the lipid-rich core of human carotid atherosclerotic plaques (12). Upon plaque rupture, LPA activates platelets and can lead to thrombus formation that is fully inhibited by antagonists of LPA GPCR (12). Several bioactive mediators, including LPA, 1-O-hexadecyl-2-azeleoyl-phosphatidylcholine (AZ-PC), and stearoyl-oxovaleryl phosphatidylcholine (SOV-PC), are generated as a result of oxidative modification of LDL (6, 7, 13). Together, these mediators affect endothelial and vascular smooth muscle cells (VSMCs) and circulating blood cells including platelets and macrophages (14–16). Recently, unsaturated acyl species of LPA have been reported to elicit neointima formation in a noninjury model of the rat carotid artery (17). This study was based on the previous observation that unsaturated acyl species of LPA caused phenotypic dedifferentiation of cultured VSMCs from the rat aorta (18).
These studies led us to investigate the effects of moxLDL on the rat carotid artery, identify the active LPA-like lipid in moxLDL, and characterize the LPA receptor–mediating neointima formation and VSMC dedifferentiation. Here we report that a brief exposure to alkyl ether analogs of LPA, products we found generated during the oxidative modification of LDL, or to unsaturated acyl forms of LPA, the predominant species produced as a result of platelet activation, induce progressive formation of neointima in vivo in a noninjury rat carotid artery model. These effects are completely inhibited by pretreatment with the PPARγ antagonist GW9662. The structure-activity relationship for neointima induction by LPA analogs in vivo is identical to that of PPARγ activation in vitro and distinct from that of G protein–coupled LPA receptor activation. Neointima were elicited by the PPARγ-specific agonists Rosiglitazone (Rosi) and AZ-PC; however, neither a PPARα-selective ligand SOV-PC, nor vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), and platelet-derived growth factor (PDGF) had such an effect. LPA activated the PPAR-regulated CD36 scavenger receptor expression in the neointimal tissue and in CV-1 cells transfected with a reporter gene containing the CD36 promoter. Activation of the CD36 reporter was activated by alkyl ether and unsaturated acyl species of LPA and required the PPAR response element (PPRE). In cultured VSMCs, Rosi and LPA 20:4 but not LPA 20:0 elicited morphological dedifferentiation and down-regulated the expression of heavy caldesmon (hCAD) mRNA, a marker of the differentiated phenotype. The morphological dedifferentiation and decrease in hCAD mRNA expression induced by these compounds was reversed by GW9662 treatment. These results suggest that activation of the nuclear transcription factor PPARγ in this model is both necessary and sufficient for neointimal lesion formation.
Materials and Methods
LPA and sphingosine-1–phosphate (S1P) were from Avanti Polar Lipids. GW9662 was from Tocris Cookson Inc. Rosi was from ARC Inc. Stereoisomers of alkyl-glycerophosphate (alkyl-GP), LPA 18:2, 18:3, 20:0, 20:4, fluorinated LPA analogs (19, 20), including 1,1-difluorodeoxy-(2R)-palmitoyl-sn-glycero-3-phosphate (XY-4), its regioisomer 1-palmitoyl-(2R)-fluorodeoxy-sn-glycero-3-phosphate (XY-8; Fig. 1), and AZ-PC (Fig. 1) were synthesized as described previously (16, 21, 22) and provided by Echelon Biosciences Inc. SOV-PC (Fig. 1) was a gift from Dr. Judy Berliner (University of California, Los Angeles, Los Angeles, CA).
Figure 1.

Structural formulas of lipid mediators used in the study.
nLDL was purchased from Sigma-Aldrich, freed of EDTA by desalting on PD10 columns (Amersham Biosciences), and oxidized using Cu2+ as a catalyst (14). Protein concentrations were determined using the BCA protein assay kit (Pierce Chemical Co.). Final LDL concentrations were adjusted to 5 mg/ml before use. The plasma LDL concentration is 1.6 mg/ml but in patients can be as high as 8 ± 6 mg/ml (23).
All procedures using animals have been reviewed and approved by the University of Tennessee Health Science Center Memphis Institutional Animal Care and Use Committee. Topical application of the test compounds was performed using the model developed and characterized recently by Yoshida et al. (17). Briefly, the right carotid artery of anesthetized adult male Sprague-Dawley rats (250–300 g) was surgically exposed. The caudal origin of the common carotid artery (CCA) was ligated using a vessel clip, followed by exposure and ligation of the internal carotid artery above the bifurcation. The external carotid artery was exposed, and a polyethylene catheter was inserted such that it never reached the CCA, thereby avoiding mechanical injury to the vessel. The clip occluding the CCA was temporarily released, and the vessel was rinsed with a retrograde injection of 500 μl physiological saline to remove residual blood. The vessel was again clipped, and 100 μl of treatment solution was injected. After 60 min of incubation, the cannula was withdrawn, the external carotid artery was ligated, and blood flow was restored. Animals were allowed to recover and were killed 7–56 d later by intracardiac perfusion of 10% buffered formaldehyde (pH 7.4). The CCA from the jugular arch to the bifurcation was dissected, embedded in paraffin, and processed for histological analysis. 5-μm-thick sections were cut and stained with hematoxylin and eosin or Masson's trichrome stain. Intima to media ratios were quantified using an image analysis system (Scion Image CMS-800).
LPA and alkyl-GP species were extracted from nLDL and moxLDL in acidic butanol followed by quantitative analysis by stable isotope dilution electrospray ionization mass spectrometry as described previously (24). CV-1 cells were plated in 96-well plates (5 × 103 cells per well) in DME supplemented with 10% FBS. The next day, the cells were transiently transfected with 125 ng of pGL3-PPRE–acyl-CoA oxidase (Acox)–renilla luciferase (Rluc), or 125 ng pGL3-CD36(-273), or pGL3-CD36(-261), 62.5 ng of pcDNAI-PPARγ, and 12.5 ng of pSV–β-galactosidase (Promega) using LipofectAMINE 2000 (Invitrogen). 24 h after transfection, cells were treated with 1% FBS-supplemented OptiMEMI (Invitrogen) containing DMSO or test compound (10 μM) in DMSO for 20 h. Luciferase and β-galactosidase activities were measured with the Steady-Glo® Luciferase Assay System (Promega) and the Galacto-Light Plus™ System (Applied Biosystems), respectively. Samples were run in quadruplicate, and the mean ± SE were calculated. Data are representative of at least three independent transfections. Student's t test was used for null hypothesis testing, and P < 0.05 was considered significant.
Rat aortic VSMC cultures were established as described by Hayashi et al. (18) in the presence of 2 ng/ml insulin-like growth factor (IGF)-1. Sister cultures were established in the presence of IGF-1 and treated with 1 μM of Rosi, LPA 20:4, or LPA 20:0 on day 2 for an additional 3 d. Half of the cultures received 200 nM GW9662 30 min prior exposure to LPA and Rosi. Cell morphology was recorded on day 5, and cells were harvested for mRNA extraction and quantitative RT-PCR.
RT-PCR for LPA GPCR expression was done as described earlier by Wang et al. (25). To quantify hCaD mRNA, quantitative PCR was performed applying the real-time SYBR Green PCR method using a Sequence Detection System Model 7700 (Applied Biosystems) instrument. The rat hCaD and GAPDH (reference control mRNA)-specific primers were designed with Primer Express Software (Applied Biosystems), and forward and reverse primers were as follows: 5′-GAACCAAAGCTGAGCAGGACA-3′ and 5′-TTCGTGCAGCCTCCATTCTT-3′ for hCaD; 5′-AAGCTCACTGGCATGGCCTT-3′ and 5′-CGGCATGTCAGATCCACAAC-3′ for GAPDH. The amplification reaction was performed with SYBR Green PCR Master Mix (Applied Biosystems) following the manufacturer's protocol. mRNA abundance calculation was based on Ct values as described previously (25). The expression level of hCAD mRNA was normalized to GAPDH mRNA. Each PCR reaction was performed at least three times, and the result was expressed as mean ± SEM. Statistical comparison of mRNA expression was evaluated by ANOVA, and P < 0.05 was considered statistically significant.
Results
To evaluate the effect of oxidatively modified LDL on vascular remodeling, we treated the common carotid artery of rats in situ for 1 h with nLDL and moxLDL using a procedure that avoided direct mechanical damage to the vessel. This model was described recently and characterized by Yoshida et al. (17). 2 wk after treatment, carotid arteries were dissected en bloc and processed for histological evaluation. moxLDL, but not nLDL, elicited pronounced and significant neointima formation as illustrated in Fig. 2, A–C.
Figure 2.

moxLDL treatment induces neointima formation in rat carotid arteries. Representative views of Masson's trichrome-stained, paraffin-embedded sections from animals treated with nLDL (A) or moxLDL (B) (5 mg LDL protein/ml) 2 wk after a 1-h treatment. Bar, 500 μm. Intima to media ratios were quantified (C, n = 5).
Minimal oxidization of LDL generates LPA-like biological activity (8). LPA elicits numerous effects on cells of the cardiovascular system including stimulation of platelet aggregation, activation of macrophages and endothelial cells, and the dedifferentiation and proliferation of VSMCs (8, 16, 18, 26–28). Many of these LPA-elicited cellular effects are implicated in the development of neointima lesions. Therefore, we hypothesized that oxidative modification of LDL increases LPA levels in atherogenic moxLDL. The concentrations of five acyl-LPA species were determined in nLDL and moxLDL after copper-mediated minimal oxidization (Fig. 3 A). Surprisingly, total acyl-LPA levels in moxLDL were not significantly different (180 ± 19 pmol/mg LDL protein, n = 4) from the nLDL control (190 ± 13 pmol/mg LDL protein, n = 4). However, there were significant decreases in the concentration of polyunsaturated acyl-LPA species in moxLDL, a finding consistent with oxidative degradation. Although the majority of LPA characterized in biological fluids and tissues is the acyl form, the alkyl ether glycerophosphate analogue, alkyl-GP, has also been detected (29). Alkyl-GP has biological properties distinct from acyl-LPA. For example, alkyl-GP is 50 times more potent than acyl-LPA in the activation of platelets (12, 30, 31). We also quantified alkyl-GP levels in the LDL preparations and found that alkyl-GP content was sixfold higher in moxLDL, with the octadecenyl (18:1) species showing a 10-fold increase over nLDL (Fig. 1 E and Fig. 3 B). Interestingly, the rank order of alkyl-GP species present in moxLDL was the same as reported for the lipid core of human atherosclerotic plaques (12).
Figure 3.
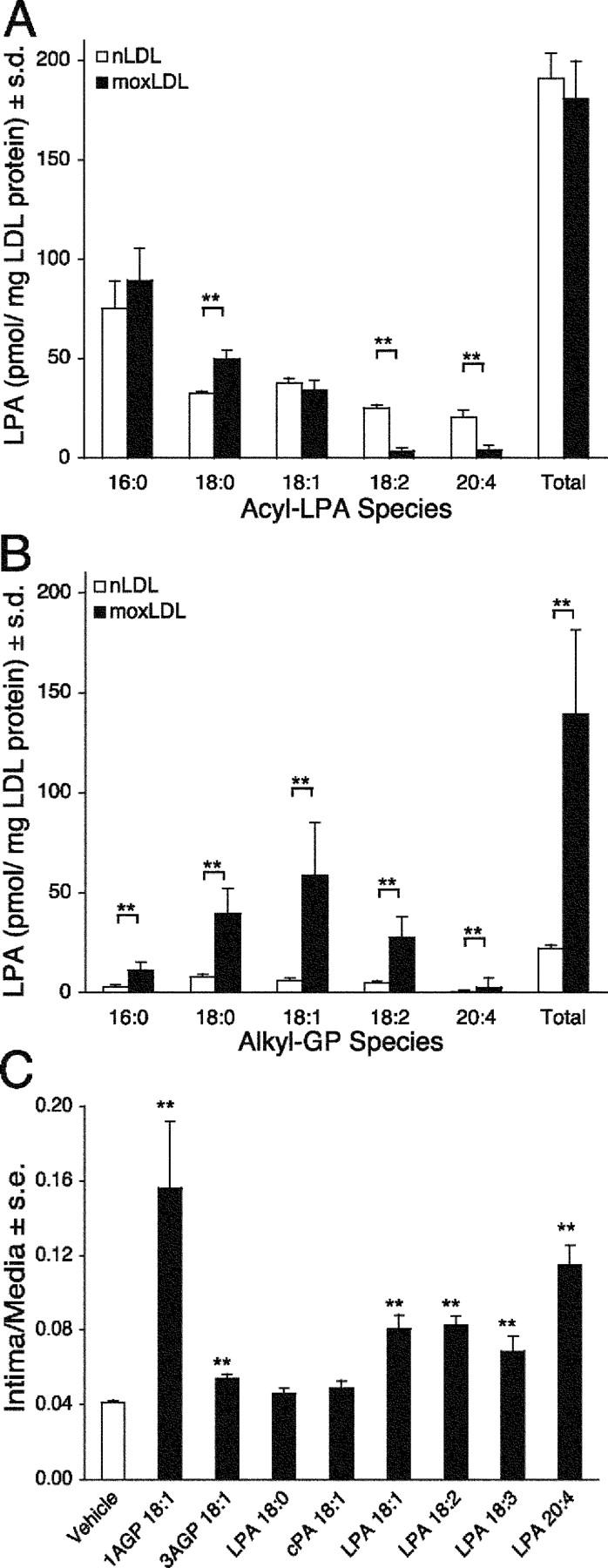
The five most abundant acyl-LPA (A) and alkyl-GP (B) species were quantified in nLDL and moxLDL using stable isotope dilution electrospray ionization mass spectrometry. The lack of difference in the total acyl-LPA content between nLDL and moxLDL is in sharp contrast to the sixfold increase in alkyl-GP levels in moxLDL (n = 4). In the batch of nLDL used in the experiments shown in Figs. 2 and 3, the alkyl-GP concentration was 0.1 μM, and the total concentration of unsaturated LPA plus alkyl-GP was 0.5 μM, whereas in moxLDL these concentrations were 0.7 and 0.9 μM, respectively. (C) Structure-activity relationship of neointimal lesion induction for various acyl-LPA (10 μM) and alkyl-GP (AGP) species (10 μM). Only select LPA species elicit neointima as the effect was stereoselective with a preference for 1AGP over 3AGP. LPA 18:0 and cPA 18:1 did not elicit detectable neointima (for structural formulas see Fig. 1).
Mild oxidation of LDL produces prothrombotic and proatherogenic moxLDL (6, 8, 12). LPA GPCR antagonist abolish platelet aggregation elicited by moxLDL, indicating that LPA plays an essential role in the thrombogenic effects of moxLDL (12). To define the contribution of LPA to the neointima-inducing potential of moxLDL, we determined the effect of various alkyl-GP and acyl-LPA species on neointima formation in the rat carotid artery model (Fig. 3 C). 1-O-octadecenyl-glycerophosphate (1AGP; the natural stereoisomer) was highly effective, whereas 3-O-octadecenyl-glycerophosphate (3AGP; the unnatural stereoisomer) was modestly effective in eliciting neointima. The ether bond in alkyl-GP is resistant to cleavage by phospholipases A. Consequently, the metabolic conversion of alkyl-GP–derived fatty alcohols can be ruled out, suggesting that intracellular phospholipases of the A1 type are not involved in generating a bioactive metabolite of alkyl-GP. 2,3-Cyclic phosphatidic acid (cPA; 18:1), an endogenous, unsaturated acyl-LPA analogue containing a cyclic phosphate, was inactive. Unlike alkyl-GP, cPA18:1 is a substrate for phospholipases A. Hence, the lack of its neointima-inducing action indicates that oleic acid, a potential hydrolysis product of cPA 18:1, is not sufficient to elicit this response. Together, these results indicate a stereospecific requirement for both an unsaturated fatty acid/fatty albyl group and a free phosphate on the glycerol backbone in order to stimulate neointima formation.
Activation of platelets results in LPA production, which is dominated by the polyunsaturated 20:4 (arachidonoyl) and 18:2 (linoleoyl) acyl species. Since LPA activates platelet aggregation, LPA production from platelets sets up a feed forward regulation mechanism that perpetuates platelet activation and the production of LPA species enriched in polyunsaturated fatty acids (24). Yoshida et al. (17) showed in the rat that LPA species containing unsaturated fatty acyl groups 16:1, 18:1, and 18:2 induced formation of neointimal lesions, whereas the saturated acyl-LPA species were inactive (Fig. 3 C). We exposed rat carotid arteries for 1 h to LPA 20:4 or 20:0 and monitored vascular remodeling for up to 8 wk posttreatment. LPA 20:4 was chosen because it is the most abundant species in human serum (∼40% of total [24, 32]) with concentrations up to 2.5 μM. In contrast, the total circulating concentration of acyl-LPA in plasma is <0.1 μM (24). This brief exposure to 2.5 μM LPA 20:4 elicited progressive neointimal growth, whereas 2.5 μM LPA 20:0 was completely inactive (Fig. 4 A). The extent of neointima development elicited by LPA 18:1 treatment was concentration dependent up to 10 μM, the highest concentration tested and reached statistical significance at 5 μM (P < 0.01; Fig. 4 B). This concentration is equivalent to the total LPA concentration found in human serum (24).
Figure 4.
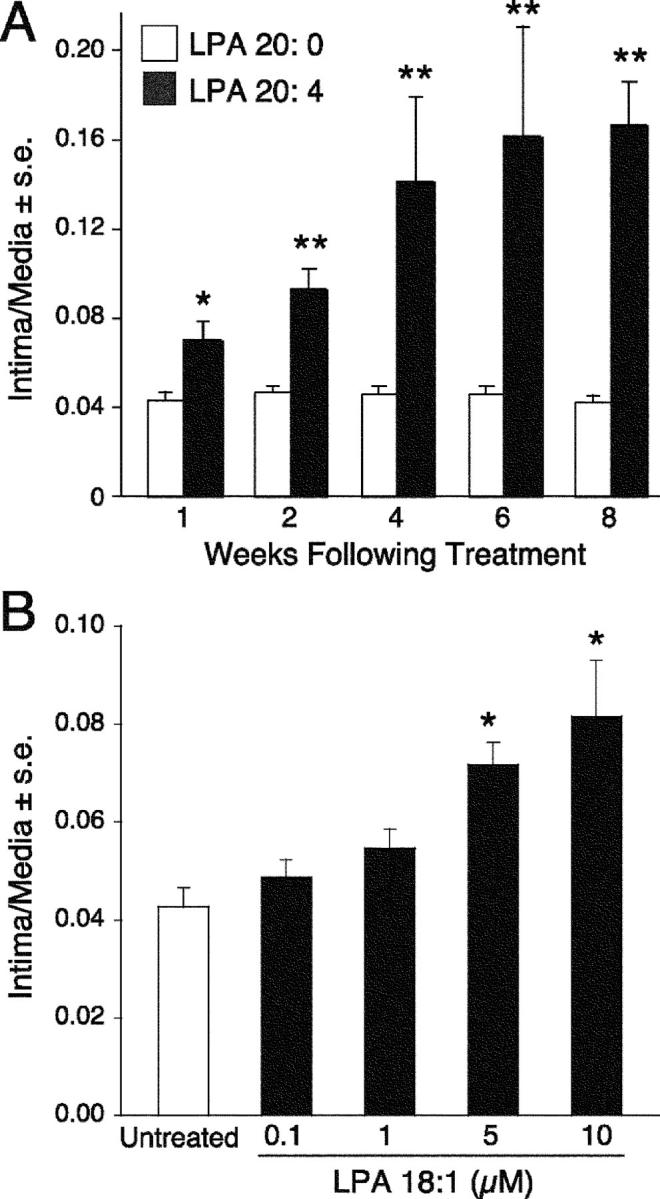
(A) Exposure of rat carotid arteries for 1 h to 2.5 μM LPA 20:4, but not to LPA, 20:0 elicited progressive growth of neointima that continued for up to 8 wk posttreatment. Quantitative morphometric analysis for groups of 5 animals. (B) Dose–response curve for LPA 18:1-elicited neointimal response. Mean (± SE) intima to media ratios were determined for groups of five animals 2 wk after treatment.
LPA is a growth factor-like phospholipid mediator that activates specific G protein–coupled plasma membrane receptors LPA1, LPA2, and LPA3 encoded by the endothelial differentiation gene family (33) and the distantly related LPA4 (34). RT-PCR analysis using gene-specific primers showed dominant expression of LPA1, low levels of LPA4, and no transcripts for LPA2 or LPA3 in untreated rat carotid arteries (Fig. 3 A and Fig. 5 A).
Figure 5.
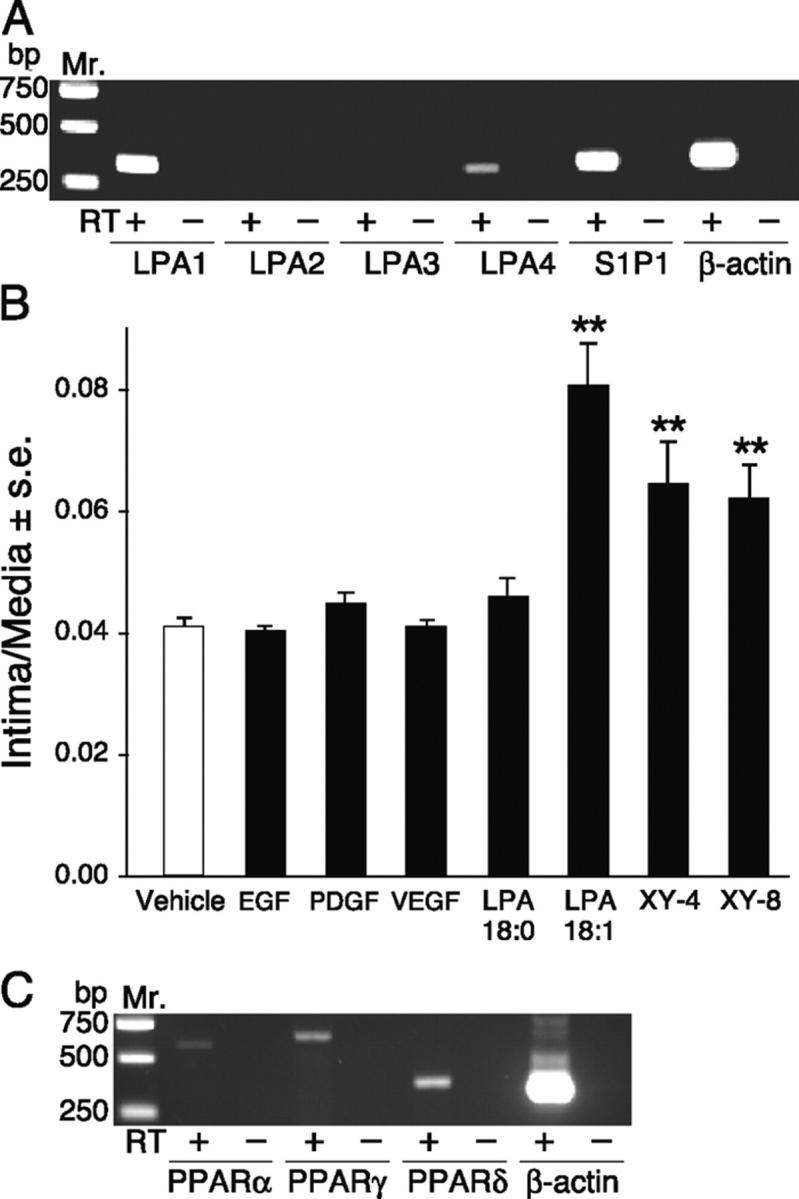
(A) RT-PCR of LPA GPCR in the rat carotid tissue. LPA1, LPA4, and S1P1 were detected in RNA extracted from the whole carotid tissue. (B) Effect of polypeptide growth factors and non-LPA GPCR ligands fluorinated LPA analogs on neointima formation. Animals treated with 10 μM LPA 18:1, XY-4, and its regioisomer XY-8 but not those treated with EGF (50 ng/ml), VEGF (10 ng/ml), PDGF-BB (10 ng/ml), or LPA 18:0 (10 μM) showed neointima formation. Groups of five animals were treated with the compounds. (C) RT-PCR analysis detected PPARα, PPARδ, and PPARγ transcripts in the normal carotid tissue.
LPA GPCRs are activated by both saturated and unsaturated acyl-LPA species with a rank order of potency of acyl-LPA>alkyl-GP>cPA (35). Moreover, LPA GPCRs do not show stereoselectivity to alkyl-GP (21). The structure-activity relationship for LPA-induced neointima formation was markedly different from that described for LPA GPCR. First, neointima formation shows a rank order of alkyl-GP>acyl-LPA, with cPA being inactive. Second, unsaturated but not saturated fatty acyl species stimulated lesion formation (Fig. 3 C). Third, formation of neointima shows a stereoselective preference for 1-O-octadecenyl-GP over 3-O-octadecenyl-GP. Fourth, in contrast to alkyl-GP and unsaturated acyl-LPA, EGF (50 ng/ml), VEGF (10 ng/ml), and PDGF (10 ng/ml) failed to induce detectable neointima in this model after a 2-wk period (Fig. 5 B). LPA GPCRs have been found to transactivate the EGF and PDGF receptors (for review see reference 9); thus, the lack of neointimal response to authentic ligands of these tyrosine kinase receptors discredits the involvement of such a mechanism. Fifth, fluorinated LPA-like PPARγ agonists that are four orders of magnitude less potent as LPA GPCR agonists than LPA 18:1 (11) were nearly as potent at inducing neointima formation in the rat model (Fig. 5 B). These results lead to the conclusion that a receptor distinct from known LPA GPCR mediates neointimal lesion formation.
LPA1-induced cell proliferation is fully blocked by pertussis toxin (PTX; 28). To examine the role of PTX-sensitive G proteins in the neointimal response elicited by LPA, vessels were treated with 100 ng/ml PTX for 30 min before and during LPA 20:4 exposure. PTX pretreatment attenuated, but did not abolish, the response to LPA 20:4 (Fig. 6 A). Among LPA GPCRs, the LPA3 receptor is unique with a preference for unsaturated LPA species (35). However, RT-PCR analysis of normal carotid tissue showed no detectable LPA3 transcript (Fig. 5 A). Likewise, dioctylglycerol pyrophosphate (DGPP), a competitive antagonist of the LPA3 and LPA1 receptors (36), produced a modest inhibition (Fig. 6 A). These observations discount, but do not completely exclude, a major role for LPA GPCRs in neointima formation.
Figure 6.

(A) PTX and DGPP, inhibitors of LPA GPCR signaling, partially attenuated neointima formation induced by LPA 20:4, whereas the PPARγ-specific antagonist GW9662 completely abolished this effect. (B) Rosi, AZ-PC, moxLDL, and unsaturated acyl forms of LPA all induced neointima formation that was completely abolished by GW9662. In contrast, SOV-PC, a PPARα-selective agonist, was ineffective in stimulating the development of neointima after 2 wk. (C) An in vitro assay using CV1 cells transfected with PPARγ and a PPRE-Acox-Rluc reporter gene showed an identical structure-activity relationship when exposed to different LPA species as found for the same set of ligands in the neointima assay in vivo (see Fig. 3 C). (D) The PPARγ antagonist GW9662 (10 μM) abolished, whereas PTX (100 ng/ml, 2 h) and DGPP (10 μM, 2 h) pretreatment and coapplication with LPA partially inhibited LPA 20:4-induced PPRE-Acox-Rluc reporter gene expression in vitro. Vehicle contained 1% DMSO, Rosi (10 μM), LPA20:0 (10 μM), or LPA 20:0 (10 μM) were applied for 20 h. Luciferase and β-galactosidase activities (mean ± SEM) were measured in the cell lysate (n = 4). (E and F) Dose–response relationship of LPA 20:4- and Rosi-induced activation of PPRE-Acox-Rluc reporter gene expression in vitro. *P < 0.05 and **P < 0.01, significant differences over vehicle control.
In addition to its plasma membrane receptors, LPA was shown recently to be an agonist of the nuclear transcription factor PPARγ (11). PPARγ has long been implicated in atherogenesis (37, 38). PPARs are lipid-activated transcription factors of the nuclear receptor superfamily that heterodimerize with the retinoic acid X receptor. PPAR/retinoic acid X receptor heterodimers bind to specific peroxisome PPREs to regulate gene expression (39). Many compounds activate PPAR, including the synthetic drug Rosi of the thiazolidinedione (TZD) family, oxidized phospholipids, fatty acids, eicosanoids, and oxidized LDL. PPARγ is expressed in macrophages/monocytes, VSMCs, endothelial cells, and is highly expressed in atherosclerotic lesions and hypertensive vascular wall (1, 37). PPARγ was detected in normal rat carotid tissue by RT-PCR (Fig. 5 C). For this reason, GW9662, a specific irreversible antagonist of PPARγ (40), was applied at a concentration of 5 μM 30 min before and was coapplied with 2.5 μM LPA 20:4. GW9662 completely abolished neointima formation elicited by LPA 20:4, indicating that PPARγ activation is required for the development of LPA-induced lesion development (Fig. 6 B).
To further evaluate the hypothesis that PPARγ activation leads to neointima formation, vessels were pretreated with GW9662 or vehicle, with subsequent coapplication of the PPARγ agonist Rosi. Rosi induced neointima formation with a time course identical to that of LPA 20:4, indicating that the activation of PPARγ is sufficient to elicit neointima formation in this model (Fig. 6 B). Lesion formation by Rosi was fully blocked by GW9662 treatment (Fig. 6 B). The endogenous PPARγ agonist AZ-PC (16), an active component of moxLDL, also caused neointima formation that was inhibited by GW9662 (Fig. 6 B). Accordingly, the neointima formed in response to moxLDL, LPA 20:4, and LPA 18:1 was also inhibited by GW9662 pretreatment. In contrast, treatment with the PPARα agonist SOV-PC (41) and saturated LPA species failed to stimulate neointima formation, and GW9662 treatment had no effect on the vessels from animals treated with these agents (Fig. 6 B). These results together with those obtained with the PPARγ agonists XY-4 and XY-8 (Fig. 5 B) support an essential role for PPARγ activation in neointima formation in our model.
The structural requirements of LPA-elicited neointima formation are distinct from those of any known LPA GPCRs. Therefore, we compared the unique in vivo neointima-eliciting LPA structure-activity relationship with that of PPARγ using an in vitro assay that utilizes an acyl-coenzyme A oxidase-luciferase (PPRE-Acox-Rluc) reporter gene construct containing PPRE (11). CV1 cells were transiently transfected with PPARγ and PPRE-Acox-Rluc and exposed to various LPA analogs for 20 h. Results from this assay (Fig. 6 C) were identical to the structure-activity relationship found in vivo (Fig. 3 C). Rosi, unsaturated acyl-LPA species, and alkyl-GP all elicited significant activation of the PPRE-Acox-Rluc reporter, whereas saturated acyl LPA species, cPA, and the related lipid mediator S1P were inactive. Interestingly, not only octadecenyl-GP but also octadecyl-GP and hexadecyl-GP activated the reporter gene, indicating the unique properties of alkyl-GP analogs in activating PPARγ. Moreover, PTX and DGPP treatment reduced but did not abolish activation of the PPRE-Acox-Rluc reporter (Fig. 6 D), results that are consistent with the in vivo experiments (Fig. 6 A). The dose–response relationship of LPA- and Rosi-elicited activation of the PPRE-Acox-Rluc reporter (Fig. 6, E and F) was similar to that of the neointimal response (Fig. 4 B), although with a lower threshold, as high nanomolar concentrations of the two PPARγ agonists were sufficient to cause a significant activation of the receptor genes.
Expression of PPARγ protein in rat carotid arteries exposed to LPA or moxLDL was examined. Immunohistological staining for the PPARγ antigen showed only a few stained nuclei in arteries from animals treated with LPA 20:0 (Fig. 7 A) and nLDL (unpublished data). In contrast, intense PPARγ immunoreactivity was observed in neointimal lesions in carotid arteries from the LPA 20:4 (Fig. 7 B) and moxLDL (unpublished data) treatment groups that strongly resembled that reported previously in atherosclerotic lesions (1, 16, 37).
Figure 7.
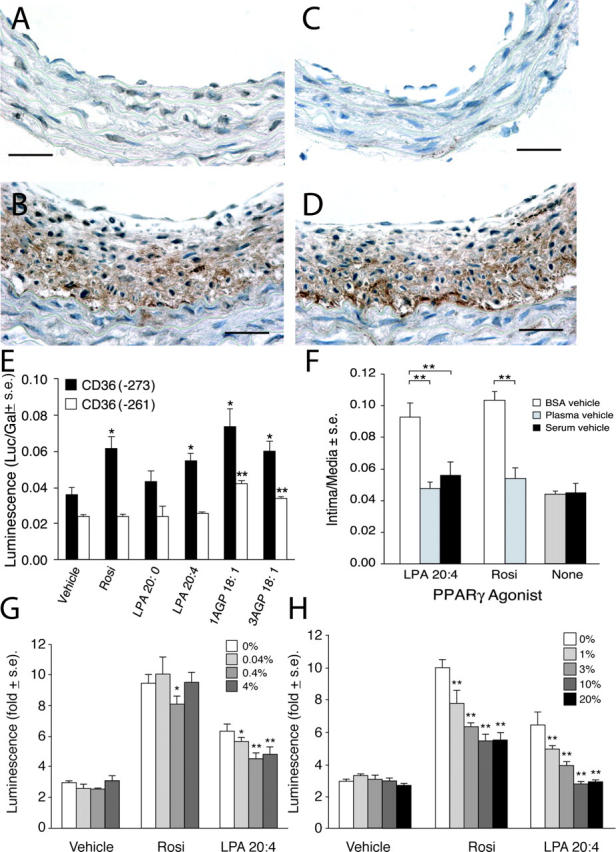
(A) Only a few nuclei show PPARγ immunoreactivity (in a carotid artery 4 wk after treatment with 2.5 μM LPA 20:0). In contrast, the multilayered neointima elicited by LPA 20:4 (B) expresses high levels of PPARγ immunoreactivity. Bars, 250 μm. Activation of PPARγ within neointima in LPA 20:4-treated carotid arteries is indicated by the strong expression of CD36 in a distribution that overlaps that of PPARγ (D). Little immunoreactivity for CD36 was noted in LPA 20:0-treated animals (C). Anti-PPARγ and anti-CD36 were from Santa Cruz Biotechnology, Inc. (E) Stimulation of CD36(−271)-Rluc and CD36(−261)-Rluc reporter genes by Rosi, LPA, and AGP in CV-1 cells. Rosi and LPA 20:4 but not LPA 20:0 (all 10 μM) elicited significant stimulation of CD36(−273)-Rluc that contains a PPRE between bp −273 and −261. Neither compound caused stimulation of the CD36(−261)-Rluc. 1AGP showed higher stimulation compared with 3AGP of the expression of the Rluc reporter. (F) Plasma and serum factors inhibit Rosi- and LPA 20:4-induced neointimal lesion formation in rat carotid arteries. Rosi (10 μM) and LPA 20:4 (2.5 μM) elicited neointima formation 2 wk after treatment when delivered as BSA complexes. In contrast, when the compounds were delivered in rat plasma or serum no neointima formation was detected (n = 5). Effect of BSA (G) or serum (H) on PPARγ activation by CPA and Rosi.
The scavenger receptor CD36, a PPARγ-regulated gene, contains a PPRE within its promoter between base pairs –273 and –261 (11). CD36 plays a critical role in lipid uptake by binding and transporting oxidized lipids, including moxLDL (42). Thus, this pathway could provide a source of ligands, such as LPA, alkyl-GP, and AZ-PC, to sustain PPARγ activation. Acyl and alkyl forms of LPA both accumulate in human atherosclerotic plaques (12). LPA activates CD36-mediated lipid uptake into macrophages through a PPARγ-PPRE–dependent mechanism (11). Immunostaining for the CD36 antigen showed no increase in immunoreactivity in vessels treated with LPA 20:0 (Fig. 3 F and Fig. 7 C) or nLDL (unpublished data). However, neointimal tissue derived from treatment with LPA 20:4 (Fig. 7 D) and moxLDL (unpublished data) showed intense CD36 immunoreactivity. The up-regulated expression of CD36 suggests that PPARγ is activated in LPA 20:4- and moxLDL-elicited neointimal lesions. When tested in vitro using CV1 cells transfected with two CD36-luc reporter constructs, one with and one without the PPRE between –273 and –261, Rosi, LPA 20:4, and alkyl-GP up-regulated the expression of a CD36-luc reporter gene, whereas LPA 20:0 was inactive (Fig. 7 E). This activation was dependent on the presence of the PPRE because neither Rosi nor LPA 20:4 activated the reporter gene with a deleted PPRE. Interestingly, whereas activation by alkyl-GP was much reduced in the PPRE deletion mutant it was not completely abolished, suggesting that in CV-1 cells other promoter elements might also become activated by this ligand but not by LPA 20:4.
There is ∼0.1 μM LPA in plasma and TZD drugs are now being used for the treatment of diabetics, yet there are no reports of increased vascular complications in these patients. To resolve this apparent controversy, we tested whether plasma factors could attenuate the neointima-inducing effects of Rosi and/or LPA. This hypothesis is based on earlier reports showing that LPA diluted in plasma and high concentrations of albumin shows diminished biological responses (32, 43). Heparinized, syngeneic plasma, or serum was compared with serum albumin (2.5 μM) as the vehicle for Rosi and LPA 20:4 delivery. Only those animals receiving Rosi or LPA 20:4 complexed with 2.5 μM albumin developed neointimal lesions, whereas those that received the compounds delivered in plasma or serum showed no significant neointima formation (Fig. 7 F). Neither plasma nor serum alone had neointima-inducing effects (Fig. 7 F). This suggests that plasma and serum factors attenuate formation of neointima and serve to mitigate widespread effects of endogenous LPA and suppress the effect of Rosi. LPA present in serum readily activates LPA GPCR-mediated biological responses (43–45); thus, the lack of activity of serum and LPA delivered in serum points to an important difference in the ligand recognition of LPA GPCR versus PPARγ. Moreover, Tokumura et al. (46) reported that the transbilayer movement of alkyl-GP is blocked by high concentrations (2%) of albumin, whereas it remains substantial at low albumin concentrations (0.05%), which is similar to what we used in our assays. To further substantiate the notion that transbilayer movement of LPA and Rosi could be affected by albumin and plasma/serum factors in a concentration-dependent manner, we applied the PPRE-Acox-Rluc reporter assay. Addition of albumin (0.04–4% wt/vol; Fig. 7 G) or serum (1–20% vol/vol; Fig. 7 H) inhibited LPA- and Rosi-induced activation of the PPRE-Acox-Rluc reporter gene in a concentration-dependent manner, providing further support to the hypothesis that carrier proteins could provide a physiological barrier to the transbilayer movement of these ligands, thus preventing/attenuating activation of PPARγ.
Hayashi et al. (18) reported that unsaturated LPA species induced phenotypic modulation of cultured VSMCs. Because the neointimal response and PPARγ display a similar selectivity for unsaturated LPA, we hypothesized that activation of PPARγ plays a role in the phenotypic dedifferentiation. We tested this hypothesis in the same experimental paradigm used by Hayashi et al. In this culture system, IGF-1 maintains the differentiated spindle shape of VSMCs, indicated by high levels of hCAD mRNA expression (18). VSMCs established in the presence of 2 ng/ml IGF-1 for 2 d were exposed to 1 μM of either Rosi, LPA 20:4, or LPA 20:0 for 3 d with or without 200 nM GW9662. Those VSMCs exposed to Rosi and LPA 20:4 developed a fibroblast-like flattened morphology (Fig. 8, E and G), whereas cultures exposed to LPA 20:0 (Fig. 8 C) maintained the spindle-like differentiated morphology seen in the IGF-1–treated controls (Fig. 8 A). GW9662 treatment reversed this effect of LPA 20:4 and Rosi (Fig. 8, F and H). Using quantitative RT-PCR, we found that hCAD mRNA expression decreased by all treatments but was most pronounced in VSMCs exposed to LPA 20:4 and Rosi (Fig. 8 I). GW treatment caused a significant increase in hCAD mRNA expression. These results support the hypothesis that PPARγ plays an essential role in the phenotypic modulation of VSMCs.
Figure 8.

PPARγ agonists elicit phenotypic modulation and dedifferentiation of VSMCs in vitro. VSMC cultures established in the presence of 2 ng/ml IGF-1 (A) were treated with 1 μM of each LPA 20:0 (C), LPA 20:4 (E), and Rosi (G) for 3 d. LPA 20:4 and Rosi treatments lead to a pronounced change in the morphology of VSMCs. Pretreatment of the cultures with 200 nM GW9662 for 30 min did not affect the spindle-like morphology of the IGF-1– (B) and LPA 20:0-treated cultures (D). In contrast, GW9662 reversed the flattened morphology into a spindle-like shape in cultures treated with LPA 20:4 (F) and Rosi (H, calibration bar 100 μm). Expression of hCAD mRNA decreased significantly by day 5 in VSMCs treated with Rosi and LPA (I, white bars) compared with the IGF-treated control cultures. This trend was reversed in cultures pretreated with 200 nM GW9662 (I, black bars) as the PPARγ antagonist caused a significant increase in the abundance of hCAD mRNA measured by quantitative RT-PCR (P < 0.01, ANOVA).
Discussion
Here we show that VSMCs and unsaturated acyl species of the phospholipid mediator LPA elicit progressive and long-lasting neointima formation in the rat carotid artery that requires activation of PPARγ. The structure-activity relationship for lesion development is distinct from that of known LPA GPCR. These data support the hypothesis that the LPA-induced neointimal response is mediated by a novel receptor. We propose that the relevant receptor is PPARγ. The synthetic PPARγ ligands Rosi, XY-4, XY-8, and AZ-PC mimic the effect of LPA both in vivo and in vitro. Accordingly, the PPARγ-specific antagonist GW9662 completely abolished the effect of Rosi, AZ-PC, moxLDL, and LPA20:4. In contrast, the PPARα receptor ligand SOV-PC, as well as EGF, VEGF, and PDGF, and saturated acyl LPA failed to stimulate neointima formation in this model. Hayashi et al. (18) reported that only unsaturated LPA species were effective in eliciting VSMC dedifferentiation in vitro. Using the same culture condition described by these authors, we confirmed that the phenotypic modulation of VSMCs was elicited by LPA 20:4 and established that Rosi mimicked this effect. Phenotypic modulation of VSMCs and the decrease in hCAD mRNA expression elicited by these PPARγ agonists was abolished by GW9662. The structure-activity relationship of the phenotypic modulation of VSMCs agrees with that of the neointimal lesions and activation of PPARγ in our experiments.
TZD drugs, including Rosi, are widely used in the treatment of type 2 diabetes, yet patients taking these drugs do not develop vascular complications above those associated with their primary disease (47). It is tempting to propose that neointima formation elicited by LPA could lead to atherogenesis; however, these two phenomena, although interrelated, depend on highly complex cellular and metabolic events that occur over long periods of time. Consequently, the present results pertain only to neointima formation elicited by topical application of LPA and PPARγ agonists in buffers that have low concentrations of carrier proteins (0.05% BSA). TZD PPARγ agonists have been reported to mitigate atherosclerosis in patients with type II diabetes (48). In this context, we emphasize that TZD have multiple effects on glucose and lipid metabolism and inflammatory cellular responses, some of which are not mediated by PPARγ (49). Due to this complexity of TZD effects, we adapt the hypothesis put forward by Chawla et al. (49) that the consequence of PPARγ activation may be atherogenic in some contexts and protective in others as determined by cellular context. The single and brief topical application of LPA and Rosi in the present model, although at physiologically and therapeutically relevant concentrations, is not representative of systemic and chronic application. However, one must take into consideration that the manifestation of atherosclerotic disease spans decades.
A pressing question remains: how does extracellularly applied LPA find its way to the nuclear transcription factor PPARγ? Transbilayer movement of phospholipids has been thoroughly investigated and established for phosphatidic acid (46, 50), platelet-activating factor (PAF) (46, 50), lyso-PAF (51), LPA, S1P (52), 1-O-alkyl-2–acetyl-GP, and alkyl-GP (46). Tokumura et al. (46) using alkyl-GP and its 2-acetyl analogue showed that within 30 min of extracellular application a steady-state condition develops with as much as 10–14% of the intact lipid present in the intracellular membrane compartment and the endoplasmic reticulum, contiguous with the nuclear membrane. Thus, the micromolar extracellular concentrations of lipid mediators required to elicit the neointimal response could sustain intracellular LPA concentrations in the hundred nanomolar range, an intracellular concentration well in excess of the nanomolar KD for LPA binding to PPARγ. Many of these studies used the albumin extraction method (53), which utilizes the high-affinity binding of these lipids to albumin for their quantitative extraction from the outer leaflet of the plasma membrane. Albumin concentrations ≥2% will quantitatively extract LPA from the outer leaflet and prevent its transbilayer movement. Based on this observation, and our results shown in Fig. 7, F–H, the inhibitory effect of plasma and serum could be due to its high albumin concentration (∼4%). Notwithstanding, the impact of plasma albumin on LPA bioactivity might be limited when LPA is either generated locally by platelets. LPA bound to other carriers than albumin, for example, LDL and moxLDL in particular, are taken up by receptor-mediated mechanisms and lead to the activation of PPARγ. Activation of GPCR signaling has been reported to augment the transbilayer movement of PAF and lyso-PAF (51) and could also enhance LPA uptake. This raises the possibility that LPA GPCR activation could facilitate the transbilayer movement of LPA, providing an explanation for the attenuating but not abrogating effect of PTX and DGPP we observed with respect to neointimal response and PPARγ reporter gene activation.
Generation of an LPA-like biological activity during oxidative modification of LDL has been documented previously, and here we provide evidence that alkyl ether analogs of LPA increase in moxLDL. LPA generated in moxLDL has the ability to activate platelet LPA GPCR (8, 12) and lead to production of unsaturated LPA species, which are likely to contribute to its thrombogenic and atherogenic effects. Thus, it appears that LPA, through the activation of platelets and up-regulation of CD36-mediated moxLDL uptake, could provide a link between two feed-forward loops; one, which leads to LPA production, and another that delivers ligands to PPARγ. These two pathways potentially participate in a synergistic mechanism that leads to neointima formation and vascular wall remodeling. We suggest that alkyl-GP, along with unsaturated acyl-LPA, in addition to oxidized phosphatidylcholines (AZ-PC), represent a novel group of bona fide endogenous PPARγ ligands that modulate vascular remodeling.
Acknowledgments
The authors thank Linda White for expert assistance with immunohistology.
This work was supported by research grants 61469 (to G. Tigyi), CA92160 (to G. Tigyi), and HL070231 (to T.M. McIntyre) from the National Institutes of Health, Human Frontier Science Program grant RG0073-2000-B (to G.D. Prestwich), and the American Heart Association 0120228B (to D.L. Baker).
Abbreviations used in this paper: 1AGP, 1-octadecenyl-glycerophosphate; 3AGP, 3-O-octadecenyl-glycerophosphate; Acox, acyl-CoA oxidase; alkyl-GP, alkyl ether glycerophosphate; AZ-PC, 1-O-hexadecyl-2-azeleoyl-phosphatidylcholine; CCA, common carotid artery; cPA, 2,3-cyclic phosphatidic acid; DGPP, dioctylglycerol pyrophosphate; EGF, epidermal growth factor; GPCR, G protein–coupled receptor; hCAD, heavy caldesmon; IGF, insulin-like growth factor; LDL, low density lipoprotein; LPA, lysophosphatidic acid; moxLDL, minimally oxidized LDL; nLDL, native LDL; PAF, platelet-activating factor; PDGF, platelet-derived growth factor; PPAR, peroxisome proliferator-activated receptor; PPRE, PPAR response element; PTX, pertussis toxin; Rluc, renilla luciferase; Rosi, Rosiglitazone; S1P, sphingosine 1-phosphate; SOV-PC, stearoyl-oxovaleryl phosphatidylcholine; TZD, thiazolidinedione; VEGF, vascular endothelial growth factor; VSMC, vascular smooth muscle cell; XY-4, 1,1-difluorodeoxy-(2R)-palmitoyl-sn-glycero-3-phosphate; XY-8, 1-palmitoyl-(2R)-fluorodeoxy-sn-glycero-3-phosphate.
C. Zhang and D.L. Baker contributed equally to this work.
References
- 1.Lusis, A.J. 2000. Atherosclerosis. Nature. 407:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinberg, D. 2002. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat. Med. 8:1211–1217. [DOI] [PubMed] [Google Scholar]
- 3.Dzau, V.J., R.C. Braun-Dullaeus, and D.G. Sedding. 2002. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat. Med. 8:1249–1256. [DOI] [PubMed] [Google Scholar]
- 4.Ross, R. 1999. Atherosclerosis is an inflammatory disease. Am. Heart J. 138:S419–S420. [DOI] [PubMed] [Google Scholar]
- 5.Libby, P. 2002. Inflammation in atherosclerosis. Nature. 420:868–874. [DOI] [PubMed] [Google Scholar]
- 6.McIntyre, T.M., G.A. Zimmerman, and S.M. Prescott. 1999. Biologically active oxidized phospholipids. J. Biol. Chem. 274:25189–25192. [DOI] [PubMed] [Google Scholar]
- 7.Berliner, J.A., G. Subbanagounder, N. Leitinger, A.D. Watson, and D. Vora. 2001. Evidence for a role of phospholipid oxidation products in atherogenesis. Trends Cardiovasc. Med. 11:142–147. [DOI] [PubMed] [Google Scholar]
- 8.Siess, W., K.J. Zangl, M. Essler, M. Bauer, R. Brandl, C. Corrinth, R. Bittman, G. Tigyi, and M. Aepfelbacher. 1999. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA. 96:6931–6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tigyi, G., and A. Parrill. 2003. Molecular mechanisms of lysophosphatidic acid action. Prog. Lipid Res. 42:498–526. [DOI] [PubMed] [Google Scholar]
- 10.Spector, A.A. 2003. Plaque rupture, lysophosphatidic acid, and thrombosis. Circulation. 108:641–643. [DOI] [PubMed] [Google Scholar]
- 11.McIntyre, T.M., A.V. Pontsler, A.R. Silva, A. St Hilaire, Y. Xu, J.C. Hinshaw, G.A. Zimmerman, K. Hama, J. Aoki, H. Arai, and G.D. Prestwich. 2003. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proc. Natl. Acad. Sci. USA. 100:131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rother, E., R. Brandl, D.L. Baker, P. Goyal, H. Gebhard, G. Tigyi, and W. Siess. 2003. Subtype-selective antagonists of lysophosphatidic acid receptors inhibit platelet activation triggered by the lipid core of atherosclerotic plaques. Circulation. 108:741–747. [DOI] [PubMed] [Google Scholar]
- 13.Berliner, J.A., and J.W. Heinecke. 1996. The role of oxidized lipoproteins in atherogenesis. Free Radic. Biol. Med. 20:707–727. [DOI] [PubMed] [Google Scholar]
- 14.Weidtmann, A., R. Scheithe, N. Hrboticky, A. Pietsch, R. Lorenz, and W. Siess. 1995. Mildly oxidized LDL induces platelet aggregation through activation of phospholipase A2. Arterioscler. Thromb. Vasc. Biol. 15:1131–1138. [DOI] [PubMed] [Google Scholar]
- 15.Subbanagounder, G., N. Leitinger, D.C. Schwenke, J.W. Wong, H. Lee, C. Rizza, A.D. Watson, K.F. Faull, A.M. Fogelman, and J.A. Berliner. 2000. Determinants of bioactivity of oxidized phospholipids. Specific oxidized fatty acyl groups at the sn-2 position. Arterioscler. Thromb. Vasc. Biol. 20:2248–2254. [DOI] [PubMed] [Google Scholar]
- 16.Davies, S.S., A.V. Pontsler, G.K. Marathe, K.A. Harrison, R.C. Murphy, J.C. Hinshaw, G.D. Prestwich, A.S. Hilaire, S.M. Prescott, G.A. Zimmerman, and T.M. McIntyre. 2001. Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor gamma ligands and agonists. J. Biol. Chem. 276:16015–16023. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida, K., W. Nishida, K. Hayashi, Y. Ohkawa, A. Ogawa, J. Aoki, H. Arai, and K. Sobue. 2003. Vascular remodeling induced by naturally occurring unsaturated lysophosphatidic acid in vivo. Circulation. 108:1746–1752. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi, K., M. Takahashi, W. Nishida, K. Yoshida, Y. Ohkawa, A. Kitabatake, J. Aoki, H. Arai, and K. Sobue. 2001. Phenotypic modulation of vascular smooth muscle cells induced by unsaturated lysophosphatidic acids. Circ. Res. 89:251–258. [DOI] [PubMed] [Google Scholar]
- 19.Xu, Y., L. Qian, and G.D. Prestwich. 2003. Synthesis of alpha-fluorinated phosphonates from α-fluorovinylphosphonates: a new route to analogues of lysophosphatidic acid. Org. Lett. 5:2267–2270. [DOI] [PubMed] [Google Scholar]
- 20.Xu, Y., L. Qian, and G.D. Prestwich. 2003. Synthesis of monofluorinated analogues of lysophosphatidic acid. J. Org. Chem. 68:5320–5330. [DOI] [PubMed] [Google Scholar]
- 21.Yokoyama, K., D.L. Baker, T. Virag, K. Liliom, H.S. Byun, G. Tigyi, and R. Bittman. 2002. Stereochemical properties of lysophosphatidic acid receptor activation and metabolism. Biochim. Biophys. Acta. 1582:295–308. [DOI] [PubMed] [Google Scholar]
- 22.Xu, Y., and G.D. Prestwich. 2002. Synthesis of chiral (α,α-difluoroalkyl)phosphonate analogues of (lyso)phosphatidic acid via hydrolytic kinetic resolution. Org. Lett. 4:4021–4024. [DOI] [PubMed] [Google Scholar]
- 23.Sprecher, D.L., E.J. Schaefer, K.M. Kent, R.E. Gregg, L.A. Zech, J.M. Hoeg, B. McManus, W.C. Roberts, and H.B.J. Brewer. 1984. Cardiovascular features of homozygous familial hypercholesterolemia: analysis of 16 patients. Am. J. Cardiol. 54:20–30. [DOI] [PubMed] [Google Scholar]
- 24.Baker, D.L., D.M. Desiderio, D.D. Miller, B. Tolley, and G.J. Tigyi. 2001. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal. Biochem. 292:287–295. [DOI] [PubMed] [Google Scholar]
- 25.Wang, D.A., H. Du, J.H. Jaggar, D.N. Brindley, G.J. Tigyi, and M.A. Watsky. 2002. Injury-elicited differential transcriptional regulation of phospholipid growth factor receptors in the cornea. Am. J. Physiol. Cell Physiol. 283:C1646–C1654. [DOI] [PubMed] [Google Scholar]
- 26.Simon, M.F., H. Chap, and L. Douste-Blazy. 1982. Human platelet aggregation induced by 1-alkyl-lysophosphatidic acid and its analogs: a new group of phospholipid mediators? Biochem. Biophys. Res. Commun. 108:1743–1750. [DOI] [PubMed] [Google Scholar]
- 27.Tokumura, A., K. Fukuzawa, J. Isobe, and H. Tsukatani. 1981. Lysophosphatidic acid-induced aggregation of human and feline platelets: structure-activity relationship. Biochem. Biophys. Res. Commun. 99:391–398. [DOI] [PubMed] [Google Scholar]
- 28.Tokumura, A., M. Iimori, Y. Nishioka, M. Kitahara, M. Sakashita, and S. Tanaka. 1994. Lysophosphatidic acids induce proliferation of cultured vascular smooth muscle cells from rat aorta. Am. J. Physiol. 267:C204–C210. [DOI] [PubMed] [Google Scholar]
- 29.Sugiura, T., S. Nakane, S. Kishimoto, K. Waku, Y. Yoshioka, A. Tokumura, and D.J. Hanahan. 1999. Occurrence of lysophosphatidic acid and its alkyl ether-linked analog in rat brain and comparison of their biological activities toward cultured neural cells. Biochim. Biophys. Acta. 1440:194–204. [DOI] [PubMed] [Google Scholar]
- 30.Tokumura, A., J. Sinomiya, S. Kishimoto, T. Tanaka, K. Kogure, T. Sugiura, K. Satouchi, K. Waku, and K. Fukuzawa. 2002. Human platelets respond differentially to lysophosphatidic acids having a highly unsaturated fatty acyl group and alkyl ether-linked lysophosphatidic acids. Biochem. J. 365:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon, M.F., A. Rey, I. Castan-Laurel, S. Gres, D. Sibrac, P. Valet, and J.S. Saulnier-Blache. 2002. Expression of ectolipid phosphate phosphohydrolases in 3T3F442A preadipocytes and adipocytes. Involvement in the control of lysophosphatidic acid production. J. Biol. Chem. 277:23131–23136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sano, T., D. Baker, T. Virag, A. Wada, Y. Yatomi, T. Kobayashi, Y. Igarashi, and G. Tigyi. 2002. Multiple mechanisms linked to platelet activation result in lysophosphatidic acid and sphingosine 1-phosphate generation in blood. J. Biol. Chem. 277:21197–21206. [DOI] [PubMed] [Google Scholar]
- 33.Fukushima, N., I. Ishii, J.J. Contos, J.A. Weiner, and J. Chun. 2001. Lysophospholipid receptors. Annu. Rev. Pharmacol. Toxicol. 41:507–534. [DOI] [PubMed] [Google Scholar]
- 34.Noguchi, K., S. Ishii, and T. Shimizu. 2003. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J. Biol. Chem. 278:25600-25666. [DOI] [PubMed] [Google Scholar]
- 35.Bandoh, K., J. Aoki, A. Taira, M. Tsujimoto, H. Arai, and K. Inoue. 2000. Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-activity relationship of cloned LPA receptors. FEBS Lett. 478:159–165. [DOI] [PubMed] [Google Scholar]
- 36.Fischer, D.J., N. Nusser, T. Virag, K. Yokoyama, D. Wang, D.L. Baker, D. Bautista, A.L. Parrill, and G. Tigyi. 2001. Short-chain phosphatidates are subtype-selective antagonists of lysophosphatidic acid receptors. Mol. Pharmacol. 60:776–784. [PubMed] [Google Scholar]
- 37.Diep, Q.N., and E.L. Schiffrin. 2001. Increased expression of peroxisome proliferator-activated receptor-alpha and -gamma in blood vessels of spontaneously hypertensive rats. Hypertension. 38:249–254. [DOI] [PubMed] [Google Scholar]
- 38.Li, A.C., and C.K. Glass. 2002. The macrophage foam cell as a target for therapeutic intervention. Nat. Med. 8:1235–1242. [DOI] [PubMed] [Google Scholar]
- 39.Tugwood, J.D., I. Issemann, R.G. Anderson, K.R. Bundell, W.L. McPheat, and S. Green. 1992. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 11:433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leesnitzer, L.M., D.J. Parks, R.K. Bledsoe, J.E. Cobb, J.L. Collins, T.G. Consler, R.G. Davis, E.A. Hull-Ryde, J.M. Lenhard, L. Patel, et al. 2002. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 41:6640–6650. [DOI] [PubMed] [Google Scholar]
- 41.Subbanagounder, G., A.D. Watson, and J.A. Berliner. 2000. Bioactive products of phospholipid oxidation: isolation, identification, measurement and activities. Free Radic. Biol. Med. 28:1751–1761. [DOI] [PubMed] [Google Scholar]
- 42.Febbraio, M., D.P. Hajjar, and R.L. Silverstein. 2001. CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Invest. 108:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hama, K., K. Bandoh, Y. Kakehi, J. Aoki, and H. Arai. 2002. Lysophosphatidic acid (LPA) receptors are activated differentially by biological fluids: possible role of LPA-binding proteins in activation of LPA receptors. FEBS Lett. 523:187–192. [DOI] [PubMed] [Google Scholar]
- 44.Tigyi, G., A. Henschen, and R. Miledi. 1991. A factor that activates oscillatory chloride currents in Xenopus oocytes copurifies with a subfraction of serum albumin. J. Biol. Chem. 266:20602–20609. [PubMed] [Google Scholar]
- 45.Tigyi, G., and R. Miledi. 1992. Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J. Biol. Chem. 267:21360–21367. [PubMed] [Google Scholar]
- 46.Tokumura, A., T. Tsutsumi, and H. Tsukatani. 1992. Transbilayer movement and metabolic fate of ether-linked phosphatidic acid (1-O-octadecyl-2-acetyl-sn-glycerol-3-phosphate) in guinea pig peritoneal polymorphonuclear leukocytes. J. Biol. Chem. 267:7275–7283. [PubMed] [Google Scholar]
- 47.Wysowski, D.K., G. Armstrong, and L. Governale. 2003. Rapid increase in the use of oral antidiabetic drugs in the United States, 1990-2001. Diabetes Care. 26:1852–1855. [DOI] [PubMed] [Google Scholar]
- 48.Rocchi, S., and J. Auwerx. 1999. Peroxisome proliferator-activated receptor-gamma: a versatile metabolic regulator. Ann. Med. 31:342–351. [DOI] [PubMed] [Google Scholar]
- 49.Chawla, A., Y. Barak, L. Nagy, D. Liao, P. Tontonoz, and R.M. Evans. 2001. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 7:48–52. [DOI] [PubMed] [Google Scholar]
- 50.Pagano, R.E., and K.J. Longmuir. 1985. Phosphorylation, transbilayer movement, and facilitated intracellular transport of diacylglycerol are involved in the uptake of a fluorescent analog of phosphatidic acid by cultured fibroblasts. J. Biol. Chem. 260:1909–1916. [PubMed] [Google Scholar]
- 51.Tokumura, A., T. Tsutsumi, J. Yoshida, and H. Tsukatani. 1990. Translocation of exogenous platelet-activating factor and its lyso-compound through plasma membranes is a rate-limiting step for their metabolic conversions into alkylacylglycerophosphocholines in rabbit platelets and guinea-pig leukocytes. Biochim Biophys Acta. 1044:91–100. [DOI] [PubMed] [Google Scholar]
- 52.Boujaoude, L.C., C. Bradshaw-Wilder, C. Mao, J. Cohn, B. Ogretmen, Y.A. Hannun, and L.M. Obeid. 2001. Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate. J. Biol. Chem. 276:35258–35264. [DOI] [PubMed] [Google Scholar]
- 53.Homma, H., A. Tokumura, and D.J. Hanahan. 1987. Binding and internalization of platelet-activating factor 1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine in washed rabbit platelets. J. Biol. Chem. 262:10582–10587. [PubMed] [Google Scholar]