

Abstract
Periodic fever syndromes (PFSs) comprise a subset of the hereditary autoinflammatory disorders that are defined by recurrent self-resolving attacks of systemic inflammatory reactions in the absence of infection or autoimmunity. Recent advances have led to the discovery that members of a new family of genes, the PYRIN family, account for several hereditary PFSs. Here we discuss new insights into the function of PYRIN proteins and the molecular basis of PFSs.
Periodic Fever Syndromes.
The genetic cause of six hereditary PFSs has been identified: Familial Mediterranean Fever (FMF), TNF receptor–associated periodic syndrome (TRAPS), hyperimmunoglobulinemia D and PFS (HIDS), familial cold autoinflammatory syndrome (FCAS)/familial cold urticaria (FCU), Muckle-Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease (NOMID)/chronic infantile neurologic cutaneous and articular syndrome (CINCA). More than 70 yr ago, the first autosomal recessive hereditary PFS, now known FMF, was defined. FMF is characterized by episodic fever spikes lasting for days, followed by attack-free periods as long as several years. The gene responsible for FMF was reported in 1997 and denoted MEFV, and the protein was named Pyrin (Marenostrin) (1, 2). The Pyrin protein consists of a conserved NH2-terminal PYRIN domain, followed by a COOH-terminal B-box zinc finger, a PRY/SPRY domain, and two overlapping nuclear localization signals (Fig. 1). Mutations in the gene encoding proline serine threonine phosphatase–interacting protein (PSTPIP1), a Pyrin-interacting protein, are associated with the dominantly inherited autoinflammatory syndrome of pyogenic arthritis, pyodermagangrenosum, and acne (PAPA). These mutations accentuate the interaction between Pyrin and PSTP1 (3). Pyrin and PSTPIP1 proteins associate with the cytoskeleton (4), but the significance of this observation is unclear.
Figure 1.

PYRIN domain and related proteins. PYRIN domain and some related proteins are presented. Domains shown include: CARD; hematopoietic interferon-inducible nuclear proteins with a 200-amino-acid repeat (HIN200/IFI20); NAIP, CIITA, HET-E, and TP1 (NACHT); LRR; baculovirus inhibitor of apoptosis repeat (BIR); Trp-Asp 40 (WD40); activation domain (AD); NACHT-associated domain (NAD), TIR; coiled-coil (CC); cysteine-rich domain (CYS); transmembrane domain (TM); zinc finger B-box (zf-B-box); Dictyostelium discoideum dual specificity kinase SplA and ryanodyne receptor (SPRY); SPRY associated (PRY); domain with function to find (FIIND).
HIDS or Dutch-type periodic fever is another autosomal recessive PFS characterized by fever attacks of up to a week, accompanied by constitutively elevated levels of serum IgD. In 1999, a candidate gene for HIDS was identified, MVK, encoding mevalonate kinase (5, 6). Indeed, patients with HIDS show increased concentrations of mevalonic acid during fever episodes, correlating with reduced mevalonate kinase activity, suggesting that HIDS represents a metabolic defect in the isoprenoid biosynthesis, presumably resulting not only in periodic production of abnormal levels of pyrogenic isoprenoids but also in increased IL-1β secretion.
The first defined PFS with an autosomal dominant mode of inheritance was TRAPS, initially referred to as Familial Hibernian Fever, which is caused by mutations in the gene encoding TNFR1 (7). Given the prominent role of TNF receptors in inflammatory responses, it is not surprising that polymorphisms in this gene are linked to a PFS.
In 1999, two PFSs, namely FCAS (or FCU) and MWS, were linked to the gene CIAS1 (8, 9). The encoded protein, Cryopyrin, is a member of the PYRIN and NACHT domain–containing (PAN) family of proteins, which has at least 15 human members (Table I). These proteins contain a characteristic combination of three domains, including a PYRIN domain, a specific nucleotide-binding fold (NACHT domain), and several tandem copies of leucine-rich repeats (LRRs) similar to the extracellular domain of Toll-like receptors (TLRs) (Fig. 1). Mutations in CIAS1 are found mainly within or in the vicinity of the NACHT domain, which is thought to mediate oligomerization of this protein. Mutations in CIAS1 are also associated with another autoinflammatory disorder, CINCA (NOMID), a disorder where short recurrent fever episodes are common, though severe arthropathic, neurologic, and dermatologic symptoms predominate (10, 11). Among the 20 human genes that encode a NACHT domain, hereditary mutations associated with inflammatory diseases have also been identified in NOD2, which encodes a protein with a similar architecture to Cryopyrin except that it contains a pair of caspase recruitment domains (CARDs) instead of the PYRIN domain. Mutations in NOD2 have been linked to kinships with familial Crohn's disease, a form of inflammatory bowel disease, and Blau syndrome, which causes granulomatous arthritis, uveitis, and skin rash (12), with mutations tending to reside in either the LRR or NACHT domain, respectively, resulting putatively in either loss of function (familial Crohn's disease) or gain of function (Blau syndrome).
Table I.
PAN Family Proteins
| PAN | Alternative name |
Chromosome | Expressiona | Inducer | Domainsb | LRRs | Length (aa)/kD |
|---|---|---|---|---|---|---|---|
| Cryopyrin | PYPAF1 NALP3 |
1q44 | C, G, T, M | TNF↑, PAMPs↑ |
P-N-A-L | 9 | 1,034/118 |
| PAN1 | PYPAF2 NALP2 NBS1 |
19q13.4 | Ubiquitous expressed; high in tumors |
Cell cycle regulated |
P-N-A-L | 8 | 1,062/121 |
| PAN2 | PYPAF4 NALP4 |
19q13.42 | B, M, F, pl, lg, li, ki, pa, sp, th |
P-N-A-L | 8 | 994/113 | |
| PAN3 | PYPAF5 NALP6 |
11p15 | T, E, G | P-N-A-L | 5 | 892/99 | |
| PAN4 | NALP8 | 19q13.4 | P-N-A-L | 7 | 1,029/117 | ||
| PAN5 | NALP10 Pynod |
11p15 | P-N-A-L | 0 | 655/75 | ||
| PAN6 | PYPAF7 NALP12 Monarch-1 |
19q13.4 | L, DC, M, E, G | NO↑ TNF↓, IFNγ↓ |
P-N-A-L | 11 | 1,061/120 |
| PAN7 | PYPAF3 NALP7 NOD12 |
11q13.42 | P-N-A-L | 9 | 980/112 | ||
| PAN8 | NALP14 | 11p15.4 | P-N-A-L | 12 | 1,093/125 | ||
| PAN9 | 19q13.4 | P-N-A-L | 9 | 1,014/115 | |||
| PAN10 | PYPAF6 NALP11 NOD17 |
19q13.42 | B | P-N-A-L | 9 | 1,033/118 | |
| PAN11 | PYPAF8 NALP5 mater |
11q13.42 | Oocyte specific | P-N-A-L | 13 | 1,200/134 | |
| PAN12 | NALP9 | 19q13.4 | P-N-A-L | 10 | 986/113 | ||
| PAN13 | NALP13 | 19q13.4 | P-N-A-L | 9 | 1,043/119 | ||
| PAN14 | NOD27 | 16q13 | M, breast cancer↑, leukemia↓, renal tumors↓ |
LPS↓ | P-N-A-L | 20 | 1,866/205 |
| NAC | DEFCAP CARD7 NALP1 |
17p12.13 | M, B | LPS↓, Ionomycin↓ PMA↓ |
P-N-L-A-F-C | 5 | 1,429/161 |
B, B cells; C, chondrocytes; E, eosinophils; G, granulocytes; M, monocytes; T, T cells; ki, kidney; li, liver; lg, lung; pa, pancreas; pl, placenta; sp, spleen; th, thymus; ↑, induction; ↓, inhibition.
P, PYRIN domain; N, NACHT; A, angiotensin receptor–related (or NAD); L, LRR; C, CARD; F, FIIND; ↑, induction; ↓, inhibition.
The PYRIN Domain: A Link To Inflammatory Pathways.
With the exception of HIDS, all characterized PFS involve proteins containing the death domain fold (DDF), consisting of a characteristic bundle of five to six α-helices. Four subfamilies have been identified, including the death domain, death effector domain, CARD, and PYRIN domain, which mediate homotypic protein–protein interactions. To date, identified downstream effectors of DDFs include caspases, a family of cysteine proteases, and proteins linked to activation of Jun NH2-terminal kinase and kinases that activate NF-κB family members.
The PYRIN domain is the most recently recognized DDF and also the most recently evolved and found only among vertebrates. The three-dimensional structure of PYRIN domains from apoptosis-associated speck-like protein containing a CARD (ASC), a small adaptor protein that contains both a PYRIN and a CARD domain, and NALP1 have been solved recently by NMR, revealing the characteristic DDF fold. The role (if any) of post-translational modifications in regulating PYRIN–PYRIN interactions is unclear, though TNF stimulation can induce phosphorylation of at least two PYRIN family proteins (13).
The PYRIN domain can be found in association with numerous other types of domains (Fig. 1) that may either serve as sensors of changes in cellular environment or effectors that trigger responses. PYRIN domains can bind indirectly to at least two proteins important in inflammation: pro–caspase-1 and the IκB kinase (IKK) complex, resulting in either activation or inhibition of these enzymes.
Caspase-1 is primarily involved in proteolytic processing and activation of proinflammatory cytokines, pro-1β and pro–IL-18, though also capable of participating in apoptosis in certain pathological contexts. Some PYRIN proteins connect to pro–caspase-1 via ASC (Fig. 1) (14). The CARD of ASC can bind pro–caspase-1, thus acting as a molecular bridge to other PYRIN proteins mediated via their PYRIN domains (15). ASC represents only one of two genes in the human genome combining CARD and PYRIN domains, the other being NALP1 (Fig. 1). NALP1 uses its CARD to bind pro–caspase-5 and the PYRIN domain to link via ASC to pro–caspase-1, resulting in a protein complex that controls activation of caspases-1 and -5, and which has been dubbed the “inflammasome” (16). Cryopyrin and NALP2 also form inflammasomes, but distinct from NALP1 (17), these PANs associate with ASC, cardinal, and pro–caspase-1 but not pro–caspase-5. The central role of ASC in host defense responses linked to caspase-1 activation was revealed recently by gene ablation studies in mice, showing a severe defect in caspase-1 activation in macrophages from Asc −/− mice in response to TLR activation and infection with intracellular pathogens and increased resistance to endotoxic shock of these mice (18). In contrast to wild-type Cryopyrin, hereditary mutants spontaneously associate with ASC and trigger caspase-dependent processing of pro–IL-1β (19). Macrophages from MWS patients display spontaneous activation of caspase-1 and secretion of active IL-1β. Similarly, patients suffering from NOMID have elevated serum cytokines (17). Peripheral blood lymphocytes isolated from PAPA patients, which produce mutant forms of the Pyrin-interacting protein PSTPIP1, also secrete elevated levels of IL-1β, correlating with increased PSTPIP1-Pyrin association in these cells (3). Also FMF-associated Pyrin mutations induce an increase in bioactive IL-1β when transfected into monocytes (20). Together, these results suggest that hereditary alterations in these PYRIN family proteins or their interacting partners represent gain of function mutations, where the proteins constitutively signal. However, macrophages from Pyrin −/− mice exhibit heightened sensitivity to LPS and produce more (not less) active caspase-1 and IL-1β (20). Thus, one cannot eliminate the possibility that wild-type versions of some of these proteins normally suppress IL-1β production, with the hereditary mutations representing loss of function mutations.
Some PYRIN proteins have been shown to associate with the IKK complex, affecting activation of NF-κB (13, 21, 22), a family of transcription factors that directly controls expression of a variety of genes involved in inflammation and host defense. How PYRIN domains bind IKK complex proteins is unclear, though one route may again involve ASC. The CARD of ASC can bind Cardiak (Rick, Rip2), which in turn binds IKKγ (15). Alternatively, ASC conceivably could connect to IKKγ via Cardinal, which contains a CARD that can bind ASC (unpublished data) and also reportedly directly binds IKKγ (23). However, other routes to IKK complex proteins probably also exist, as suggested for the PAN2 protein (22). Both Cryopyrin and Pyrin utilize ASC to link to NF-κB activation pathways. However, at least in overexpression situations, Pyrin has also been reported to interfere with the interaction of ASC and Cryopyrin (13, 21, 24). Moreover, hereditary mutations found in Cryopyrin also show enhanced NF-κB activation in collaboration with ASC compared with wild-type Cryopyrin, whereas Cryopyrin expressed in the absence of ASC blocks activation of NF-κB (19, 25). Thus, complex networks of interactions among PYRIN domain–containing proteins may set thresholds for NF-κB induction, assuring that inflammation is induced only if the proper stoichiometry ratios of components are present in protein complexes containing PYRIN family proteins.
Although PAN family proteins are capable of regulating NF-κB activity, the physiological importance of this function of PYRIN domain–containing proteins is unknown. In this regard, recent clinical experience with use of recombinant IL-1 receptor antagonists has demonstrated marked benefits in patients harboring Cryopyrin mutations (26), attesting to the importance of excessive IL-1 production in this PAN family protein disease. It would be interesting to compare this result with drugs such as IKK inhibitors or proteasome inhibitors that suppress NF-κB induction.
A Network of PYRIN Domain Interactions.
Another important goal of future research is to delineate which of the ∼21 PYRIN domain–containing proteins encoded in the human genome interact with each other and what the functional consequences of those interactions are. One insightful example of regulatory interactions among PYRIN domain–containing proteins is found in POP1, a recently identified small protein that consists only of a PYRIN domain (Fig. 1). POP1 binds ASC and interferes with its ability to collaborate with Pyrin or Cryopyrin in NF-κB activation (13). However, in contrast to its dominant-negative effect on ASC-mediated NF-κB induction POP1 enhances ASC-mediated caspase-1 activation, at least when overexpressed. By binding the PYRIN domain of ASC, POP1 may induce multimerization of ASC, leaving its CARD free for interaction with pro–caspase-1. Multiple other examples of regulatory interactions among PYRIN domain–containing proteins are likely to emerge as research in this field progresses. These interactions certainly could involve PYRIN–PYRIN interactions, but other domains associated with these proteins could also mediate their interactions, especially NACHT domains, which are known for their propensity to oligomerize. Interestingly, recent evidence suggests that NACHT domains from different PAN family proteins can associate, implying complex networks of protein heterooligomerization mediated by NACHT–NACHT interactions (27). Interestingly, the human genome is predicted to encode proteins that combine a NACHT and LRRs but lacking the NH2-terminal effector domain region. Such proteins conceivably could act as dominant-negative regulators of PANs, relying on NACHT–NACHT interactions to thwart assembly of functional signaling complexes.
A Role of PYRIN Proteins in Apoptosis?
Several PYRIN proteins have been implicated in apoptosis, though the mechanisms remain poorly defined. Overexpression of ASC for example triggers apoptosis of some types of cells (28), whereas antisense-mediated suppression of ASC expression reportedly reduces cell death in myeloid leukemia cells (14). Coexpressing Pyrin with ASC can overcome its proapoptotic action (29), suggesting that PYRIN–PYRIN domain interactions are important determinants of whether ASC modulates apoptosis. One likely mechanism for ASC-induced apoptosis is caspase-1 activation, inasmuch as its excessive activation can trigger activation of apoptotic caspases, and caspase-1–dependent apoptosis was impaired in macrophages from Asc −/− mice (18). In this regard, both the apical protease in the mitochondrial pathway, caspase-9, and caspase-8, the pinnacle protease in the death receptor pathway, have been implicated in ASC-induced apoptosis (28, 30). Also, macrophages from Pyrin −/− mice display impaired caspase-8 activity along with reduced susceptibility to apoptosis (20). Recently, ASC was suggested to bind and activate the proapoptotic Bcl-2 family member Bax, invoking a mitochondrial apoptosis pathway involving cytochrome c release. This ASC/Bax mechanism for cell death was implicated in a DNA damage response pathway, where ASC expression is induced by activated p53 (31). NALP1 involvement in the mitochondrial pathway for apoptosis has also been reported (32). Similarly, TLRs, well known for their role in innate immunity, are capable of triggering caspase-dependent apoptosis in some contexts (33). Further work is required to ascertain the circumstances where PYRIN domain–containing proteins participate in apoptosis regulation and the repertoire of apoptosis-relevant proteins with which they interact. Like CARDs and DDs, the PYRIN domains may operate at cross-roads of inflammation and apoptosis.
PANs As Intracellular Pathogen-sensing Proteins.
Most PAN family proteins, with the exception of PAN5 (Pynod), contain LRRs similar to the extracellular LRR domains of the TLRs (Fig. 1). The LRR domains of TLRs bind a variety of microbe-derived molecules and transduce signals into cells via their intracellular Toll/IL-1 receptor (TIR) domains (Fig. 1). Similarly, the genomes of plants, such as Arabidopsis, contain as many as hundreds of resistance (R) genes that encode host defense proteins combining pathogen-sensing LRRs with a NB-ARC domain, (a NACHT-like fold) and various effector domains comprised of TIR-, coiled-coil–, or leucine zipper–like domains (Fig. 1). One of the downstream responses elicited via these R gene products is the hypersensitive response, where altruistic cell death is induced to limit pathogen spread. R-gene products are also implicated in regulation of a multiprotein complex that may be likened to the IKK complex and the SCF ubiquitin–ligase complex that targets IκBα for destruction. Their domain arrangement is reminiscent of PANs. Thus, PAN family proteins represent excellent candidates for the long-sought sensors of intracellular pathogens.
In plants, the LRRs have been shown to suppress activation of the R-gene protein, operating as autoinhibitory domains with inhibition relieved upon binding pathogen-derived molecules (34). Similarly deleting the LRRs from mammalian PAN family proteins seems to boost their ability to activate caspase-1 or NF-κB. Thus, the LRRs may act as a switch, converting PANs from inactive to active conformations. Thus, one mechanism underlying periodic fever syndromes might be uncontrolled formation of proinflammatory signaling complexes, independently of sensing pathogens. However, data complicating this simple interpretation have come from the observation that Pynod (PAN5/NALP10), which is the only PAN family member lacking LRRs, blocks Cryopyrin and Ipaf (CLAN)–mediated activation of caspase-1 and NF-κB (35).
Though no definitive evidence exists to date that the LRRs of PANs bind microbial products, LRRs of the closely related proteins, Nod1 and Nod2, have recently been reported to bind bacterial muropeptides, leading to NF-κB induction (36). Nod1, Nod2, and Ipaf are highly similar to PANs, except that they contain CARDs instead of PYRIN domains at their NH2 termini (Fig. 1). They are also implicated in pro–caspase-1 and NF-κB activation, undergoing oligomerization in response to invading gram-negative bacteria (36). Consistent with this idea, macrophages from ipaf −/− mice are defective in caspase-1 activation in response to intracellular pathogens but not TLR agonists (18). NAIP and CIITA, which are structurally similar to PAN proteins, containing NACHT and LRR domains but lacking DDFs, are also implicated in host defense: they have been shown to regulate host susceptibility to the intracellular pathogen Listeria and class I histocompatibility antigen expression, respectively (Fig. 1) (36).
Future research, therefore, should seek to define the exogenous or endogenous ligands for the LRRs of PAN family proteins. Given that precedence exists for the LRRs of TLRs binding nucleic acids (e.g., CpG DNA; poly I:C), one topic worthy of exploration concerns the proinflammatory effects observed for some antisense oligonucleotides and synthetic small interfering RNAs. Knowledge of the sequence requirements for interactions of oligonucleotides with LRRs of PANs might allow design of more side effect–free reagents. Interestingly, one of the PYRIN family proteins, IFI16, has been implicated recently in DNA damage and repair signaling (37), suggesting additional links between PYRIN domain–containing proteins and sensing of altered nucleic acids.
Diverse Intracellular Locations of PYRIN Proteins.
The location of a protein often says a lot about its function. PYRIN proteins exhibit some interesting patterns of subcellular location. Pyrin was originally predicted to function as a RoRet family transcription factor. However, only one alternative spliced isoform of Pyrin localizes to the nucleus, whereas the full-length protein associates with tubulin and actin, leading to speculation that Pyrin might regulate cytoskeletal organization in leukocytes (38). Interestingly, hereditary Pyrin mutants do not show altered cellular distribution and still colocalize with ASC (39). In contrast, Cryopyrin displays granular cytoplasmic localization (17). Proteins of another branch of the PYRIN family, the HIN200 proteins, such as human IFI16, AIM2, and MNDA (Fig. 1), localize exclusively to the nucleus and associate with various transcription factors via their HIN200 domains, suggesting a role for some PYRIN proteins as modulators of gene expression (40). IFI16 was identified recently as a BRCA1-associated protein enhancing p53-mediated apoptosis in response to DNA damage (37). The PAN-related protein, CIITA, also operates in the nucleus to control expression of MHC class I genes (41).
The adaptor protein ASC derives its name, “apoptotic-associated speck-like protein containing a caspase recruitment domain,” in part from its tendency to localize to cytosolic specks when overexpressed or transcriptionally induced (14). In gene transfection experiments, several ASC-binding PYRIN proteins, including Pyrin, Cryopyrin, POP1, PAN6, PAN3, and zCaspy are recruited into these specks via PYRIN–PYRIN interactions. Conceivably therefore, ASC and certain ASC-binding PYRIN proteins, including Pyrin, might participate in cytosolic macromolecular complexes (signalosomes or inflammasomes) that serve as scaffolds for achieving activation of caspases, NF-κB–activating kinases, and perhaps other proteins involved in host defense, inflammation, or apoptosis.
Regulation of PYRIN Family Gene Expression.
Although expression of some PYRIN domain–containing proteins is ubiquitous (PAN2, ASC), others are produced predominantly in cells directly involved in host defense, such as macrophages, neutrophils, lymphocytes, and cells that play pathogen barrier functions, such as intestinal epithelial cells (Table I). The stimuli that modulate expression of PYRIN proteins are only just beginning to be defined but include inflammatory cytokines and LPS. Expression of some PYRIN domain–containing proteins is controlled by interferons, including the HIN200 family members, Pyrin, and ASC. Inflammatory mediators such as LPS and TNFα up-regulate expression of Cryopyrin, and its expression is also increased at sites of inflammation (25). Likewise, Pyrin is induced as an immediate-early gene by proinflammatory molecules (e.g., TNFα, LPS, and IFNs) but inducibility is inhibited by antiinflammatory cytokines (e.g., IL-4, IL-10, and TGFβ) (42). However, in mice the strongest expression of Pyrin apparently occurs in response to IL-4 and IL-10 (20). Moreover, evidence of diseases-associated alterations in PYRIN family gene expression has begun to emerge. For example, ASC gene silencing in association with DNA hypermethylation has been observed in breast cancers, melanomas, and lung cancers (43). ASC silencing might be advantageous for cancers due to its ability to: (a) induce apoptosis in the p53/Bax pathway after DNA damage and (b) modulate activation of NF-κB family transcription factors that control expression of proteins involved in tumor recognition by cytolytic T cells (44). Regardless of the explanation, studies of PYRIN protein expression in cancer are likely to provide new insights into tumor biology, particularly given the close association recognized between inflammation and malignancy.
Concluding Remarks.
Recent discovery of a large family of genes encoding PYRIN domain proteins in mammalian genomes opens a new window into innate immunity, inflammatory diseases, and host defense. At present, our understanding is relatively poor of the physiological roles, molecular mechanisms, and modes of regulation of networks of PYRIN proteins. Only two PYRIN family genes, Pyrin and Asc, have been knocked out in mice thus far (18, 20), and thus there is a need for more such experiments aimed at uncovering the fundamental roles of these genes in host defense and inflammation in vivo. However, because of the divergent expansion and genetic drift of these genes in mice relative to humans, mouse models of relevance to human biology may be limited. Analysis of serum and inflammatory cells derived from PFS patients suggests that increased production of IL-1β represents a common underlying mechanism (Fig. 2), the importance of which is documented by the encouraging clinical responses of MWS and PAPA patients to treatment with recombinant IL-1 receptor antagonist (Anakinra) (3, 26). Thus, inappropriate production of IL-1 (also known as endogenous pyrogen) appears to be tightly linked to PFS, playing a causative role in the symptoms associated with these disorders. The full nuances of the disturbances in regulatory networks of PYRIN–PYRIN and NACHT–NACHT interactions caused by hereditary mutations in FPS proteins, such as Cryopyrin, remain to be elucidated.
Figure 2.
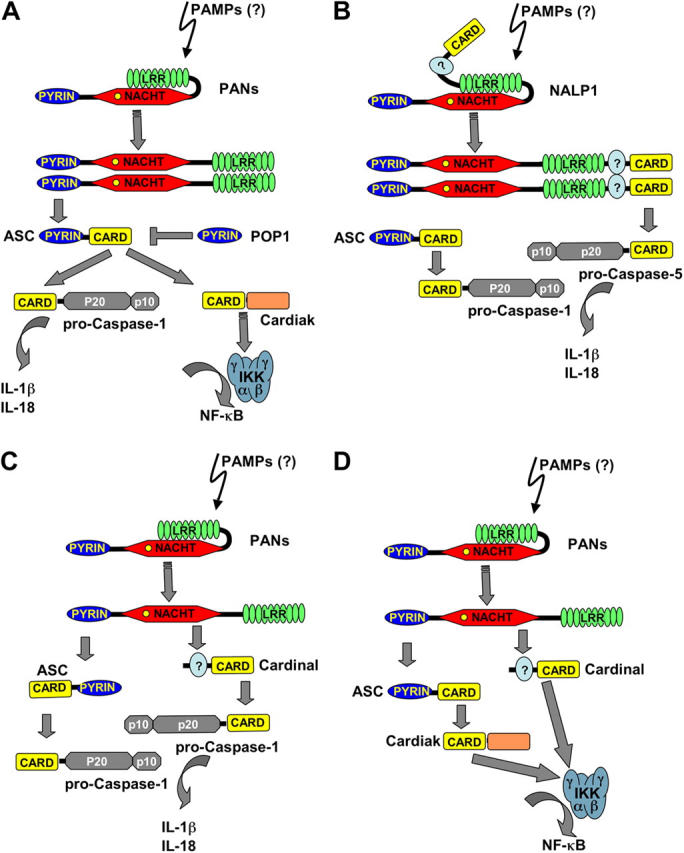
Potential molecular mechanisms underlying PFSs. (A) PAN family proteins are thought to sense pathogens via their LRR domains, triggering NACHT domain–mediated oligomerization, resulting in association with the adaptor protein ASC and activation of downstream effectors, including caspase-1 and IKK complex by the induced proximity mechanism. The ASC-associated Ser/Thr kinase Cardiak might potentially provide the link to IKKγ. Through PYRIN–PYRIN interactions, family members such as POP1 may prevent spontaneous activation of these downstream effectors. (B) NALP1, which is a unique PAN in that it contains COOH-terminal FIIND and CARD domains, has been suggested to connect via PYRIN–PYRIN interaction to ASC, which then links to pro–caspase-1 by CARD–CARD interaction. NALP1 further utilizes its CARD to also recruit pro–caspase-5 into the inflammasome (45), resulting in activation of both caspases. (C) PANs, including Cryopyrin and NALP2 were recently demonstrated to form inflammasomes that also recruit ASC by PYRIN–PYRIN interaction, which then links to pro–caspase-1 by CARD–CARD association. In addition, the CARD containing protein Cardinal, which resembles the COOH-terminal FIIND and CARD domain structure of NALP1, associates with these PANs via FIIND–NACHT interaction and then recruits caspase-1 by CARD–CARD association, leading to activation of this caspase and secretion of bioactive IL-1β. Consistent with an inhibitory role for the LRR domain, this domain suppresses association of PANs and Cardinal, suggesting that LRR ligands might be required to induce this interaction. (D) Theoretically, PANs might also link via Cardinal to the IKK complex.
Although PYRIN proteins have been linked to certain known mediators of immune regulation, namely caspase-1 and NF-κB, additional downstream efforts may await discovery, particularly given the nuclear location of some PYRIN proteins. Also, the adaptor proteins that link PYRIN proteins to downstream effectors of host defense certainly extend beyond ASC, given that only some of the PYRIN proteins bind this bipartite adaptor. The prospect that PANs with their LRRs might provide a mechanism for sensing the presence of intracellular pathogens, thus complementing the TLR system, is another exciting possibility, but direct evidence is so far lacking that PAMPs bind and activate these PANs. Thus, a high priority for future research is elucidation of the identity of the ligands of LRRs of PANs, including determination of whether endogenous ligands may exist for these proteins, analogous to some of the TLRs that can be stimulated by heat shock proteins released from dying cells at times of cellular stress and tissue injury. Finally, the identification of genes encoding PYRIN domain proteins offers new avenues of exploration for attempts to understand the origins of other inflammatory and autoimmune diseases.
Acknowledgments
We thank J. Valois for manuscript assistance and the National Institutes of Health (AI 56324, GM 61694) and the Department of Defense (DAMD17-01-1-0171) for generous support.
References
- 1.Consortium, T.F.F. 1997. A candidate gene for familial mediterranean fever. Nat. Genet. 17:25–31. [DOI] [PubMed] [Google Scholar]
- 2.Consortium, T.I.F. 1997. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial mediterranean fever. Cell. 90:797–807. [DOI] [PubMed] [Google Scholar]
- 3.Shoham, N.G., M. Centola, E. Mansfield, K.M. Hull, G. Wood, C.A. Wise, and D.L. Kastner. 2003. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. USA. 100:13501–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wise, C.A., J.D. Gillum, C.E. Seidman, N.M. Lindor, R. Veile, S. Bashiardes, and M. Lovett. 2002. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum. Mol. Genet. 11:961–969. [DOI] [PubMed] [Google Scholar]
- 5.Drenth, J.P., L. Cuisset, G. Grateau, C. Vasseur, S.D. van de Velde-Visser, J.G. de Jong, J.S. Beckmann, J.W. van der Meer, and M. Delpech. 1999. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International hyper-IgD study group. Nat. Genet. 22:178–181. [DOI] [PubMed] [Google Scholar]
- 6.Houten, S.M., W. Kuis, M. Duran, T.J. de Koning, A. van Royen-Kerkhof, G.J. Romeijn, J. Frenkel, L. Dorland, M.M. de Barse, W.A. Huijbers, et al. 1999. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat. Genet. 22:175–177. [DOI] [PubMed] [Google Scholar]
- 7.McDermott, M.F., I. Aksentijevich, J. Galon, E.M. McDermott, B.W. Ogunkolade, M. Centola, E. Mansfield, M. Gadina, L. Karenko, T. Pettersson, et al. 1999. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 97:133–144. [DOI] [PubMed] [Google Scholar]
- 8.Cuisset, L., J.P. Drenth, J.M. Berthelot, A. Meyrier, G. Vaudour, R.A. Watts, D.G. Scott, A. Nicholls, S. Pavek, C. Vasseur, et al. 1999. Genetic linkage of the Muckle-Wells syndrome to chromosome 1q44. Am. J. Hum. Genet. 65:1054–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffman, H.M., F.A. Wright, D.H. Broide, A.A. Wanderer, and R.D. Kolodner. 2000. Identification of a locus on chromosome 1q44 for familial cold urticaria. Am. J. Hum. Genet. 66:1693–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aksentijevich, I., M. Nowak, M. Mallah, J.J. Chae, W.T. Watford, S.R. Hofmann, L. Stein, R. Russo, D. Goldsmith, P. Dent, et al. 2002. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 46:3340–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feldmann, J., A.M. Prieur, P. Quartier, P. Berquin, S. Certain, E. Cortis, D. Teillac-Hamel, A. Fischer, and G. de Saint Basile. 2002. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 71:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girardin, S.E., J.P. Hugot, and P.J. Sansonetti. 2003. Lessons from Nod2 studies: towards a link between Crohn's disease and bacterial sensing. Trends Immunol. 24:652–658. [DOI] [PubMed] [Google Scholar]
- 13.Stehlik, C., M. Krajewska, K. Welsh, S. Krajewski, A. Godzik, and J.C. Reed. 2003. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated NF-κB and pro-caspase-1 regulation. Biochem. J. 373:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masumoto, J., S. Taniguchi, K. Ayukawa, H. Sarvotham, T. Kishino, N. Niikawa, E. Hidaka, T. Katsuyama, T. Higuchi, and J. Sagara. 1999. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 274:33835–33838. [DOI] [PubMed] [Google Scholar]
- 15.Stehlik, C., S.H. Lee, A. Dorfleutner, A. Stassinopoulos, J. Sagara, and J.C. Reed. 2003. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J. Immunol. 171:6154–6163. [DOI] [PubMed] [Google Scholar]
- 16.Martinon, F., K. Burns, and J. Tschopp. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell. 10:417–426. [DOI] [PubMed] [Google Scholar]
- 17.Agostini, L., F. Martinon, K. Burns, M.F. McDermott, P.N. Hawkins, and J. Tschopp. 2004. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 20:319–325. [DOI] [PubMed] [Google Scholar]
- 18.Mariathasan, S., K. Newton, D.M. Monack, D. Vucic, D.M. French, W.P. Lee, G.M. Roose, S. Erickson, and V.M. Dixit. 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 430:213-218. [DOI] [PubMed] [Google Scholar]
- 19.Dowds, T.A., J. Masumoto, L. Zhu, N. Inohara, and G. Nunez. 2004. Cryopyrin induced IL-1 secretion in monocytic cells: Enhanced activity of disease-associated mutants and requirement for ASC. J. Biol. Chem. 279:21924–21928. [DOI] [PubMed] [Google Scholar]
- 20.Chae, J.J., H.D. Komarow, J. Cheng, G. Wood, N. Raben, P.P. Liu, and D.L. Kastner. 2003. Targeted disruption of Pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell. 11:591–604. [DOI] [PubMed] [Google Scholar]
- 21.Stehlik, C., L. Fiorentino, A. Dorfleutner, J.M. Bruey, E.M. Ariza, J. Sagara, and J.C. Reed. 2002. The PAAD/PYRIN family protein ASC is a dual regulator of a conserved step in nuclear factor kappaB activation pathways. J. Exp. Med. 196:1605–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiorentino, L., C. Stehlik, V. Oliveira, M.E. Ariza, A. Godzik, and J.C. Reed. 2002. A novel PAAD-containing protein that modulates NF-kappa B induction by cytokines tumor necrosis factor-alpha and interleukin-1beta. J. Biol. Chem. 277:35333–35340. [DOI] [PubMed] [Google Scholar]
- 23.Inohara, N., T. Koseki, J. Lin, L. del Peso, P.C. Lucas, F.F. Chen, Y. Ogura, and G. Nunez. 2000. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J. Biol. Chem. 275:27823–27831. [DOI] [PubMed] [Google Scholar]
- 24.Dowds, T.A., J. Masumoto, F.F. Chen, Y. Ogura, N. Inohara, and G. Nunez. 2003. Regulation of cryopyrin/Pypaf1 signaling by pyrin, the familial Mediterranean fever gene product. Biochem. Biophys. Res. Commun. 302:575–580. [DOI] [PubMed] [Google Scholar]
- 25.O'Connor, W., Jr., J.A. Harton, X. Zhu, M.W. Linhoff, and J.P. Ting. 2003. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/ CATERPILLER 1.1 Is an inducible inflammatory mediator with NF-kappaB suppressive properties. J. Immunol. 171:6329–6333. [DOI] [PubMed] [Google Scholar]
- 26.Hawkins, P.N., H.J. Lachmann, E. Aganna, and M.F. McDermott. 2004. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 50:607–612. [DOI] [PubMed] [Google Scholar]
- 27.Damiano, J., V. Oliveira, K. Welsh, and J.C. Reed. 2004. Heterotypic interactions among NACHT domains: implications for regulation of innate immune responses. Biochem. J. 381:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McConnell, B.B., and P.M. Vertino. 2000. Activation of a caspase-9-mediated apoptotic pathway by subcellular redistribution of the novel caspase recruitment domain protein TMS1. Cancer Res. 60:6243–6247. [PubMed] [Google Scholar]
- 29.Richards, N., P. Schaner, A. Diaz, J. Stcukey, E. Shelden, A. Wadhwa, and D.L. Gumucio. 2001. Interaction between Pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J. Biol. Chem. 276:39320–39329. [DOI] [PubMed] [Google Scholar]
- 30.Masumoto, J., T.A. Dowds, P. Schaner, F.F. Chen, Y. Ogura, M. Li, L. Zhu, T. Katsuyama, J. Sagara, S. Taniguchi, et al. 2003. ASC is an activating adaptor for NF-kappaB and caspase-8-dependent apoptosis. Biochem. Biophys. Res. Commun. 303:69–73. [DOI] [PubMed] [Google Scholar]
- 31.Ohtsuka, T., H. Ryu, Y.A. Minamishima, S. Macip, J. Sagara, K.I. Nakayama, S.A. Aaronson, and S.W. Lee. 2004. ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat. Cell Biol. 6:121–128. [DOI] [PubMed] [Google Scholar]
- 32.Chu, Z.-L., F. Pio, Z. Xie, K. Welsh, M. Krajewska, S. Krajewski, A. Godzik, and J.C. Reed. 2001. A novel enhancer of the Apaf1 apoptosome involved in cytochrome c-dependent caspase activation and apoptosis. J. Biol. Chem. 276:9239–9245. [DOI] [PubMed] [Google Scholar]
- 33.Aliprantis, A.O., R.B. Yang, M.R. Mark, S. Suggett, B. Devaux, J.D. Radolf, G.R. Klimpel, P. Godowski, and A. Zychlinsky. 1999. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 285:736–739. [DOI] [PubMed] [Google Scholar]
- 34.Moffett, P., G. Farnham, J. Peart, and D.C. Baulcombe. 2002. Interaction between domains of a plant NBS-LRR protein in disease resistance-related cell death. EMBO J. 21:4511–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang, Y., M. Hasegawa, R. Imamura, T. Kinoshita, C. Kondo, K. Konaka, and T. Suda. 2004. PYNOD, a novel Apaf-1/CED4-like protein is an inhibitor of ASC and caspase-1. Int. Immunol. 16:777–786. [DOI] [PubMed] [Google Scholar]
- 36.Chamaillard, M., S.E. Girardin, J. Viala, and D.J. Philpott. 2003. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell. Microbiol. 5:581–592. [DOI] [PubMed] [Google Scholar]
- 37.Aglipay, J.A., S.W. Lee, S. Okada, N. Fujiuchi, T. Ohtsuka, J.C. Kwak, Y. Wang, R.W. Johnstone, C. Deng, J. Qin, et al. 2003. A member of the Pyrin family, IFI16, is a novel BRCA1-associated protein involved in the p53-mediated apoptosis pathway. Oncogene. 22:8931–8938. [DOI] [PubMed] [Google Scholar]
- 38.Mansfield, E., J.J. Chae, H.D. Komarow, T.M. Brotz, D.M. Frucht, I. Aksentijevich, and D.L. Kastner. 2001. The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood. 98:851–859. [DOI] [PubMed] [Google Scholar]
- 39.Cazeneuve, C., S. Papin, I. Jeru, P. Duquesnoy, and S. Amselem. 2004. Subcellular localisation of marenostrin/pyrin isoforms carrying the most common mutations involved in familial Mediterranean fever in the presence or absence of its binding partner ASC. J. Med. Genet. 41:e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnstone, R.W., and J.A. Trapani. 1999. Transcription and growth regulatory functions of the HIN-200 family of proteins. Mol. Cell. Biol. 19:5833–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ting, J.P., and J. Trowsdale. 2002. Genetic control of MHC class II expression. Cell. 109:S21–S33. [DOI] [PubMed] [Google Scholar]
- 42.Centola, M., G. Wood, D.M. Frucht, J. Galon, M. Aringer, C. Farrell, D.W. Kingma, M.E. Horwitz, E. Mansfield, S.M. Holland, et al. 2000. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 95:3223–3231. [PubMed] [Google Scholar]
- 43.Conway, K.E., B.B. McConnell, C.E. Bowring, C.D. Donald, S.T. Warren, and P.M. Vertino. 2000. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 60:6236–6242. [PubMed] [Google Scholar]
- 44.Williams, L.K., J.D. Taxman, W.M. Linhoff, W. Reed, and J.P. Ting. 2003. Cutting Edge: Monarch-1: A Pyrin/Nucleotide-binding domain/Leucine-rich repeat protein that controls classical and nonclassical MHC class I genes. J. Immunol. 170:5354–5358. [DOI] [PubMed] [Google Scholar]
- 45.Martinon, F., K. Burns, and J. Tschopp. 2002. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell. 10:417–426. [DOI] [PubMed] [Google Scholar]