

Abstract
The prevention and treatment of prevalent infectious diseases and tumors should benefit from improvements in the induction of antigen-specific T cell immunity. To assess the potential of antigen targeting to dendritic cells to improve immunity, we incorporated ovalbumin protein into a monoclonal antibody to the DEC-205 receptor, an endocytic receptor that is abundant on these cells in lymphoid tissues. Simultaneously, we injected agonistic α-CD40 antibody to mature the dendritic cells. We found that a single low dose of antibody-conjugated ovalbumin initiated immunity from the naive CD4+ and CD8+ T cell repertoire. Unexpectedly, the αDEC-205 antigen conjugates, given s.c., targeted to dendritic cells systemically and for long periods, and ovalbumin peptide was presented on MHC class I for 2 weeks. This was associated with stronger CD8+ T cell–mediated immunity relative to other forms of antigen delivery, even when the latter was given at a thousand times higher doses. In parallel, the mice showed enhanced resistance to an established rapidly growing tumor and to viral infection at a mucosal site. By better harnessing the immunizing functions of maturing dendritic cells, antibody-mediated antigen targeting via the DEC-205 receptor increases the efficiency of vaccination for T cell immunity, including systemic and mucosal resistance in disease models.
Keywords: dendritic cell, DEC-205 receptor, vaccination, CD8 T cell, immunotherapy
Introduction
For many diseases that lead to high mortality and morbidity, such as AIDS and malaria, it is likely that vaccines will need to elicit strong T cell–mediated immunity composed of IFN-γ secreting CD4+ helper and CD8+ cytolytic T lymphocytes (for reviews see references 1–4). To induce such responses, it would be valuable to harness the DC system of antigen-presenting cells (5, 6). At least three sets of DC functions are pertinent. First, DCs efficiently process antigens, including complex microbes and tumor cells, and display these on both MHC class I and II products to CD8+ and CD4+ T cells, respectively (7, 8). Second, DCs become potent stimulators of immunity after undergoing a complex differentiation or maturation program in response to a panel of stimuli including microbial ligands for toll-like receptors (9, 10), innate lymphocytes (11, 12), and CD40 ligation (13). Third, DCs localize to the T cell areas of lymphoid organs (14, 15), where they expand antigen-specific T cells (16–18) and when mature, induce IFN-γ–producing helper and killer T cells (19, 20).
We set out to marshal these features of DCs to improve vaccination. Our strategy was to target antigens to the DEC-205 endocytosis receptor. It is expressed at high levels on lymphoid tissue DCs (21–23) and greatly enhances the efficiency of antigen presentation (24, 25). We followed the consequences of DEC-205 antigen targeting in naive mice with a polyclonal T cell repertoire. We will show that a single low s.c. dose of a protein-based vaccine is able to charge DCs with antigen systemically and for long periods, particularly on MHC class I products. In parallel, naive mice develop immunity, including CD8+ T cell–mediated immunity, which is considerably enhanced relative to prior methods of immunization with 1,000-fold higher doses of antigen and is associated with stronger protection in anti-viral and anti-tumor models.
Materials and Methods
Antibodies and Reagents.
Alexa488-conjugated αDEC-205 (NLDC-145), αOVA (3A11.1), and isotype control (III/10) antibodies were prepared using the Alexa Fluor® 488 protein labeling kit (Molecular Probes).
Mice.
Adult female C57BL/6 (B6) mice, and CD4−/− and CD8−/− B6 knockouts, were purchased from Jackson ImmunoResearch Laboratories. Ovalbumin (OVA)-specific, TCR-transgenic CD45.1+ OT-I and CD45.1+ OT-II mice were used as described previously (20). DEC-205−/− mice were provided by Dr. M. Nussenzweig (The Rockefeller University, New York, NY).
Conjugation of OVA to Monovalent Monoclonal Antibodies.
Monovalent IgG's were conjugated to LPS-free OVA (Seikagaku Corp.) that had been activated with succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC; Pierce Chemical Co.) according to the manufacturer's protocol. In brief, the antibodies were reduced using 100 mM 2-mercaptoethanesulfonic acid sodium salt (MESNA; Sigma-Aldrich) for 30 min at 37°C and separated from the reducing agent over a desalting column. Then the activated OVA was mixed with the reduced antibodies overnight at 4°C. The antibody:OVA conjugates were passed over a protein G column to remove unconjugated OVA, concentrated by spin columns, and evaluated by spectrophotometry and SDS-PAGE. Monovalent IgG:OVA conjugates were characterized by SDS-PAGE and Western blotting. Quantification of the OVA content of the conjugates was achieved by comparison with known quantities of OVA on the same blot detected with an HRP-conjugated polyclonal rabbit anti-OVA antibody (Research Diagnostics, Inc.).
Purification of DCs and Antigen-specific T Cells.
Single cell suspensions were prepared from lymph nodes or spleen with 400 U/ml collagenase D (Roche) for 25 min and CD11c+ cells purified by MACS®. OVA-specific transgenic CD8+ or CD4+ T cells were prepared from lymph node or spleen cell suspensions of OT-I or OT-II mice using negative selection with hybridoma supernatants directed against MHC class II, F4/80, B220, NK 1.1, and CD4 or CD8 and goat anti–rat Dynabeads® (Dynal) at a ratio of four beads to one target cell.
Antigen Targeting and Maturation of DCs In Vivo.
Mice were injected s.c. in the paws with OVA protein, or Ig conjugates of OVA protein, without or with a stimulus for DC maturation, which was the 1C10 agonistic αCD40 antibody (26) injected i.p. at 25–50 μg/mouse as described previously (20).
Assays with TCR-transgenic T Cells to Monitor Antigen Presentation on MHC Class I and II Products.
In vitro antigen presentation assays were performed by adding CD11c+ DCs, selected from lymph nodes and spleens of OVA-treated mice, to 105 OT-I or OT-II T cells in round bottom 96-well plates (1 DC:3 T cell ratio). At 48 h, [3H]thymidine (1 μCi; Amersham Biosciences) was added for 12 h to detect incorporation into DNA. In vivo assays were performed by injecting 106 CD45.1+ OT-I or OT-II T cells that had been labeled at 107 cells/ml with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes) for 10 min at 37°C.
Assays for OVA Immunization.
Proliferation of primed CD4+ or CD8+ T cells was evaluated by labeling bulk spleen suspensions with CFSE as above (but at 1 μM) and restimulating with LPS-free OVA (500 μg/ml) for 5 d in 24-well dishes at 2.5 × 106 cells/well. Cultures were then washed, stained for CD4 and CD8 and evaluated for proliferation by flow cytometry. ELISPOT assays were performed by restimulating spleen suspensions for 2 d with H-2Kb-restricted peptide (SIINFEKL; 1.0 μM) or an I-Ab-restricted peptide (LSQAVHAAHAEINEAGR; 1.0 μM). The in vivo response of OVA-specific CD8+ T cells was evaluated by staining with Kb-SIINFEKL–PE tetramers (provided by Dr. E. Pamer, Memorial Sloan Kettering Institute, New York, NY) and CD62L for 1 h at 4°C. Also IFN-γ–producing effector cells were evaluated by culturing 5 × 106 lymph node or spleen cells with SIINFEKL peptide (1.0 μM) for 6 h in the presence of brefeldin A (5 μg/ml; Sigma-Aldrich). Cells were then harvested, stained for extracellular CD8, and then stained for cytokines with the BD intracellular cytokine staining starter kit (Becton Dickinson). In vivo CTL assays were performed as described (27) by injecting 1:1 mixtures of peptide-pulsed and -unpulsed syngeneic splenocytes (7 × 106 each) and, 12–18 h later, specific lysis quantified as {[1 - (ratio unprimed / ratio primed)] × 100}, with ratio determined as % CFSElo/% CFSEhi (28).
Vaccine-induced Resistance Assays.
Tumor challenges were performed with 5 × 106 MO4, OVA-bearing B16 melanoma cells injected s.c. on the right flank either 7 d before, or 30–90 d after, immunization. Nontransduced B16 melanoma cells were used as controls to show that immunity was OVA-dependent. Challenge with recombinant vaccinia–OVA virus was performed with 105 PFU applied intranasally as described (29). 7 d later, lungs were harvested, extracts prepared by physical disruption, and viral titers evaluated by plaque forming assay on CV-1 cells. Tumor data are expressed as average tumor size from groups of at least five mice, where vaccinia titers as average ± 1 SD for groups of at least five mice.
Results
Preparation of Monovalent αDEC-205:OVA Conjugates That More Efficiently Harness the Antigen-presenting Activity of DCs In Vivo.
We first modified a prior strategy to conjugate an antigen to a monoclonal antibody to the DEC-205 receptor (20). This antibody selectively targets to lymph node DCs after s.c. injection (19, 20). With the mild-reducing agent MESNA to cleave interheavy chain disulfide bonds, we produced monovalent fragments of the antibody (Fig. 1 A). The exposed sulfhydryls of nearly all the antibody molecules could then be cross-linked with SMCC-activated OVA (see Conjugation of OVA to Monovalent Monoclonal Antibodies section in Materials and Methods), yielding 132-kD conjugates containing OVA and rat IgG (Fig. 1 B). The αDEC-205:OVA conjugates, as well as conjugates produced with an isotype matched nonreactive antibody called III/10, were subjected to Western blotting along side known quantities of OVA protein to quantify the amount of OVA in the conjugates, generally ∼10% of the total protein (unpublished data). When the monovalent αDEC-205:OVA conjugates were injected s.c., the OVA was presented to MHC class I– and MHC class II–restricted T cells in vivo, as assessed with OVA-specific reporter T cells from CD8+ OT-I and CD4+ OT-II, TCR-transgenic mice. Both types of T cells, which were labeled with CFSE before injection, proliferated vigorously (five to seven division cycles) in response to αDEC-205:OVA but not to isotype matched III/10–OVA conjugates (Fig. 1 C). When we compared the efficacy of achieving antigen presentation in vivo by monovalent antibody targeted OVA and soluble OVA, the conjugated OVA was >1,000 times more effective for MHC class I presentation and >50 times greater for MHC class II presentation (Fig. 1 C). For example, 2,500 ng of soluble OVA did not elicit a proliferative response from CD8+ OT-I T cells, but 2 ng of OVA in αDEC-205:OVA caused most of the T cells to enter multiple cycles of division (Fig. 1 D). In DEC-205 knockout mice (DEC-205−/−), presentation of αDEC-205:OVA, but not soluble unconjugated OVA, was abolished (Fig. 1 C), proving that presentation of αDEC-205:OVA was strictly dependent upon this endocytic receptor. Thus the injection of antigen conjugated to a monovalent αDEC-205 antibody markedly enhances the efficiency of antigen presentation in vivo.
Figure 1.

Characterization of monoclonal IgG:OVA conjugates. (A) IgG:OVA conjugates at various stages of conjugation. Nonreduced gel (left) of the 80-kD monovalent IgG after MESNA treatment, and reduced and boiled (right) to show heavy and light chains. (B) Western analysis of antibody (DEC-205 and III/10 isotype control) OVA conjugates. (C) C57BL/6 or DEC-205−/− mice were injected i.v. with 106 CFSE-labeled OT-I or OT-II T cells and 24 h later with either antibody conjugates (DEC-205 and III/10 isotype control at the same doses) containing 50 ng of OVA or 25 μg soluble OVA s.c. 3 d later, proliferation in lymph nodes was evaluated by flow cytometry. (D) As in C, but graded doses of OVA conjugated to IgG- or endotoxin-free OVA were used. For the III/10 isotype control, the highest dose of conjugate with 250 ng of OVA was used. Representative of two or more experiments.
Immunization of CD4+ and CD8+ T Cells with a Combination of OVA Targeting to DCs and a CD40-based Maturation Stimulus.
Previous studies had demonstrated the capacity for DEC-205–targeted antigens to immunize large numbers of TCR-transgenic T cells in vivo, as long as antigen was given together with an agonistic αCD40 monoclonal antibody to mature the lymph node DCs (19, 20). We therefore tested if we could prime the endogenous naive repertoire that contains a low frequency of antigen-specific T cells. We monitored the induction of immunity to graded doses of antigen with two standard assays for immune priming: T cell proliferation in response to antigen and IFN-γ–secreting ELISPOTS. We first injected 500 ng of OVA conjugated to αDEC-205 (5 μg of antibody conjugate injected s.c. in four paws), in combination with 25 μg of αCD40. 7 d later, both CD4+ and CD8+ T cells proliferated after in vitro restimulation with OVA protein (Fig. 2 A). The proliferation was weak to undetectable in control mice primed with αDEC-205:OVA alone, αCD40 alone, or a mixture of OVA and αCD40 (Fig. 2 A). The mice also developed OVA-specific IFN-γ–secreting effector cells, with the CD8+ response being more vigorous than the CD4+ response (Fig. 2 B). When we used the ELISPOT assay to compare αDEC-205:OVA to OVA (each together with αCD40), the targeted antibody was >1,000 times more effective for immunizing naive mice (Fig. 2 C). Therefore antigen targeting to DCs via DEC-205, coupled with αCD40, greatly increases the efficiency with which a protein initiates T cell–mediated immunity from a polyclonal naive repertoire.
Figure 2.

αDEC-205:OVA with αCD40 primes both CD4+ and CD8+ T cells in vivo. (A) αDEC-205:OVA containing 500 ng of OVA was administered to naive C57BL/6 mice s.c. with 25 μg of αCD40. 7 d later, spleen cell suspensions were CFSE labeled and restimulated in vitro for 5 d with LPS-free OVA (500 μg/ml) to evaluate proliferation by flow cytometry. (B) As in A, but the cells were restimulated with either SIINFEKL (1.0 μM) or LSQAVHAAHAEINEAGR (2.0 μM) peptides for 2 d and IFN-γ secretion evaluated by ELISPOT. (C) Mice were immunized with grade doses of OVA as a soluble protein or conjugated to αDEC-205. IFNγ secretion was evaluated after 7 d in the lymph nodes and spleen as in B. Representative of at least two experiments.
The Durability of the Effector CD8+ T Cell Response When Antigen Is Targeted to DCs.
We concentrated our subsequent studies on the CD8+ response, because it is a special challenge to be able to present nonreplicating antigens to CD8+ T cells in vivo and this would be valuable for the design of safe nonreplicating and subunit vaccines. We gave a single dose of 50–100 ng of OVA conjugated to αDEC-205 (i.e., 0.5–1.0 μg of total antibody:OVA protein per mouse) together with 25 μg of agonistic αCD40 s.c., and then we monitored the development of effector T cells using assays for cytokine secretion and cytolytic activity. Antibody targeting to maturing DCs was able to elicit vigorous IFN-γ secretion by CD8+ T cells in both the lymph nodes and spleen, but in addition, the response was long lived (Fig. 3 A). At all time points tested (14, 21, 60, and 90 d) after administration of a single dose αDEC-205:OVA with αCD40, the CD8+ splenocytes had been primed to secrete IFN-γ upon peptide restimulation (Fig. 3 A). Administration of either the antigen (αDEC-205:OVA) or DC maturation stimulus (αCD40) alone failed to elicit any response (Fig. 3 A, left panels). To verify that the CD8+ response included cells with in vivo cytolytic function, we injected a mixture of peptide pulsed and unpulsed syngeneic splenocytes (7 × 106 cells each) 14 d after immunization. Effective and specific CTLs were observed in the lymph nodes (Fig. 3 B) and spleen (unpublished data), with nearly all of the peptide-pulsed targets being eradicated from these organs. The CTL responses were undiminished in a CD4−/− mouse, but completely absent in CD8−/− mice and DEC-205−/− mice (Fig. 3 B). CTL activity remained vigorous 60 d after immunization (Fig. 3 C, 77% lysis at day 60, compared with 93% lysis in Fig. 3 B at day 14), and even 90 d after immunization, CTLs were still detected, although at lower levels (30% lysis; unpublished data). These results indicate that a single immunization with αDEC-205:OVA and αCD40 leads to the durable formation of effector memory T cells.
Figure 3.

αDEC-205:OVA in combination with αCD40 induces durable and strong OVA-specific responses by CD8+ T cells. (A) αDEC-205:OVA containing 50 ng of OVA was administered to naive C57BL/6 mice s.c. with 25 μg of αCD40. 14, 21, 60, and 90 d later, intracellular IFN-γ staining was evaluated by flow cytometry without or with OVA peptide restimulation. Indicated percentages are percent IFN-γ+ CD8+ cells. (B) Wild-type, DEC-205−/−, CD8−/−, and CD4−/− mice were treated as in A. 14 d later, 7 × 106 of each, CFSE-labeled syngeneic splenocytes pulsed with peptide (CFSEhi) or not (CFSElo), were injected i.v. to detect active killer cells in the lymph nodes. (C) As in B, but mice were evaluated after 60 d. Data are representative of two or more experiments.
The Immune Response to αDEC-205:OVA Is Greater Than with Other Immunization Strategies.
To compare the DC-targeting strategy with other immunization approaches that are commonly used to induce strong T cell–mediated immunity to proteins, we studied (a) splenic DCs matured and pulsed ex vivo with OVA (30, 31), as well as (b) free antigens (OVA protein, OVA peptide, and αDEC-205:OVA) suspended in CFA (32) or given together with αCD40. 7 and 30 d after immunization, we evaluated the expansion of OVA-specific T cells by MHC class I tetramer staining in lymph node and spleen. At both time points, the combination of αDEC-205:OVA with αCD40 was much more effective, especially if one examined the spleen, a site for the accumulation of effector memory T cells (Fig. 4 A). The frequency of antigen-binding CD8+ cells was much higher in response to 50 ng of OVA conjugated to αDEC-205 (5.4%) relative to 50 μg soluble OVA, injected along with either αCD40 (1.2%) or CFA (0.3%); 50 μg preprocessed OVA peptide with αCD40 was even less effective (Fig. 4 A). On day 7, the tetramer-positive cells in the αDEC-205:OVA treated mice had down-regulated CD62L confirming that these T cells were effectors (Fig. 3) with the potential to migrate into peripheral tissues. The degree of expansion of tetramer-positive cells correlated closely with the production of functioning effector cells assayed by IFN-γ secretion, which again was much higher after αDEC-205:OVA targeting relative to other forms of antigen delivery (Fig. 4 B). These results indicate that direct in vivo delivery of protein antigens to DCs is more effective than several existing approaches for vaccine priming of antigen-specific CD8+ T cells.
Figure 4.

Enhanced efficacy of αDEC-205:OVA plus αCD40 relative to other immunization approaches. (A) C57BL/6 mice were immunized s.c. with several methods: Spleen DC pulsed ex vivo with 10 μg/ml each of αDEC-205:OVA and αCD40; 500 μg OVA in CFA; 50 μg OVA with 25 μg αCD40; 50 μg of SIINFEKL peptide with 25 μg αCD40; or 50 ng of OVA in αDEC-205:OVA with 25 μg of αCD40. 7 or 30 d later, lymph nodes were harvested and T cell expansion evaluated by Kb-SIINFEKL–PE tetramer and CD62L staining. The gate for the y-axis was placed relative to the CD62L-negative tetramer binding cells in the right panel. Indicated percentages are percent of CD8+ lymphocytes. (B) As in A, but IFN-γ secretion evaluated by intracellular cytokine staining. Data are means of three experiments.
Systemic and Prolonged Distribution of OVA after αDEC-205 Targeting to DCs.
To determine how DEC-205 targeting improves antigen delivery in situ, the rate and persistence of antibody loading of DCs in lymphoid tissues were evaluated over time. The isotype-matched control III/10 antibody bound weakly if at all to DCs at all time points (Fig. 5 A). In contrast, within 30 min of s.c. injection, Alexa488 ®-conjugated αDEC-205 began to load a sizable fraction of the CD11c+ DCs in the draining lymph nodes, consistent with the direct movement of antibody from the skin injection site via the protein-rich afferent lymph to the lymph node. Unexpectedly, the αDEC-205 quickly appeared on all of the CD8+ DCs of the spleen (the CD8+ DC subset is also the DEC-205 high subset in spleen although in lymph nodes, DEC-205 and CD8 expression are not coordinate on certain DC subsets; 33, 34), indicating that antibody was gaining access to the blood stream (Fig. 5 A, arrows). Considerable loading in the mesenteric lymph node also was detectable, but at longer times after injection (Fig. 5 A). By 6 h, αDEC-205 loaded at least 50% of the draining lymph node DCs and ∼40% and ∼30% in the distal lymph node and spleen DCs, respectively. Interestingly, αDEC-205 persisted on the DCs in all the organs for at least 3 d after injection (Fig. 5 A, bottom). The presence of OVA in the DCs of a draining lymph node and spleen was also evident by intracellular staining for OVA (Fig. 5 B). Isolation of the CD11c+ DCs from spleen and lymph nodes 15 h after injection of αDEC-205:OVA with or without αCD40 confirmed that these DCs could present the captured OVA to TCR-transgenic T cells (Fig. 5 C). When αDEC-205:OVA was compared with a 1,000-fold higher dose of soluble OVA (each given together with αCD40), the former was presented much more vigorously by DCs from systemic lymphoid tissues (Fig. 5 D). These results indicate that low doses of intracutaneous anti-DC antibodies rapidly target along with an associated antigen systemically to DCs in lymphoid tissues for days.
Figure 5.
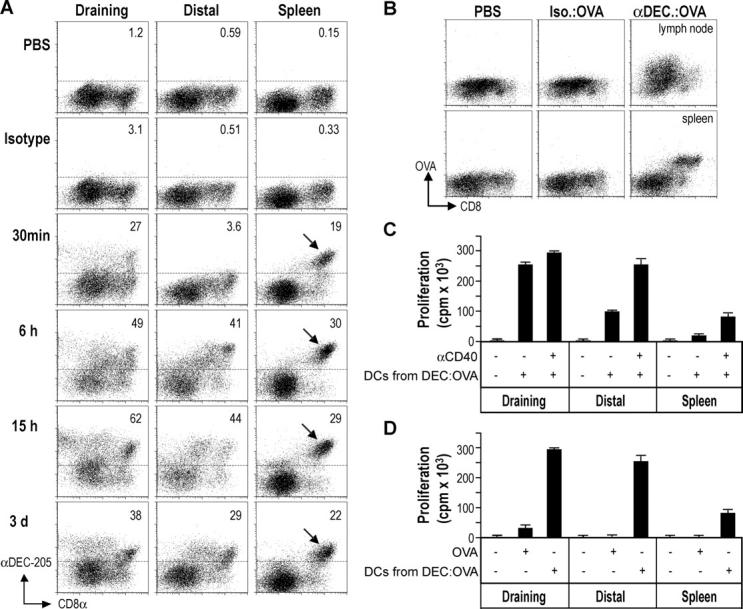
Systemic antigen presentation after DEC-205 targeting in situ. (A) C57BL/6 mice were given 10 μg of Alexa488-conjugated antibodies s.c. At the indicated time points, CD11c+ cells were enriched from the draining or distal lymph nodes or spleen for evaluation by flow cytometry. The frequencies of DCs capturing the injected Igs are shown, and the DEC-205 and CD8 high subset of splenic DCs arrowed. (B) C57BL/6 mice were given 10 μg of αDEC-205:OVA, isotype:OVA, or PBS s.c. and, after 18 h, CD11c+ cells were enriched from draining or distal lymph nodes or spleen. The presence of OVA was evaluated by intracellular staining with Alexa488-conjugated αOVA and flow cytometry. (C) 15 h after s.c. treatment with 5 μg of αDEC-205:OVA or the isotype conjugate ± αCD40, CD11c+ lymph node or spleen DCs were selected and used to stimulate OT-I T cells without further addition of OVA. (D) As in C, but mice were treated with αCD40 and either αDEC-205:OVA (5 μg), OVA (500 μg), or PBS. Data are representative of at least two experiments.
Prolonged Presentation of MHC Class I–Peptide Complexes on Antigen-targeted DCs.
To investigate the persistence of MHC–OVA peptide complexes in vivo, we pretreated mice with αDEC-205:OVA or OVA, each with or without αCD40, for 1, 3, 7, 15, or 30 d before transferring CFSE-labeled OT-I OVA-specific T cells. Surprisingly, given the evidence that the half life of DCs in lymph nodes is ∼1.5–2 d (35, 36), presentation was still vigorous in the lymph nodes 15 d (but not 30 d; unpublished data) after immunization with just 50 ng of OVA in αDEC-205:OVA conjugates (Fig. 6 A, top left). Co-administration of αCD40 slightly increased the presentation, especially at day 15. In contrast, proliferation elicited by administration of 50 μg of soluble OVA or 50 μg of preprocessed peptide (unpublished data), was minimally detectable at 7 d after injection, even if coadministered with αCD40 (Fig. 6 A, bottom left). Likewise, when mice were primed with ex vivo–loaded αCD40-matured splenic DCs, presentation was not detectable beyond 3 d after injection (Fig. 6 A, top right). If a high dose of OVA protein (500 μg) was administered in CFA, proliferation also was detectable 15 d after administration (Fig. 6 A, bottom right; as was the case for 50 ng of DEC-205–targeted OVA in CFA; unpublished data), probably because the oily CFA emulsion allows the depot of injected antigen to persist. In contrast to MHC class I, MHC class II–peptide complexes were no longer detectable at 7 d after injection of αDEC-205:OVA (Fig. 6 B). To test if the superiority of MHC class I presentation was due to an expanded OVA-specific CD8+ T cell repertoire, we immunized mice with preprocessed MHC class I and II binding OVA peptides. If anything the MHC class II–restricted response was greater (Fig. 6 C), suggesting that αDEC-205 targeting seems to prioritize presentation on MHC class I products. The results in Figs. 5 and 6 indicate that the local injection of a single low dose of DC-targeted antigen recreates a situation analogous to a systemic infection, with prolonged presentation of antigen in most lymphoid tissues.
Figure 6.

Prolonged MHC class I, but not MHC class II, presentation after DEC-205 targeting in situ. (A) C57BL/6 mice were immunized to OVA under the conditions listed above each panel for 15, 7, 3, or 1 d before transferring 106 CFSE-labeled OT-I T cells. Proliferation in the lymph nodes was monitored by flow cytometry 3 d later. (B) As in A, but CFSE-labeled OT-I or OT-II T cells were transferred. (C) C57BL/6 mice were treated with 50 ug MHC class I binding peptide (SIINFEKL) in CFA, 50 μg MHC class II binding peptide (LSQAVHAAHAEINEAGR) in CFA, CFA alone, or PBS. IFN-γ secretion was evaluated after 12 d in the lymph nodes as in Fig. 2 B. Data are representative of at least two experiments.
DEC-205 Antigen Targeting As a Potential Vaccination Strategy for Resistance to Tumors.
We first studied resistance to a B16 melanoma stably transduced with OVA (termed MO4). We began with protection studies in which vaccinated mice were challenged at a distal site with MO4 cells s.c., but this was done 2–3 mo after a single vaccination to assess vaccine memory. The mice that received αDEC-205:OVA conjugate in conjunction with αCD40 were protected against a subsequent administration of 5 × 106 tumor cells 2–3 mo later (Fig. 7 A), while mice that received only one component of the vaccine (antigen or adjuvant) or the isotype conjugate were not (unpublished data). This protection was specific for OVA, as the vaccinated mice were not protected against an identical tumor line (B16) that did not express OVA (unpublished data). Studies with knockout mice determined that protection required DEC-205 expression, CD8+ T cells and, to a lesser extent, CD4+ T cells (Fig. 7 A). We have not identified the basis for the decreased resistance in CD4-depleted mice, since there was no measurable difference in the frequency of cytokine producing effectors between wild-type and CD4−/− mice (unpublished data). We then tested DC targeting in a more demanding therapeutic assay, in which the OVA-bearing MO4 tumor cells were allowed to develop into 0.5–1.0-cm-diameter tumors for 7 d before treatment with different strategies. The combination of αDEC-205:OVA in conjunction with αCD40 was able to induce a therapeutic effect, and this was much superior to other strategies, such as OVA in complete Freund's adjuvant and ex vivo–loaded DCs (Fig. 7 B).
Figure 7.

Immunization with a single low dose of αDEC-205:OVA and αCD40 elicits resistance to OVA-modified pathogens. (A) C57BL/6 mice were vaccinated as described in Fig. 3 A. 60 d later, mice were challenged with 5 × 106 MO4 cells s.c. and tumor growth evaluated. (B) C57BL/6 mice were inoculated with MO4 tumor cells as in (A). 7 d later, mice were treated as in Fig. 4 A and tumor growth evaluated. (C) C57BL/6 mice were treated as in Fig. 3 A. 30 d after vaccination, mice were challenged with 105 PFU of vaccinia–OVA intranasally. 7 d later, lungs were harvested and virus titer evaluated by a plaque-forming assay. (D) As in C, but mice were weighed daily after viral challenge. Data are representative of at least two experiments.
DEC-205 Targeting of Antigens As a Potential Vaccination Strategy for Mucosal Resistance.
To evaluate if mucosal immunity could be established by this new systemic vaccination approach, mice were immunized with 50 ng of OVA conjugated to αDEC-205 together with αCD40 s.c., and 2 wk later, the animals were challenged with intranasal recombinant vaccinia OVA. Protection was observed at a mucosal surface by measuring virus titres in the lung (Fig. 7 C), but in addition, the mice did not lose weight as a result of infection (Fig. 7 D). In contrast, no protection was observed relative to the PBS control if the animals had been vaccinated with either the isotype conjugate or αDEC-205:OVA or αCD40 alone (Fig. 7 C). Therefore a single intracutaneous dose of only 50 ng of DC-targeted antigen is effective in generating protective immunity, including at a mucosal surface.
Discussion
Endocytic receptors are valuable targets to probe the function of DCs within lymphoid tissues (19, 20). DCs have a number of potential receptors for antigen uptake. One, DEC-205/CD205, is known to be expressed in abundance in situ (21–23), although more prominently on certain DC subsets (33, 34). Antigens can be introduced into αDEC-205 antibodies by genetic engineering (19) or by chemical conjugation (20). The antigens then target selectively to DCs, which in turn present peptides to CD4+ and CD8+ T cells. Previous work with this approach has focused on TCR-transgenic T cells, so that the number and function of antigen-specific T cells could be more readily followed (19, 20). Here we have used OVA as a model to determine the consequences of antigen targeting to DCs in naive mice with a polyclonal T cell repertoire, including protective systemic immunity. In addition to showing the efficacy of the antigen-targeting approach in naive mice, we made some surprising findings with respect to underlying mechanisms, and we found that antigen targeting produces much stronger immunity than the standard use of much higher doses (1,000-fold) of soluble antigen with complete Freund's adjuvant, including therapeutic tumor immunity in an experimental system.
Our initial observation was that very small amounts of antigen targeted to DCs were capable of inducing combined CD4+ and CD8+ immunity, as long as a DC maturation stimulus was also administered (Fig. 2). A subset of DCs in lymph nodes already have the properties of mature cells in the steady-state, including an inability to process a new offering of a protein antigen (37). However, we showed previously that some DEC-205+ cells processed and presented antigens in the steady-state but required exposure to agonistic αCD40 antibody to induce immunity, in keeping with their designation as “immature” (19, 20). With respect to the stimuli for DC maturation in vivo, additional experiments will be required to determine the relative value of CD40 ligation and other stimuli, such as ligands for toll-like receptors. Nevertheless, when αCD40 was used with αDEC-205:OVA, immunity developed and at high levels, particularly when compared with αCD40 and soluble OVA (Fig. 2). The strong and durable formation of effector memory cells with the combination of αDEC-205:OVA and αCD40 is relevant for therapeutic vaccination, but for protective vaccines, further studies of central memory will be needed. Nevertheless, the enhanced immunity we observed with antigen-targeting provides additional evidence for a pivotal role of maturing DCs in initiating immunity.
Interestingly, within 30 min of injection into the skin, antibody gained access to many of its targets in the draining lymph node, and also, the antibody moved into the efferent lymph and blood to reach the large reservoir of spleen DCs. Over slightly longer time periods (hours), the αDEC-205 antibodies reached distal sites, such as the DCs in mesenteric and mediastinal lymph nodes. In contrast, antigen carriage by peripheral DCs would take >6 h, and these DCs would not move past the draining lymph node to reach other organs. The rapid antibody-mediated targeting of antigen to DCs systemically contrasts with nontargeted soluble protein, which primarily was presented in draining lymphoid organs (Fig. 5 D). Antireceptor antibodies are therefore valuable antigen-targeting vehicles, because they impart specificity to antigen delivery and importantly, they exploit the protein retrieval function of the lymphatic system to distribute the antibody systemically to large numbers of DCs in lymphoid tissues.
We also noted that DEC-205–targeted antigens were presented for much longer times than expected (Fig. 6 A), but primarily in the case of peptides complexed to MHC class I rather than class II products (Fig. 6 B). The greater sensitivity for class I presentation may not be due to inherent differences in priming OVA-specific CD8+ T cells relative to CD4+ T cells, because when we injected preprocessed MHC class I and II binding peptides with CFA, the latter were more effective immunogens (Fig. 6 C). Therefore DEC-205 targeting is more prominently enhancing MHC class I presentation (Fig. 1) and for prolonged periods (Fig. 6). We speculate that this reflects certain features of DEC-205. This receptor may be specialized to target antigens to the exogenous pathway for antigen processing and presentation on MHC class I (20), which likely entails the fusion of elements of the rough endoplasmic reticulum with endocytic vesicles (38, 39). Antigen also may persist in these compartments for long periods and even be reprocessed by other DCs when the initial cells die (40). An additional possibility is that there may be longer lived DEC-205+ DCs in intact lymph nodes. The half life of many DCs is short, <2 d (35, 36), but previous turnover studies may not have included these longer-lived DCs if they were present but not isolated from lymph nodes. For example, Garg et al. have recently shown that certain DEC-205 high DCs derived from the skin can live for 2 wk after migration to the lymph nodes (41). Importantly, a single s.c. injection of DC-targeted antigen seems to recreate what would be seen in many systemic infections, the presence and presentation of antigen in multiple lymphoid sites including mesenteric lymph nodes and spleen.
We would like to suggest that the appropriate targeting of antigen to DCs has the potential to improve vaccine design. Antibody-mediated targeting of antigens to these cells, coupled with an effective maturation stimulus, provides an alternative to more empirical approaches to protein and subunit vaccines. To date, only antibodies to the DEC-205 antigen uptake receptor (24, 25) have been tested, so it is unclear whether targeting to endocytic receptors, and this receptor in particular, is essential. We are finding that it is possible to engineer protein antigens into the α-DEC-205 antibody (here we have used protein chemically conjugated to monovalent DEC-205 antibody fragments), which should make it feasible to vaccinate with a group of antibodies to deliver a mixture of proteins from microbial agents and tumors. Because of the ability of this vaccination approach to prime cytolytic CD8+ T cells, and because DCs in vivo should efficiently present additional antigens from targets killed by these T cells (40, 42, 43), the immunity may be broadened even further, especially in the setting of tumor immunotherapy.
The development of vaccines capable of more effective induction of T cell–mediated immunity represents a major challenge for global health. More effective harnessing of DCs, which are nature's adjuvants for inducing immunity, is one strategy to improve vaccine design. Targeting of antigens via αDEC-205 antibodies to maturing DCs results in CD8+ T cell immunity that is stronger than other approaches (Figs. 3 and 4), and it is associated with efficacy in relatively demanding systemic and mucosal models of protective immunity (Fig. 7), possibly because of the systemic and prolonged presentation of antigens. The data at this juncture pertain to an experimental protein antigen, OVA, but this new approach to vaccination markedly enhances immunity and protection relative to alternative, actively used approaches. The latter involve DCs charged ex vivo with antigen and antigen administered together with complete Freund's adjuvant. We are now engineering clinically relevant antigens into the α-DEC-antibody to further address the proposal that appropriately targeted vaccines, by directly harnessing large numbers of maturing DCs, will greatly improve vaccine efficacy.
Acknowledgments
The authors thank Judy Adams for help with the graphics.
This work was supported by National Institute of Allergy and Infectious Diseases grants AI 13013, AI 40874, and CA84512 to R.M. Steinman, AI 41111 and AI 48204 to T. Moran, and a fellowship from the Instituto Mexicano del Seguro Social to L. Bonifaz.
Abbreviations used in this paper: CFSE, carboxyfluorescein diacetate succinimidyl ester; MESNA, 2-mercaptoethanesulfonic acid sodium salt; OVA, ovalbumin.
References
- 1.Seder, R.A., and J.R. Mascola. 2003. Basic Immunology of Vaccine Development. Academic Press, Boston. 51–72.
- 2.McMichael, A.J., and T. Hanke. 2003. HIV vaccines 1983-2003. Nat. Med. 9:874–880. [DOI] [PubMed] [Google Scholar]
- 3.Reed, S.G., and A. Campos-Neto. 2003. Vaccines for parasitic and bacterial diseases. Curr. Opin. Immunol. 15:456–460. [DOI] [PubMed] [Google Scholar]
- 4.Finn, O.J. 2003. Cancer vaccines: between the idea and the reality. Nat. Rev. Immunol. 3:630–641. [DOI] [PubMed] [Google Scholar]
- 5.Lu, W., X. Wu, Y. Lu, W. Guo, and J.M. Andrieu. 2003. Therapeutic dendritic-cell vaccine for simian AIDS. Nat. Med. 9:27–32. [DOI] [PubMed] [Google Scholar]
- 6.Steinman, R.M., and M. Pope. 2002. Exploiting dendritic cells to improve vaccine efficacy. J. Clin. Invest. 109:1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jung, S., D. Unutmaz, P. Wong, G.-I. Sano, K. De los Santos, T. Sparwasser, S. Wu, S. Vuthoori, K. Ko, F. Zavala, E.G. Pamer, D.R. Littman, and R.A. Lang. 2002. In vivo depletion of CD11c+ dendritic cells abrogation priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 17:211-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thery, C., and S. Amigorena. 2001. The cell biology of antigen presentation in dendritic cells. Curr. Opin. Immunol. 13:45–51. [DOI] [PubMed] [Google Scholar]
- 9.Janeway, C.A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. [DOI] [PubMed] [Google Scholar]
- 10.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335–376. [DOI] [PubMed] [Google Scholar]
- 11.Bendelac, A., and R. Medzhitov. 2002. Adjuvants of immunity: harnessing innate immunity to promote adaptive immunity. J. Exp. Med. 195:F19–F23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujii, S., K. Shimizu, C. Smith, L. Bonifaz, and R.M. Steinman. 2003. Activation of natural killer T cells by α-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a co-administered protein. J. Exp. Med. 198:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caux, C., C. Massacrier, B. Vanbervliet, B. Dubois, C. Van Kooten, I. Durand, and J. Banchereau. 1994. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 180:1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Witmer, M.D., and R.M. Steinman. 1984. The anatomy of peripheral lymphoid organs with emphasis on accessory cells: light microscopic, immunocytochemical studies of mouse spleen, lymph node and Peyer's patch. Am. J. Anat. 170:465–481. [DOI] [PubMed] [Google Scholar]
- 15.Austyn, J.M., J.W. Kupiec-Weglinski, D.F. Hankins, and P.J. Morris. 1988. Migration patterns of dendritic cells in the mouse. Homing to T cell-dependent areas of spleen, and binding within marginal zone. J. Exp. Med. 167:646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ingulli, E., A. Mondino, A. Khoruts, and M.K. Jenkins. 1997. In vivo detection of dendritic cell antigen presentation to CD4+ T cells. J. Exp. Med. 185:2133–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bousso, P., and E. Robey. 2003. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat. Immunol. 4:579–585. [DOI] [PubMed] [Google Scholar]
- 18.von Andrian, U.H., and T.R. Mempel. 2003. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 3:867–878. [DOI] [PubMed] [Google Scholar]
- 19.Hawiger, D., K. Inaba, Y. Dorsett, K. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonifaz, L., D. Bonnyay, K. Mahnke, M. Rivera, M.C. Nussenzweig, and R.M. Steinman. 2002. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kraal, G., M. Breel, M. Janse, and G. Bruin. 1986. Langerhans cells, veiled cells, and interdigitating cells in the mouse recognized by a monoclonal antibody. J. Exp. Med. 163:981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Witmer-Pack, M.D., W.J. Swiggard, A. Mirza, K. Inaba, and R.M. Steinman. 1995. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. II. Expression in situ in lymphoid and nonlymphoid tissues. Cell. Immunol. 163:157–162. [DOI] [PubMed] [Google Scholar]
- 23.Guo, M., S. Gong, S. Maric, Z. Misulovin, M. Pack, K. Mahnke, M. Nussenzweig, and R.M. Steinman. 2000. A monoclonal antibody to the DEC-205 endocytosis receptor on human dendritic cells. Hum. Immunol. 61:729–738. [DOI] [PubMed] [Google Scholar]
- 24.Jiang, W., W.J. Swiggard, C. Heufler, M. Peng, A. Mirza, R.M. Steinman, and M.C. Nussenzweig. 1995. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 375:151–155. [DOI] [PubMed] [Google Scholar]
- 25.Mahnke, K., M. Guo, S. Lee, H. Sepulveda, S.L. Swain, M. Nussenzweig, and R.M. Steinman. 2000. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 151:673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heath, A.W., W.W. Wu, and M.C. Howard. 1994. Monoclonal antibodies to murine CD40 define two distinct functional epitopes. Eur. J. Immunol. 24:1828–1834. [DOI] [PubMed] [Google Scholar]
- 27.Hernandez, J., S. Aung, W.L. Redmond, and L.A. Sherman. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194:707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong, P., and E.G.P. Am. 2003. Feedback regulation of pathogen-specific T cell priming. Immunity. 18:499–511. [DOI] [PubMed] [Google Scholar]
- 29.Brimnes, M.K., L. Bonifaz, R.M. Steinman, and T.M. Moran. 2003. Influenza virus-induced dendritic cell maturation is associated with the induction of strong T cell immunity to a coadministered, normally nonimmunogenic protein. J. Exp. Med. 198:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayordomo, J.I., T. Zorina, W.J. Storkus, L. Zitvogel, C. Celluzzi, L.D. Falo, C.J. Melief, S.T. Ilstad, W.M. Kast, A.B. DeLeo, and M.T. Lotze. 1995. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat. Med. 1:1297–1302. [DOI] [PubMed] [Google Scholar]
- 31.Ludewig, B., S. Ehl, U. Karrer, B. Odermatt, H. Hengartner, and R.M. Zinkernagel. 1998. Dendritic cells efficiently induce protective antiviral immunity. J. Virol. 272:3812–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Bon, A., G. Schiavoni, G. D'Agostinio, I. Gresser, F. Belardelli, and D.F. Tough. 2001. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 14:461–470. [DOI] [PubMed] [Google Scholar]
- 33.Vremec, D., and K. Shortman. 1997. Dendritic cells subtypes in mouse lymphoid organs. Cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 159:565–573. [PubMed] [Google Scholar]
- 34.Inaba, K., M. Pack, M. Inaba, H. Sakuta, F. Isdell, and R.M. Steinman. 1997. High levels of a major histocompatibility complex II–self peptide complex on dendritic cells from lymph node. J. Exp. Med. 186:665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamath, A.T., J. Pooley, M.A. O'Keeffe, D. Vremec, Y. Zhan, A. Lew, A. D'Amico, L. Wu, D.F. Tough, and K.S. Shortman. 2000. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 165:6762–6770. [DOI] [PubMed] [Google Scholar]
- 36.Kamath, A.T., S. Henri, F. Battye, D.F. Tough, and K. Shortman. 2002. Developmental kinetics and lifespan of dendritic cells in mouse lymphoid organs. Blood. 100:1734–1741. [PubMed] [Google Scholar]
- 37.Wilson, N.S., D. El-Sukkari, G.T. Belz, C.M. Smith, R.J. Steptoe, W.R. Heath, K. Shortman, and J.A. Villadangos. 2003. Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood. 102:2187–2194. [DOI] [PubMed] [Google Scholar]
- 38.Houde, M., S. Bertholet, E. Gagnon, S. Brunet, G. Goyette, A. Laplante, M.F. Princiotta, P. Thibault, D. Sacks, and M. Desjardins. 2003. Phagosomes are competent organelles for antigen cross-presentation. Nature. 425:402–406. [DOI] [PubMed] [Google Scholar]
- 39.Guermonprez, P., L. Saveanu, M. Kleijmeer, J. Davoust, P. Van Endert, and S. Amigorena. 2003. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 425:397–402. [DOI] [PubMed] [Google Scholar]
- 40.Inaba, K., S. Turley, F. Yamaide, T. Iyoda, K. Mahnke, M. Inaba, M. Pack, M. Subklewe, B. Sauter, D. Sheff, et al. 1998. Efficient presentation of phagocytosed cellular fragments on the MHC class II products of dendritic cells. J. Exp. Med. 188:2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garg, S., A. Oran, J. Wajchman, S. Sasaki, C.H. Maris, J.A. Kapp, and J. Jacob. 2003. Genetic tagging shows increased frequency and longevity of antigen-presenting, skin-derived dendritic cells in vivo. Nat. Immunol. 4:907–912. [DOI] [PubMed] [Google Scholar]
- 42.Albert, M.L., B. Sauter, and N. Bhardwaj. 1998. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 392:86–89. [DOI] [PubMed] [Google Scholar]
- 43.Liu, K., T. Iyoda, M. Saternus, K. Kimura, K. Inaba, and R.M. Steinman. 2002. Immune tolerance after delivery of dying cells to dendritic cells in situ. J. Exp. Med. 196:1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]