

Abstract
Thrombopoietin (Tpo) is the primary cytokine regulating megakaryocyte development and platelet production. Tpo signaling through its receptor, c-mpl, activates multiple pathways including signal transducer and activator of transcription (STAT)3, STAT5, phosphoinositide 3-kinase–Akt, and p42/44 mitogen-activated protein kinase (MAPK). The adaptor protein Lnk is implicated in cytokine receptor and immunoreceptor signaling. Here, we show that Lnk overexpression negatively regulates Tpo-mediated cell proliferation and endomitosis in hematopoietic cell lines and primary hematopoietic cells. Lnk attenuates Tpo-induced S-phase progression in 32D cells expressing mpl, and Lnk decreases Tpo-dependent megakaryocyte growth in bone marrow (BM)–derived megakaryocyte culture. Consistent with this result, we found that in both BM and spleen, Lnk-deficient mice exhibited increased numbers of megakaryocytes with increased ploidy compared with wild-type mice. In addition, Lnk-deficient megakaryocytes derived from BM and spleen showed enhanced sensitivity to Tpo during culture. The absence of Lnk caused enhanced and prolonged Tpo induction of STAT3, STAT5, Akt, and MAPK signaling pathways in CD41+ megakaryocytes. Furthermore, the Src homology 2 domain of Lnk is essential for Lnk's inhibitory function. In contrast, the conserved tyrosine near the COOH terminus is dispensable and the pleckstrin homology domain of Lnk contributes to, but is not essential for, inhibiting Tpo-dependent 32D cell growth or megakaryocyte development. Thus, Lnk negatively modulates mpl signaling pathways and is important for Tpo-mediated megakaryocytopoiesis in vivo.
Keywords: hematopoiesis, megakaryocytes, cytokine receptors, cell proliferation, endomitosis
Introduction
Thrombopoietin (Tpo), the primary cytokine regulating megakaryocyte development and platelet production, signals through the mpl receptor (1, 2). Both Tpo−/− and mpl−/− mice are severely thrombocytopenic with 10–15% of the normal platelet levels (3, 4). In addition, Tpo and mpl nullizygous mice exhibit markedly reduced megakaryocyte and CFU-megakaryocyte progenitor numbers as well as decreased megakaryocyte ploidy (5). Interestingly, in the absence of Tpo or mpl, the residual megakaryocytes and platelets are morphologically and functionally normal (6, 7). Therefore, Tpo/mpl is important for controlling megakaryocyte numbers by promoting their survival and/or proliferation as well as differentiation, but is not essential for megakaryocyte cytoplasmic maturation or platelet formation.
Mpl belongs to the type I cytokine receptor family, which includes the erythropoietin receptor, growth hormone receptor, and prolactin receptor. They share homology in the membrane proximal region, which includes the Box1 and Box2 domains (8). Ligand binding induces activation of the JAK2 that is associated with the receptor through Box1 and Box2 (9). JAK2-deficient fetal livers fail to respond to Tpo in forming megakaryocyte colonies, which reveals its essential role in mpl signaling (10).
Activated JAK2 phosphorylates tyrosine residues on cytokine receptors, thereby providing docking sites for several Src homology 2 (SH2) domain–containing downstream signaling proteins. Mpl activates many signaling pathways in hematopoietic cell lines and primary megakaryocytes and platelets, including STAT3 and 5, Shc–Ras–mitogen-activated protein kinase (MAPK), and SHP2–Gab–phosphoinositide-3 kinase–Akt pathways (2). These signaling molecules have important roles in hematopoiesis, and dysregulation of these signaling pathways is implicated in leukemogenesis. STAT5a5b nullizygous mice exhibit cytopenias affecting multiple peripheral blood lineages, including reduced platelet numbers (11). Increased apoptosis, rather than decreased cell proliferation, accounts for the intrinsic defect in early progenitors and stem cells observed in STAT5a−/−5b−/− mice (12). In addition, massive apoptosis due to the lack of STAT5 blocks terminal myeloid differentiation (13).
Transgenic overexpression of a dominant negative form of STAT3 did not affect megakaryocytopoiesis or thrombopoiesis; however, platelet recovery from myelosuppression after 5-fluorouracil treatment was significantly delayed (14). These data indicate that STAT3 is dispensable for megakaryocyte and platelet development, but is required for effective expansion of early megakaryocyte progenitors. The phosphoinositide 3-kinase–Akt pathway is necessary, but not sufficient, for Tpo-induced proliferation of megakaryocytic progenitors (15), and the p42/p44–MAPK pathway is important for Tpo-induced endomitosis in primary megakaryocytes (16).
Lnk is a member of a newly discovered adaptor protein family that also contains APS and SH2-B. These three share common interaction domains: a proline-rich amino-terminus, a pleckstrin homology (PH) domain, an SH2 domain, and a conserved tyrosine near the carboxyl-terminus (17). Mice nullizygous for Lnk revealed an essential role for Lnk in B cell lymphopoiesis (18). The expansion of pro–/pre– and immature B cells in Lnk−/− mice is due to hyperproliferation of immature B cells (18). In the absence of Lnk, BM-derived cells are hypersensitive to stem cell factor (SCF) activation, confirming that Lnk normally negatively regulates c-Kit signaling pathways in immature B cells. Indeed, Lnk becomes phosphorylated and interacts with c-Kit upon SCF binding (19). Lnk nullizygous mice on a KitW/+ background exhibit marginally restored B cell abnormalities, supporting the notion that Lnk negatively regulates the SCF–c-Kit signaling pathway (19). Overexpression of Lnk in aorta-gonad-mesonephros (AGM) primary cultures suppresses the emergence of CD45+ hematopoietic cells via inhibition of the SCF–c-Kit signaling pathways (20).
In addition to lymphoid and AGM region developmental defects, Lnk-deficient mice also display a fivefold elevation of circulating platelets (21). Significant increased cell numbers of the megakaryocytic and erythroid lineages as well as increased B cells account for the splenomegaly in the Lnk-deficient mice (21). Megakaryocytic (CFU-Meg) progenitor numbers are significantly increased in the spleens of Lnk−/− mice, and the colony sizes derived from those Lnk-deficient progenitor cells are significantly larger than WT controls (21). This indicates that Lnk-deficient hematopoietic progenitor cells are hypersensitive to the growth-promoting effects of several cytokines. Moreover, the absence of Lnk causes abnormal modulation of IL-3 and SCF–c-Kit signaling pathways (21). However, whether Lnk directly modulates Tpo–mpl-mediated signaling pathway has not been reported.
Here, we studied the function of Lnk in Tpo-mediated signaling. We showed that overexpression of Lnk inhibits Tpo-induced cell proliferation in the hematopoietic progenitor cell line, 32D, by blocking cell cycle progression. Lnk attenuates the sustained STAT5 and p42/44MAPK activation normally observed after Tpo stimulation. We further investigated the role of Lnk in primary hematopoietic cells. Lnk overexpression attenuates Tpo-dependent BM-derived megakaryocyte development by reducing megakaryocyte size and ploidy. The SH2 domain of Lnk is essential for its inhibitory function in 32D cells and also in primary BM cells. In vivo analysis recapitulated our results using cell cultures; Lnk-deficient BM and spleen displayed elevated numbers of megakaryocytes with increased ploidy. In addition, Lnk-deficient BM or spleen-derived megakaryocytes displayed enhanced sensitivity to Tpo during in vitro culture. This is shown to be a consequence of enhanced STAT3, STAT5, Akt, and MAPK signaling pathways in Lnk-deficient megakaryocytes after Tpo stimulation. Thus, Lnk negatively modulates mpl signaling pathways and is important for Tpo-mediated megakaryocytopoiesis in vivo.
Materials and Methods
Mice, Cell Lines, and Media.
Lnk-deficient mice were provided by Dr. T. Pawson (Samuel Lunenfeld Research Institute, Toronto, Canada). Lnk nullizygous mice were backcrossed onto the C57/BL6 background for seven generations. WT controls were littermates or purchased from Jackson ImmunoResearch Laboratories. IL-3–dependent 32D clone 3 cells were purchased from the American Type Culture Collection. 32D cells were maintained in RPMI 1640 containing 10% heat-inactivated FBS (JRH Biosciences) and 10% WEHI-3B cell supernatant as a source of IL-3 (WEHI media). Tpo growth media consisted of RPMI 1640 containing 10% FBS supplemented with 1 ng/ml of megakaryocytes growth and differentiation factor (MGDF). MGDF, a pegylated and truncated human recombinant Tpo, was provided by Amgen, Inc.
Retroviral Constructs.
MSCV-IRES-GFP (MIG) vector was a gift from Dr. K. Humphries (Terry Fox Laboratories, Vancouver, Canada). pcDNA-Lnkwt, Lnk (R354E), and Lnk (Y536F) constructs were provided by Dr. S. Takaki (University of Tokyo, Tokyo, Japan). Lnk cDNA was digested with BamHI and EcoRI, and subsequently blunt-end ligated to the MIG vector linearized by HpaI. The MIG-Lnk (W191A) construct was generated through site-directed mutagenesis using the following oligos: 5′-cagcaatggacagcggcgcacgcGCgcagcggggtcgcctggtgc-3′, and 5′-gcaccaggcgaccccgctgcGCgcgtgcgccgctgtccattgctg-3′ (Stratagene). 5′-HA-tagged mpl was generated by PCR and inserted into the EcoRI and XhoI sites of MSCV-PGKneo vector (CLONTECH Laboratories, Inc.).
Retroviral Transduction.
3 μg of each retroviral construct was cotransfected with 1 μg pCL-Eco (22) into one 100-cm plate of 50% confluent Phoenix packaging cells (provided by G. Nolan, Stanford University, Stanford, CA), using Fugene 6 reagent (Roche Applied Science). The resulting supernatant was collected 48 h later. The viral titer was determined by serial dilution of the viral supernatant and infection of 3T3 cells; 2–10 × 106 PFU/ml was routinely obtained. Supernatants with equal viral titers were used in each experiment. 32D/mpl cells or lineage negative (Lin−) BM cells (see Lin− BM-derived Megakaryocyte Culture and Polylipid Analysis) were spin-infected with the desired viral supernatant containing 10 μg/ml polybrene (Sigma-Aldrich) at 2,000 revolutions/min (Sorvall RT7) for 1 h at 30°C.
Cell Proliferation Assay.
To measure 32D cell proliferation, we seeded triplicate samples of cells (200 μl/well; 2 × 105/ml) in 96-well plates, and cultured them for 3 d with different concentrations of cytokines. We added 15 μl of 3-(4,5-dimethylthiazole-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT; Promega) to each well and measured live cell numbers according to the manufacturer's instructions. Cell growth assays were performed in 24-well plates with 1 ml of cells per well at a concentration of 2 × 105 cells/ml. Every day a portion of the cells was removed and counted in the presence of trypan blue to exclude dead cells.
Cell Cycle Analysis.
2 d after retroviral transduction, 32D/mpl cells were washed and switched to Tpo media. After overnight culture, 1-ml aliquots of cells from each sample were incubated at 37°C in the presence of 10 μg/ml Hoechst 33342 (Sigma-Aldrich) for 0.5 h. 2 μg/ml propidium iodide (PI) was added, and the cells were subjected to flow cytometry using the LSR II flow cytometer (BD Biosciences). Collected data were analyzed using ModFit software (Verity Software).
Lin− BM-derived Megakaryocyte Culture and Polyploidy Analysis.
8–12-wk-old C57Bl6/J or BALB/cJ mice obtained from The Jackson Laboratory were used to isolate BM cells. BM cells from the tibia and femurs were dissociated by passing through 18-gauge needles and filtered through a 70-micron cell strainer. The BM cells were labeled with a biotinylated cocktail of lineage-specific antibodies followed by a tetrameric antibody complex and magnetic colloids (StemCell Technologies Inc.). Lin− BM cells were purified using a 0.3-inch StemStep column following the manufacturer's protocol (StemCell Technologies Inc.). Lin− BM cells were plated in 24-well tissue culture plates at 5 × 105 cells/well and spin-infected with desired viral supernatant for 1 h. Subsequently, the Lin− BM cells were allowed to expand and express transduced genes by culturing in the following media: IMDM containing 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin and streptomycin, 50 μM β-mercaptoethanol, 50 ng/ml SCF, 50 ng/ml Flt3 ligand, and 20 ng/ml IL-6 (PeproTech). 2 d later, the GFP+ cells were purified using flow cytometric sorting, and plated at 0.5 × 105 cells/well in megakaryocyte culture media in an eight-well chamber coverglass (Nunc). The megakaryocyte culture media is composed of IMDM containing 1% Nutridoma-SP (Roche Applied Science), 2 mM l-glutamine, 100 U/ml penicillin and streptomycin, and 50 μM β-mercaptoethanol, supplemented with 50 ng/ml Tpo. 4 d later, the megakaryocytes were imaged using a Nikon TE300 inverted microscope equipped with a Hamamatsu Orca digital camera, and the images were acquired using OpenLab software (Improvision). The sizes of the megakaryocytes were manually measured in OpenLab; thousands of megakaryocytes were manually measured in each group in each experiment. Median cell sizes were obtained in each experiment, and the mean of the cell size from at least three independent experiments was calculated.
To determine ploidy, cultured megakaryocytes were labeled in 50 μg/ml PI in 0.1% sodium citrate solution (hypotonic PI) overnight or longer, and subjected to flow cytometry using flow cytometer FACSCalibur (BD Biosciences; reference 23).
Megakaryocyte Quantification and Culture.
8–16-wk-old WT and Lnk-deficient mice were killed by cervical dislocation. BM and spleen cells were dissociated in CATCH buffer (Ca2+ and Mg2+-free PBS containing 3.5% BSA, 1 mM adenosine, 2 mM theophylline, and 0.38% sodium citrate), and passed through a 100-micron cell strainer. A portion of the cells was enumerated in the presence of 3% acetic acid to exclude enucleated red blood cells. The remaining cells were incubated with Fc blocker to block nonspecific binding and stained with FITC–CD41 antibody (1:200; BD Biosciences). A small portion of the cells was subjected to FACS analysis to measure the percentage of CD41+ cells. The remaining cells were labeled with hypotonic PI solution as mentioned before. 50 μg/ml RNase A was added and incubated at room temperature for 0.5 h before FACS analysis. Cells from one femur and one tibia, or from a quarter of a spleen were used from each mouse. All the cells were subjected to FACS analysis, and CD41+ cells were gated for DNA content analysis. Cells with DNA content ≥8N were quantified. At least four to six mice of each genotype were used.
For megakaryocyte culture, BM or spleen cells were dissociated in IMDM with 5% FBS. Total BM or spleen cells were enumerated, washed in serum-free media, and divided into five groups. Each group of cells was cultured in the serum-free media (as mentioned before) containing different concentrations of Tpo (0, 0.1, 1, 10, and 50 ng/ml Tpo), at 106 cells/ml. 4 d later, the cells were harvested in CATCH buffer, and stained with FITC–CD41 antibodies. The DNA were labeled with hypotonic-PI solution and polyploidy were quantified as mentioned before. These experiments were repeated independently four times.
The mean megakaryocytes ploidy was calculated according to the following equation:
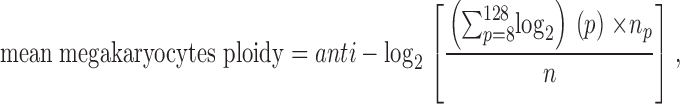 |
where np is the number of megakaryocytes in ploidy class p (p = 8, 16, 32, 64, 128), and n is the total number of megakaryocytes in all ploidy classes (24).
Protein Lysates and Western Blot Analysis.
After sorting 32D–mpl cells for the GFP+ population expressing either vector alone or a vector encoding Lnk, we allowed them to recover in WEHI (IL-3) media overnight. Cells were starved in RPMI 1640 containing 1% BSA for 4 h. We subsequently stimulated the cells with 20 ng/ml Tpo for 0, 10, 30, and 120 min, and directly lysed them in SDS sample buffer (BioRad Laboratories) (100 μl lysis buffer per 106 cells). The samples were Western blotted with the indicated antibodies: anti-STAT5 (C-17) antibody (1:500; Santa Cruz Biotechnology, Inc.), anti–p-STAT5 (pTyr 694), p-STAT3 (pTyr 705), STAT3, p-MAPK (pThr202/Tyr204), and p42/44 MAPK antibody (1:1,000, Cell Signaling Technology).
BM cells were labeled with FITC–CD41, and CD41+ megakaryocytes were sorted using a MoFlo high-speed sorter. In time course studies, megakaryocytes were labeled with FITC–CD41 and first purified by EasyStep magnetic beads (StemCell Technologies), and further purified by flow cytometric sorting. Purified CD41+ cells were starved in IMDM with 1% BSA for 1–2 h, and stimulated with 1 or 10 ng/ml Tpo for 10 min in IMDM plus 0.1% BSA. Time course studies were performed in the presence of 10 ng/ml Tpo for 0, 10, 30, 60, and 120 min. Equal numbers of cells (2–3 × 105) were used for each time point. The protein lysates were resolved on NuPAGE Novex Bis-Tris gels and Western blotted with anti-pAkt (Ser 473) or Akt (Cell Signaling Technology), or STAT5 and p42/44 MAPK antibodies as described before. These experiments were performed independently three to five times.
Results
The Lnk SH2 Domain Is Required for Inhibiting the Growth of 32D Cells Expressing mpl.
IL-3–dependent 32D hematopoietic cells were used to establish a stable cell line expressing the Tpo receptor (mpl), designated as 32D–mpl. Although the parental cells did not respond to Tpo, 32D–mpl cells proliferated in a dose-dependent manner with maximal growth at 1–10 ng/ml Tpo (Fig. 1 a). At those concentrations, Tpo-induced cell proliferation was as robust as that of IL-3. Therefore, we used 1 ng/ml Tpo as the optimal concentration in all subsequent experiments.
Figure 1.

Lnk inhibits 32D cell growth via blocking S-phase progression, and the SH2 domain of Lnk is essential for its inhibitory function. (a) Establishment of mpl-expressing 32D cells. Parental 32D cells and cells stably expressing mpl were cultured in IL-3 or different concentrations of Tpo. Live cell numbers in the presence of Tpo relative to that in IL-3 after 3 d of culture were determined by MTT absorbance. (b) Overexpression of Lnk in 32D/mpl cells inhibits cell growth in response to Tpo. We introduced either vector alone or WT Lnk into 32D/mpl cells and determined the proportion of infected cells as those that express GFP 2 d later. We measured the GFP+ fraction every 3 d, and the percentage of GFP+ cells relative to the infection rate 2 d after infection was plotted. Results are representative of more than five independent experiments. (c) 2 d after the infection, total cell numbers were counted and GFP+ percentages were determined daily. The numbers of vector- or Lnk-expressing GFP+ cells were calculated and plotted (mean ± SD). (d) Cells infected with either vector alone or Lnk were subjected to cell cycle analysis after overnight culture in the presence of Tpo; only GFP+ cells were gated in both plots. Results are representative of three independent experiments. (e) We introduced the WT or the mutant Lnk cDNA constructs into 32D/mpl cells and measured the GFP+ fraction every 3 d as described in b. The cells were maintained in the presence of 1 ng/ml Tpo, and the percentage of GFP+ cells relative to the infection rate 2 d after infection was plotted. Results are representative of more than three independent experiments.
The effect of Lnk in Tpo-dependent 32D cell growth was examined by overexpression of WT Lnk using the MIG vector. MIG is a bicistronic vector containing GFP downstream of an IRES. As GFP expression is tightly correlated with the expression of the gene cloned upstream of the IRES (25), we were able to identify cells expressing Lnk by analyzing GFP fluorescence. We introduced either vector alone or Lnk into 32D/mpl cells and determined the fraction of GFP+ infected cells 2 d later. We measured the GFP+ fraction every 3 d, as the cells divide, relative to that 2 d after infection (Fig. 1 b). When the cells were grown in Tpo, the Lnk expressing fraction was dramatically reduced to 20% after 3 d and to 3% after 6 d, whereas the fraction of control vector-expressing GFP+ cells was unchanged (100%) over the 9-d experiment. Lnk expression also inhibited cell growth in the presence of IL-3, but to a lesser extent (Fig. 1 b). To confirm that Lnk overexpression blocked cell proliferation, cell numbers of vector-expressing and Lnk-expressing cells were counted and calculated daily. As shown in Fig. 1 c, Lnk expressing 32D–mpl cells did not increase in number, whereas the vector control cells exhibited exponential growth. Lnk did not inhibit the growth of nonhematopoietic cell lines, such as 3T3 or 293T cells, indicating that Lnk is not toxic to most cell lines (unpublished data).
Next, we set out to study whether Lnk inhibits the cell cycle, and if so, at which stage. 2 d after infecting 32D–mpl cells with MIG vector alone or MIG-Lnk, we plated them in Tpo media. After overnight culture, portions of cells from both groups were subjected to cell cycle analysis. GFP+ cells expressing vector alone grew exponentially, demonstrating a cell cycle profile of 47% S-phase, 44% G1, and 9% G2/M phase (Fig. 1 d, left). In contrast, GFP+ cells expressing Lnk showed a markedly decreased S-phase population (16%), and an increased proportion of G1 (61%) and G2/M (23%) cells (Fig. 1 d, right). Therefore, overexpression of Lnk blocked S-phase progression, and led to accumulation of the cells in the G1 and G2/M phases.
To investigate the importance of different domains of Lnk in growth inhibition, we generated point mutations in the Lnk cDNA to ablate individual domains and overexpressed these mutant forms of Lnk in 32D–mpl cells (Fig. 1 e). When cultured in the presence of 1 ng/ml Tpo, the fraction of cells expressing WT Lnk became reduced dramatically, whereas mutation of the SH2 domain Lnk (Lnk R364E) completely abolished the inhibitory function of Lnk on cell proliferation (Fig. 1 e). In contrast, mutation of the conserved tyrosine at the COOH terminus (Y536F) did not affect Lnk's inhibitory function (Fig. 1 e). The PH domain mutation (W191A) of Lnk moderately compromised the inhibitory function of Lnk (Fig. 1 e).
These findings indicate the SH2 domain of Lnk is essential for its inhibitory function, whereas the PH domain plays a minor role and the conserved tyrosine at the COOH terminus is dispensable for this Lnk-dependent function.
The SH2 Domain of Lnk Is Essential for Inhibiting the Sustained Activation of STAT5 and p42/44 MAPK Induced by Tpo.
To investigate the mechanism by which Lnk inhibits 32D cell proliferation, we studied the activation of downstream signaling molecules. By flow cytometry, we purified 32D–mpl cells transduced with the vector alone or WT Lnk or Lnk mutants, and subjected them to Western blotting analysis after Tpo stimulation. In vector-transduced 32D/mpl cells, Tpo induced maximal STAT5 phosphorylation at 10 min, and the level of phosphorylated STAT5 slowly dropped to 30% at 30 min, and 15% at 120 min (Fig. 2 a, lanes 1–4). In Lnk-transduced cells, Tpo activated maximal STAT5 phosphorylation at 10 min to a level similar to that in control cells; however, the level of phosphorylated STAT5 dropped rapidly thereafter, with 15% remaining at 30 min and only 4% at 120 min (Fig. 2 a, lanes 5–8). Similarly, in control cells, Tpo activated maximal p42/44MAPK phosphorylation at 10–30 min, and the level of phosphorylated MAPK slowly dropped to 50% of the maximum at 120 min (Fig. 2 b, lanes 1–4). Lnk expression only moderately attenuated maximal activation of MAPK (60% of the control cells) at 10 min. However, Lnk inhibited prolonged activation of p44/42MAPK induced by Tpo, resulting in undetectable levels of MAPK activation at 120 min (Fig. 2 b, lanes 5–8). Total STAT5 or MAPK protein levels were unaffected (Fig. 2, bottom). Thus, overexpression of Lnk attenuated the prolongation of STAT5 and MAPK phosphorylation induced by Tpo.
Figure 2.

The SH2 domain of Lnk is critical for inhibiting sustained STAT5 and p42/44 MAPK phosphorylation induced by Tpo. 32D/mpl cells infected with either vector alone or the WT or the mutant forms of Lnk were purified, stimulated with Tpo, and lysed at indicated intervals followed by Western blotting analysis. (a) STAT5 phosphorylation and protein levels after Tpo administration. The triangles indicate the 120-min time points. (b) P42/44 MAPK phosphorylation and protein levels after Tpo administration. White lines indicate that intervening lanes have been spliced out. Representatives of three independent experiments are shown.
Fig. 2 also shows that the Lnk SH2 domain is essential for Lnk inhibition of STAT5 and MAPK activation. Lnk (R364E)-transduced cells showed similar STAT5 activation kinetics to vector controls (Fig. 2 a, lanes 9–12). In contrast, Lnk (W191A) and Lnk (Y536F)-transduced cells attenuated prolonged activation of STAT5 to a similar extent as seen in WT Lnk-transduced cells (Fig. 2 a, lanes 13–20). Similar to the inhibition of sustained STAT5 activation, Lnk inhibited sustained MAPK activation induced by Tpo (Fig. 2 b, lanes 1–8). The R364E mutation abolished Lnk inhibition of prolonged MAPK activation (Fig. 2 b, lanes 9–12), whereas the W191A and Y536F mutations did not affect Lnk inhibitory function (Fig. 2 b, lanes 13–20). Thus, the SH2 domain of Lnk is essential for inhibiting mpl signaling in 32D cells, whereas neither the phosphorolatable tyrosine at the COOH terminus nor the PH domain is essential for these inhibitory functions of Lnk.
The SH2 Domain of Lnk Is Required to Reduce the Growth, Size, and Ploidy of BM-derived Megakaryocytes.
To study Lnk function in Tpo/mpl signaling in primary cells, we analyzed BM-derived megakaryocytes. Lin− hematopoietic progenitor BM cells were infected either with MIG vector or MIG-Lnk, and GFP+ cells were purified by flow cytometry and equal numbers of cells were plated onto coverglasses in serum-free medium containing Tpo as the only cytokine. 3–4 d later, the megakaryocytes were imaged (Fig. 3 b) and cell sizes were quantified (Fig. 3 a). Lnk expression reduced megakaryocyte size to ∼60% of vector-transduced controls (19 μm vs. 33 μm; Fig. 3 a). However, they both displayed similar gross morphologies examined by light microscopy (Fig. 3 b). Most nonmegakaryocytes that did not respond to Tpo were condensed and dead by the end of 4 d of culture (double-lined arrows); only large megakaryocytes were apparent (Fig. 3 b). White arrows indicate immature megakaryocytes with a smooth cell membrane before extensive changes in morphology; short wide arrows point to megakaryocytes beginning to undergo cell membrane and cytoplasmic maturation; and long thin arrows indicate mature megakaryocytes that have undergone advanced stages of cytoplasmic maturation (Fig. 3 b). Therefore, Lnk overexpression reduced megakaryocyte sizes without grossly blocking cytoplasmic morphology changes.
Figure 3.
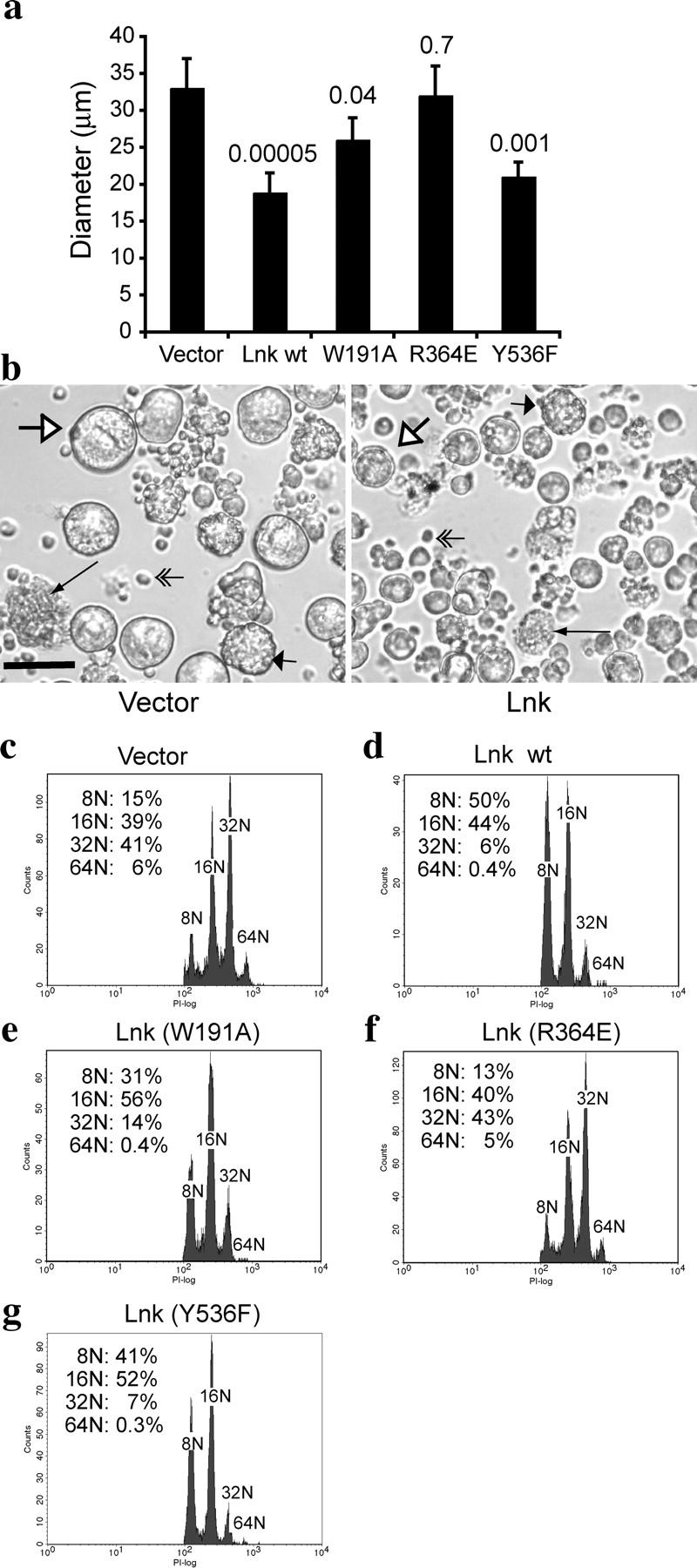
The SH2 domain of Lnk is required to reduce the size and ploidy of megakaryocytes. Lin− BM cells were transduced with either vector alone, or with WT or mutant forms of Lnk, and GFP+ cells were purified and cultured in serum-free media containing only Tpo (see Materials and Methods). After 4 d of growth and differentiation, the diameters of the megakaryocytes were measured and the median diameters (±SD) are shown in a. Student's t tests were performed on each form of Lnk compared to the vector control, and p-values are shown on the top of each bar. (b) Differential interference contrast images of megakaryocytes derived from vector-transduced Lin− progenitor cells (left) and megakaryocytes expressing Lnk (right). White arrows, long thin arrows, and short wide arrows indicate megakaryocytes at different developmental stages (see Results). Double-lined arrows point to dead cells that do not respond to Tpo. Bar, 50 μm. (c–g) Ploidy analysis of the megakaryocytes derived from vector-transduced (c), Lnk-transduced (d), Lnk (W191A)-transduced (e), Lnk (R364E)-transduced (f), and Lnk (Y536F)-transduced (g) Lin− BM cells. GFP+ cells were gated, and cells with 8N and greater ploidy were analyzed.
Megakaryocyte size often correlates with DNA content; therefore, we analyzed the ploidy of the megakaryocytes derived from these BM cultures. We considered megakaryocytes to be those cells with DNA content exceeding 4N. Lnk overexpression reduced megakaryocyte numbers, causing a 37 ± 5% reduction in the number of cells with DNA content of 8N and greater. Megakaryocytes derived from vector-transduced Lin− BM cells showed an array of DNA distribution, with 16N (39%) and 32N (41%) being the most predominant populations, and 8N (15%) and 64N (6%) as minor ones (Fig. 3 c). Megakaryocytes derived from the Lnk-transduced Lin− BM culture exhibited a dramatic decrease in DNA ploidy. The most predominant populations were 8N (50%) and 16N (44%), whereas the 32N (6%) population was minor and the 64N (<1%) peak was almost invisible (Fig. 3 d). Therefore, Lnk overexpression in primary Lin− BM cells reduced both megakaryocyte number and ploidy.
Next, we examined the individual domains of Lnk responsible for reducing megakaryocyte size and ploidy. The SH2 domain mutant that abolished Lnk function in 32D cells, R364E, did not affect megakaryocyte size or ploidy when overexpressed in Lin− BM cells (Fig. 3, a and f). In contrast, overexpression of Lnk (Y536F) reduced megakaryocyte size and ploidy to a similar extent as WT Lnk (Fig. 3, a and g). Consistently, the PH domain mutation (W191A) moderately affected Lnk function in reducing megakaryocyte size and ploidy (Fig. 3, a and e). Thus, the SH2 domain of Lnk is crucial for reducing Tpo-mediated megakaryocyte development, whereas the PH domain and conserved tyrosine are not essential for Lnk-mediated inhibition of Tpo signaling.
Increased BM and Splenic Megakaryocyte Numbers and Ploidy in Lnk Nullizygous Mice.
Both BM and spleens of Lnk-deficient mice possess increased numbers of megakaryocytes. To investigate megakaryocytopoiesis in Lnk-deficient mice, first we analyzed both in BM and spleen the total number of megakaryocytes as well as the number of mature megakaryocytes with ≥8N ploidy (Table I and Fig. 4). CD41, also called glycoprotein IIb, is expressed in all megakaryocytes with DNA contents form 2N to 128N, and is commonly used as a megakaryocyte marker. Thus, we determined total megakaryocyte numbers by quantifying cells expressing the CD41 surface marker. We also quantified mature megakaryocytes as those that are CD41+ and have 8N or greater ploidy. As shown in Table I, in Lnk-deficient mice total BM cells are only slightly increased in number (30%, line 1), whereas the percentage of CD41+ megakaryocytes increased twofold (line 2). Mature megakaryocytes with 8N and greater ploidy increased 2.6-fold, compared with age- and sex-matched WT controls (line 3). Spleens from Lnk-deficient animals displayed more profound differences than did BM. Although the total number of spleen cells in Lnk-deficient mice increased approximately twofold (line 4), the total number of CD41+ megakaryocytes increased fivefold (line 5). Remarkably, the number of mature megakaryocytes in the spleen increased 10-fold compared with WT controls (Line 6). Therefore, Lnk-deficient BM and spleens had increased numbers of megakaryocytes and proportionally more mature megakaryocytes (≥8N) compared with WT controls.
Table I.
CD41+ Megakaryocytes from the BM and Spleens of WT and Lnk Nullizygous Mice
| Line | Genotype | WT | Lnk | Fold increasea |
|---|---|---|---|---|
| 1 | Total BM cells per mouse | 58 ± 9 × 106 | 78 ± 10 × 106 | 1.3 (P < 0.01) |
| 2 | Percentage of CD41+ cells in BM | 1.1 ± 0.2% | 2.2 ± 0.5% | 2.0 (P < 0.005) |
| 3 | Megakaryocytes per BM (CD41+, ≥8N) | 25 ± 7 × 103 | 66 ± 24 × 103 | 2.6 (P < 0.01) |
| 4 | Total spleen cells per mouse | 173 ± 27 × 106 | 345 ± 76 × 106 | 2.0 (P < 0.001) |
| 5 | Percentage of CD41+ cells in spleen | 2.1 ± 0.5% | 10.7 ± 2.4% | 5.1 (P < 0.005) |
| 6 | Megakaryocytes per spleen (CD41+, ≥8N) | 4.3 ± 0.5 × 103 | 45 ± 6 × 103 | 10 (P < 0.0005) |
8–16-wk-old mice were used to isolate BM and spleen cells. Total BM and spleen cell numbers (mean ± SD) were enumerated by hemoctocytometer counting. Percentages of CD41+ cells (mean ± SD) were determined by flow cytometry. The numbers of CD41+ megakaryocytes with 8N and greater ploidy were determined by flow cytometry.
Student's t test was performed, and the p-values from each group are shown.
Figure 4.

Increased megakaryocyte ploidy in Lnk nullizygous mice. CD41+ megakaryocytes from WT BM (a) and Lnk-deficient BM (b) were analyzed for DNA content. Only mature megakaryocytes with 8N and greater ploidy are shown. (c) Quantification of the percentages of individual ploidy classes (≥8N) of BM megakaryocytes (mean ± SD). Mean ploidy was calculated as stated in Materials and Methods. Student's t test was performed. P < 0.001.
We further investigated the ploidy distribution of mature megakaryocytes (Fig. 4). Lnk-deficient megakaryocytes exhibited a higher ploidy distribution than WT controls. Compared with WT BM, Lnk-deficient BM had a lower percentage of megakaryocytes with low ploidy (8N and 16N), whereas the percentages of cells with high ploidy (32N and 64N) doubled (Fig. 4 c). The mean ploidy of Lnk-deficient BM megakaryocytes increased to 18N, compared with 15N for WT counterparts (Fig. 4 c). Lnk-deficient spleens also exhibited an array of megakaryocytes with different DNA contents, however, WT spleens contained too few mature megakaryocytes (≥8N) to be quantified (unpublished data).
Enhanced Sensitivity to Tpo in Lnk-deficient Megakaryocyte Culture.
Because Tpo is the most important cytokine regulating megakaryocytopoiesis and is involved in every step of this process, we investigated whether Lnk is a physiological inhibitor of Tpo-dependent megakaryocytopoiesis. Increases in megakaryocyte number result from cell proliferation (mitosis), whereas polyploidy is a result of endomitosis, a process that is characterized by continued DNA replication in the absence of cytokinesis (26). To assess megakaryocyte proliferation and endomitosis in response to Tpo, we cultured BM and spleen cells in serum-free media with Tpo as the only cytokine. As shown in Fig. 5, Tpo stimulated megakaryocyte production in a dose-dependent manner, reaching maximal levels at 10–50 ng/ml Tpo. The number of CD41+ megakaryocytes with 8N and greater ploidy derived from WT BM cultures increased 1.6-fold at the maximal Tpo concentration (Fig. 5 a). In contrast, the number of megakaryocytes derived from Lnk-deficient BM increased nearly threefold in the presence of Tpo (Fig. 5 a). This resulted in fivefold more megakaryocytes produced at high Tpo concentrations (50 ng/ml), relative to WT BM. Similarly, spleen cultures from WT mice exhibited a minimal increase in megakaryocyte production (1.5-fold induction) in response to Tpo. In contrast, the number of megakaryocytes derived from Lnk-deficient spleen cultures increased threefold (Fig. 5 b). This led to a 16-fold increase in megakaryocyte production in the presence of high concentration of Tpo (50 ng/ml), compared with WT spleen cells. These data indicate that the absence of Lnk enhanced the proliferation of megakaryocytes in response to Tpo.
Figure 5.
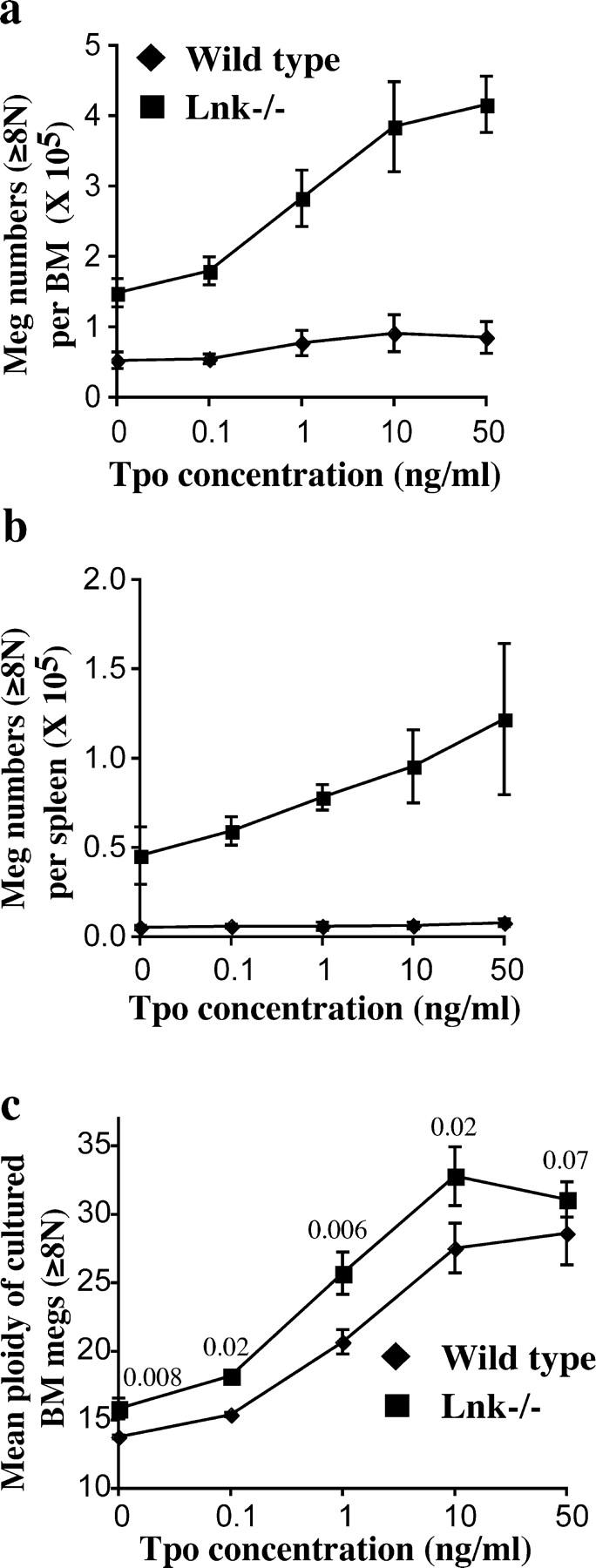
Enhanced megakaryocyte growth and ploidy in response to Tpo during in vitro megakaryocyte culture. Total BM (a) or spleen (b) cells from WT mice or Lnk nullizygous mice were cultured in different concentrations of Tpo for 4 d, the numbers of CD41+ megakaryocytes with 8N and greater ploidy were enumerated (mean ± SD). (c) Mean ploidies (±SD) of BM-derived megakaryocytes were plotted. A Student's t test was performed, and p-values are shown on top of each pair of points.
During in vitro culture Tpo, in a dose-dependent manner, induces megakaryocyte proliferation and maturation and increases megakaryocyte ploidy. As shown in Fig. 5 c, Tpo increased megakaryocyte ploidy in both WT and Lnk-deficient BM cells. Importantly, Lnk-deficient megakaryocytes showed a higher ploidy than WT controls at all Tpo concentrations (Fig. 5 c). Interestingly, in the presence of a maximal Tpo concentration (50 ng/ml), the difference of megakaryocyte ploidy between these two groups was not statistically significant (Fig. 5 c). Similarly, in the megakaryocyte culture derived from Lnk-deficient spleens, Tpo also increased megakaryocyte ploidy (unpublished data). However, cultures from WT spleens generated insufficient numbers of megakaryocytes to determine the ploidy.
Enhanced Tpo/mpl Signaling Pathways in Lnk-deficient Megakaryocytes.
To investigate the mechanism by which Lnk deficiency enhances Tpo-dependent megakaryocytopoiesis, we used purified CD41+ megakaryocytes and measured the four following signaling pathways induced by Tpo after 10 min of stimulation: p44/42MAPK, Akt, STAT 3, and STAT5 (Fig. 6, a–c). The absence of Lnk enhanced the extent of activation of all four signaling molecules stimulated by either 1 or 10 ng/ml Tpo (Fig. 6, a–c, top). As expected, the total levels of these signaling proteins remained similar (Fig. 6, a–c, bottom). Therefore, Lnk-deficient megakaryocytes displayed enhanced activation of four major signaling pathways induced by Tpo.
Figure 6.

Enhanced Tpo-induced signaling pathways in Lnk-deficient megakaryocytes. (a–c) CD41+ BM megakaryocytes from WT mice or Lnk nullizygous mice were stimulated with 0, 1, or 10 ng/ml of Tpo for 10 min. Protein lysates from equal numbers of cells were subjected to Western blotting analysis. Phosphorylation and total protein levels of p42/44MAPK and Akt (a), STAT5 (b), and STAT3 (c) are shown. (d and e) Purified CD41+ BM megakaryocytes from WT mice or Lnk nullizygous mice were stimulated with 10 ng/ml of Tpo, and lysed at indicated intervals followed by Western blotting analysis. Phosphorylation and total protein levels of Akt (d) and p42/44MAPK (e) are shown.
To investigate whether Lnk deficiency causes alterations in the kinetics of Tpo-induced signaling pathways, we analyzed the duration of Akt and MAPK activation after Tpo stimulation. In WT megakaryocytes, 10 ng/ml Tpo induced maximal Akt phosphorylation at 10 min and the level of Akt phosphorylation was down-regulated by 60 min. In contrast, Lnk-deficient megakaryocytes showed sustained Akt activation for at least 60 min (Fig. 6 d, top). Similarly, in WT megakaryocytes Tpo induced maximal p44/42MAPK phosphorylation at 10 min, and activation declined thereafter (Fig. 6 e, top). In contrast, Lnk-deficient megakaryocytes exhibited prolonged activation of MAPK for as long as 120 min (Fig. 6 e, top). The total levels of these signaling proteins remained similar (Fig. 6, d and e, bottom). Therefore, the absence of Lnk caused enhanced and prolonged activation of Tpo-induced signaling pathways.
Discussion
Lnk Is a Physiological Negative Regulator of Tpo-mediated Signaling and Megakaryocytopoiesis.
Our most important result is that physiological levels of the adaptor protein Lnk negatively regulate Tpo-mediated signaling both for megakaryocyte proliferation and endomitosis. This was evidenced by our finding that both BM and spleen of Lnk-deficient mice contained increased total numbers of megakaryocytes that had a significantly increased ploidy, compared with WT mice. In addition, Lnk-deficient megakaryocytes derived from both BM and spleen showed enhanced sensitivity to Tpo during in vitro culture. A principal, if not the only, action of Lnk is direct inhibition of mpl signal transduction because, after Tpo administration, CD41+ megakaryocytes from Lnk-deficient mice exhibited increased activation of STAT3, STAT5, Akt, and MAPK proteins, as well as prolonged activation of the two signaling proteins we tested, MAPK and Akt.
Conversely, we showed that Lnk overexpression negatively regulates Tpo-mediated cell proliferation in hematopoietic cell lines by attenuating Tpo-induced S-phase progression and causing cells to accumulate in the G1 and G2/M stages. In addition, Lnk overexpression decreased Tpo-dependent megakaryocyte numbers and ploidy in BM-derived megakaryocyte cultures. In parallel, we showed that Lnk overexpression reduces the duration of activation of STAT5 and MAPK signaling proteins. Together, our work establishes that the level of Lnk protein tightly regulates signal transduction by the mpl receptor protein and, thus, both the number and ploidy of megakaryocytes.
Regulation of Cytokine Receptor Signaling by the Lnk Family of Adaptor Proteins.
Negative regulators of receptor signaling are important for quantitatively controlling a cell's intrinsic sensitivity to cytokines. As examples, protein tyrosine phosphatase SHP-1 docks to phosphorylated erythropoietin receptor, resulting in dephosphorylation of JAK2 (27). The SH2-containing inositol-5-phosphatase inactivates the phosphoinositide 3-kinase–Akt pathway (28). Several suppressor of cytokine signaling proteins are induced by JAK–STAT pathways upon cytokine stimulation, and act within a classical negative feedback loop both to inhibit activation of signaling pathways and to target signaling components for proteasomal degradation (29, 30). As a result, loss of these negative regulators causes elevated and prolonged signaling to multiple cytokines.
The Lnk family of adaptor proteins is structurally distinct from the aforementioned negative regulators in that they do not possess any enzymatic activity. We found that, in 32D–mpl cells, overexpression of Lnk does not significantly inhibit the initial activation of mpl-mediated signaling pathways such as STAT5 or p42/44MAPK. Rather, Lnk diminishes the time these pathways remain active after a single application of a high concentration of Tpo to cytokine-deprived cells. Consistent with this finding, we showed that loss of Lnk enhances four Tpo-induced signaling pathways (STAT3, STAT5, MAPK, and Akt) in CD41+ megakaryocytes. In addition, loss of Lnk prolongs the activation of the two signaling pathways tested (MAPK and Akt) in CD41+ megakaryocytes. Similarly, BM-derived mast cells from Lnk−/− mice show prolonged activation of MAPK in response to IL-3 or SCF, compared with WT controls (21).
We found that in both hematopoietic cell lines and megakaryocyte progenitors, the SH2 domain of Lnk is essential for Lnk's function. Overexpression of an SH2 mutant Lnk does not inhibit mpl signaling either in 32D–mpl cells or Tpo-mediated megakaryocytopoiesis. Similarly, this SH2 domain mutation abolishes the binding of Lnk to c-Kit upon stimulation with SCF, leading to loss of Lnk-dependent inhibition of hematopoietic cell development in AGM culture (20). The conserved tyrosine near the COOH terminus of Lnk, Y536, is phosphorylated by c-Kit upon SCF binding. However, transgenic animals expressing Lnk Y536F under the control of lymphocyte-specific promoters reveal that this tyrosine is dispensable for the negative regulatory roles of Lnk in lymphocyte development (31). In agreement, we found that Y536 is not important for Lnk's function in modulating Tpo-dependent 32D cell proliferation or megakaryocyte development. Interestingly, the PH domain of Lnk is important, but not required, for inhibiting 32D cell growth or Tpo-mediated megakaryocytopoiesis, whereas it is dispensable for inhibiting hematopoietic differentiation in AGM culture (20). Together, these findings suggest that one or more tyrosine-phosphorylated proteins interact with the Lnk SH2 domain and are crucial for Lnk inhibitory functions. The fact that loss of Lnk enhances four Tpo-induced signaling pathways (STAT3, STAT5, MAPK, and Akt) in CD41+ megakaryocytes suggests that Lnk may inhibit JAK2 activation or mpl phosphorylation. We are currently investigating whether Lnk directly binds to phosphotyrosines in mpl or JAK2, whether this affects JAK2 kinase activity, and whether this is part of the mechanism by which Lnk down-regulated downstream signaling pathways.
Regulation of Hematopoiesis by Lnk.
Among mice nullizygous for Lnk family members, Lnk-deficient mice show the most profound perturbation in hematopoiesis. In addition to abnormal expansion of immature B cells (18) and enhanced stem cell repopulating activity (19), Lnk-deficient animals exhibit elevated platelet numbers in the peripheral blood, and increased numbers of CFU-megakaryocyte progenitors in the BM and spleen (21). Here, we found that BM and spleen of Lnk-deficient mice display an abnormal accumulation of both total CD41+ megakaryocytes and mature megakaryocytes with 8N or greater ploidy. The latter phenotype is more profound in the spleen (10-fold increase in cell number) than the BM (2.6-fold increase). The extensive extramedullary hematopoiesis observed in Lnk-deficient mice includes abnormal expansion of erythroid cells and B lymphocytes, as well as megakaryocytes in the spleen. Thus, Lnk likely regulates signaling by multiple cytokine receptors in multiple types of hematopoietic cells.
Megakaryocyte lineage commitment, CFU-megakaryocyte and megakaryocyte proliferation, megakaryocyte polyploidization, cytoplasmic maturation, and platelet fragmentation are related but separable processes. Through mutagenesis in mice many genes have been found to be important for various steps of megakaryocyte and platelet development (32). Polyploidization, uniquely occurring during megakaryocyte maturation, results from endomitosis; that is, continued DNA replication in the absence of cytokinesis (26). Megakaryocyte proliferation (mitosis) and differentiation (endomitosis) are separable processes, though both are dependent on Tpo. Exogenous expression of the c-myc oncogene drives overproduction of megakaryocytes with decreased ploidy (33), indicating that c-myc is important for megakaryocyte proliferation, but not endomitosis. In contrast, transgenic mice overexpressing cyclin D3 display large megakaryocytes with high ploidy (34), whereas antisense oligonucleotides to cyclin D3 inhibit megakaryocytic endomitosis in BM culture (35). MAPK also is important for megakaryocyte endomitosis (16); MAPK is also localized in the platelet-yielding demarcation membrane system (36), suggesting it may be also involved in platelet release.
Many cytokines regulate aspects of the complex megakaryocyte development process, including IL-3, -6, -11, SCF, LIF, and erythropoietin (1). However, Tpo is the major physiological regulator of megakaryocytopoiesis and thrombopoiesis (1). Tpo alone can promote normal megakaryocyte development and platelet formation in vitro from CD34+ stem cells (37). Thus, our studies have focused on the modulation of Tpo-mediated hematopoiesis by overexpression or loss of Lnk. Overexpression of Lnk in primary BM cells cultured in the presence of Tpo leads to reduced megakaryocyte numbers, sizes, and ploidy. Using in vitro BM and spleen cultures, we demonstrated that loss of Lnk renders megakaryocytes hypersensitive to Tpo, reflected in augmented proliferation and polyploidization. Consistent with this finding, lack of Lnk causes elevated megakaryocyte numbers and ploidy both in the BM and spleen. Therefore, Lnk attenuates Tpo-induced cell proliferation and endomitosis.
We found that overexpression of Lnk in primary BM progenitor cells inhibits megakaryocyte proliferation, resulting in smaller numbers of megakaryocytes. Lnk also inhibits endomitosis, thereby decreasing the extent of polyploidization. Consistent with this data, Lnk overexpression attenuates sustained activation of MAPK induced by Tpo in 32D cells, and Lnk-deficient megakaryocytes exhibit enhanced Tpo-induced MAPK activation. Further experiments are required to address whether Lnk inhibition of Tpo-induced mitosis and endomitosis is mediated through MAPK and/or cyclin D3.
Together, our data show that Lnk is a physiological negative regulator for Tpo-mediated megakaryocytopoiesis. Although overexpression of Lnk reduces the number and ploidy of megakaryocytes, their morphology appears grossly normal, and the fact that Lnk-deficient mice have functional platelets indicates that Lnk may not qualitatively affect platelet formation. Because Tpo is also not essential for megakaryocyte cytoplasmic maturation or platelet formation, this result suggests that Lnk acts in megakaryocytes primarily by restricting mpl signaling. Interestingly, Lnk heterozygous (Lnk+/−) mice display a phenotype intermediate between that of Lnk homozygous (Lnk−/−) mice and WT mice (reference 21 and unpublished data), suggesting that Lnk protein level is tightly regulated and critical for its function in regulating cytokine receptor signaling.
Acknowledgments
We thank Dr. T. Pawson for Lnk-deficient mice and Dr. S. Takaki for Lnk cDNA constructs. We thank G. Paradis for help with flow cytometry analysis and sorting, N. Watson for assistance in microscopy, and F. Reinhardt for animal maintenance.
H.F. Lodish is supported by National Institutes of Health grant HL32262 and National Science Foundation grant EEC-9843342/67983 from the Biotechnology Process Engineering Center at Massachusetts Institute of Technology. W. Tong is supported by a postdoctoral fellowship from the Damon Runyon Cancer Research Foundation and a Cancer Research Institute fellowship.
The authors have no conflicting financial interests.
Abbreviations used in this paper: AGM, aorta-gonad-mesonephros; IRES, internal ribosomal entry site; Lin−, lineage negative; MAPK, mitogen-activated protein kinase; MIG, MSCV-IRES-GFP; PH, pleckstrin homology; PI, propidium iodide; SCF, stem cell factor; SH2, Src homology 2; Tpo, thrombopoietin.
References
- 1.Kaushansky, K. 1995. Thrombopoietin: the primary regulator of platelet production. Blood. 86:419–431. [PubMed] [Google Scholar]
- 2.Kaushansky, K. 2003. Thrombopoietin: a tool for understanding thrombopoiesis. J. Thromb. Haemost. 1:1587–1592. [DOI] [PubMed] [Google Scholar]
- 3.Gurney, A.L., K. Carver-Moore, F.J. de Sauvage, and M.W. Moore. 1994. Thrombocytopenia in c-mpl-deficient mice. Science. 265:1445–1447. [DOI] [PubMed] [Google Scholar]
- 4.de Sauvage, F.J., K. Carver-Moore, S.M. Luoh, A. Ryan, M. Dowd, D.L. Eaton, and M.W. Moore. 1996. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J. Exp. Med. 183:651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander, W.S., A.W. Roberts, N.A. Nicola, R. Li, and D. Metcalf. 1996. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood. 87:2162–2170. [PubMed] [Google Scholar]
- 6.Bunting, S., R. Widmer, T. Lipari, L. Rangell, H. Steinmetz, K. Carver-Moore, M.W. Moore, G.A. Keller, and F.J. de Sauvage. 1997. Normal platelets and megakaryocytes are produced in vivo in the absence of thrombopoietin. Blood. 90:3423–3429. [PubMed] [Google Scholar]
- 7.Alexander, W.S. 1999. Thrombopoietin and the c-Mpl receptor: insights from gene targeting. Int. J. Biochem. Cell Biol. 31:1027–1035. [DOI] [PubMed] [Google Scholar]
- 8.Watowich, S.S., H. Wu, M. Socolovsky, U. Klingmuller, S.N. Constantinescu, and H.F. Lodish. 1996. Cytokine receptor signal transduction and the control of hematopoietic cell development. Annu. Rev. Cell Dev. Biol. 12:91–128. [DOI] [PubMed] [Google Scholar]
- 9.Drachman, J.G., and K. Kaushansky. 1995. Structure and function of the cytokine receptor superfamily. Curr. Opin. Hematol. 2:22–28. [DOI] [PubMed] [Google Scholar]
- 10.Parganas, E., D. Wang, D. Stravopodis, D.J. Topham, J.C. Marine, S. Teglund, E.F. Vanin, S. Bodner, O.R. Colamonici, J.M. van Deursen, et al. 1998. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 93:385–395. [DOI] [PubMed] [Google Scholar]
- 11.Snow, J.W., N. Abraham, M.C. Ma, N.W. Abbey, B. Herndier, and M.A. Goldsmith. 2002. STAT5 promotes multilineage hematolymphoid development in vivo through effects on early hematopoietic progenitor cells. Blood. 99:95–101. [DOI] [PubMed] [Google Scholar]
- 12.Bradley, H.L., T.S. Hawley, and K.D. Bunting. 2002. Cell intrinsic defects in cytokine responsiveness of STAT5-deficient hematopoietic stem cells. Blood. 100:3983–3989. [DOI] [PubMed] [Google Scholar]
- 13.Kieslinger, M., I. Woldman, R. Moriggl, J. Hofmann, J.C. Marine, J.N. Ihle, H. Beug, and T. Decker. 2000. Antiapoptotic activity of Stat5 required during terminal stages of myeloid differentiation. Genes Dev. 14:232–244. [PMC free article] [PubMed] [Google Scholar]
- 14.Kirito, K., M. Osawa, H. Morita, R. Shimizu, M. Yamamoto, A. Oda, H. Fujita, M. Tanaka, K. Nakajima, Y. Miura, et al. 2002. A functional role of Stat3 in in vivo megakaryopoiesis. Blood. 99:3220–3227. [DOI] [PubMed] [Google Scholar]
- 15.Geddis, A.E., N.E. Fox, and K. Kaushansky. 2001. Phosphatidylinositol 3-kinase is necessary but not sufficient for thrombopoietin-induced proliferation in engineered Mpl-bearing cell lines as well as in primary megakaryocytic progenitors. J. Biol. Chem. 276:34473–34479. [DOI] [PubMed] [Google Scholar]
- 16.Rojnuckarin, P., J.G. Drachman, and K. Kaushansky. 1999. Thrombopoietin-induced activation of the mitogen-activated protein kinase (MAPK) pathway in normal megakaryocytes: role in endomitosis. Blood. 94:1273–1282. [PubMed] [Google Scholar]
- 17.Rudd, C.E. 2001. Lnk adaptor: novel negative regulator of B cell lymphopoiesis. Sci. STKE. 2001:PE1. [DOI] [PubMed]
- 18.Takaki, S., K. Sauer, B.M. Iritani, S. Chien, Y. Ebihara, K. Tsuji, K. Takatsu, and R.M. Perlmutter. 2000. Control of B cell production by the adaptor protein lnk. Definition of a conserved family of signal-modulating proteins. Immunity. 13:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takaki, S., H. Morita, Y. Tezuka, and K. Takatsu. 2002. Enhanced hematopoiesis by hematopoietic progenitor cells lacking intracellular adaptor protein, Lnk. J. Exp. Med. 195:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nobuhisa, I., M. Takizawa, S. Takaki, H. Inoue, K. Okita, M. Ueno, K. Takatsu, and T. Taga. 2003. Regulation of hematopoietic development in the aorta-gonad-mesonephros region mediated by Lnk adaptor protein. Mol. Cell. Biol. 23:8486–8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velazquez, L., A.M. Cheng, H.E. Fleming, C. Furlonger, S. Vesely, A. Bernstein, C.J. Paige, and T. Pawson. 2002. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J. Exp. Med. 195:1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naviaux, R.K., E. Costanzi, M. Haas, and I.M. Verma. 1996. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70:5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson, C.W., L.K. Brown, B.C. Somerville, S.A. Lyles, and A.T. Look. 1984. Two-color flow cytometric measurement of DNA distributions of rat megakaryocytes in unfixed, unfractionated marrow cell suspensions. Blood. 63:768–778. [PubMed] [Google Scholar]
- 24.Arriaga, M., K. South, J.L. Cohen, and E.M. Mazur. 1987. Interrelationship between mitosis and endomitosis in cultures of human megakaryocyte progenitor cells. Blood. 69:486–492. [PubMed] [Google Scholar]
- 25.Liu, X., S.N. Constantinescu, Y. Sun, J.S. Bogan, D. Hirsch, R.A. Weinberg, and H.F. Lodish. 2000. Generation of mammalian cells stably expressing multiple genes at predetermined levels. Anal. Biochem. 280:20–28. [DOI] [PubMed] [Google Scholar]
- 26.Ravid, K., J. Lu, J.M. Zimmet, and M.R. Jones. 2002. Roads to polyploidy: the megakaryocyte example. J. Cell. Physiol. 190:7–20. [DOI] [PubMed] [Google Scholar]
- 27.Klingmuller, U., U. Lorenz, L.C. Cantley, B.G. Neel, and H.F. Lodish. 1995. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 80:729–738. [DOI] [PubMed] [Google Scholar]
- 28.Krystal, G., J.E. Damen, C.D. Helgason, M. Huber, M.R. Hughes, J. Kalesnikoff, V. Lam, P. Rosten, M.D. Ware, S. Yew, and R.K. Humphries. 1999. SHIPs ahoy. Int. J. Biochem. Cell Biol. 31:1007–1010. [DOI] [PubMed] [Google Scholar]
- 29.Inagaki-Ohara, K., T. Hanada, and A. Yoshimura. 2003. Negative regulation of cytokine signaling and inflammatory diseases. Curr. Opin. Pharmacol. 3:435–442. [DOI] [PubMed] [Google Scholar]
- 30.Krebs, D.L., and D.J. Hilton. 2000. SOCS: physiological suppressors of cytokine signaling. J. Cell Sci. 113:2813–2819. [DOI] [PubMed] [Google Scholar]
- 31.Takaki, S., Y. Tezuka, K. Sauer, C. Kubo, S.M. Kwon, E. Armstead, K. Nakao, M. Katsuki, R.M. Perlmutter, and K. Takatsu. 2003. Impaired lymphopoiesis and altered B cell subpopulations in mice overexpressing Lnk adaptor protein. J. Immunol. 170:703–710. [DOI] [PubMed] [Google Scholar]
- 32.Shivdasani, R.A. 2001. Molecular and transcriptional regulation of megakaryocyte differentiation. Stem Cells. 19:397–407. [DOI] [PubMed] [Google Scholar]
- 33.Thompson, A., Y. Zhang, D. Kamen, C.W. Jackson, R.D. Cardiff, and K. Ravid. 1996. Deregulated expression of c-myc in megakaryocytes of transgenic mice increases megakaryopoiesis and decreases polyploidization. J. Biol. Chem. 271:22976–22982. [DOI] [PubMed] [Google Scholar]
- 34.Zimmet, J.M., D. Ladd, C.W. Jackson, P.E. Stenberg, and K. Ravid. 1997. A role for cyclin D3 in the endomitotic cell cycle. Mol. Cell. Biol. 17:7248–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang, Z., Y. Zhang, D. Kamen, E. Lees, and K. Ravid. 1995. Cyclin D3 is essential for megakaryocytopoiesis. Blood. 86:3783–3788. [PubMed] [Google Scholar]
- 36.Sun, S., C.W. Jackson, and K. Ravid. 2000. MAP kinase localizes to the platelet-yielding demarcation membrane system in megakaryocytes. Blood. 95:1511. [PubMed] [Google Scholar]
- 37.Guerriero, R., U. Testa, M. Gabbianelli, G. Mattia, E. Montesoro, G. Macioce, A. Pace, B. Ziegler, H.J. Hassan, and C. Peschle. 1995. Unilineage megakaryocytic proliferation and differentiation of purified hematopoietic progenitors in serum-free liquid culture. Blood. 86:3725–3736. [PubMed] [Google Scholar]