

Abstract
The mechanisms of homing of endothelial progenitor cells (EPCs) to sites of ischemia are unclear. Here, we demonstrate that ex vivo–expanded EPCs as well as murine hematopoietic Sca-1+/Lin− progenitor cells express β2-integrins, which mediate the adhesion of EPCs to endothelial cell monolayers and their chemokine-induced transendothelial migration in vitro. In a murine model of hind limb ischemia, Sca-1+/Lin− hematopoietic progenitor cells from β2-integrin–deficient mice are less capable of homing to sites of ischemia and of improving neovascularization. Preactivation of the β2-integrins expressed on EPCs by activating antibodies augments the EPC-induced neovascularization in vivo. These results provide evidence for a novel function of β2-integrins in postnatal vasculogenesis.
The term vasculogenesis was originally introduced to describe the de novo formation of new vessels from angioblasts during embryonic development (1). Accumulating evidence suggests that vasculogenesis, mediated by circulating bone marrow–derived endothelial progenitor or hematopoietic stem cells, plays an important role in postnatal neovascularization of adult ischemic tissues (2–7). Human endothelial progenitor cells (EPCs) were initially characterized by the expression of the VEGF receptor 2 (VEGF R2; Flk-1) and a hematopoietic marker such as CD133 (6). EPCs are mobilized from the bone marrow during ischemia (8, 9) or exogenously by stimulation with cytokines such as VEGF and contribute to neovascularization of ischemic tissues (4, 8, 10) or tumors (11). Infusion of EPCs or isolated hematopoietic progenitor cells (e.g., murine Sca-1+/Lin− cells) augmented neovascularization of ischemic myocardium and limbs and improved left ventricular function after myocardial ischemia (12–15). EPCs are preferentially recruited to sites of ischemia and incorporated into vascular structures (2, 4, 8, 12, 16). The mechanisms of EPC homing to sites of ischemia are still unclear. Because integrins are mediating the homing of transplanted hematopoietic stem cells to the bone marrow (17) as well as the recruitment of inflammatory cells to sites of inflammation, we investigated the contribution of integrins and especially of β2-integrins for homing and neovascularization capacity of EPCs and hematopoietic stem cells to areas of ischemia.
Recruitment of inflammatory cells requires a coordinated sequence of multistep adhesive and signaling events, including selectin-mediated rolling, leukocyte activation by chemokines, integrin-mediated firm adhesion and diapedesis (18–22). During firm adhesion of leukocytes to the endothelium, members of the β2-integrin family, LFA-1 (αLβ2, CD11a/CD18), Mac-1 (αMβ2, CD11b/CD18), and p150,95 (αXβ2, CD11c/CD18), as well as β1-integrins on leukocytes interact with endothelial counterligands such as ICAM-1, VCAM-1, and surface-associated fibrinogen. Mac-1 also regulates leukocyte adhesion to provisional matrix substrates including fibrinogen, which is deposited at sites of inflammation and injury upon increased vascular permeability and damage (19, 20, 23). Because β2-integrins are strongly expressed on EPCs, we studied the role of the β2-integrins for homing and neovascularization capacity of peripheral blood–derived cultivated human EPCs, bone marrow–derived murine hematopoietic Sca-1+/Lin− as well as VEGF R2+/Lin− progenitor cells. Our results show that β2-integrins mediate the adhesive interactions of EPCs to mature endothelial cells and to extracellular matrix proteins and are critical for chemokine-induced transendothelial migration of EPCs in vitro. In a mouse model of hind limb ischemia, using murine Sca-1+/Lin− hematopoietic progenitor cells from β2-integrin–deficient (β2−/−) mice, we demonstrate that β2-integrins are involved in the homing of hematopoietic progenitor cells to sites of ischemia and are critical for their neovascularization capacity. Alternately, preactivation of the β2-integrins on EPCs by activating antibodies significantly augments the in vivo neovascularization capacity of EPCs, indicating a new therapeutic approach to promote homing of EPCs.
Results
EPCs express active β2-integrins
To characterize the expression of adhesion receptors on EPCs, we used a microarray assay comparing EPCs and human umbilical vein endothelial cells (HUVECs). The endothelial phenotype of the ex vivo–cultivated EPCs was confirmed by immunostaining, FACS analysis, and functional response to shear stress as described previously (12, 24, 25). Strikingly, EPCs expressed mRNA for the β2-integrin subunit and for the corresponding CD11a, CD11b, and CD11c subunits, whereas mature endothelial cells showed only a very low mRNA expression of the β2-integrins (Fig. 1 A). FACS analysis confirmed the surface expression of the β2-integrin (CD18) and the CD11a, CD11b, and CD11c subunits (Fig. 1 B). Coexpression of the endothelial markers von Willebrand factor (vWF) and CD31 on β2-integrin positive EPCs was demonstrated by FACS analysis (Fig. 1 B and not depicted). mRNA expression of eNOS and VE-cadherin confirmed the endothelial phenotype of the EPCs (Fig. 1 C and not depicted). In contrast with EPCs, mature endothelial cells (HUVECs) do not express the β2-integrin subunit (CD18) on the cell surface (unpublished data).
Figure 1.
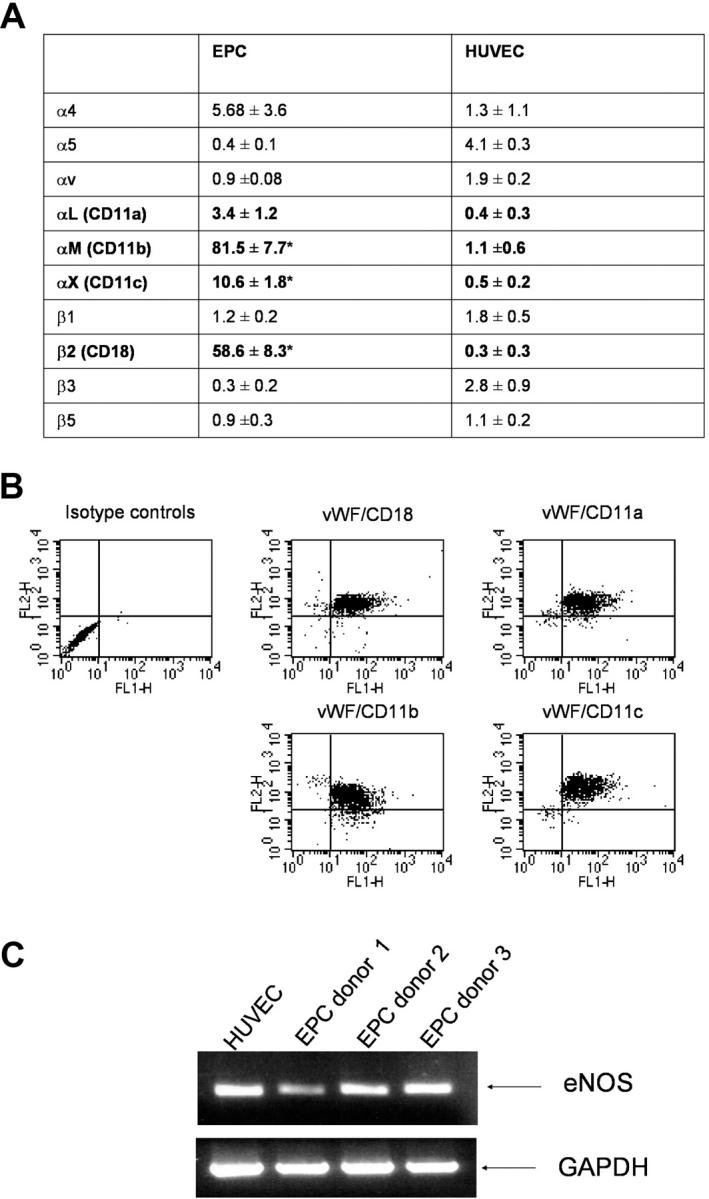
Expression of β2-integrins in human EPCs. (A) mRNA expression profile of integrins in EPCs and HUVECs was assessed by microarray (n = 3 each). *, P < 0.05 versus HUVECs. (B) FACS analysis of EPCs. FL1-H: vWF; FL2-H: integrins (n = 3 each). (C) mRNA expression of eNOS and GAPDH was assessed in EPCs from three different donors.
Because β2-integrins undergo a conformational activation in the presence of divalent cations, such as Mn2+ (26), we investigated whether Mn2+ is capable of activating β2-integrins on the surface of EPCs. For this purpose, we detected activated β2-integrins by using specific antibodies, which recognize activation-dependent epitopes of the CD11a (mAb24) and CD11b (CBRM1/5) subunits (27, 28). Although the stimulation of EPCs with Mn2+ had no effect on the total protein expression of CD11a or CD11b subunits, it enhanced the expression of the activation-dependent epitopes on the CD11a and CD11b subunits (Fig. 2 A). These results demonstrate that EPCs express functionally active β2-integrins on their surface.
Figure 2.

Integrins and EPC adhesion. (A) EPCs were activated with 2 mM MnCl2 for 30 min. Activation of integrins was determined by CBRM1/5 and mAb24 antibodies (left) and expression of CD11a and CD11b was controlled by FACS analysis (right; n = 3). (B) Adhesion of EPCs to 10 ng/ml TNF-α–prestimulated HUVECs was stimulated where indicated with MnCl2. 105 EPCs/well (in 100 μl adhesion buffer) were added to the HUVEC monolayers in the absence or presence of 30 μg/ml of blocking monoclonal anti–β2-integrin antibodies (clone IB4 or mAb 60.3), 30 μg/ml of murine isotype control antibodies, 50 μM of the inhibitory agents cyclic RGD peptide or 50 μM of VLA-4-inhibitor. The total number of EPCs added to the well is set as 100%. *, P < 0.01 versus MnCl2 + isotype antibody; #, P < 0.01 versus isotype control. (C and D) EPCs were treated with 2 mM MnCl2 or 10 μg/ml β2-integrin–activating antibody. Adhesion to fibrinogen- or ICAM-1–coated plates was detected after 20 min in the presence or absence of β2-integrin–inhibitory antibody or isotype control antibodies (n = 4). *, P < 0.05 versus MnCl2 + isotype IgG; #, P < 0.05 versus activating anti-β2 + isotype IgG.
β2-integrins mediate adhesion of EPCs on endothelial cell monolayers and extracellular matrix proteins
To assess the functional capacity of β2-integrins expressed by EPCs, we investigated their role in the adhesion of EPCs on mature endothelial cell monolayers (HUVECs) and on recombinant human ICAM-1 and fibrinogen in a static adhesion assay. Stimulation of EPCs with Mn2+ significantly increased the adhesion of EPCs to TNF-α–prestimulated HUVECs. Addition of an inhibitory β2-integrin antibody significantly blocked EPC adhesion to HUVECs (Fig. 2 B). VLA–4 as well as αVβ3-integrin can also mediate intercellular adhesive interactions by binding to VCAM-1 and PECAM-1, respectively (23, 29). Therefore, a synthetic VLA-4 inhibitor and a cyclic RGD peptide (established inhibitor of the αVβ3- and αVβ5-integrin) were engaged in these studies. However, these inhibitors did not significantly inhibit EPC adhesion to HUVECs (Fig. 2 B). An inhibitory VLA-4 antibody (clone HP2/1), as well as an inhibitory β1-integrin antibody (clone 6S6), also had no effect on the adhesion of EPCs to HUVECs (unpublished data). These results demonstrate that EPC adhesion to endothelial cells is predominantly mediated by β2-integrins expressed on EPCs.
Endothelial ICAM-1 and extracellular matrix-associated fibrinogen are established ligands for the β2-integrins (30–33). Therefore, we investigated whether EPCs are capable of binding to immobilized recombinant human ICAM-1 and fibrinogen via β2-integrins. Indeed, stimulation with either Mn2+ or an activating β2-integrin antibody (KIM185) induced adhesion of EPCs to immobilized human ICAM-1 and fibrinogen (Fig. 2, C and D). Adhesion induced by both stimuli was completely abolished in the presence of an inhibitory β2-integrin antibody (Fig. 2, C and D). In contrast, an inhibitory β1-integrin antibody had no effect on the adhesion of EPCs to human ICAM-1 (unpublished data). These results demonstrate that EPCs bind to fibrinogen and endothelial ICAM-1 in a β2-integrin–dependent manner.
Role of β2-integrins for transmigration of EPCs
We investigated the involvement of β2-integrins in the transendothelial migration of EPCs in a transwell transmigration assay. Chemoattraction of EPCs by MCP-1, SDF-1α, and VEGF significantly increased the transmigration rate of EPCs through HUVEC monolayers (Fig. 3). Addition of an inhibitory β2-integrin antibody (anti-CD18) significantly reduced EPC transmigration, whereas an inhibitory β1-integrin antibody (anti-CD29) and RGD peptides had no effect (Fig. 3). Moreover, an inhibitory VLA-4 antibody did not affect chemokine-induced transendothelial migration of EPCs (unpublished data). Thus, β2-integrins, but not β1-integrins, mediate chemokine and VEGF-induced transendothelial migration of EPCs (Fig. 3).
Figure 3.
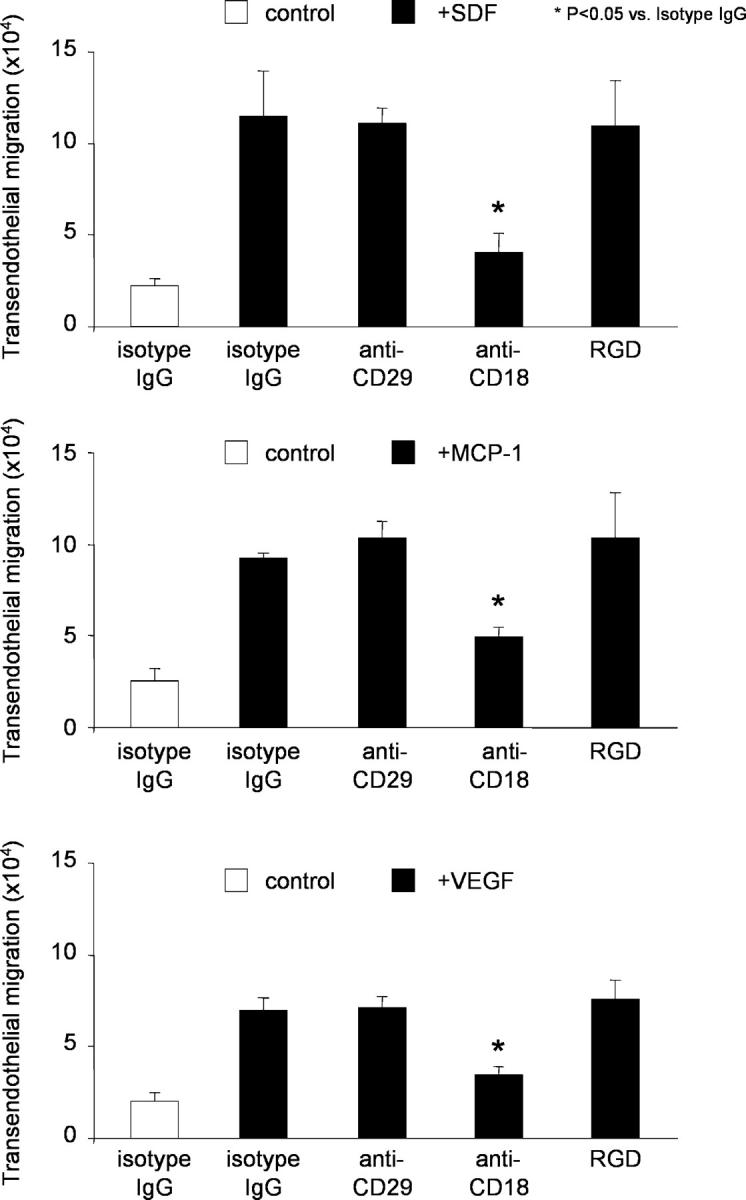
β2-Integrins mediate the chemokine-induced transendothelial migration of EPCs. Transendothelial migration of EPCs was stimulated where indicated with 50 ng/ml MCP-1, 100 ng/ml SDF-1, or 50 ng/ml VEGF for 18 h. EPCs transmigrated in the presence or absence of indicated blocking antibodies or isotype control antibodies (30 μg/ml each) or 10 μg/ml RGD peptides. Data of a typical experiment (triplicates) are presented. Similar results were obtained in at least three separate experiments. *, P < 0.05 versus MCP-1, SDF-1, or VEGF + isotype control IgG.
Role of β2-integrins for neovascularization after ischemia
Next, we determined whether β2−/− mice have an impaired neovascularization capacity. Hind limb ischemia was induced in WT and β2−/− mice. Perfusion was assessed 14 d after induction of ischemia by laser Doppler imaging. β2−/− mice showed a significantly impaired recovery in limb perfusion as compared with WT controls (Fig. 4, A and B), suggesting that β2-integrins contribute to neovascularization after ischemia.
Figure 4.
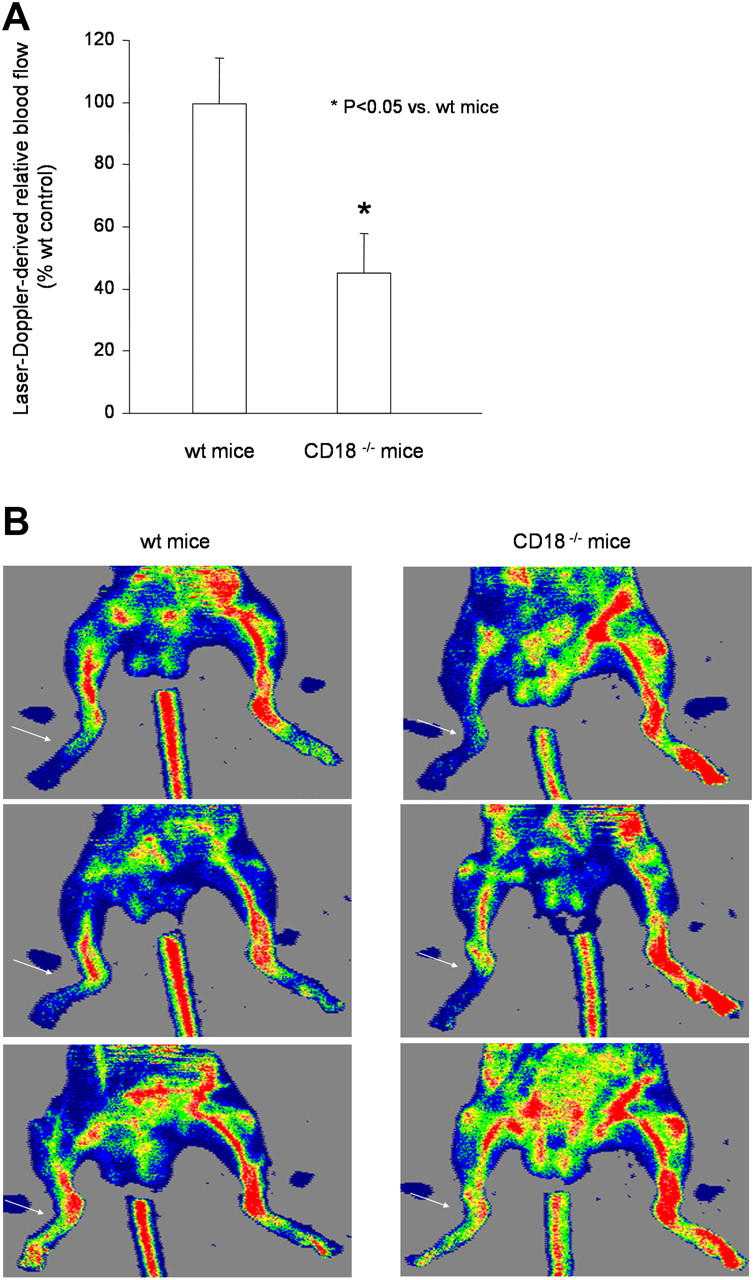
Neovascularization of WT versus CD18 −/− mice after hind limb ischemia. 14 d after induction of hind limb ischemia, perfusion of the ischemic limb was determined by laser Doppler imaging. Quantification (n = 6 per group) (A) and representative images (B). Arrows indicate ischemic linbs.
Role of β2-integrins for the in vivo homing and neovascularization capacity of Sca-1+/Lin− bone marrow cells
To assess the in vivo relevance of β2-integrins for progenitor cell homing to sites of ischemia and progenitor cell–induced neovascularization, we used bone marrow–derived stem/progenitor cells in a mouse model of hind limb ischemia. First, we detected the expression of CD18 on peripheral blood Sca-1+/c-kit+ cells. The β2-integrin subunit CD18 was expressed on >95% of the Sca-1+/c-kit+ cells (Fig. 5 A). Peripheral blood cells from β2−/− mice were used as negative control. As expected, no CD18 staining was detectable (Fig. 5 A). Second, we isolated Sca-1+/Lin− progenitor cells from the bone marrow of wild-type mice by magnetic beads and detected the expression of β2-integrin by FACS analysis. The majority of isolated Sca-1+/Lin− cells (98.5 ± 0.2%) expressed the CD18 surface antigen. Moreover, adhesion of murine bone marrow Lin− progenitor cells to TNF-α–pretreated mature endothelial cells was significantly blocked by β2-blocking antibodies (unpublished data).
Figure 5.
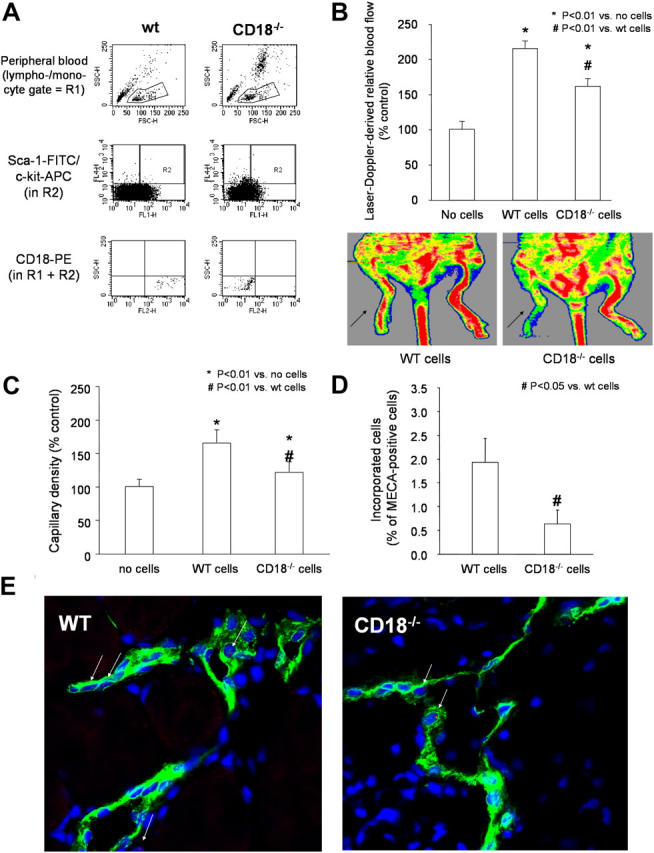
Role of β2-integrins for in vivo homing of Sca-1 + /Lin − stem/progenitor cells and neovascularization capacity. (A) Peripheral blood was gated on the lymphocyte/monocyte fraction (R1) and stained for Sca-1–FITC (FL1) and c-Kit–allophycocyanin (APC; FL4). Gate R2 defines cells, which are Sca-1+c-Kit+. Cells detected in both R1 and R2 are analyzed for expression of CD18-PE (FL2). Expression of CD18 in Sca-1+/c-Kit+ cells was deficient in CD18−/− mice. A representative FACS analysis is shown. (B and C) A total of 105 male Sca-1+/Lin− cells were isolated from bone marrow and infused intravenously into female nude mice 24 h after induction of hind limb ischemia. Laser Doppler-derived blood flow (arrows indicate ischemic limbs) (B) and capillary density (C) was determined after 2 wk (n ≥ 5 per group). (D and E) Incorporation of Y-chromosome positive injected murine Sca-1+/Lin− cells isolated from male wild-type or CD18−/− mice in female recipients after hind limb ischemia (n = 4 per group; n = 10 images per mouse were analyzed). Representative images (E) stained for the panendothelial marker MECA-32 (FITC, green), nuclei (Sytox; blue), and Y-chromosome (Cy3-labeled murine probe, red). Arrows indicate Y-chromosome positive cells.
The functional activity of the stem/progenitor cells to improve neovascularization was assessed by intravenous infusion of Sca-1+/Lin− bone marrow cells derived from either wild-type or β2−/− mice into athymic mice 24 h after induction of limb ischemia. After 14 d, transplantation of Sca-1+/Lin− bone marrow cells from wild-type mice significantly enhanced the recovery of blood flow of the ischemic hind limbs of athymic mice as compared with ischemic hind limbs from untreated athymic mice (Fig. 5 B). In contrast, β2−/− Sca-1+/Lin− bone marrow cells were significantly less effective for improving recovery of limb perfusion as compared with wild-type Sca-1+/Lin− bone marrow cells (Fig. 5 B).
Moreover, histological evaluation of ischemic hind limbs of athymic mice 14 d after cell infusion revealed a significantly lower capillary density in mice receiving β2−/− Sca-1+/Lin− bone marrow cells compared with mice receiving wild-type cells (Fig. 5 C). Furthermore, the number of incorporated male Sca-1+/Lin− cells in female recipients was determined by fluorescence in situ hybridization for the murine Y-chromosome of the infused male cells. The number of incorporated Y-chromosome positive cells was significantly lower for the infusion of β2−/− cells as compared with the infusion of wild-type cells (Fig. 5, D and E).
Similar results were obtained when using isolated VEGF R2+/Lin− bone marrow–derived cells. The majority of VEGF R2+/Lin− cells expressed CD18 (89 ± 9.6%). Moreover, VEGF R2+/Lin− cells derived from CD18−/− mice cells showed a significantly reduced capacity to augment blood flow after ischemia as compared with WT cells (WT 180 ± 15% of untreated control mice; CD18−/− cells: 125 ± 7% of untreated control mice). These results indicate that the β2-integrins are involved in the homing of progenitor cells to ischemic tissues and their neovascularization capacity.
Activation of the β2-integrins improves in vivo homing and neovascularization capacity of EPCs
Because β2-integrins are involved in the homing of EPCs, we investigated whether preactivation of β2-integrins may improve homing and neovascularization capacity of human EPCs in the mouse model of hind limb ischemia. Ex vivo–expanded human EPCs isolated from peripheral blood were pretreated with the β2-integrin–activating antibody (KIM 185), which was shown before to enhance β2-integrin–dependent adhesion of EPCs to endothelial ICAM-1 or fibrinogen (Fig. 2), and were subsequently infused into athymic mice. To be able to detect an increase in progenitor cell–mediated neovascularization, we used a reduced number of EPCs (105 cells), which is lower than previously published numbers (5 × 105 EPCs; reference 25), to yield a 50% improvement of neovascularization as compared with untreated mice.
Preincubation of the EPCs with the activating β2-integrin antibody resulted in a significantly enhanced neovascularization capacity of infused EPCs in comparison with control antibody-treated EPCs as assessed by laser Doppler imaging (Fig. 6 A). Incorporation of human EPCs was detected by confocal microscopy using antibodies directed against human HLA and the endothelial marker protein vWF (Fig. 6, B–D). Incorporation of β2-activating antibody-treated EPCs into the ischemic muscle was increased in comparison with control antibody-treated EPCs (Fig. 6, B and C). Moreover, the numbers of capillaries and small arterioles (20–50 μm) were significantly augmented in mice treated with preactivated EPCs (Fig. 6, E and F; capillary density: EPC + control mAb: 0.77 ± 0.10 capillaries per myocyte; EPC + activating mAb: 1.32 ± 0.09; P = 0.003). Thus, an external activation of the β2-integrins by an activating antibody before infusion is capable of improving the neovascularization capacity of EPCs.
Figure 6.
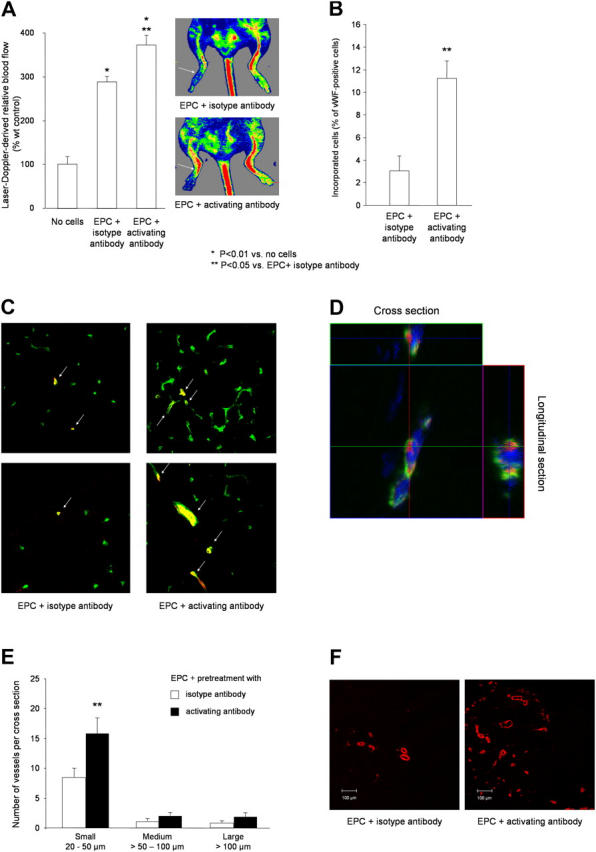
Activation of β2-integrins improves in vivo homing and neovascularization capacity of EPCs. (A) EPCs were pretreated with β2-integrin–activating antibody or isotype control antibody (20 μg/ml each) for 30 min, and 105 cells were injected intravenously in nude mice 24 h after induction of hind limb ischemia. Perfusion was determined by laser Doppler imaging after 2 wk (n = 6 per group). (B–D) Incorporation of human EPCs (stained for HLA-ABC; Alexa 555, red) into vessels (endothelial marker CD31-FITC, green) after pretreatment with isotype control antibody or activating β2-integrin antibody. Double positive cells were quantified (B) and representative overviews (C) and high power images (D) are shown from 10 sections provided by five mice per group. (E and F) Conductance vessels were identified by staining for smooth muscle α-actin using a Cy3-labeled mouse monoclonal antibody (red). The number of small (20–50 μm), medium (>50–100 μm), and large vessels (>100 μm) was counted separately. Data are mean ± SEM (n = 5). (A, B, and E) *, P < 0.05 versus no cells; **, P < 0.05 versus isotype Ab-treated EPCs.
Discussion
The data of the present paper underscore the importance of β2-integrins for the proangiogenic activity of EPCs and bone marrow–derived progenitor cells. Specifically, our investigations revealed the following: (a) EPCs as well as hematopoietic stem/progenitor cells express β2-integrins; (b) β2-integrins expressed on EPCs can be activated by Mn2+ and can mediate the adhesion of EPCs to mature endothelial cells, to recombinant human ICAM-1, and to fibrinogen and the chemokine-induced transendothelial migration of EPCs; (c) β2−/− animals display a neovascularization defect in the model of hind limb ischemia; (d) β2-integrins are involved in the in vivo homing of progenitor cells to sites of ischemia and their vascular integration and significantly contributed to the neovascularization capacity of infused bone marrow Sca-1+/Lin− or VEGF R2+/Lin− progenitor cells in the mouse model of hind limb ischemia as demonstrated using the β2−/− progenitor cell populations; (e) stimulation of ex vivo–expanded human EPCs by preincubation with an activating β2-integrin antibody significantly enhanced the homing of EPCs to sites of ischemia and EPC-induced neovascularization. Therefore, the present paper unravels a novel function of the β2-integrin subunit CD18 for neovascularization exceeding its well-known function in innate and adaptive immune responses.
Increasing evidence suggests that β2-integrins are not only expressed on differentiated leukocytes but also on hematopoietic stem/progenitor cells (34, 35). Our in vitro data suggest that β2-integrins expressed on EPCs mediate homing functions such as endothelial adhesion and transmigration. Moreover, β2-integrins contribute to the in vivo homing of bone marrow–derived progenitor cells to ischemic tissue. In line with these data, it has been reported previously that β2-integrins mediate adhesion and transmigration of hematopoietic stem/progenitor cells (36–38). In a recent paper assessing in vivo homing of embryonic EPCs derived from cord blood, the circulating cells arrested within tumor microvessels extravasated into the interstitium and incorporated into neovessels, suggesting that adhesion and transmigration are involved in the recruitment of EPCs to sites of tumor angiogenesis (39). Thus, it is conceivable to speculate that ex vivo–expanded adult EPCs and hematopoietic stem/progenitor cells may engage similar pathways for recruitment to sites of ischemia and incorporation into newly forming vessels.
Our in vivo data provide the first evidence for a direct participation of β2-integrins in neovascularization processes and particularly in stem/progenitor cell–mediated, ischemia-induced vasculogenesis. Adamis and colleagues previously highlighted the role of β2-integrins for corneal and choroidal angiogenesis induced by injury (40, 41). In both studies, β2−/− mice displayed a reduced inflammation-associated angiogenic response after injury and these effects were associated with reduced inflammatory cell infiltrates (40, 41). Yet, no incorporation of leukocytes into new vessels was reported in these studies. Moreover, the same group demonstrated that, in the case of retinal ischemia, leukocyte–endothelial cell interactions contribute to the development of ischemia by inducing vascular obliteration via Fas ligand–mediated endothelial cell apoptosis (42). In contrast with these findings, our data provide evidence that, during hind limb ischemia, intravenous infusion of bone marrow hematopoietic progenitor cells leads to incorporation of the transplanted cells in newly formed vessels and to improvement of neovascularization in an at least partially β2-integrin–dependent manner. As opposed to EPCs, infusion of inflammatory cells, such as monocytes/macrophages, had only a slight if any effect on the neovascularization of ischemic limbs in the model of hind limb ischemia in athymic mice (25). Thus, our results support a novel direct function of β2-integrins in progenitor cell–induced vasculogenesis during ischemia, which is distinct from the indirect role of β2-integrins in the inflammation-associated angiogenesis described by Adamis and colleagues (40, 41). Because the β2−/− mice display no defect in the mobilization of progenitor cells (43), the neovascularization defect in the β2−/− mice in the model of hind limb ischemia is most conceivably mediated by a homing defect of progenitor cells into ischemic tissue.
Interestingly, our present data indicate that the recruitment of hematopoietic progenitor cells to sites of ischemia is mediated at least in part by different mechanisms compared with the homing of infused cells into the bone marrow of lethally irradiated recipient mice, which is predominantly mediated via α4β1 (17, 43). In this context, β2-integrins only act in a synergistic manner together with the α4β1-integrin. Our finding that β2-integrin deficiency does not completely block homing and neovascularization improvement after infusion of Sca-1+/Lin− bone marrow cells suggests that other mechanisms may additionally be involved in these processes. We cannot exclude that α4β1-integrin partially compensates for the lack of β2-integrin during in vivo homing of Sca-1+/Lin− bone marrow cells. Interestingly, the homing of inflammatory cells during pneumonia or myocardial ischemia in β2−/− mice is mediated by the α4β1-integrin (44, 45). Moreover, the initial cell arrest of embryonic progenitor cell homing during tumor angiogenesis was suggested to be mediated by E- and P-selectin and P-selectin glycoprotein ligand-1 (39). Yet, it is important to underscore that this work was performed with embryonic EPCs, whereas we used adult EPCs and bone marrow stem/progenitor cells. It is likely that different cell types may use distinct mechanisms for homing to sites of ischemia. In addition, it is well established that interactions of selectins with selectin–ligands mediate the rolling of cells on the surface of endothelial cells as the initial step of homing (21). Further studies are needed to elucidate a potentially synergistic role of other adhesion molecules and their counterligands for the multistep recruitment process of adult endothelial progenitor and stem cells to ischemic tissue.
Regardless of potentially additive mechanisms involved in the recruitment of stem/progenitor cells to areas of ischemia, our data clearly demonstrate that preincubation of EPCs with a β2-integrin–activating antibody markedly enhanced the incorporation of transplanted EPCs in vessels and the neovascularization of ischemic limbs. The peripheral blood–derived EPCs used in the present paper are already used in clinical trials to improve neovascularization in patients with ischemic heart diseases (15). Thus, our results could have important clinical implications as they disclose a mechanism to enhance homing of EPCs and, thereby, improve neovascularization capacity of infused EPCs.
In summary, the present paper demonstrates for the first time a critical role of β2-integrins in vitro and in vivo for homing and neovascularization capacity of endothelial progenitor and hematopoietic progenitor cells. Moreover, our results show that activation of β2-integrins appears to be a feasible and promising tool to improve the efficacy of EPC-induced neovascularization. A better understanding of the homing mechanisms of EPCs may lead to the development of new therapeutic strategies for improvement of vasculogenesis in patients with ischemic diseases.
Materials and Methods
Cell culture
Mononuclear cells (MNCs) were isolated by density–gradient centrifugation with Ficoll from peripheral blood of healthy human volunteers as described previously (46). Immediately after isolation, total MNCs (8 × 106 cells/ml medium; cell density 2.5 × 106 cells/cm2) were plated on culture dishes coated with 10 μg/ml human fibronectin (Sigma-Aldrich) and maintained in endothelial basal medium (Cambrex) supplemented with 1 μg/ml hydrocortisone, 12 μg/ml bovine brain extract, 50 μg/ml gentamycin, 50 ng/ml amphotericin B, 10 ng/ml epidermal growth factor, and 20% FCS. After 3 d, nonadherent cells were removed, and adherent cells were incubated in medium for another 24 h before initiation of the experiments. EPCs were characterized by dual staining for 1,1′–dioctadecyl–3,3′,3′–tetramethylindo-carbocyanine–labeled acetyl low-density lipoprotein, lectin, and expression of endothelial markers KDR, VE-cadherin, and vWF (25).
Sca-1+/Lin− cells were purified from BM MNCs from wild-type and CD18−/− mice by negative selection using a cocktail of biotinylated antibodies to lineage markers (Lineage cell depletion kit, mouse; Miltenyi Biotec) for 10 min at 4°C followed by antibiotin microbeads for 15 min (Miltenyi Biotec). The Lin− BM cells were incubated with anti–Sca-1 microbeads (Miltenyi Biotec) for 15 min and Sca-1+/Lin− BM cells were collected (7). To obtain VEGFR2+ Lin− cells, Lin− cells were incubated with biotinylated Flk-1 antibodies (DSB-X biotin protein labeling kit; Molecular Probes; antibody was obtained from BD Biosciences) for 30 min at 4°C followed by antibiotin microbeads for 15 min.
HUVECs were purchased from Cambrex and cultured in endothelial basal medium supplemented with 1 μg/ml hydrocortisone, 12 μg/ml bovine brain extract, 50 μg/ml gentamycin, 50 ng/ml amphotericin-B, 10 ng/ml epidermal growth factor, and 10% FCS until the third passage. After detachment with trypsin, cells (4 × 105 cells) were grown in 6-cm cell culture dishes or 96-well plates for at least 18 h as described previously (47).
Oligonucleotide microarrays, FACS
10 μg of total RNA was hybridized to the HG-U95Av2 microarray (9670 human genes; Affymetrix, Inc.). The standard protocol used for sample preparation and microarray processing is available from Affymetrix, Inc. Expression data were analyzed using Microarray Suite version 5.0 (Affymetrix, Inc.) and GeneSpring version 4.2 (Silicon Genetics). 3 × 105 human EPCs, peripheral blood, or isolated Sca-1+/Lin− cells were incubated for 30 min at 4°C with FITC- or PE-labeled antibodies (anti-CD11a, -CD11b, -CD11c, -CD18, –Sca-1, and –c-kit; BD Biosciences; anti-vWF was obtained from Acris) or CBRM1/5-antibody (BD Biosciences) for 30 min at 37°C. The mAb24 antibody (provided by N. Hogg, Cancer Research UK London Research Institute, London, England, UK) was incubated for 30 min at 4°C and detected with a secondary FITC-labeled goat anti–mouse antibody (DakoCytomation). Surface expression was quantified using a FACS Calibur (BD Biosciences).
Adhesion, transmigration experiments
Cell–cell adhesion
Cell–cell adhesion was performed as described previously (48, 49). Confluent HUVEC monolayers were stimulated with TNF-α (Sigma-Aldrich) for 24 h. Ex vivo–expanded EPCs were stained with Cell Tracker green-CMFDA (Molecular Probes) and were resolved in adhesion buffer (150 mM NaCl, 20 mM Hepes, 2 mM MgCl2, 0.05% BSA, pH 7.4). A total of 105 EPCs/well (in 100 μl adhesion buffer) was added to the HUVEC monolayers in the absence or presence of blocking monoclonal β2-integrin antibodies (clone IB4; Qbiogene; or mAb 60.3; J. Harlan, University of Washington, Seattle, WA), murine isotype control antibodies (Qbiogene), inhibitory agents cyclic RGD peptide or VLA-4 inhibitor (4-[{2-methyl-phenyl}aminocarbonyl]aminophenyl)acetyl-fibronectin-CS-1 fragment (1980–1983). After 20 min of incubation (37°C), the plates were washed twice with adhesion buffer at room temperature to remove nonadherent cells. Adherent cell tracker green-labeled EPCs were quantified in triplicates on a fluorescence plate reader (Fluostat; BMG Lab Technologies).
Cell–matrix adhesion
Cell–matrix adhesion was performed as described previously (48, 49). 96-well plates were coated overnight (4°C) with 10 μg/ml human fibrinogen (Enzyme Research Laboratories) or soluble recombinant human ICAM-1 (Bender MedSystems) and blocked with 1% (wt/vol) BSA for 1 h at room temperature. Ex vivo–expanded human EPCs in adhesion buffer were seeded at 1.2 × 105 cells/well in 100 μl in the absence or presence of 2 mM MnCl2 or activating human β2-integrin antibody (clone KIM185; M. Robinson, Celltech Ltd., Slough, England, UK) and were incubated with blocking β2-integrin mAb (clone IB4 or mAb 60.3) or murine isotype control antibodies (Qbiogene) for 20 min at 37°C. After removal of nonadherent cells by two washing steps, adhesion was quantified in triplicates by counting adherent cells in five randomly selected fields per well (magnification, 20; Axiovert 100; Carl Zeiss MicroImaging, Inc.).
Transmigration
Transendothelial migration was performed as described previously (50) using 6.5-mm transwell filters with 8-μm pore size (Costar). After inserts were coated with 0.2% gelatin (Sigma-Aldrich), HUVECs were seeded on transwell filters and cultivated for 48 h before the experiments were performed in a humidified atmosphere (37°C, 5% CO2). At the beginning of the experiment, 600 μl of migration assay medium (serum-free RPMI 1640 in the absence or presence of MCP-1, SDF1α, or VEGF; R&D Systems) was added to the lower compartment of the transwell system. EPCs (5 × 105 in 100 μL) were added to the top compartment in the presence or absence of 30 μg/ml of blocking anti–β2-integrin antibody (mAb 60.3), anti–β1-integrin antibody (clone 6S6; Chemicon), anti–α4-integrin antibody (clone HP2.1; Immunotech), murine isotype control antibodies (Qbiogene), or RGD peptides. After 18 h at 37°C, the number of cells transmigrated to the bottom compartment was quantified in duplicates with a cell counter (CASY-Counter; Schärfe-System). All inserts were fixed and stained to confirm the confluence of the endothelial monolayer.
Animal experiments
Mice.
8-wk-old β2−/− mice and their age-matched wild-type littermates (either 129/Sv or C57BL/6J) were generated as described previously (51). All mice were genotyped by Southern blot analysis (51) and maintained under pathogen-free conditions. Athymic NMRI nude mice (6–8 wk) were obtained from The Jackson Laboratory. The animal experiments were approved from the Regional Board of Land Hessen, Germany.
Model of hind limb ischemia
The proximal femoral artery, including the superficial and the deep branch as well as the distal saphenous artery, were ligated. In transplantation experiments, progenitor cells were intravenously injected in nude mice 24 h after induction of limb ischemia. Human EPCs were pretreated with 20 μg/ml activating β2-integrin antibody (clone KIM 185) or isotype control antibody for 30 min at 37°C and washed twice to remove unbound antibodies before injection (105 EPCs/mouse). In some experiments, sex-mismatched murine Sca-1+/Lin− or Flk-1+/Lin− bone marrow cells from male β2−/− or wild-type mice were used. After 2 wk, we calculated the ischemic (right) versus normal (left) limb blood flow ratio using a Laser Doppler blood flow imager (Moor Instruments).
Histology.
The capillary density and the number and size of conductant vessels in the semimembraneous and adductor muscles were determined using 8-μm cryosections. Endothelial cells were identified with the panendothelial marker MECA-32 followed by donkey anti–rat IgG Alexa488 or CD31-FITC (BD Biosciences). Injected human EPCs were identified by costaining for HLA-ABC (allophycocyanin labeled; BD Biosciences) and vWF (Acris). Male murine BM-derived cells were identified by fluorescence in situ hybridization for the murine Y-chromosome (Cy3-labeled probe: Cambio; reference 7). Nuclei were stained with Sytox (Molecular Probes). Images were obtained by confocal microscopy (LSM 510; Carl Zeiss MicroImaging, Inc.).
Statistical analysis
Continuous variables are expressed as mean ± SD or SEM. Comparisons between groups were analyzed by Student's t test (two-sided) or analysis of variance with Bonferroni adjustment for experiments with more than two subgroups (SPSS 11.5 software). p-values <0.05 were considered as statistically significant.
Acknowledgments
We thank M. Muhly-Reinholz, T. Röxe, and M. Näher for their excellent technical assistance. Moreover, we thank Drs. N. Hogg, J. Harlan, and M. Robinson for providing the antibodies mAb24, mAb60.3, and KIM185, respectively.
This work was supported by the Forschergruppe 501 (He 3044/2-2 to C. Heeschen) and the Alfried Krupp-Stiftung (to S. Dimmeler). K. Sasaki was in part supported by the Japan Heart Foundation/Bayer Yakuhin Research Grant Abroad. The work of K. Scharffetter-Kochanek was funded in party by the Collaborative Research Center SFB497-C7, Ulm.
The authors have no conflicting financial interests.
T. Chavakis' present address is Experimental Immunology Branch, National Cancer Institute/National Institutes of Health, Bethesda, MD 20892.
Abbreviations used: β2−/−, β2-integrin–deficient; EPC, endothelial progenitor cell; HUVEC, human umbilical vein endothelial cell; MNC, mononuclear cell; vWF, von Willebrand factor.
References
- 1.Carmeliet, P. 2000. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6:389–395. [DOI] [PubMed] [Google Scholar]
- 2.Asahara, T., T. Murohara, A. Sullivan, M. Silver, R. van der Zee, T. Li, B. Witzenbichler, G. Schatteman, and J.M. Isner. 1997. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 275:964–967. [DOI] [PubMed] [Google Scholar]
- 3.Shi, Q., S. Rafii, M.H. Wu, E.S. Wijelath, C. Yu, A. Ishida, Y. Fujita, S. Kothari, R. Mohle, L.R. Sauvage, et al. 1998. Evidence for circulating bone marrow-derived endothelial cells. Blood. 92:362–367. [PubMed] [Google Scholar]
- 4.Asahara, T., H. Masuda, T. Takahashi, C. Kalka, C. Pastore, M. Silver, M. Kearne, M. Magner, and J.M. Isner. 1999. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 85:221–228. [DOI] [PubMed] [Google Scholar]
- 5.Grant, M.B., W.S. May, S. Caballero, G.A. Brown, S.M. Guthrie, R.N. Mames, B.J. Byrne, T. Vaught, P.E. Spoerri, A.B. Peck, and E.W. Scott. 2002. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat. Med. 8:607–612. [DOI] [PubMed] [Google Scholar]
- 6.Rafii, S., and D. Lyden. 2003. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat. Med. 9:702–712. [DOI] [PubMed] [Google Scholar]
- 7.Aicher, A., C. Heeschen, C. Mildner-Rihm, C. Urbich, C. Ihling, K. Technau-Ihling, A.M. Zeiher, and S. Dimmeler. 2003. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med. 9:1370–1376. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi, T., C. Kalka, H. Masuda, D. Chen, M. Silver, M. Kearney, M. Magner, J.M. Isner, and T. Asahara. 1999. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat. Med. 5:434–438. [DOI] [PubMed] [Google Scholar]
- 9.Shintani, S., T. Murohara, H. Ikeda, T. Ueno, T. Honma, A. Katoh, K. Sasaki, T. Shimada, Y. Oike, and T. Imaizumi. 2001. Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation. 103:2776–2779. [DOI] [PubMed] [Google Scholar]
- 10.Asahara, T., T. Takahashi, H. Masuda, C. Kalka, D. Chen, H. Iwaguro, Y. Inai, M. Silver, and J.M. Isner. 1999. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 18:3964–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyden, D., K. Hattori, S. Dias, C. Costa, P. Blaikie, L. Butros, A. Chadburn, B. Heissig, W. Marks, L. Witte, et al. 2001. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 7:1194–1201. [DOI] [PubMed] [Google Scholar]
- 12.Kalka, C., H. Masuda, T. Takahashi, W.M. Kalka-Moll, M. Silver, M. Kearney, T. Li, J.M. Isner, and T. Asahara. 2000. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc. Natl. Acad. Sci. USA. 97:3422–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kocher, A.A., M.D. Schuster, M.J. Szabolcs, S. Takuma, D. Burkhoff, J. Wang, S. Homma, N.M. Edwards, and S. Itescu. 2001. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat. Med. 7:430–436. [DOI] [PubMed] [Google Scholar]
- 14.Kawamoto, A., H.C. Gwon, H. Iwaguro, J.I. Yamaguchi, S. Uchida, H. Masuda, M. Silver, H. Ma, M. Kearney, J.M. Isner, and T. Asahara. 2001. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation. 103:634–637. [DOI] [PubMed] [Google Scholar]
- 15.Assmus, B., V. Schachinger, C. Teupe, M. Britten, R. Lehmann, N. Dobert, F. Grunwald, A. Aicher, C. Urbich, H. Martin, et al. 2002. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE-AMI). Circulation. 106:3009–3017. [DOI] [PubMed] [Google Scholar]
- 16.Aicher, A., W. Brenner, M. Zuhayra, C. Badorff, S. Massoudi, B. Assmus, T. Eckey, E. Henze, A.M. Zeiher, and S. Dimmeler. 2003. Assessment of the tissue distribution of transplanted human endothelial progenitor cells by radioactive labeling. Circulation. 107:2134–2139. [DOI] [PubMed] [Google Scholar]
- 17.Papayannopoulou, T., G.V. Priestley, B. Nakamoto, V. Zafiropoulos, and L.M. Scott. 2001. Molecular pathways in bone marrow homing: dominant role of alpha(4)beta(1) over beta(2)-integrins and selectins. Blood. 98:2403–2411. [DOI] [PubMed] [Google Scholar]
- 18.Muller, W.A., S.A. Weigl, X. Deng, and D.M. Phillips. 1993. PECAM-1 is required for transendothelial migration of leukocytes. J. Exp. Med. 178:449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- 20.Carlos, T.M., and J.M. Harlan. 1994. Leukocyte-endothelial adhesion molecules. Blood. 84:2068–2101. [PubMed] [Google Scholar]
- 21.Muller, W.A. 2002. Leukocyte-endothelial cell interactions in the inflammatory response. Lab. Invest. 82:521–533. [DOI] [PubMed] [Google Scholar]
- 22.Schenkel, A.R., Z. Mamdouh, and W.A. Muller. 2004. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat. Immunol. 5:393–400. [DOI] [PubMed] [Google Scholar]
- 23.Plow, E.F., T.A. Haas, L. Zhang, J. Loftus, and J.W. Smith. 2000. Ligand binding to integrins. J. Biol. Chem. 275:21785–21788. [DOI] [PubMed] [Google Scholar]
- 24.Kawamoto, A., T. Asahara, and D.W. Losordo. 2002. Transplantation of endothelial progenitor cells for therapeutic neovascularization. Cardiovasc. Radiat. Med. 3:221–225. [DOI] [PubMed] [Google Scholar]
- 25.Urbich, C., C. Heeschen, A. Aicher, E. Dernbach, A.M. Zeiher, and S. Dimmeler. 2003. Relevance of monocytic features for neovascularization capacity of circulating endothelial progenitor cells. Circulation. 108:2511–2516. [DOI] [PubMed] [Google Scholar]
- 26.Takagi, J., and T.A. Springer. 2002. Integrin activation and structural rearrangement. Immunol. Rev. 186:141–163. [DOI] [PubMed] [Google Scholar]
- 27.Dransfield, I., and N. Hogg. 1989. Regulated expression of Mg2+ binding epitope on leukocyte integrin alpha subunits. EMBO J. 8:3759–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diamond, M.S., and T.A. Springer. 1993. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 120:545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piali, L., P. Hammel, C. Uherek, F. Bachmann, R.H. Gisler, D. Dunon, and B.A. Imhof. 1995. CD31/PECAM-1 is a ligand for alpha v beta 3 integrin involved in adhesion of leukocytes to endothelium. J. Cell Biol. 130:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altieri, D.C., R. Bader, P.M. Mannucci, and T.S. Edgington. 1988. Oligospecificity of the cellular adhesion receptor Mac-1 encompasses an inducible recognition specificity for fibrinogen. J. Cell Biol. 107:1893–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marlin, S.D., and T.A. Springer. 1987. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1). Cell. 51:813–819. [DOI] [PubMed] [Google Scholar]
- 32.Loike, J.D., B. Sodeik, L. Cao, S. Leucona, J.I. Weitz, P.A. Detmers, S.D. Wright, and S.C. Silverstein. 1991. CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the A alpha chain of fibrinogen. Proc. Natl. Acad. Sci. USA. 88:1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diamond, M.S., D.E. Staunton, A.R. de Fougerolles, S.A. Stacker, J. Garcia-Aguilar, M.L. Hibbs, and T.A. Springer. 1990. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18). J. Cell Biol. 111:3129–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becker, P.S., S.K. Nilsson, Z. Li, V.M. Berrios, M.S. Dooner, C.L. Cooper, C.C. Hsieh, and P.J. Quesenberry. 1999. Adhesion receptor expression by hematopoietic cell lines and murine progenitors: modulation by cytokines and cell cycle status. Exp. Hematol. 27:533–541. [DOI] [PubMed] [Google Scholar]
- 35.Orschell-Traycoff, C.M., K. Hiatt, R.N. Dagher, S. Rice, M.C. Yoder, and E.F. Srour. 2000. Homing and engraftment potential of Sca-1(+)lin(−) cells fractionated on the basis of adhesion molecule expression and position in cell cycle. Blood. 96:1380–1387. [PubMed] [Google Scholar]
- 36.Kollet, O., A. Spiegel, A. Peled, I. Petit, T. Byk, R. Hershkoviz, E. Guetta, G. Barkai, A. Nagler, and T. Lapidot. 2001. Rapid and efficient homing of human CD34(+)CD38(−/low)CXCR4(+) stem and progenitor cells to the bone marrow and spleen of NOD/SCID and NOD/SCID/B2m(null) mice. Blood. 97:3283–3291. [DOI] [PubMed] [Google Scholar]
- 37.Peled, A., V. Grabovsky, L. Habler, J. Sandbank, F. Arenzana-Seisdedos, I. Petit, H. Ben-Hur, T. Lapidot, and R. Alon. 1999. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34(+) cells on vascular endothelium under shear flow. J. Clin. Invest. 104:1199–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peled, A., O. Kollet, T. Ponomaryov, I. Petit, S. Franitza, V. Grabovsky, M.M. Slav, A. Nagler, O. Lider, R. Alon, et al. 2000. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 95:3289–3296. [PubMed] [Google Scholar]
- 39.Vajkoczy, P., S. Blum, M. Lamparter, R. Mailhammer, R. Erber, B. Engelhardt, D. Vestweber, and A.K. Hatzopoulos. 2003. Multistep nature of microvascular recruitment of ex vivo–expanded embryonic endothelial progenitor cells during tumor angiogenesis. J. Exp. Med. 197:1755–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakurai, E., H. Taguchi, A. Anand, B.K. Ambati, E.S. Gragoudas, J.W. Miller, A.P. Adamis, and J. Ambati. 2003. Targeted disruption of the CD18 or ICAM-1 gene inhibits choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 44:2743–2749. [DOI] [PubMed] [Google Scholar]
- 41.Moromizato, Y., S. Stechschulte, K. Miyamoto, T. Murata, A. Tsujikawa, A.M. Joussen, and A.P. Adamis. 2000. CD18 and ICAM-1-dependent corneal neovascularization and inflammation after limbal injury. Am. J. Pathol. 157:1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishida, S., K. Yamashiro, T. Usui, Y. Kaji, Y. Ogura, T. Hida, Y. Honda, Y. Oguchi, and A.P. Adamis. 2003. Leukocytes mediate retinal vascular remodeling during development and vaso-obliteration in disease. Nat. Med. 9:781–788. [DOI] [PubMed] [Google Scholar]
- 43.Papayannopoulou, T., G.V. Priestley, B. Nakamoto, V. Zafiropoulos, L.M. Scott, and J.M. Harlan. 2001. Synergistic mobilization of hemopoietic progenitor cells using concurrent beta1 and beta2 integrin blockade or beta2-deficient mice. Blood. 97:1282–1288. [DOI] [PubMed] [Google Scholar]
- 44.Bowden, R.A., Z.M. Ding, E.M. Donnachie, T.K. Petersen, L.H. Michael, C.M. Ballantyne, and A.R. Burns. 2002. Role of alpha4 integrin and VCAM-1 in CD18-independent neutrophil migration across mouse cardiac endothelium. Circ. Res. 90:562–569. [DOI] [PubMed] [Google Scholar]
- 45.Tasaka, S., S.E. Richer, J.P. Mizgerd, and C.M. Doerschuk. 2002. Very late antigen-4 in CD18-independent neutrophil emigration during acute bacterial pneumonia in mice. Am. J. Respir. Crit. Care Med. 166:53–60. [DOI] [PubMed] [Google Scholar]
- 46.Dimmeler, S., A. Aicher, M. Vasa, C. Mildner-Rihm, K. Adler, M. Tiemann, H. Rutten, S. Fichtlscherer, H. Martin, and A.M. Zeiher. 2001. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J. Clin. Invest. 108:391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chavakis, E., E. Dernbach, C. Hermann, U.F. Mondorf, A.M. Zeiher, and S. Dimmeler. 2001. Oxidized LDL inhibits vascular endothelial growth factor-induced endothelial cell migration by an inhibitory effect on the Akt/endothelial nitric oxide synthase pathway. Circulation. 103:2102–2107. [DOI] [PubMed] [Google Scholar]
- 48.Chavakis, T., A. Bierhaus, N. Al-Fakhri, D. Schneider, S. Witte, T. Linn, M. Nagashima, J. Morser, B. Arnold, K.T. Preissner, and P.P. Nawroth. 2003. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J. Exp. Med. 198:1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chavakis, T., M. Hussain, S.M. Kanse, G. Peters, R.G. Bretzel, J.I. Flock, M. Herrmann, and K.T. Preissner. 2002. Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat. Med. 8:687–693. [DOI] [PubMed] [Google Scholar]
- 50.Rohnelt, R.K., G. Hoch, Y. Reiss, and B. Engelhardt. 1997. Immunosurveillance modelled in vitro: naive and memory T cells spontaneously migrate across unstimulated microvascular endothelium. Int. Immunol. 9:435–450. [DOI] [PubMed] [Google Scholar]
- 51.Scharffetter-Kochanek, K., H. Lu, K. Norman, N. van Nood, F. Munoz, S. Grabbe, M. McArthur, I. Lorenzo, S. Kaplan, K. Ley, et al. 1998. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 188:119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]