

Abstract
Neutrophils are key effector cells of the innate immune response and are required to migrate and function within adverse microenvironmental conditions. These inflammatory sites are characterized by low levels of oxygen and glucose and high levels of reductive metabolites. A major regulator of neutrophil functional longevity is the ability of these cells to undergo apoptosis. We examined the mechanism by which hypoxia causes an inhibition of neutrophil apoptosis in human and murine neutrophils. We show that neutrophils possess the hypoxia-inducible factor (HIF)-1α and factor inhibiting HIF (FIH) hydroxylase oxygen-sensing pathway and using HIF-1α–deficient myeloid cells demonstrate that HIF-1α is directly involved in regulating neutrophil survival in hypoxia. Gene array, TaqMan PCR, Western blotting, and oligonucleotide binding assays identify NF-κB as a novel hypoxia-regulated and HIF-dependent target, with inhibition of NF-κB by gliotoxin or parthenolide resulting in the abrogation of hypoxic survival. In addition, we identify macrophage inflammatory protein-1β as a novel hypoxia-induced neutrophil survival factor.
As neutrophils migrate from the circulation to sites of inflammation, they are required to adapt to and function within oxygen tensions much lower than those encountered in the circulatory system. Moreover, the physiological oxygen gradient that normally exists between the alveolus and mitochondria (1) is often greatly exaggerated in disease settings. Indeed, in rheumatoid synovium and empyema cavities, neutrophils have to function at oxygen tensions as low as 0–3 kPa (2, 3). Although granulocytes contain abundant mitochondria and have the capacity for aerobic metabolism, they rely almost exclusively on anaerobic glycolysis for the generation of ATP (4). Hence, these cells are intrinsically well adapted to operate in oxygen challenged environments. This is of critical importance for the resolution of inflammation because both excessive neutrophil activation and prolonged survival have been implicated in a number of disease settings, including the acute respiratory distress syndrome, bronchiectasis, and nonresolving pneumonias.
Historically work has focused on the ability of the body to regulate O2 delivery to tissues through hypoxic regulation of specialized cells within the carotid body and kidney. However, we now recognize that oxygen sensing is a more universal cellular event. This response is mediated by the oxygen-sensitive regulation of the α dimers of hypoxia-inducible factor (HIF; references 1–3) transcriptional complex. The regulation of HIF-α subunits occurs at both a protein and transcriptional level through the action of a novel class of prolyl and asparaginyl hydroxylase enzymes. In the presence of oxygen, iron, and 2-oxoglutarate, prolyl hydroxylase (1–3) enzymes hydroxylate Pro402 and 564 residues within the oxygen-dependent domain of HIF enabling it's binding to von Hippel-Lindau tumor suppressor protein (pVHL; references 5–8). This initiates ubiquitination and subsequent degradation; as a consequence, HIFα is normally maintained at very low levels under normoxic conditions. In addition, the asparaginyl hydroxylase factor–inhibiting HIF (FIH) hydroxylates the COOH-terminal of HIF at Asn803, thus blocking the recruitment of the p300 coactivator needed for HIF transcriptional activity (9–11). Therefore, reduction of hydroxylase activity by hypoxia results in increased protein stability and transcriptional activity of the HIF complex, which in turn, regulates the transcription of an array of hypoxia-responsive genes including key glycolytic enzymes (12), erythropoietin (12), adrenomedullin (13), SM20 (14) and VEGF (15). With regard to the inflammatory response, further levels of complexity have been revealed with the identification of PHD (prolyl hydroxylase domain–containing enzyme)-dependent HIF-independent oxygen-sensitive pathways, and the characterization of distinct target gene specificities for HIF-1α and 2α (16, 17).
The vital nature of the HIF-1α oxygen-sensing pathway in myeloid cell function has recently been revealed using murine HIF-1α conditional knockouts. In this model, the absence of HIF-1α in myeloid cells results in the depletion of intracellular ATP pools and a profound impairment of neutrophil aggregation, motility, bacterial killing, and invasion (18). In vivo, this is manifest as a loss of inflammatory response to tissue damage at pauci-vascular sites, namely the skin and articular cartilage. This study established for the first time the critical role for HIF-1α in the regulation of myeloid cell glycolysis and its requirement for granulocytic inflammation, and provided a direct link between oxygen sensing mechanisms and neutrophil activity.
A further and major regulator of neutrophil function is the ability of these cells to undergo constitutive apoptosis (19). This event triggers the phagocytosis and clearance of apoptotic neutrophils by local macrophages and is vital for the limitation of tissue damage in vivo (20, 21). Our in vitro studies have shown that the intrinsic apoptotic thresholds in these cells can be modified by an array of extracellular cytokine and physiological environmental stimuli (19, 22) and that the transcription factor NF-κB is critically involved in enhancing neutrophil survival (23). We have previously demonstrated that hypoxia causes a profound, concentration-dependent and reversible inhibition of neutrophil apoptosis that is not mimicked by other forms of cellular stress including glucose deprivation or heat shock (24). Moreover, this survival effect was inhibited by two structurally discrete iron chelators (24) desferrioxamine (DFO) and hydroxypyridone. These data implicate a role for a ferroprotein in the regulation of neutrophil apoptosis. Given the similarity of this to the iron dependency of the hydroxylase pathway and our demonstration of the importance of HIF in granulocyte-mediated inflammation, we sought to establish the role of HIF in the regulation of neutrophil apoptosis.
Here, we confirm that human peripheral blood neutrophils are resistant to apoptosis when cultured at reduced oxygen tensions (24). Unlike the survival effect of certain cytokines and growth factors, this response was phosphoinositide 3-kinase (PI3-kinase) independent, indicating involvement of a discrete survival pathway. The hypoxic survival effect was mimicked by the competitive inhibition of hydroxylase enzymes and associated with marked stabilization of HIF-1α. A direct role for HIF-1α in the hypoxic inhibition of neutrophil apoptosis was provided by experiments in HIF-1α–deficient murine neutrophils, which displayed markedly reduced cell survival after anoxic challenge. The ability of hypoxia to increase NF-κB p65 transcript abundance, protein expression and activity, together with the ablation of hypoxic survival by the NF-κB inhibitors gliotoxin and parthenolide, and the inhibition of hypoxic induction of NF-κB in HIF-1α knockout murine neutrophils suggests that HIF-1α-dependent regulation of the NF-κB pathway mediates the survival response observed. Furthermore, we identified macrophage inflammatory protein-1β (MIP-1β) as a novel hypoxia-stimulated granulocyte survival factor, which provides alternative and indirect facilitation of the direct hypoxic survival response.
Results
Hypoxia inhibits neutrophil apoptosis
Human peripheral blood neutrophils after 20 h in culture displayed constitutive levels of apoptosis matching those previously reported (25). This was confirmed using both morphological analysis and FACS quantification of AV and PI staining (Fig. 1 A). This level of apoptosis was maintained with culture at 10 kPa, but significantly inhibited when cells were cultured under either anoxic (0 kPa) or hypoxic (3 kPa) conditions. Using a colorimetric substrate reaction, caspase 3 activity in whole cell lysates was also significantly inhibited at 20 h under reduced oxygen tensions (Fig. 1 B). Concurrent electron microscopy analysis of neutrophils ageing in vitro revealed classic appearance and progression of apoptosis (Fig. 1 C) with early loss of euchromatin heterochromatin differentiation followed by later changes of nuclear condensation and capping. In low oxygen tensions, the cells appeared either normal or showed only minor changes in euchromatin/heterochromatin differentiation compared with cells cultured under normoxic conditions (percent cells with nuclear condensation and capping at 20 h: 53% normoxia compared with 16% hypoxia).
Figure 1.
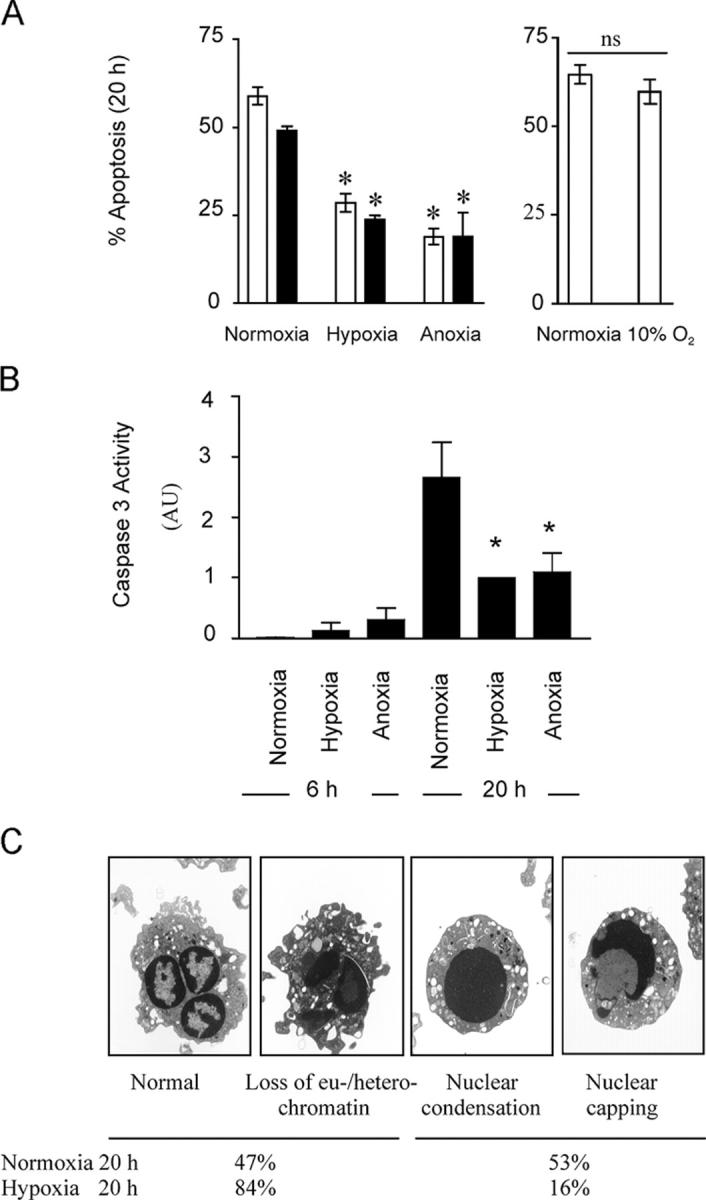
Hypoxia inhibits constitutive neutrophil apoptosis. (A) Apoptosis. After in vitro culture of human peripheral blood neutrophils for 20 h, cells were assessed for apoptosis by morphology (open bars) or FACS analysis of AV/PI staining (shaded bars). Results represent mean ± SEM (n = 3), *, P < 0.05 compared with normoxic controls. (B) Caspase 3 activity. At the indicated times, caspase 3 activity (arbitrary units) was measured in neutrophil whole cell lysates after culture under different oxygen tensions. Results represent mean ± SEM (n = 4); *, P < 0.05 compared with time-matched normoxic controls. (C) Electron microscopy of neutrophil apoptosis. Electron microscopy appearance of neutrophils aged for 20 h in vitro displaying progressive changes associated with apoptosis; this progression was inhibited in cells incubated under hypoxic or anoxic conditions.
Hypoxic survival of neutrophils is PI3-kinase independent
Supernatants obtained from neutrophils cultured at reduced oxygen tensions had a survival effect (assessed at 20 h) on freshly isolated cells. This transferable survival effect was time dependent, unrelated to the oxygen tension of the supernatants because they were reoxygenated before use, and was only observed in supernatants harvested after 12 h of hypoxic incubation (Fig. 2, A and B). Furthermore, there was no stabilization of HIF-1α protein in freshly isolated neutrophils cultured in supernatants obtained from neutrophils incubated under hypoxia. 10 μM LY294002, which we have previously demonstrated to cause a selective inhibition of neutrophil PI3-kinase activity (26, 27), fully inhibited the supernatant-induced survival (Fig. 2 B). In marked contrast, hypoxia-mediated neutrophil survival was unmodified by 10 μM LY294002 or 100 nM wortmannin (Fig. 2 C). These data indicate that although hypoxia causes the release of a soluble factor with the capacity to operate in an autocrine survival manner, this does not account for the direct hypoxia-mediated survival effect, which is PI3-kinase independent.
Figure 2.
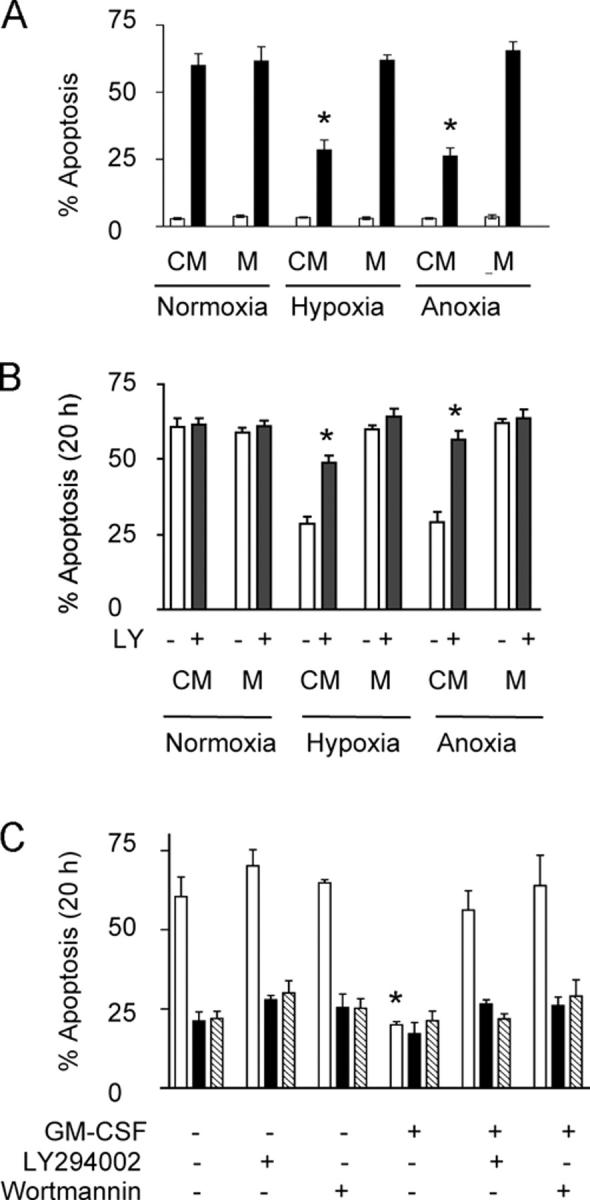
Hypoxia induces the release of a PI3-kinase–dependent survival factor. (A) Survival factor secretion. Conditioned medium (CM) obtained from normoxic, hypoxic, or anoxic neutrophils was transferred to freshly isolated cells with subsequent analysis of apoptosis at 6 h (open bars) and 20 h (shaded bars). Results represent mean ± SEM (n = 3); *, P < 0.05 compared with medium (M)-only controls. (B) Survival effect of CM is PI3-kinase dependent. Neutrophils were cultured with CM in the presence of the PI3-kinase inhibitor LY294002 (shaded bars), and apoptosis was assessed by morphology. Results represent mean ± SEM (n = 3); *, P < 0.05 compared with medium (M)-only controls. (C) PI3-kinase– independent hypoxic survival. Neutrophils were cultured in normoxia (open bars), hypoxia (shaded bars), or anoxia (hatched bars) in the presence of PI3-kinase inhibitors LY294002 or wortmannin or the survival cytokine GM-CSF, and apoptosis was assessed morphologically. Results represent mean ± SEM (n = 3); *, P < 0.05 compared with normoxic control.
MIP-1β is a novel hypoxia-stimulated granulocyte survival factor
Pretreatment of hypoxic supernatants with trypsin (1:250 [wt/vol]) (at a concentration that blocked GM-CSF [100 ng/ml]–mediated survival) completely abrogated the survival effect of the supernatants obtained from hypoxic neutrophils (Fig. 3 A). However, this survival effect was not modified by heat inactivation at 56°C (Fig. 3 B), which is characteristic of chemokines. Luminex analysis of supernatants subsequently identified a significant time-dependent increase in secreted MIP-1β after hypoxic stimulation (not depicted), a finding which was independently verified by ELISA (Fig. 3 C). No increase in supernatant levels of the known neutrophil survival cytokines IL-8, IL-6, IL-1β, GM-CSF, TNFα, or migration inhibitory factor (MIF), was detected after hypoxic stimulation. Critically, preincubation of the hypoxia-generated supernatants with a specific MIP-1β neutralizing antibody at a concentration that blocked ELISA detection of MIP-1β in hypoxic supernatants (100 μg/ml; Fig. 3 D) completely blocked the survival effect.
Figure 3.
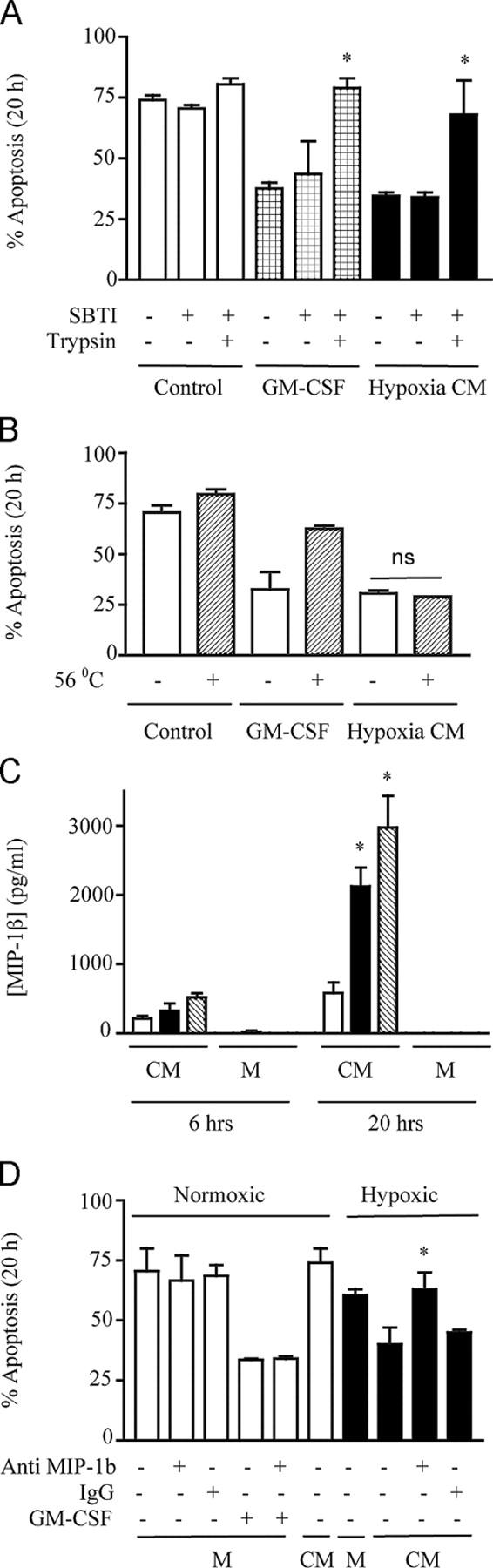
MIP-1β is a novel hypoxia-stimulated granulocyte survival factor. (A) Trypsin sensitivity. Freshly isolated cells were incubated in conditioned medium (CM) obtained from normoxic neutrophils (control) or hypoxic neutrophils (hypoxia CM). These media were either untreated or treated for 2 h with trypsin (1:250 wt/vol), followed by treatment with soya bean trypsin inhibitor (SBTI) or SBTI alone. The effects of trypsin on GM-CSF (100 ng/ml)–mediated neutrophil survival was examined in parallel. Apoptosis was subsequently analyzed at 20 h, and the results represent mean ± SD (n = 2); *, P < 0.05 compared with matched trypsin-untreated conditions. (B) Heat insensitivity. Neutrophils were incubated with CM obtained from normoxic neutrophils (control), hypoxic neutrophils (hypoxia CM), or GM-CSF (100 ng/ml)–supplemented monofeed that was previously heated to 56°C for 45 min. Apoptosis was subsequently analyzed by morphology at 20 h. Results represent mean ± SEM (n = 3). (C) MIP-1β secretion. MIP-1β released into the CM obtained from normoxic (open bar), hypoxic (shaded bar), or anoxic (hatched bar) neutrophils or unconditioned medium (M) was measured by ELISA at 6 and 20 h. Results represent mean ± SEM (n = 3); *, P < 0.05 compared with time-matched normoxic CM. (D) MIP-1β antibody blocks transferable survival. CM obtained from normoxic or hypoxic neutrophils or medium alone (M) was incubated in the presence (+) or absence (−) of 100 μg/ml anti–MIP-1β antibody or 100 μg/ml of total goat IgG isotype control for 30 min at room temperature, before being added to freshly isolated cells. GM-CSF (100 ng/ml) in the presence (+) or absence (−) of anti–MIP-1β or IgG controls were run in parallel, and apoptosis was assessed by 20-h morphology. Results represent mean ± SD (n = 2); *, P < 0.05 compared with matched MIP-1β antibody–untreated conditions.
Competitive inhibition of hydroxylase enzymes mimics hypoxic survival
Using a series of immunoprecipitation and inhibitor assays, we looked for the presence of classical oxygen-sensing regulators in human neutrophils. FIH was demonstrated in neutrophil lysates under all oxygen tensions and in the presence of both the iron chelator DFO (1 mM) and the competitive hydroxylase inhibitor dimethyloxaloylglycine (DMOG; 1 mM) (Fig. 4 A). DMOG was able to fully mimic the inhibition of neutrophil apoptosis observed at low oxygen tensions (8 ± 3% apoptosis 20 h, 1 mM DMOG, P < 0.05) (Fig. 4 B). Moreover, DMOG was unable to enhance the survival effect of hypoxia and was effective at 100 μM, which compares with maximal effects in tissue culture cells at 1 mM (28, 29). Accumulation of HIF-1α was observed both in hypoxia and anoxia, and after culture with 1 mM DFO or 1 mM DMOG (Fig. 4 C). Of note, we were unable to detect HIF-2α (unpublished data), supporting our previous work describing a lack of HIF-2α RNA in neutrophils (24).
Figure 4.
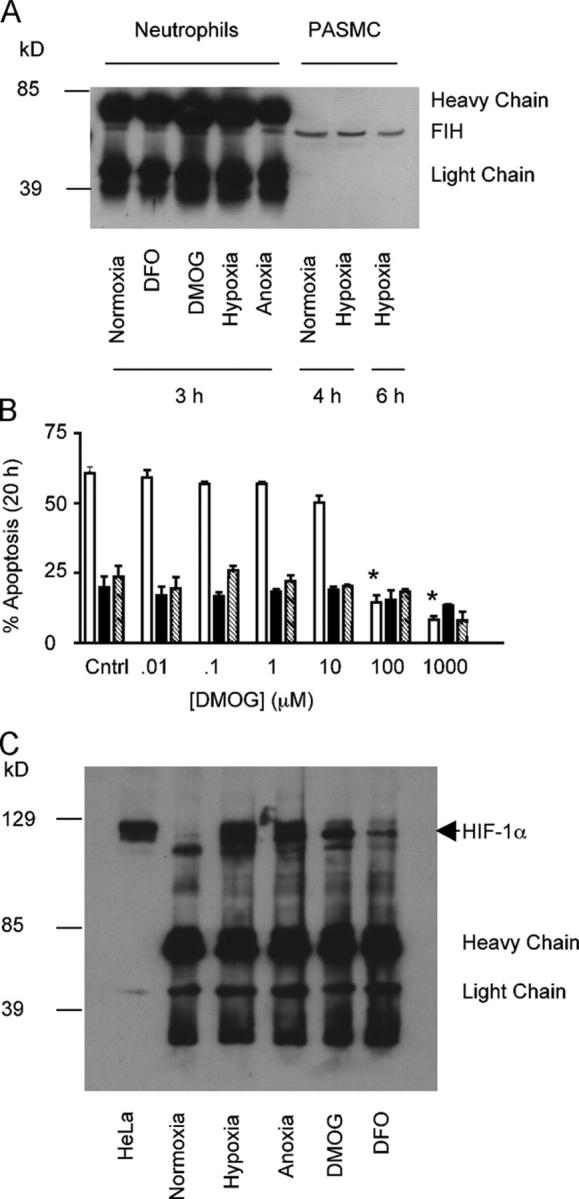
Competitive inhibition of hydroxylase enzymes mimics hypoxic survival. (A) Human neutrophils possess FIH. After culture, under each oxygen tension or in the presence of either the iron chelator DFO or the hydroxylase inhibitor DMOG, whole cell lysates were prepared from peripheral blood neutrophils; immunoprecipitation was performed with an antibody to FIH and separated on SDS-PAGE. Control lysates were also prepared from pulmonary artery smooth muscle cells and run directly on SDS-PAGE without immunoprecipitation. (B) DMOG inhibits human neutrophil apoptosis. Neutrophils were cultured with increasing concentrations of DMOG under normoxia (open bars), hypoxia (shaded bars), and anoxia (hatched bars), and apoptosis was assessed by morphology. Results represent mean ± SEM (n = 3); *, P < 0.05 compared with normoxic control. (C) Hypoxia stabilizes HIF-1α in human neutrophils. Lysates were prepared from neutrophils after culture in hypoxia, anoxia, normoxia or in the presence of DMOG or DFO. Lysates were immunoprecipitated with an antibody to HIF-1α and separated by SDS-PAGE. Both blots are representative of n = 3.
HIF-1α regulates neutrophil survival at reduced oxygen tensions
To establish a more direct link between HIF-1α and the regulation of neutrophil apoptosis, we used a recently established murine HIF-1α conditional knockout model (18). Compared with wild-type controls, bone marrow–derived neutrophils obtained from lysMcre HIF-1α–targeted animals demonstrated a marked increase in ghost cells and cells displaying pyknotic nuclei and nuclear fragmentation after a 20-h culture in anoxia (P < 0.05; Fig. 5 A). This was matched by a decrease in neutrophil survival, as indicated by FACS-quantified PI staining (Fig. 5 B). The lack of HIF-1α did not, however, influence the extent of apoptosis observed under normoxic conditions (20-h normoxic survival: for knockout animals, 76 ± 5%; and for wild-type animals, 77 ± 5%) (Fig. 5 C). In contrast to human and murine peripheral blood neutrophils, a significant annexin V–negative and PI-positive population of cells were evident in our cultured murine bone marrow–derived cells. This is common to many studies using immature bone marrow–derived neutrophils (30). Together, these results demonstrate that the presence of HIF-1α is essential for murine neutrophils to survive in an oxygen deficient environment.
Figure 5.

HIF-1α regulates neutrophil survival at reduced oxygen tensions. (A) Apoptotic morphology. Murine bone marrow–derived neutrophils were cultured for 20 h under normoxic or anoxic conditions. Apoptosis was assessed by morphology. (B) Annexin V/ Pi. Identical sets of cells were analyzed by FACS for AV/PI staining. (C) Reduced survival in HIF-1α knockout cells under anoxic conditions. Percent survival was calculated after AV/PI staining. Results represent mean ± SEM (n = 3); *, P < 0.05 compared with HIF-1α +/+ control.
Hypoxia regulates neutrophil transcript abundance
To identify potential downstream targets of HIF-1α in human neutrophils, we performed a series of gene array experiments looking at changes in transcript abundance with reduced oxygen tension. Radioactively labeled complex cDNA probes were prepared from neutrophil RNA and hybridized to a 988–cDNA sequence verified the apoptosis-targeted nylon filter gene array generated in our laboratories (31). After an initial culture period of 3 h, we saw no detectable changes in the relative transcript abundance among normoxic, hypoxic, or anoxic conditions (unpublished data). However, with a more prolonged culture (6 h), glyceraldehyde 3-phosphate dehydrogenase (G3PDH), MIF, triosephosphate isomerse-1, and NF-κB (p65) showed a twofold or more relative increase in expression in both hypoxia and anoxia compared with normoxia (Fig. 6 A). This was shown to be significant by Bayesian t tests (Fig. 6 A, table). We subsequently confirmed the changes for G3PDH, MIF, and NF-κB (p65) using TaqMan quantitative PCR; in these experiments, the expression of each transcript was estimated relative to granulocyte-specific CSF3R identified on the gene array as a nonchanging, nonoxygen-regulated endogenous control (Fig. 6 B). The G3PDH response offers an appropriate internal positive control in these experiments because of its well-characterized regulation by oxygen tension (32) and, more specifically, HIF (33, 34).
Figure 6.


Hypoxia regulates human neutrophil transcript abundance. (A) Regulation of transcript abundance. Log (base 10) mean signal intensities were calculated and compared from array filters prepared using RNA from human peripheral blood neutrophils cultured under the oxygen tensions shown. Points represent the mean (±SEM bars) of n = 3 experiments; each gene is presented in duplicate. Outer lines represent a twofold change between conditions. All significant transcripts are individually named. The table shows the statistical analysis of the gene array data. Cyber-T tests were performed to calculate the Bayesian p values shown. (B) TaqMan confirmation. Array findings (open bars) were validated for hypoxia (H), and anoxia (A) by TaqMan analysis of duplicate RNA samples (shaded bars) using the nonchanging endogenous control CSF3R (granulocytes). Data represent mean ± SEM of n = 3 experiments.
NF-κB activity is required for hypoxic survival
After the identification of NF-κB p65 as a hypoxia-regulated transcript, we examined the effects of hypoxia on total NF-κB p65 expression relative to IkBα expression, and overall effects of hypoxia on NF-κB activity. At early time points (3–12 h), we observe progressive and near complete loss of constitutive NF-κB expression with a parallel loss in IκBα (Fig. 7, A and B). In normoxia, this early decline remained unchecked and there was no subsequent rebound in detectable NF-κB or IκBα protein levels. However, at reduced oxygen tensions, we observed a later (12–20 h) recovery in NF-κB p65 and IκBα levels, which followed the early hypoxia-driven increase in p65 transcript abundance. NF-κB activity assays at these times confirmed the capacity of the newly expressed NF-κB to bind appropriate oligonucleotide sequences (Fig. 7 C). Moreover culture of neutrophils in reduced oxygen tensions with the structurally and mechanistically discrete NF-κB inhibitors gliotoxin and parthenolide, at concentrations that are specific and do not modify constitutive apoptotic rates (23), resulted in the ablation of the hypoxic survival effect (Fig. 8, A and B). These inhibitors also attenuated the hypoxic survival effect if added 12 h into the incubation period (unpublished data).
Figure 7.
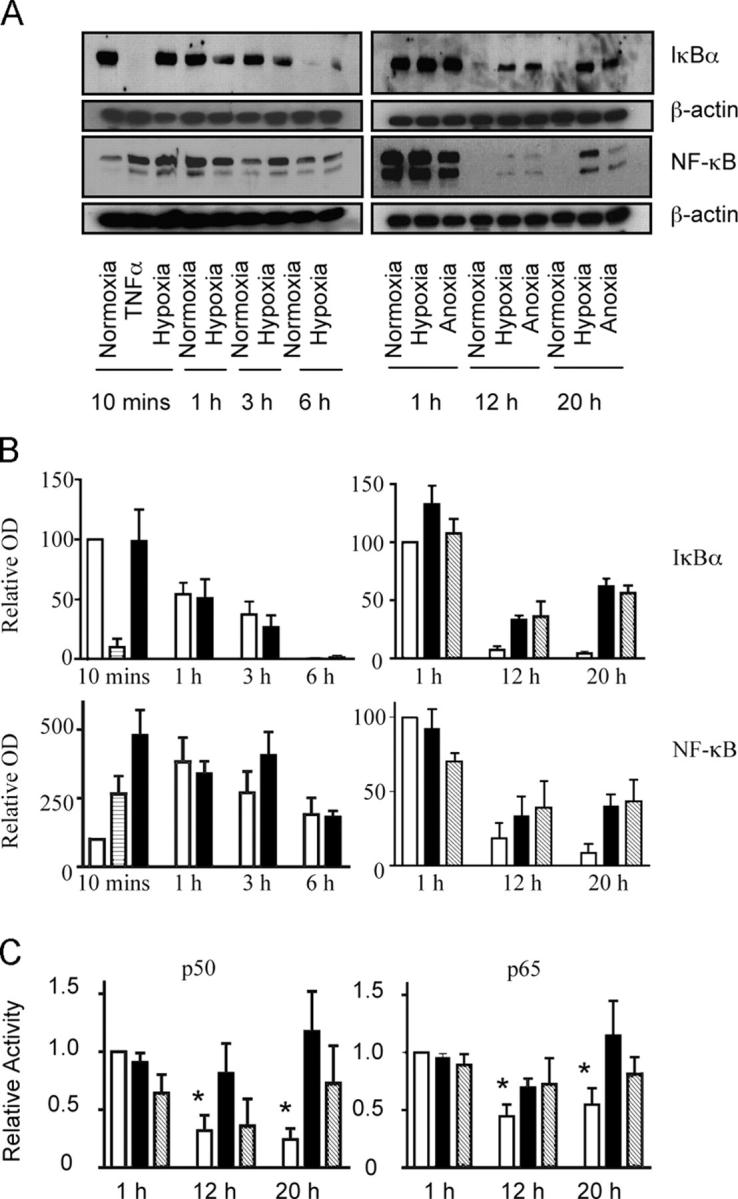
Hypoxia stimulates the reexpression of NF-κB protein and maintains activity. (A) Reexpression of IκBα and NF-κB protein with prolonged oxygen deprivation. Human neutrophil lysates were prepared after culture in normoxia ± TNF, hypoxia, or anoxia for 10 min–20 h, and the total protein was measured by Western blot. Blots shown are representative of n = 3–9 experiments. (B) OD quantification of Western blots. Optical densities of IκBα and NF-κB Western blots were quantified for normoxia (open bars), hypoxia (shaded bars), anoxia (hatched bars) and TNF-α (striped bars) using Scion corporation software and normalized to normoxia 10 min (10 min-6 h blots) or normoxia 1 h (1–20 h blots). Data represent mean ± SEM (n = 4). (C) Decreased p50 and p65 activity with prolonged normoxia. p50 or p65 DNA binding activity was measured by ELISA in normoxic (open bars), hypoxic (shaded bars) or anoxic (hatched bars) neutrophil lysates at the time points shown. Data represent mean ± SEM for n = 3 experiments, * P < 0.05 compared with normoxia 1 h.
Figure 8.
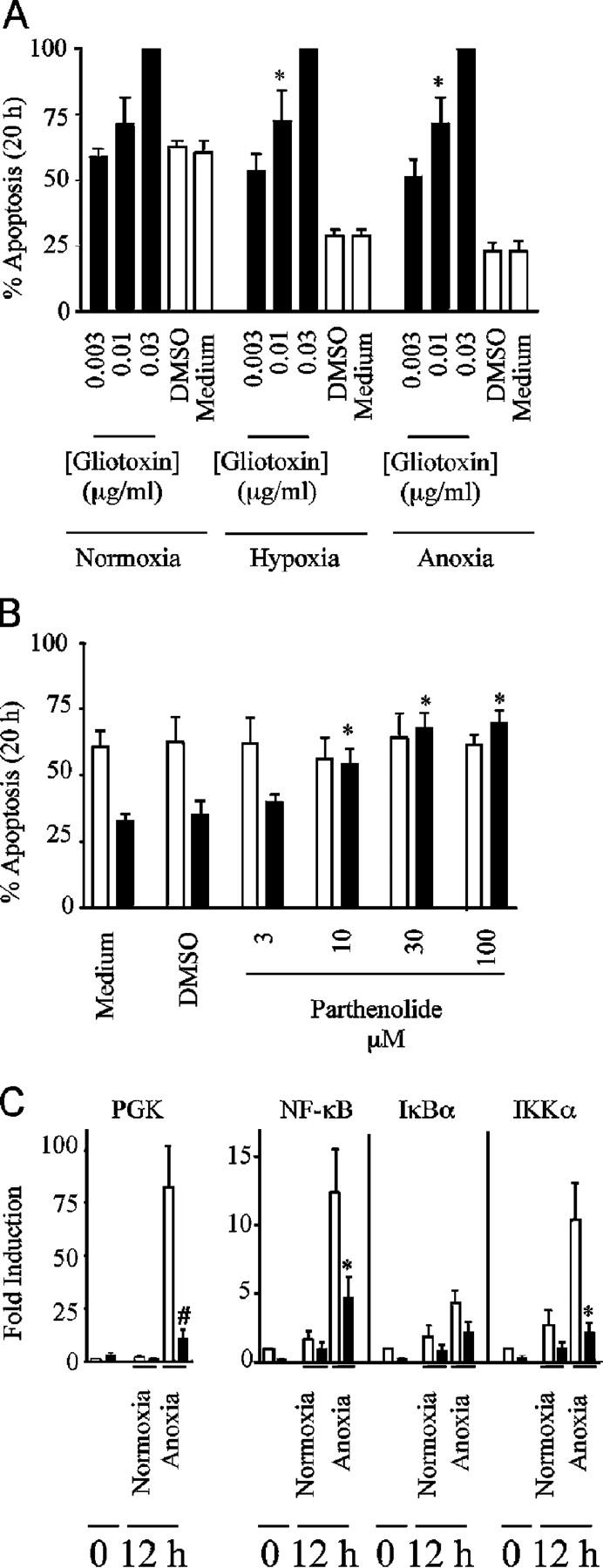
NF-κB activity is required for hypoxic neutrophil survival and dependent on HIF-1α expression. (A) Inhibition of hypoxic survival with gliotoxin. Neutrophils were incubated under the oxygen tensions shown in the presence or absence of gliotoxin for 20 h. Apoptosis was assessed by morphology. Results represent mean ± SEM of n = 3 experiments; *, P < 0.05 compared with oxygen-matched medium only controls. (B) Inhibition of hypoxic survival with parthenolide. Neutrophils were incubated in normoxia (open bars) or hypoxia (shaded bars) in the presence or absence of parthenolide for 20 h. Apoptosis was assessed by morphology. Results represent mean ± SEM of n = 3 experiments; *, P < 0.05 compared with oxygen-matched medium-only controls. (C) Hypoxic induction of PGK, NF-κB, and IKKα message is inhibited in HIF-1α knockout mice. TaqMan analysis of PGK, NF-κB, IκBα, and IKKα transcript abundance relative to β-actin was performed on cDNA isolated from bone marrow–derived murine neutrophils from wild-type (open bars) and HIF-1α knockout mice (shaded bars) after culture in normoxia and anoxia. Data represent mean fold changes ± SD from pooled cDNA from 16 mice in two independent experiments; *, P < 0.05 and #, P < 0.005 compared with matched wild-type controls.
We have previously demonstrated that NF-κB is a key survival transcription factor in cytokine-stimulated neutrophils (23) and, hence, the hypoxic-driven stabilization and activation of NF-κB provides a direct link between oxygen deprivation and a recognized antiapoptotic pathway. To address more specifically the link between HIF-1α and NF-κB, we performed a series of real-time PCR experiments looking at changes in transcript abundance in HIF-1α wild-type and knockout murine neutrophils. After 12 h in culture, we saw a significant hypoxic induction of NF-κB, and IKKα, an alternative regulator of NF-κB, in addition to PGK, a known HIF-1α–regulated glycolytic enzyme (Fig. 8 C). This induction was significantly abrogated in the HIF-1α knockout mice and implicates a HIF-1α–dependent regulation of both NF-κB and IKKα.
Discussion
Our results describe for the first time the presence of the functionally active HIF-1α hydroxylase oxygen-sensing pathway in human peripheral blood neutrophils. The absence of HIF-2α expression demonstrates the differential expression of key members of this pathway between various myeloid lineage cells (35, 36). Because of the nonredundant function between different HIF α subunits (16), this expression pattern may have a significant impact on how granulocytes sense and respond to changes in oxygenation.
The ability of hypoxia to regulate apoptotic thresholds is a cell-specific phenomenon. Hence, in neuronal cells, adenocarcinoma HT 29 cells, certain oncogenically transformed cells and cardiac myocytes hypoxia clearly acts to induce rather than inhibit apoptotic cell death (37, 38). This may reflect the different abilities of cells to adapt to anaerobic metabolism. In neutrophils, we show that the inhibition of apoptosis by hypoxia is accompanied by a time-dependent induction of transcripts for G3PDH and triosephosphate isomerse-1. The induction of these key glycolytic enzymes provides a mechanism for the continued generation of ATP, which is an essential requirement for neutrophil functional responses to inflammatory stimuli (39). With respect to GAPDH, this also provides indirect evidence for HIF-1α transcriptional activation caused by the presence of functional hypoxia-responsive elements within the GAPDH promoter region (33, 34).
Initial work in the myeloid-targeted HIF-1α knockout mice has provided a further link between in vivo inflammatory responses and HIF-1α–dependent maintenance of intracellular energy homeostasis (18). In addition to the ATP-dependent loss of function and diminished migratory potential, our data suggest that a second mechanism, namely decreased neutrophil survival, may also contribute to the impaired inflammatory response observed in these animals. Hence, we show a marked reduction in cell survival after anoxic challenge in murine bone marrow–derived neutrophils lacking HIF-1α. Because we see no modification of constitutive apoptosis in the HIF−/− cells, the reduced survival would only be unmasked at sites of low oxygenation. This would be consistent with the normal total neutrophil count, but diminished inflammation seen in the HIF-1α myeloid-targeted knockout mice and the exaggerated inflammatory responses described in the VHL knockout animals (18). Although murine bone marrow–derived neutrophils display different AV/PI staining properties and lower 20-h levels of constitutive cell death, the above data clearly support a role for HIF-1α the regulation of human neutrophil survival under hypoxic conditions. This conclusion is further supported by preliminary data obtained in patients with von Hippel-Lindau disease where enhanced survival of peripheral blood neutrophils is observed (unpublished data). Contaminating bone marrow monocytes are unlikely to influence these observations because human peripheral blood monocyte depletion does not modify the rate of apoptosis under either normoxia or hypoxia (40). The involvement of a hydroxylase-dependent pathway is confirmed in human granulocytes by the antiapoptotic properties of the competitive hydroxylase inhibitor DMOG.
The mechanism by which HIF regulates neutrophil apoptosis remains to be fully characterised. However, the inhibition of hypoxic survival by gliotoxin and parthenolide implies that NF-κB is an important downstream effector of the HIF-1α–dependent response. Temporally, this is supported by the early stabilization of HIF-1α that is followed by the increase at 6 h in p65 transcript abundance and subsequent (12–20 h) reexpression of NF-κB and the persistence in NF-κB p65 and p50 DNA binding activity. The regulation of NF-κB by hypoxia is already described for different cell types, including J774.1 murine macrophage and RF/6A retinal lines (41, 42), yet interactions between HIF and NF-κB are poorly understood. Studies looking at the nonhypoxic induction of HIF have shown a number of indirect links between the HIF and NF-κB transcription pathways. In neuronal cells, a HIF-dependent up-regulation of erythropoietin has been described to prevent excitotoxin-induced apoptosis through Jak2 NF-κB cross-talk (43), whereas in human embryonic kidney cells, TNFα stimulates the accumulation of ubiquitinated HIF via an NF-κB–dependent pathway (44). Furthermore, the essential modulator of NF-κB NEMO/IKKγ has recently been shown to be a binding partner for HIF-2α (Peet, D.J., and C. Braken, personal communication). To date, no direct association between HIF and NF-κB has been reported after stimulation of cells with hypoxia. Using a series of real-time PCR reactions in HIF-1α knockout and wild-type neutrophils, we show for the first time the hypoxic induction of NF-κB transcription to be dependent on the presence of HIF-1α. Moreover, we show a HIF-1α–dependent up-regulation of the alternative NF-κB regulator IKKα. IKKα is recognized both to facilitate NF-κB transcriptional activity through the alternative pathway and itself be critical for the histone phosphorylation required for the activation of NF-κB–directed gene expression (45). Thus HIF-1α not only facilitates an increase in NF-κB message but also facilitates the pathways required for enhanced transcriptional activity.
A key regulator of the NF-κB pathway in neutrophils is IκBα. After neutrophil stimulation with pro-inflammatory molecules, IκBα is degraded resulting in unopposed NF-κB activity (46, 47). Conversely, the nuclear localization of IκBα results in increased neutrophil apoptosis (48). Given the persistent expression of IκBα protein with prolonged hypoxic culture and the lack of HIF-1α–dependent changes in murine transcript abundance, it is unlikely that HIF mediates the stimulation of NF-κB through the transcriptional inhibition of IκBα, although an indirect regulation of IκBα nuclear trafficking remains possible.
It is important to note that the inhibition of the PI3-kinase pathway has no effect on the direct inhibition of neutrophil apoptosis by hypoxia. This is in direct contrast to the survival effect of the supernatants derived from hypoxically cultured cells. Using heat inactivation and trypsin digestion, we first identify this factor to be a protein and possible cytokine. Luminex analysis, ELISA, and blocking antibody experiments subsequently support the identity of this cytokine as MIP-1β. Although the identification of MIP-1β as an oxygen-sensitive granulocyte survival factor is novel, MIP-1β is recognized to be up-regulated in Bacillus Calmette-Guerin–stimulated neutrophils along with MCP-1 and MIP-1α (49). Interestingly, these cells were also noted to have diminished rates of apoptosis. In addition, MIP-1β secretion by murine alveolar macrophages has been shown to be oxygen sensitive (50). These studies further support the role of MIP-1β as a novel hypoxia-stimulated granulocyte survival factor. In parallel, gene array analysis of human neutrophils identified the hypoxic induction of MIF mRNA. MIF has previously been described both as a neutrophil survival factor through its inhibition of Bax/Bid cleavage and caspase 3 activity (51) and as a hypoxia-regulated cytokine (52). However, we saw no induction of MIF protein release into the culture media of hypoxia-treated neutrophils. Although this does not exclude an intracellular up-regulation of MIF protein and the subsequent down-regulation of Bax–Bid pathways, this does exclude MIF as a transferable survival factor in our system.
In summary, we have revealed that the hypoxic inhibition of neutrophil apoptosis is regulated by the HIF-1α hydroxylase oxygen-sensing pathway and NF-κB reexpression, and indirectly regulated by the release of the novel survival factor MIP-1β. Although we have identified the transcriptional and functional activation of NF-κB after hypoxia and the HIF-1α–dependent regulation of NF-κB and Iκκα transcript, the precise mechanism of this interaction remains to be elucidated. HIF-1α would appear to be the critical upstream regulator of this hypoxic survival pathway because deletion of HIF-1α in murine neutrophils results in both a reduction in NF-κB and Iκκα message and anoxia-stimulated cell death.
Materials and Methods
Isolation and culture of peripheral blood neutrophils from healthy human volunteers
Neutrophils were purified by dextran sedimentation and discontinuous plasma-Percoll gradients (25). Purified cells were resuspended at 5 × 106 cells/ml in IMDM supplemented with 10% autologous serum and 50 U/ml streptomycin and penicillin. Cells were cultured in the presence or absence of 1 mM DFO, 1 mM DMOG (unless otherwise stated; a gift from C. Pugh, University of Oxford, Oxford, UK), 200 U/ml TNFα, 100 ng/ml GM-CSF, 100 μg/ml anti–human MIP-1β, 100 μg/ml of total goat IgG, 1:250 (wt/vol) trypsin, or 1:250 (wt/vol) soya bean trypsin inhibitor in normoxic (19 kPa), hypoxic (3 kPa) or anoxic (0 kPa) environments. Normoxia was controlled by using a humidified 5% CO2/air incubator, and hypoxia, by pregasing Dulbecco's medium for 30 min in a sealed hypoxic work station with 5% CO2/balance N2 gas mix and subsequent culture in a humidified hypoxic (CO2/N2) incubator. Anoxia was maintained with a MACS500 Don Whitley catalyst–dependent anaerobic incubator using 5% CO2/10% H2/balance N2 gas mix. Supernatants were heat inactivated by warming to 56°C for 45 min. Trypsin inactivation was performed with a 2-h incubation with trypsin that was followed by the addition of an equimolar amount of soya bean trypsin inhibitor. All other inhibitor experiments were performed with a 30-min preincubation step.
Conditional knockouts and harvesting of bone marrow–derived neutrophils
Targeted myeloid deletions of HIF-1α were created via crosses into a background of lysozyme M–driven cre (lysMcre) expression (18). This resulted in an approximate 75% HIF-1α deletion rate in neutrophils (18). Mice were killed by CO2 asphyxia and bone marrow-derived neutrophils were harvested from knockout and cre wild-type animals using discontinuous 82, 62, and 51% HBSS-Percoll gradients. Cell purity, assessed using air-dried cytospins, was routinely above 70%. Cells were cultured as for human cells, replacing autologous serum with 10% FBS. Mice were handled according to protocols approved by University of California San Diego Animal Care Program, which follows the National Institutes of Health guidelines for ethical animal treatment.
Apoptosis assays
Neutrophils were harvested at the time points indicated, cytocentrifuged, fixed in methanol, and stained with May-Grünwald-Giemsa (Merck Ltd.), and the morphology was examined by oil immersion light microscopy (20). Apoptotic neutrophils were defined as those with darkly stained pyknotic nuclei. Apoptosis was also assessed by flow cytometry with fluorescein isocyanate–labeled recombinant human annexin V (AV) and propidium iodide (PI) staining. Caspase 3 activity was measured by colorimetric substrate reaction (Calbiochem) according to the manufacturer's instructions. For electron microscopy analysis (CM 100 transmission electron microscope; Philips) cells were fixed in glutaraldehyde and osmium tetroxide, dehydrated in ethanol, and embedded in Spurr's resin for sectioning.
Quantification of HIF-1α, FIH, IκBα and NF-κB protein
Whole cell extracts were prepared by resuspending 25 × 106 neutrophils in 1 ml of ice-cold hypotonic lysis buffer (10 mM Tris-HCl, pH 7.8, 1.5 mM EDTA, 10 mM KCl, 0.5 mM DTT, 1 mM sodium orthovanadate, 2 mM levamisole, 0.5 mM benzamidine, and 0.05% NP-40 and Complete protease inhibitor cocktail; Roche Applied Science); this was followed by sonication (setting: 15, 15 s) (model Sanyo Soniprep-150; Fisson's Instruments). HIF-1α and FIH were immunoprecipitated with anti–HIF-1α mouse monoclonal (Ab1; Ab Cam) and anti-FIH rabbit polyclonal (a gift from D. Peet, The University of Adelaide, Adelaide, Australia), respectively. Protein recovery for all samples was assessed by immunoblotting with primary antibodies against HIF-1α (BD Transduction Laboratories), FIH (gift from D. Peet), IκBα (New England Biolabs Cell Signaling Technology), and NF-κB p65 (Santa Cruz Biotechnology Inc.). All samples were protein corrected, and equal loading confirmed by β-actin expression.
Gene array analysis
Neutrophils (100 × 106/condition) were lysed with 2 ml TRIzol, and RNA was extracted using chloroform phase partitioning and isopropanol. Samples were DNase digested (Ambion) and UV quantified, and the integrity was measured by 2100 Bioanalyser (Agilent Technologies). cDNA probes labeled with α-[33P]dCTP were synthesized from RNA by reverse transcription, purified, and hybridized to nylon gene array filters. Each filter composed duplicate spots of 988 unique and sequence-verified cDNAs with nonhomologous DNA controls (poly A, salmon sperm DNA) and spiked Arabidopsis cDNA (reference 31; GEO database accession GSE968). Filters were read by phosphoimager, spots were quantified using Imagene 5.1, and after normalization for each blood donor, the data were analyzed with Genespring software. All experiments were performed in triplicate, each time neutrophils derived from a new blood donor and RNA replicates hybridized once to the array were used.
TaqMan real-time PCR of human peripheral blood neutrophils
cDNAs were synthesized from duplicate aliquots of RNA (as described in the previous paragraph) using random hexamers and run at a final concentration of 100 ng/μl. Assays-on-Demand Gene expression TaqMan MGB 6FAM dye-labeled products (Applied Biosystems) were used for macrophage; MIF, GAPDH (G3PDH), NF-κB (p65), and CD27-binding protein (SIVA) target assays were performed according to the manufacturer's (Applied Biosystems) instructions. The reactions were quantified relative to the threshold cycle for the highly expressed nonchanging endogenous control CSF3R (granulocyte). Partial primer sequences were obtained from Applied Biosystems.
TaqMan real-time PCR of murine bone marrow neutrophils
Neutrophils (5 × 106/condition) were lysed with 1 ml TRIzol and RNA extracted using BCP phase partitioning and isopropanol. Samples were DNase digested (Ambion), and cDNA was synthesized using random hexamers and run at a final concentration of 100 ng/ml. The following 6FAM dye-labeled probes and primers were designed: for the murine NF-κB probe, 5′-6[FAM]CGCTTTCGGAGGTGCTTTCGCAG[BHQ], primer forward, 5′ GGCGGCACGTTTTACTCTTT 3′, and primer reverse, 5′ CCGTCTCCAGGAGGTTAATGC 3′; for the IκBα probe, 5′-6[FAM]TGCACTTGGCAATCATCCACGAAGA[BHQ] 3′, primer forward, 5′ CGGAGGACGGAGACTCGTT 3′, and primer reverse, 5′ CTTCCATGGTCAGCGGCTT 3′; for the IKKα probe, 5′-6[FAM]CGTGTTCTCAAGGAGCTGTTTGGTCACC[BHQ] 3′, primer forward, 5′ TGCACACCGTGCAGAGTCA 3′, and primer reverse, 5′ TGCTTGCAGCCCAACAACT 3′; for the PGK primer forward, 5′ CTGTGGTACTGAGAGCAGCAAGA 3′, and primer reverse, 5′ CAGGACCATTCCAAACAATCTG 3′; and for the β-actin primer forward, 5′ AGGCCCAGAGCAAGAGAGG 3′, primer reverse: 5′ TACATGGCTGGGGTGTTGAA 3′. Afterwards they were run at 250 nM (probe) and 900 nM (forward/reverse primers) per reaction. Threshold cycles were quantified relative to β-actin.
NF-κB activity assays
Whole cell lysates were prepared as described earlier (Quantification of HIF-1α…protein section) and NF-κB p50 and p65 activities were measured using the Active Motif TransAm NF-κB Family ELISA, with bound oligonucleotide containing the NF-κB consensus binding site, as recommended by the manufacturer (Active Motif Europe).
Cytokine analysis
Supernatants were collected at 6 and 20 h by centrifugation, and MIP-1β, IL-8, IL-6, IL-1β, GM-CSF, TNF-α, and MIF releases were measured using duo set ELISA kits according to the manufacturer's instructions (R&D Systems). Luminex analysis of supernatants was performed by Rules-Based Medicine, INC.
Statistical analysis
Gene array data were analyzed for statistical significance after normalization using a Bayesian prior modified t test (31). All other data are expressed as mean ± SEM, and significance determined by one-way analysis of variance with a post-test Tukey of P < 0.05.
Acknowledgments
We thank C. Pugh for providing the DMOG, D. Peet for providing the FIH antibody, A. Evans for support with the gene array experiments, J. Skepper for EM preparation, and J. Deighton for help with ELISA conditions. We are also grateful to Dr. K. Smith for helpful discussions.
This work was supported by the Medical Research Council (to S.R. Walmsley), Raymond and Beverly Sackler Studentship Fund (to S.R. Walmsley), Wellcome Trust (to E.R. Chilvers and C. Peyssonnaux), Wellbeing (to C. Peyssonnaux), and Papworth Hospital National Health Service Trust.
E.R. Chilvers is a recipient of noncommercial grants from AstraZeneca, UK and Boehringer Ingelheim, UK; all other authors have no conflicting financial interests.
Abbreviations used: DFO, desferrioxamine; DMOG, dimethyloxaloylglycine; FIH, factor inhibiting HIF; GP3DH, glyceraldehyde 3-phosphate dehydrogenase; HIF, hypoxia- inducible factor; IKKα, IκB kinase-α; IκBα, inhibitor of κB; MIF, macrophage migration inhibitory factor; MIP-1β, macrophage inflammatory protein-1β; PI3-kinase, phosphoinositide 3-kinase.
References
- 1.Nunn, J.F. 1993. Nunn's Applied Respiratory Physiology. Cambridge University Press, Cambridge, UK. 658 pp.
- 2.Ellis, G.A., S.E. Edmonds, K. Gaffney, R.B. Williams, and D. Blake. 1994. Synovial tissue oxygenation profile in inflamed and non-inflamed knee joints. Br. J. Rheumatol. 33:172. [Google Scholar]
- 3.Niinikoski, J., T.K. Hunt, and J. Englebert Dunphy. 1972. Oxygen supply in healing tissue. Am. J. Surg. 123:247–252. [DOI] [PubMed] [Google Scholar]
- 4.Levene, P.A., and G.M. Meyer. 1912. The action of leucocytes on glucose. J. Biol. Chem. 11:361–370. [Google Scholar]
- 5.Masson, N., C. William, P. Maxwell, C.W. Pugh, and P.J. Ratcliffe. 2001. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 20:5197–5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ivan, M., K. Kondo, H. Yang, W. Kim, J. Valiando, M. Ohh, A. Salic, J.M. Asara, W.S. Lane, and W.G. Kaelin Jr. 2001. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 292:464–468. [DOI] [PubMed] [Google Scholar]
- 7.Jaakkola, P., D.R. Mole, Y.M. Tian, M.I. Wilson, J. Gielbert, S.J. Gaskell, A.V. Kriegsheim, H.F. Hebestreit, M. Mukherji, C.J. Schofield, et al. 2001. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2 regulated prolyl hydroxylation. Science. 292:468–472. [DOI] [PubMed] [Google Scholar]
- 8.Yu, F., S.B. White, Q. Zhao, and F.S. Lee. 2001. Dynamic, site-specific interaction of hypoxia-inducible factor-1α with the von Hippel-Lindau tumour suppressor protein. Cancer Res. 61:4136–4142. [PubMed] [Google Scholar]
- 9.Mahon, P.C., K. Hirota, and G.L. Semenza. 2001. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15:2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lando, D., D.J. Peet, D.A. Whelan, J.J. Gorman, and M.L. Whitelaw. 2002. Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science. 295:858–861. [DOI] [PubMed] [Google Scholar]
- 11.Sang, N., J. Fang, V. Srinivas, I. Leshchinsky, and J. Caro. 2002. Carboxyl-terminal transactivation activity of hypoxia-inducible factor 1α is governed by a von Hippel-Lindau protein-independent, hydroxylation-regulated association with p300/CBP. Mol. Cell. Biol. 22:2984–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semenza, G.L. 1999. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15:551–578. [DOI] [PubMed] [Google Scholar]
- 13.Cormier-Regard, S., S.V. Nguyen, and W.C. Claycomb. 1998. Adrenomedullin gene expression is developmentally regulated and induced by hypoxia in rat ventricular cardiac myocytes. J. Biol. Chem. 273:17787–17792. [DOI] [PubMed] [Google Scholar]
- 14.Metzen, E., U. Berchner-Pfannschmidt, P. Stengel, J.H. Marxsen, I. Stolze, M. Klinger, W.Q. Huang, C. Wotzlaw, T. Hellwig-Burgel, W. Jelkmann, et al. 2003. Intracellular localisation of human HIF-1 alpha hydroxylases: implications for oxygen sensing. J. Cell Sci. 116:1319–1326. [DOI] [PubMed] [Google Scholar]
- 15.Wenger, R.H. 2002. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2 regulated gene expression. FASEB J. 16:1151–1162. [DOI] [PubMed] [Google Scholar]
- 16.Kotch, L.E., N.V. Iyer, E. Laughner, and G.L. Semenza. 1999. Defective vascularisation of HIF-1α-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev. Biol. 209:254–267. [DOI] [PubMed] [Google Scholar]
- 17.Iyer, N.V., L.E. Kotch, F. Agani, S.W. Leung, E. Laughner, R.H. Wenger, M. Gassmann, J.D. Gearhart, A.M. Lawler, A.Y. Yu, and G.L. Semenza. 1998. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 12:149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cramer, T., Y. Yamanishi, B.E. Clausen, I. Forster, R. Pawlinski, N. Mackman, V.H. Haase, R. Jaenisch, M. Corr, V. Nizet, et al. 2003. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 112:645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haslett, C. 1992. Resolution of acute inflammation and the role of apoptosis in the tissue fate of granulocytes. Clin. Sci. 83:639–648. [DOI] [PubMed] [Google Scholar]
- 20.Savill, J.S., A.H. Wyllie, J.E. Henson, M.J. Walport, P.M. Henson, and C. Haslett. 1989. Macrophage phagocytosis of ageing neutrophils in inflammation: programmed cell death in the neutrophil leads to recognition by macrophages. J. Clin. Invest. 83:865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown, S., I. Heinisch, E. Ross, K. Shaw, C.D. Buckley, and J. Savill. 2002. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 418:200–203. [DOI] [PubMed] [Google Scholar]
- 22.Cox, G., J. Gauldie, and M. Jordana. 1992. Bronchial epithelial cell-derived cytokines (G-CSF and GM-CSF) promote the survival of peripheral blood neutrophils in vitro. Am. J. Respir. Cell Mol. Biol. 7:507–513. [DOI] [PubMed] [Google Scholar]
- 23.Ward, C., E.R. Chilvers, M.F. Lawson, J.G. Pryde, S. Fujihara, S.N. Farrow, C. Haslett, and A.G. Rossi. 1999. NF-κB activation is a critical regulator of human granulocyte apoptosis in vitro. J. Biol. Chem. 274:4309–4318. [DOI] [PubMed] [Google Scholar]
- 24.Mecklenburgh, K.I., S.R. Walmsley, A.S. Cowburn, M. Wiesener, B.J. Reed, P.D. Upton, J. Deighton, A.P. Greening, and E.R. Chilvers. 2002. Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood. 100:3008–3016. [DOI] [PubMed] [Google Scholar]
- 25.Haslett, C., L.A. Guthrie, M.M. Kopamiak, R.B. Johnson, and P.M. Henson. 1985. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am. J. Pathol. 119:101–110. [PMC free article] [PubMed] [Google Scholar]
- 26.Klein, J.B., M.J. Rane, J.A. Scherzer, P.Y. Coxon, R. Kettritz, J.M. Mathiesen, A. Buridi, and K.R. McLeish. 2000. Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J. Immunol. 164:4286–4299. [DOI] [PubMed] [Google Scholar]
- 27.Cowburn, A.S., K. Cadwallader, B. Reed, N. Farahi, and E.R. Chilvers. 2002. Role of PI3-kinase-dependent Bad phosphorylation and altered transcription in cytokine-mediated neutrophil survival. Blood. 100:2607–2616. [DOI] [PubMed] [Google Scholar]
- 28.Hanson, E.S., M.L. Rawlins, and E.A. Leibold. 2003. Oxygen and iron regulation of iron regulatory protein 2. J. Biol. Chem. 278:40337–40342. [DOI] [PubMed] [Google Scholar]
- 29.Mole, D.R., I. Schlemminger, L.A. McNeill, K.S. Hewitson, C.W. Pugh, P.J. Ratcliffe, and C.J. Schofield. 2003. 2-oxoglutarate analogue inhibitors of HIF prolyl hydroxylase. Bioorg. Med. Chem. Lett. 13:2677–2680. [DOI] [PubMed] [Google Scholar]
- 30.Villunger, A., C. Scott, P. Bouillet, and A. Strasser. 2003. Essential role for the BH3-only protein BIM but redundant roles for Bax, Bcl-2, and Bcl-w in the control of granulocyte survival. Blood. 101:2393–2400. [DOI] [PubMed] [Google Scholar]
- 31.Evans, A.L., A.S. Sharkey, S.A. Saidi, C.G. Print, R.D. Catalano, S.K. Smith, and D.S. Charnock-Jones. 2003. Generation and use of a tailored gene array to investigate vascular biology. Angiogenesis. 6:93–104. [DOI] [PubMed] [Google Scholar]
- 32.Graven, K.K., R.F. Troxler, H. Kornfield, M.V. Panchenko, and H.W. Farber. 1994. Regulation of endothelial cell glyceraldehydes-3-phosphate dehydrogenase expression by hypoxia. J. Biol. Chem. 269:24446–24453. [PubMed] [Google Scholar]
- 33.Graven, K.K., Q. Yu, D. Pan, J.S. Roncarati, and H.W. Farber. 1999. Identification of an oxygen responsive enhancer element in the glyceraldehydes-3-phosphate dehydrogenase gene. Biochim. Biophys. Acta. 1447: 208–218. [DOI] [PubMed] [Google Scholar]
- 34.Lu, S., X. Gu, S. Hoestje, and D.E. Epner. 2002. Identification of an additional hypoxia responsive element in glyceraldehydes-3-phosphate dehydrogenase gene promoter. Biochim. Biophys. Acta. 1574:152–156. [DOI] [PubMed] [Google Scholar]
- 35.Walmsley, S.R., A.S. Cowburn, A. Sobolewski, J. Murray, N. Farahi, I. Sabroe, and E.R. Chilvers. 2004. Characterisation of the survival effect of tumour necrosis factor-α in human neutrophils. Biochem. Soc. Trans. 32:456–460. [DOI] [PubMed] [Google Scholar]
- 36.Leek, R.D., K.L. Talks, F. Pezzella, H. Turley, L. Campo, N.S. Brown, R. Bicknell, M. Taylor, K.C. Gatter, and A.L. Harris. 2002. Relation of hypoxia-inducible factor-2 alpha (HIF-2 alpha) expression in tumour-infiltrative macrophages to tumour angiogenesis and the oxidative thymidine phosphorylase pathway in human breast cancer. Cancer Res. 62:1326–1329. [PubMed] [Google Scholar]
- 37.Talks, K.L., H. Turley, K.C. Gatter, P.H. Maxwell, C.W. Pugh, P.J. Ratcliffe, and A.L. Harris. 2000. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumour-associated macrophages. Am. J. Pathol. 157:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenbaum, D.M., M. Michaelson, D.K. Batter, P. Doshi, and J.A. Kessler. 1994. Evidence for hypoxia-induced, programmed cell death of cultured neurons. Ann. Neurol. 36:864–870. [DOI] [PubMed] [Google Scholar]
- 39.Graeber, T.G., C. Osmanian, T. Jacks, D.E. Housman, C.J. Koch, S.W. Lowe, and A.J. Giaccia. 1996. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 379:88–91. [DOI] [PubMed] [Google Scholar]
- 40.Borregaard, N., and T. Herlin. 1982. Energy metabolism of human neutrophils during phagocytosis. J. Clin. Invest. 70:550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chandel, N.S., W.C. Trzyna, D.S. McClintock, and P.T. Schumacker. 2000. Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin. J. Immunol. 165:1013–1021. [DOI] [PubMed] [Google Scholar]
- 42.Lukiw, W.J., A. Ottlecz, G. Lambrou, M. Grueninger, J. Finley, H.W. Thompson, and N.G. Bazan. 2003. Coordinate activation of HIF-1 and NF-κB DNA binding and COX-2 and VEGF expression in retinal cells by hypoxia. Invest. Ophthalmol. Vis. Sci. 44:4163–4170. [DOI] [PubMed] [Google Scholar]
- 43.Digicaylioglu, M., and S.A. Lipton. 2001. Eythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-κB signalling cascades. Nature. 412:641–647. [DOI] [PubMed] [Google Scholar]
- 44.Zhou, J., T. Schmid, and B. Brune. 2003. Tumour necrosis factor-α causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-κB-dependent pathway. Mol. Biol. Cell. 14:2216–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamamoto, Y., U.N. Verma, S. Prajapati, Y.-T. Kwak, and R.B. Gaynor. 2003. Histone H3 phosphorylation by IKK-α is critical for cytokine-induced gene expression. Nature. 423:655–659. [DOI] [PubMed] [Google Scholar]
- 46.Vancurova, I., V. Miskolci, and D. Davidson. 2001. NF-κB activation in tumour necrosis factor α-stimulated neutrophils is mediated by protein kinase Cδ. J. Biol. Chem. 276:19746–19752. [DOI] [PubMed] [Google Scholar]
- 47.Choi, M., S. Rolle, M. Wellner, M.C. Cardoso, C. Scheidereit, F.C. Luft, and R. Kettritz. 2003. Inhibition of NF-κB by a TAT-NEMO-binding domain peptide accelerates constitutive apoptosis and abrogates LPS-delayed neutrophil apoptosis. Blood. 102:2259–2267. [DOI] [PubMed] [Google Scholar]
- 48.Castro-Alcaraz, S., V. Miskolci, B. Kalasapudi, D. Davidson, and I. Vancurova. 2002. NF-κB regulation in human neutrophils by nuclear IκBα: correlation to apoptosis. J. Immunol. 169:3947–3953. [DOI] [PubMed] [Google Scholar]
- 49.Suttmann, H., N. Lehan, A. Bohle, and S. Brandau. 2003. Stimulation of neutrophil granulocytes with Mycobacterium bovis bacillus Calmette-Guerin induces changes in phenotype and gene expression and inhibits spontaneous apoptosis. Infect. Immun. 71:4647–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.VanOtteren, G.M., T.J. Standiford, S.L. Kunkel, J.M. Danforth, and R. Streiter. 1995. Alterations of ambient oxygen tension modulate the expression of tumour necrosis factor and macrophage inflammatory protein alpha from murine alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 13:399–409. [DOI] [PubMed] [Google Scholar]
- 51.Baumann, R., C. Casaulta, S. Dagmar, S. Conus, S. Yousefi, and S. Hans-Uwe. 2003. Macrophage migration inhibitory factor delays apoptosis in neutrophils by inhibiting the mitochondria-dependent death pathway. FASEB J. 17:2221–2230. [DOI] [PubMed] [Google Scholar]
- 52.Bacher, M., J. Schrader, N. Thompson, K. Kuschela, D. Gemsa, G. Weaber, and J. Schlegel. 2003. Up regulation of macrophage migration inhibitory factor gene and protein expression in glial tumour cells during hypoxic and hypoglycaemic stress indicates a critical role for angiogenesis in glioglastoma multiforme. Am. J. Pathol. 162:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]