

Abstract
The yellow fever vaccine 17D (17D) is safe, and after a single immunizing dose, elicits long-lasting, perhaps lifelong protective immunity. One of the major challenges facing delivery of human vaccines in underdeveloped countries is the need for multiple injections to achieve full efficacy. To examine 17D as a vector for microbial T cell epitopes, we inserted the H-2Kd–restricted CTL epitope of the circumsporozoite protein (CS) of Plasmodium yoelii between 17D nonstructural proteins NS2B and NS3. The recombinant virus, 17D-Py, was replication competent and stable in vitro and in vivo. A single subcutaneous injection of 105 PFU diminished the parasite burden in the liver by ∼70%. The high level of protection lasted between 4 and 8 wk after immunization, but a significant effect was documented even 24 wk afterwards. Thus, the immunogenicity of a foreign T cell epitope inserted into 17D mimics some of the remarkable properties of the human vaccine. Priming with 17D-Py followed by boosting with irradiated sporozoites conferred sterile immunity to 90% of the mice. This finding indicates that the immune response of vaccine-primed individuals living in endemic areas could be sustained and magnified by the bite of infected mosquitoes.
Many human vaccines that are currently in use require multiple injections to be fully effective over the long term. This requirement is a severe hurdle for their utilization, particularly in underdeveloped countries where they are much needed. One notable exception is the yellow fever vaccine using the attenuated YF17D strain (17D). Developed in the 1930s, this live, highly attenuated viral vaccine has been given to hundreds of millions of individuals with minimal risk of severe side effects. Administration of a single dose of this genetically stable vaccine confers excellent protection that persists for 30 or more years, perhaps lifelong (1). The mechanisms of protection are not well understood, but they involve the production of neutralizing antibodies and probably the production of T cell–mediated immune mechanisms (2, 3).
In recent years, the remarkable properties of 17D vaccine have been exploited to create candidate vaccines against other flavivirus-mediated diseases by exchanging the premembrane and envelope genes of 17D with those of Japanese encephalitis, dengue types 1–4, and West Nile viruses. These chimeric viruses replicate to high titers in cell culture, elicit protective immunity in rodents and monkeys (4, 5), and retain or surpass the monkey neurovirulence safety standards set for 17D (6). Phase I human trials for some of these hybrid vaccines have been initiated.
However, the 17D vaccine has not been used as a vector to deliver epitopes from unrelated microbial pathogens. The only exception has been the successful insertion of the immunodominant protective B cell epitope of the circumsporozoite (CS) protein of the human malaria parasite, Plasmodium falciparum, into the envelope protein of 17D. The genomic stability of that recombinant virus, named 17D/8, was confirmed by serial in vitro passages. Immunization of mice with 17D/8 led to long lasting production of antibodies to P. falciparum sporozoites. In addition, immune serum neutralized the infectivity of wild-type YF virus (7).
Here, we studied the immunogenicity of a recombinant 17D virus expressing the protective H-2Kd–restricted CTL epitope of the CS protein of P. yoelii, a rodent malaria parasite (8). In this experimental model, a rapid and quantitative assessment of protective immunity is feasible upon sporozoite challenge of immunized mice. Moreover, the efficacy of different viral vaccine constructs can be compared. This experimental model has provided the basis for the development of various human malaria vaccine candidates that underwent clinical trials (9, 10), as well as some currently undergoing human trials.
Results
Insertion of the malaria CD8+ T cell epitope in the genome of 17D
We inserted a 10-amino acid-long H-2Kd CTL epitope of the CS protein of P. yoelii (SYVPSAEQIL) in 17D polyprotein in frame between the NS2B and NS3 nonstructural proteins (Fig. 1 A). The foreign epitope sequence was flanked by viral protease recognition sites in order to release the epitope in the cytoplasm of the host cell leaving unaltered functional viral replicase proteins. This strategy and the insertion site were chosen based on a previous observation showing that an ovalbumin epitope, inserted in the same site, yielded viable recombinant virus that elicited the production of ova-specific CTL and protection in a rodent tumor model (11).
Figure 1.

Construction and viability of 17D-Py virus. (A) Schematic representation of P. yoelii CTL SYVPSAEQIL epitope insertion into the YF17D genome. The NS2B/NS3 protease recognition sequence was duplicated flanking the CTL epitope. (B) Infectious center assay from SW-13 cells transfected with 17D or 17D-Py RNAs. Plaques were fixed in 7.5% formaldehyde and stained by crystal violet. (C) One-step growth curve of 17D or 17D-Py in SW-13 cells infected at low (0.1 PFU/cell) or high (5 PFU/cell) multiplicity. Virus titers were measured by plaque assay on SW-13 cells as described in Materials and methods. The graphs represent the mean of three independent infections. (D) RT-PCR products of the 2B/3 region amplified from viral RNA recovered after infection of naive SW-13 cells with 17D or 17D-Py1 (in vitro) or from brain homogenate of suckling mice injected i.p. with each virus (in vivo). The fragments were digested with SspI and run on a 5% acrylamide gel. The P. yoelii CTL insert contains an SspI site not present in 17D.
The recombinant construct was designated 17D-Py and the parental 17D2B/3 (referred to as 17D in this paper). The infectivity of RNAs transcribed from linearized plasmid templates was tested by electroporation of human SW-13 cells. Both the 17D-Py and the parental 17D RNAs had similar specific infectivities of ∼5 × 105 PFU/μg RNA (Fig. 1 B). 17D-Py plaques were slightly larger than those of the parental virus. We compared the growth kinetics of 17D-Py and 17D after infecting SW-13 cells at either low (0.1 PFU/cell) or high (5 PFU/cell) multiplicities of infection (MOIs). Interestingly the 17D-Py recombinant exhibited faster growth kinetics at both MOIs than the 17D parent, although similar plateau titers were obtained for both viruses. To assess the stability of the inserted sequence in cell culture, transfection supernatants were used to infect naive SW-13 cells at low MOI (0.1 PFU/cell). The resulting virus-containing supernatant was harvested and analyzed for the presence of the CTL epitope by RT-PCR, restriction digestion, and sequence analysis. As shown in Fig. 1 D, the RT-PCR products were of the expected sizes and the fragment from 17D-Py was digested to completion with SspI, which is present in the inserted P. yoelii sequence. Sequence analysis of the RT-PCR products revealed the expected sequences.
In vivo stability of 17D-Py
Young mice are highly permissive for flavivirus infection and succumb to virus-induced paralysis. To examine the stability of the recombinant virus in vivo, we injected 105 PFU of the recombinant 17D-Py or parental 17D viruses i.p. into suckling mice. All of the mice eventually became paralyzed by 10–14 d after infection. The infectious virus could be recovered from brain homogenates of the moribund animals, with high viral titers ranging from 5 × 105 to 5 × 106 PFU per brain. Viral RNA was recovered from clarified brain homogenates and assayed for the presence of the insert as described above. As found in cell culture, the expected RT-PCR products were obtained, and the 17D-Py product was digested with SspI (Fig. 1 D). In addition, sequencing of the RT-PCR products revealed the expected input sequences. These analyses indicate that the P. yoelii insert is still present in the recombinant virus after multiple cycles of replication in vivo.
A single dose of 17D-Py elicits long-lasting immunity against malaria
Groups of 6–10-wk-old BALB/c (H-2Kd) mice were immunized with a single viral dose by the s.c. route, using 20-fold dilutions of viral doses, from 102 PFU to 106 PFU of 17D-Py per mouse. 2 wk later, the mice were challenged intravenously with 20,000 sporozoites. After 40–42 h, close to the near completion of the exoerythrocytic stage (EEF) cycle, the animals were killed, and the efficacy of immunization was evaluated by enumerating IFNγ-secreting CS-specific CD8+ T cells in the spleen, and by measuring the parasite burden in the liver, as compared with controls immunized with parental 17D (105 PFU). As shown in Fig. 2, and in several subsequent experiments, immunization with 105 PFU elicited ∼67% inhibition of EEF development. Notably, a considerable degree of inhibition (52%) was obtained with viral doses as low as 5 × 103 PFU (Fig. 2).
Figure 2.

Effect of viral dose on vaccine efficacy. Inhibition of development of parasite liver stages in groups of five mice immunized with 20-fold increasing concentrations of 17D-Py. Control mice received the 17D virus (5 × 105 PFU). Results are expressed as the mean ± SE of five mice per group.
To test the longevity of the protective immune response induced by vaccination with 17D-Py, groups of mice were injected s.c. at weekly intervals for 24 wk, with a single dose of 105 PFU of the recombinant virus. 1 wk after the last mice had been immunized, all mice were challenged as described earlier in the paper. Inhibition of liver stage parasite burden peaked between 4 and 8 wk after immunization, but was still appreciable for at least 24 wk (Fig. 3 A). The number of CS-specific CD8+ T cells of these mice followed a similar course (Fig. 3 B).
Figure 3.

Persistence of protection and CS-specific CD8 + T cell–mediated responses in mice immunized with 17D-Py. (A) Percentage of inhibition of development of parasite liver stages in groups of five mice that were challenged with P. yoelii sporozoites, at different times after immunization with 17D-Py. (B) Number of IFNγ-secreting spleen cells after in vitro stimulation with the P. yoelii CS CD8+ T cell epitope. In this and subsequent experiments, splenocytes were harvested 40 h after challenge. Results are expressed as the average ± SE of five mice per group.
Heterologous prime/boost regimens can achieve sterilizing immunity
We first tried a series of homologous boost regimens with 17D-Py. No significant differences in parasite burden were seen in mice given either one dose or five consecutive daily injections of 104 or 105 PFU of 17D-Py (Fig. 4 A, groups A and B, respectively). In mice boosted with 105 PFU 3 wk after priming, the EEF burden was significantly reduced, which correlated with an increase in the number of IFNγ-secreting CS-specific CD8+ T cells (Fig. 4, A and B). The highest degree of protection (close to 90% reduction in parasite burden) was obtained when the smallest viral dose (103 PFU) was used for priming (Fig. 4, group E).
Figure 4.

Effect of a homologous boost with 17D-Py. Mice in groups A and B were immunized with 105 or 104 PFU of 17D-Py1, respectively, on five consecutive days, and challenged 2 wk later with 2 × 104 sporozoites. Mice in groups C, D, and E were primed with decreasing doses of 17D-Py (105, 104, or 103 PFU respectively), and boosted with 105 PFU 2 wk later. All animals in these groups were challenged with 2 × 104 sporozoites. In this and subsequent figures, the number of background spots in nonvaccinated control samples (naive) ranged between 0 and 4 (*). Results are expressed as the mean ± SE of five mice per group. (A) Inhibition of development of parasite liver stages. (B) Number of IFNγ-secreting spleen cells after in vitro stimulation with the P. yoelii CS CD8+ T cell epitope.
It is well documented that the prime/heterologous boost strategy elicits very strong immune responses when using viral vaccine vectors. In previous experiments, the vaccinia virus or the modified vaccinia Ankara virus (MVA) expressing the CS of P. yoelii greatly enhanced primary responses to influenza or Sindbis virus expressing the same CD8+ T cell malaria epitope (12–15). We found that priming mice with different doses (102 –105 PFU) of 17D-Py, and boosting 2 wk later with a single intravenous dose (107 PFU) of MVA-PyCS elicited large numbers of CS-specific IFNγ-secreting CD8+ T cells, antibodies to sporozoites (IFA titers, 1,600-3,200), and very effective protection (Fig. 5, A and B). Close to a 100% decrease in parasite burden in the liver was seen when the challenge was given 2–16 wk after priming with 105 PFU. This protection lasted for at least 16 wk after boosting, which is when the experiment was terminated (Fig. 6, A and B).
Figure 5.

Effect of a heterologous boost with MVA-PyCS. Groups of five mice were primed with different doses of 17D-Py and boosted i.v. 2 wk later with 107 PFU of MVA-PyCS. All mice were challenged 2 wk later with 2 × 104 sporozoites. Results are expressed as the mean ± SE of five mice per group. (A) Inhibition of development of parasite liver stages. (B) Number of IFNγ-secreting spleen cells after in vitro stimulation with the P. yoelii CS CD8+ T cell epitope.
Figure 6.

Persistence of protection after a heterologous MVA-PyCS boost. Groups of five mice were primed with 105 17D-Py and boosted 2 wk later i.v. with 107 PFU of MVA-PyCS. The groups of mice were challenged with sporozoites between 2 and 16 wk after the boost. Results are expressed as the mean ± SE of five mice per group. (A) Inhibition of development of parasite liver stages. (B) Number of IFNγ-secreting spleen cells after in vitro stimulation with the P. yoelii CS CD8+ T cell epitope.
To further document the high level of protection, another group of animals was primed with 105 PFU of 17D-Py and boosted with 107 PFU of MVA-PyCS. These animals were challenged at various times after the prime/boost immunization with 75 live P. yoelii sporozoites and the peripheral blood was examined for 2 wk for the presence of blood stages of the parasite. As shown in Table I, sterile immunity was observed in 80% of the animals. Even when the challenge was performed 16 wk after the boost, 40% of the mice still failed to develop detectable parasitemia.
Table I.
Sterile immunity induced by 17D-Py prime/MVA-PyCS boost
| Weeks after boost |
Protected/ challenged |
Sterile immunity |
|---|---|---|
| % | ||
| 2 | 8/10 | 80 |
| 4 | 8/10 | 80 |
| 8 | 6/10 | 60 |
| 16 | 4/10 | 40 |
| Controla | 0/10 | 0 |
Groups of mice were immunized as described in the legend of Fig. 5. After challenge with 75 sporozoites, the peripheral blood of the mice was examined daily for 2 wk for the presence of blood stages of the parasite.
The control group consisted of naive mice challenged and analyzed in the same way.
In malaria-endemic areas, vaccinated individuals are likely to be frequently bitten by infected mosquitoes. To determine the effect of a sporozoite boost in our model, mice primed with 105 PFU of 17D-Py were injected i.v. with 104 irradiated P. yoelii sporozoites (Fig. 7) or were subjected to the bites of 20–40 irradiated, infected mosquitoes (not depicted). 2 wk after the boost, these mice were challenged with 2 × 104 live sporozoites. As shown in Fig. 7 A, boosting with sporozoites substantially increased protection, inhibiting EEF development by 99%.
Figure 7.

Effect of a sporozoite boost. Groups of mice were primed with 105 17D-Py and, 2 wk later, were injected i.v. with 104 irradiated sporozoites. 2 wk later, the mice were challenged with 2 × 104 nonirradiated sporozoites. Controls were immunized with either 17D-Py or just with irradiated sporozoites, and challenged at the same time. Results are expressed as the mean ± SE of five mice per group. (A) Inhibition of development of parasite liver stages. (B) Number of IFNγ-secreting spleen cells after in vitro stimulation with the P. yoelii CS CD8+ T cell epitope.
In another series of experiments, we sought to determine whether the prime/sporozoite boost regimen led to sterile immunity. The animals were challenged i.v. with 75 sporozoites, and we examined them daily for parasitemia for 2 wk. Sterile immunity was achieved in 9 out of 10 sporozoite-boosted animals. This protection lasted at least 16 wk after the sporozoite booster (Table II). In contrast, all control mice primed with either 17D-Py or vaccinated once with irradiated sporozoites became parasitemic.
Table II.
Sterile immunity induced by 17D-Py prime/iSP boost
| Group | Weeks after last immunization |
Protected/ challenged |
Sterile immunity |
|---|---|---|---|
| % | |||
| Naive | N/A | 0/5 | 0 |
| ISP | 2 | 0/5 | 0 |
| 17D-Py | 2 | 0/5 | 0 |
| 17D-Py/iSP | 2 | 9/10 | 90 |
| 17D-Py/iSP | 16 | 6/10 | 60 |
One group of mice that had been primed with 17D-Py was boosted 2 wk later with 104 irradiated sporozoites. 2 wk later, the last immunization all mice were infected with 75 sporozoites i.v. For controls, groups of mice were either not immunized (naive), primed with 104 irradiated sporozoites (iSP), or with 5 × 105 PFU 17D-Py (17D-Py). The peripheral blood of the mice was examined daily for 2 wk for the presence of blood stages of the parasite. The complete absence of detectable blood infection was taken as evidence for sterile immunity.
Mechanism of protection
As aforementioned, the prime/boosting regimen led to the production of antibodies to CS as well as IFNγ-secreting CD8+ T cells. To determine the protective mechanisms, groups of mice were injected with antibodies to CD4+ or CD8+ to deplete these T cell compartments just before challenge. After ablation of CD8+ T cells, protection was abolished, but depletion of CD4+ T cells had no effect as determined by ELISPOT and tetramer assays (Fig. 8).
Figure 8.
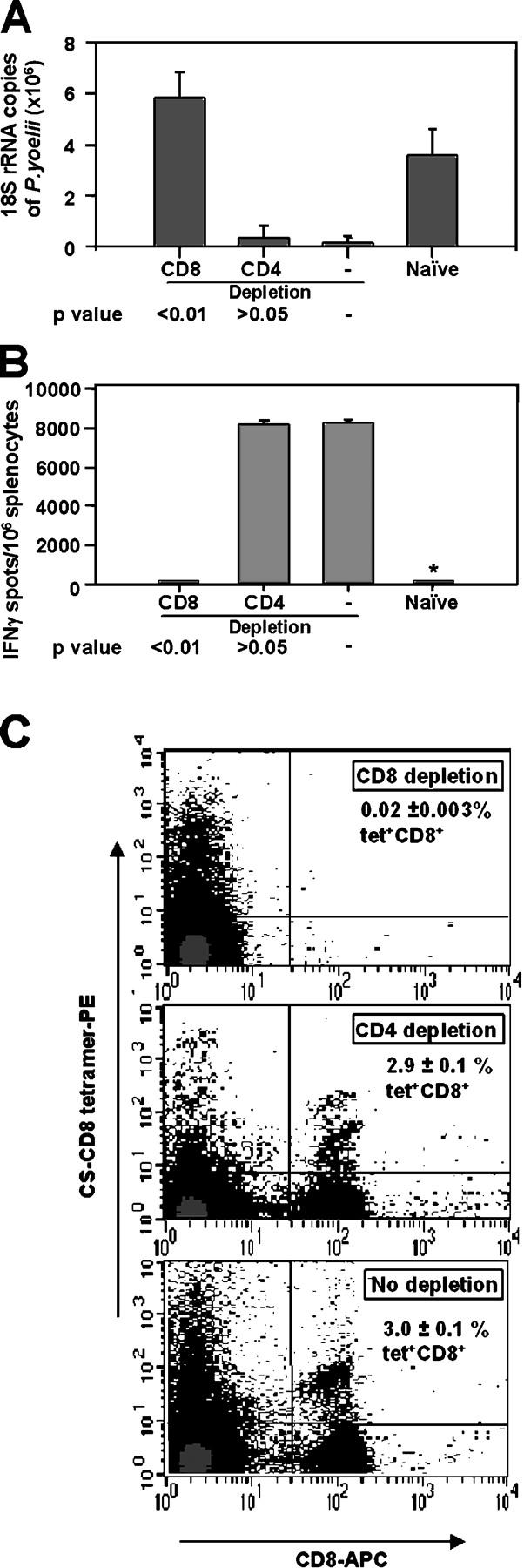
CD8 + T cells mediate protection. Groups of mice were immunized by the prime/boost regimen as described in the legend of Fig. 5. 3 d before the challenge with 2 × 104 sporozoites and daily thereafter, the mice received i.p. injections of 0.1 mg of antibodies specific for CD4+ or CD8+ T cells. Results are expressed as the mean ± SE of five mice per group. (A) Inhibition of development of parasite liver stages before and after depletion of T cells. (B) Number of IFNγ-secreting spleen cells after in vitro stimulation with the P. yoelii CS CD8+ T cell epitope. (C) Frequency of tetramer-stained SYVPSAEQI-specific T cells.
Discussion
Here, we use for the first time the highly attenuated YF17D as a vaccine vector to deliver a microbial T cell epitope. The YF17D virus, like other flaviviruses, is encoded by a single positive stranded RNA molecule. Translation of the viral message produces a polyprotein precursor that is cleaved by viral and cellular proteases (16). We inserted a CTL epitope of the CS protein of P. yoelii between the two nonstructural 17D viral proteins, NS2B and NS3, and generated the recombinant virus 17D-Py. The most important observation of this paper is that the injection of a single dose of 105 PFU of 17D-Py elicited a long lasting, high level of protection against challenge with highly infective sporozoites of P.yoelii. A significant degree of protection was seen with doses as small as 103 PFU. Other viral vectors bearing the same CTL epitope of CS has been used in rodent malaria models. However, like in other human vaccines currently in use, the T cell response after priming alone was of short duration. More importantly, protection against sporozoite challenge usually required one or more booster injections. Therefore, it appears that the immunogenicity in mice of T cell epitope inserted in the YF17D vaccine is distinctive and mimics the immunogenicity of the 17D vaccine in humans; i.e., it is long lasting and effective after priming.
Our findings provide strong support for using 17D as a general vaccine vector, even though there is scant information on the molecular mechanisms leading to the long-term memory that it evokes in humans. It is generally believed that protection in vaccinated humans is mediated in large part by neutralizing antibodies against the envelope protein E and complement-fixing antibodies to NS1, but the functional epitopes have not been identified. Human CTL responses to the vaccine have been documented (2), but there is no evidence that these vaccine-induced T cell responses have a role in protection. However, in mice, T cell epitopes in the envelope protein E and NS3 proteins are targets of antiviral responses (3). Regardless of the protective mechanism, vaccination with 17D seems to be sufficient for triggering the programs of expansion and differentiation of long-lived memory cells.
The mechanism of induction of primary CD8+ T cell responses to the malaria epitope in 17D-Py is not known but currently under investigation. The small CTL epitope is inserted between NS2B and NS3 and should be released in the cytoplasm of the infected cell. In vivo, the 17D vaccine is administrated subcutaneously and may infect professional APCs (pAPCs), such as dendritic cells, and/or parenchymal cells. However, CTL responses can only be generated by pAPCs. If the virus multiplies in a parenchymal cell, its remnants and viral antigens need to enter the pAPCs to initiate a primary response via cross-presentation. However, several independent lines of evidence show that short-lived peptides such as the malaria epitope cannot be cross-presented (17). The implication is that only the 17D-infected pAPCs participate in the initial sensitization of T cells. It will be of interest to determine the nature of the cells that are initially infected by 17D. A preference for pAPCs would provide an explanation for its unusually strong immunogenicity. In fact, dengue virus, which belongs to the same genus as YF, preferentially targets immature dendritic cells (18).
Another important finding is that the resulting recombinant virus 17D-Py is not only replication competent, but also stable in vitro and in vivo, thus minimizing the concern that the heterologous insert would be eliminated or modified during viral replication. However, if 17D is to be used as a general vaccine vector for multiple CTL epitopes, it will be necessary to understand the size and sequence constraints for inserting several microbial sequences in the same or in other positions in the 17D genome. We have recently shown that three tandem repeats of the same P. yoelii epitope can be inserted between NS2B and NS3 (unpublished data). This 17D recombinant replicates with kinetics similar to the 17D parent and is also stable in vitro and in vivo. Therefore, the potential already exists for vaccination with mixtures of a few recombinant viruses, each containing strings of three different protective T cell epitopes, to elicit immune responses in a large proportion of humans.
The present results are of particular relevance to the development of preerythrocytic human malaria vaccines. Their targets are the sporozoites that enter the host during the mosquito bite, and the liver stages (EEFs). P. falciparum vaccines that target CS, the main surface protein of sporozoites, are undergoing human trials. One of them named RTS,S protects 50% or more of naive volunteers (19), or individuals living in an endemic area in Africa. However, protection lasts only for a couple of months (20). The mechanism of protection involves neutralizing antibodies and effector CD4+ T cells (21). CS vaccines that elicit additional effector CD8+ T cells would be highly desirable. In the rodent models of malaria, there is compelling evidence that CTL can destroy the early EEFs by releasing IFNγ (22). In addition, production of specific antibodies, such as after MVA-PyCS injection, can only reduce the number of EEF in the liver. Furthermore, CS is produced in large amounts during EEF development and is processed and presented on the surface of the infected liver cells (23, 24). As shown here CS is an excellent target for CTL. Indeed, priming mice with 17D-Py that contains a single CS CTL epitope leads to the elimination of 60–70% of liver stages. This is particularly noteworthy because priming of mice using other recombinant viral vectors such as influenza or vaccinia expressing the same malaria CD8+ CTL epitope do not lead to protective responses without boosting (12, 15). In the case of Sindbis vectors, protection is optimal 12 d after priming (14), but decays quickly afterwards (Tsuji, M., personal communication). Another observation important for malaria vaccine development is the finding that the protection elicited by 17D-Py can be boosted with sporozoites and that the boosting leads to sterile immunity. It is therefore plausible that in endemic areas the immune response of vaccinated individuals will be magnified by the bite of infected mosquitoes.
This is particularly encouraging considering that the vaccines actually undergoing clinical trials for malaria in endemic areas are based on a prime/boost strategy involving two priming immunizations 3 wk apart, a boost 3 wk later, and finally another boost after 12 mo (25). In contrast, YF17D is able to induce neutralizing antibodies in 90% of vaccine recipients 10 d after vaccination, and the percentage increases to 99% after 4 wk. Moreover, the fact that YF vaccination has been included since 1993 in the World Health Organization (WHO)–sponsored Expanded Program on Immunization for children in developing countries (26) could facilitate the introduction of a recombinant YF/malaria vaccine in those countries where it is most needed. Our results showing that such recombinant viruses can be viable, stable in vivo, and are able to induce an effective antimalarial immunity in the P. yoelii-mouse model are encouraging for future attempts targeting P. falciparum. Although such vaccine candidates will not be readily testable for protection against malaria short of human clinical trials, their safety, immunogenicity, and protective efficacy against virulent YF can be tested in rhesus macaques.
Yellow fever 17D is a remarkably safe and effective vaccine, but adverse effects have been noted. Over 60 yr, an estimated 400 million doses have been administered worldwide with 2 deaths due to encephalitis. From 1996 to 2001, 150 million doses were administered and seven cases of vaccine-associated viscerotropic disease (including six deaths) occurred. The cause of this rare, but severe, reaction is unknown; but, it seems to involve an atypical host response rather than reversion of the vaccine virus to virulence (27–29) and the WHO guidelines regarding the vaccine have remained unchanged (30). This would certainly not contraindicate vaccination in malaria-endemic countries.
Finally, one can envision modernizing the 17D vaccine or 17D recombinant vaccine candidates. Currently, a standardized seed lot inoculum is amplified in embryonated chicken eggs and a crude homogenate stored frozen after lyophilization. This vaccine is difficult to manufacture, expensive, and requires refrigeration. Recombinant plasmid DNAs can now be used as a stable repository for the vaccine with seed stocks and vaccine lots generated by amplification in cell culture. Beyond this, recent work has shown that flaviviruses can be launched directly from transfected plasmid DNA (31), making it possible to eliminate cell culture propagation completely. Such new approaches offer the advantages of the DNA vaccines, ease of manufacture, and the possibility of bypassing the cold chain.
Materials and Methods
Plasmids and recombinant viruses.
For 17D2B/3, we modified the 17D genome (32) to introduce a BssHII and a BstEII cleavage site at the recognition site of the viral serine protease NS2B-NS3, as described by McAllister and colleagues (11).
For 17D-Py, we engineered the CTL epitope of Plasmodium yoelii (SYVPSAEQIL) into the 17D genome by assembly PCR using two specific oligonucleotides containing the P.yoelii CTL sequence (forward, GGAGCGCGCAGAAGTTCTTATGTCCCAAGCGCAGAACAAATATTAGGTCACCGGAGAA; and reverse, CCGGTGACCTAATATTTGTTCTGCGCTTGGGACATAAGAACTTCTGCGCGCTCCCCTGACAT) and two 17D-specific primers to generate two PCR fragments containing the P. yoelii CTL and a portion of the NS2B or NS3 gene, respectively. 1 μl out of 50 μl of each PCR reaction was mixed together and was used as template for a PCR with 17D-specific primers lying in NS2B and in NS3. The final PCR product was digested with BssHII and a BstEII and cloned into 17D2B/3.
MVA-PyCS is an MVA recombinant that expresses the entire CS protein of P.yoelii (12) and was used for heterologous boosting in some experiments.
Preparation of 17D and 17D-Py virus stocks.
The parental 17D and recombinant 17D-P.yoelii plasmids were purified by banding on CsCl and linearized by digestion with XhoI. The linearized template was transcribed by SP6 RNA polymerase in the presence of cap analogue, purified, and used to electroporate SW-13 cells as described previously (33). The specific infectivity of the viral RNA was measured by seeding serial dilutions of transfected cells on monolayers of untransfected SW-13 cells, overlaying with 0.6% agarose in medium, and counting viral plaques after 96 h. Virus stocks were harvested at 48 h after electroporation with typical yields of 107–108 PFU/ml, as determined by a plaque assay on SW-13 cells. Single use aliquots were stored frozen at –80°C until use.
17D. growth and stability in cell culture.
SW-13 cell monolayers were infected with parental 17D or recombinant 17D-Py at low (0.1 PFU/cell) or high (5 PFU/cell) multiplicity. Supernatants were collected at various times after infection and frozen at −80°C. For plaque titration, serial 10-fold dilutions were used to infect monolayers of SW-13 cells. After 1 h at 37°C, cells were overlayed with 0.6% agarose in medium and the plaques counted at 96 h after fixation with formaldehyde and staining with crystal violet (33).
To assess the stability of the inserted sequence, virus in the supernatants was concentrated by PEG precipitation (34), and the RNA was isolated by TRIzol extraction according to the manufacturer's instructions. First-strand cDNA was synthesized using SuperScript II reverse transcriptase (Invitrogen) and random hexamers and amplified by PCR using 17D-specific oligonucleotides flanking the 2B/3 insertion site. Products were sized by acrylamide gel electrophoresis, cleaved with a restriction enzyme diagnostic of the P. yoelii epitope sequence (SspI) or sequenced as a population.
Animals and parasites.
P. yoelii (17X NL strain) was maintained by alternate cyclic parasite passage in Anopheles stephensi and infected blood transfer to mice. Sporozoites were obtained by dissection of salivary glands of A. stephensi and collected in medium RPMI 1640 at 4°C.
BALB/c and C57BL/6 mice were obtained from Charles River Laboratories. Animals were maintained in the animal facility of the Department of Medical and Molecular Parasitology. All animal experimental procedures were reviewed and approved by the Institutional Animal Review committee.
Immunization, challenge, and in vivo stability of recombinant viruses.
Except when stated otherwise in the figure legends, mice were primed with 5 × 105 PFU of 17D-Py injected s.c. When given a boost, this consisted of either the recombinant 17D-Py virus used at the same dose and route, MVA-PyCS (107 PFU), or irradiated sporozites injected i.v. Immunized mice were challenged i.v. with 2 × 104 sporozoites 2 wk after immunization, except when otherwise noted.
Newborn C57BL/6 mice were injected in the peritoneum with 105 PFU of 17D (seven mice) or 17D-Py (five mice). All mice developed hind limb paralysis 10–14 d after infection. Virus was recovered from spleen and brain homogenates, and titers were determined by plaque assay (33). Virus in clarified homogenates was concentrated by PEG precipitation and RNA isolated by TRIzol extraction. The presence of Py insert was assessed by RT-PCR amplification, restriction digestion, and sequencing as described above.
Evaluation of immunogenicity and protective immunity.
In most experiments, after each vaccination regimen, we evaluated the protective immunity (inhibition of the EEF development) and malaria-specific CD8+ T cell responses. The degree of protection against sporozoite challenge elicited by the vaccination was evaluated by two criteria: the level of inhibition of development of liver stages (EEFs), and the absence of parasites in the peripheral blood of immunized and sporozoite-challenged mice (sterile immunity).
To measure the number of EEFs, livers were harvested 40–42 h after i.v. challenge with 2 × 104 sporozoites. This is the time when EEFs are almost fully developed, and the newly formed merozoites are nearly ready to exit the hepatocytes, and enter the blood circulation. Quantification of the EEFs was done by real-time PCR analysis of parasite specific rRNA. In most instances, data are presented as the average parasite RNA copies in the liver ±SE in immunized mice, compared with controls vaccinated with 17D virus (35).
Cellular immune responses were evaluated by enumerating IFNγ-producing, epitope-specific CD8+ T cells in the spleen (by ELISPOT), as described (36). Splenocytes were harvested at 40–42 h after challenge. There was no obvious difference in the spleen size in any of the experimental or control groups of mice when the animals were killed. In some experiments we determined SYVPSAEQI-CS-specific CD8+ T cells using tetramers. FACS analyses were done using FACSCalibur and CellQUEST software.
To determine whether sterile immunity had been elicited, the immunized animals were challenged i.v. with 75 sporozoites. Peripheral blood smears were made daily and were examined for the presence of blood stages, starting from the third day after challenge, for a total of 2 wk. In the P. yoelii model, blood infections result from the injection of 10 or less sporozoites, and the prepatent period never exceeded 2 wk.
Detection of antibodies to CS.
Anti-CS antibodies were detected by an indirect immunofluorescence assay, using live or air-dried P. yoelii sporozoites as described previously (37).
In vivo depletion of CD8+ or CD4+ T cells.
Each vaccinated mouse received daily 0.1 mg anti-CD4 mAb (from cell line −GK1.5) or anti-CD8 mAb (from cell line −YTS 169; Harlan Bioproducts For Science) by i.p injection, for three consecutive days. The mice were challenged with P. yoelii sporozoites 2 d after receiving the last dose of mAb. The corresponding protection and CS-specific T cells were evaluated by real-time PCR and ELISPOT.
Acknowledgments
This work was supported by grants from the Starr Foundation and the Greenberg Medical Research Institute.
The authors have no conflicting financial interests.
Abbreviations used: 17D, yellow fever vaccine 17D; CS, circumsporozoite protein; EEF, exoerythrocytic stage; MVA, modified vaccinia virus Ankara; MOI, multiplicity of infection.
D. Tao and G. Barba-Spaeth contributed equally to this paper.
References
- 1.Lefeuvre, A., P. Marianneau, and V. Deubel. 2004. Current assessment of yellow fever and yellow fever vaccine. Curr. Infect. Dis. Rep. 6:96–104. [DOI] [PubMed] [Google Scholar]
- 2.Co, M.D., M. Terajima, J. Cruz, F.A. Ennis, and A.L. Rothman. 2002. Human cytotoxic T lymphocyte responses to live attenuated 17D yellow fever vaccine: identification of HLA-B35-restricted CTL epitopes on nonstructural proteins NS1, NS2b, NS3, and the structural protein E. Virology. 293:151–163. [DOI] [PubMed] [Google Scholar]
- 3.van der Most, R.G., L.E. Harrington, V. Giuggio, P.L. Mahar, and R. Ahmed. 2002. Yellow fever virus 17D envelope and NS3 proteins are major targets of the antiviral T cell response in mice. Virology. 296:117–124. [DOI] [PubMed] [Google Scholar]
- 4.Monath, T.P., F. Guirakhoo, R. Nichols, S. Yoksan, R. Schrader, C. Murphy, P. Blum, S. Woodward, K. McCarthy, D. Mathis, et al. 2003. Chimeric live, attenuated vaccine against Japanese encephalitis (ChimeriVax-JE): phase 2 clinical trials for safety and immunogenicity, effect of vaccine dose and schedule, and memory response to challenge with inactivated Japanese encephalitis antigen. J. Infect. Dis. 188:1213–1230. [DOI] [PubMed] [Google Scholar]
- 5.Guirakhoo, F., K. Pugachev, Z. Zhang, G. Myers, I. Levenbook, K. Draper, J. Lang, S. Ocran, F. Mitchell, M. Parsons, et al. 2004. Safety and efficacy of chimeric yellow fever-dengue virus tetravalent vaccine formulations in nonhuman primates. J. Virol. 78:4761–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marchevsky, R.S., M.S. Freire, E.S. Coutinho, and R. Galler. 2003. Neurovirulence of yellow fever 17DD vaccine virus to rhesus monkeys. Virology. 316:55–63. [DOI] [PubMed] [Google Scholar]
- 7.Bonaldo, M.C., R.C. Garratt, P.S. Caufour, M.S. Freire, M.M. Rodrigues, R.S. Nussenzweig, and R. Galler. 2002. Surface expression of an immunodominant malaria protein B cell epitope by yellow fever virus. J. Mol. Biol. 315:873–885. [DOI] [PubMed] [Google Scholar]
- 8.Romero, P., J.L. Maryanski, G. Corradin, R.S. Nussenzweig, V. Nussenzweig, and F. Zavala. 1989. Cloned cytotoxic T cells recognize an epitope in the circumsporozoite protein and protect against malaria. Nature. 341:323–326. [DOI] [PubMed] [Google Scholar]
- 9.Herrington, D.A., D.F. Clyde, G. Losonsky, M. Cortesia, J.R. Murphy, J. Davis, S. Baqar, A.M. Felix, E.P. Heimer, D. Gillessen, et al. 1987. Safety and immunogenicity in man of a synthetic peptide malaria vaccine against Plasmodium falciparum sporozoites. Nature. 328:257–259. [DOI] [PubMed] [Google Scholar]
- 10.Stoute, J.A., M. Slaoui, D.G. Heppner, P. Momin, K.E. Kester, P. Desmons, B.T. Wellde, N. Garcon, U. Krzych, and M. Marchand. 1997. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS,S Malaria Vaccine Evaluation Group. N. Engl. J. Med. 336:86–91. [DOI] [PubMed] [Google Scholar]
- 11.McAllister, A., A.E. Arbetman, S. Mandl, C. Pena-Rossi, and R. Andino. 2000. Recombinant yellow fever viruses are effective therapeutic vaccines for treatment of murine experimental solid tumors and pulmonary metastases. J. Virol. 74:9197–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Aseguinolaza, G., Y. Nakaya, A. Molano, E. Dy, M. Esteban, D. Rodriguez, J.R. Rodriguez, P. Palese, A. Garcia-Sastre, and R.S. Nussenzweig. 2003. Induction of protective immunity against malaria by priming-boosting immunization with recombinant cold-adapted influenza and modified vaccinia Ankara viruses expressing a CD8+-T-cell epitope derived from the circumsporozoite protein of Plasmodium yoelii. J. Virol. 77:11859–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li, S., M. Rodrigues, D. Rodriguez, J.R. Rodriguez, M. Esteban, P. Palese, R.S. Nussenzweig, and F. Zavala. 1993. Priming with recombinant influenza virus followed by administration of recombinant vaccinia virus induces CD8+ T-cell-mediated protective immunity against malaria. Proc. Natl. Acad. Sci. USA. 90:5214–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsuji, M., C.C. Bergmann, Y. Takita-Sonoda, K. Murata, E.G. Rodrigues, R.S. Nussenzweig, and F. Zavala. 1998. Recombinant Sindbis viruses expressing a cytotoxic T-lymphocyte epitope of a malaria parasite or of influenza virus elicit protection against the corresponding pathogen in mice. J. Virol. 72:6907–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodrigues, M., S. Li, K. Murata, D. Rodriguez, J.R. Rodriguez, I. Bacik, J.R. Bennink, J.W. Yewdell, A. Garcia-Sastre, R.S. Nussenzweig, et al. 1994. Influenza and vaccinia viruses expressing malaria CD8+ T and B cell epitopes. Comparison of their immunogenicity and capacity to induce protective immunity. J. Immunol. 153:4636–4648. [PubMed] [Google Scholar]
- 16.Lindenbach, B.D., and C.M. Rice. 2001. Flaviviridae: The viruses and their replication. Fields Virology. D.M. Knipe and P.M. Howley, editors. Lippincott Williams & Wilkins, Philadelphia. 991–1042.
- 17.Ploegh, H.L. 2004. Immunology. Nothing ‘gainst time’s scythe can make defense. Science. 304:1262–1263. [DOI] [PubMed] [Google Scholar]
- 18.Tassaneetrithep, B., T.H. Burgess, A. Granelli-Piperno, C. Trumpfheller, J. Finke, W. Sun, M.A. Eller, K. Pattanapanyasat, S. Sarasombath, D.L. Birx, et al. 2003. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 197:823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lalvani, A., P. Moris, G. Voss, A.A. Pathan, K.E. Kester, R. Brookes, E. Lee, M. Koutsoukos, M. Plebanski, M. Delchambre, et al. 1999. Potent induction of focused Th1-type cellular and humoral immune responses by RTS,S/SBAS2, a recombinant Plasmodium falciparum malaria vaccine. J. Infect. Dis. 180:1656–1664. [DOI] [PubMed] [Google Scholar]
- 20.Bojang, K.A., P.J. Milligan, M. Pinder, L. Vigneron, A. Alloueche, K.E. Kester, W.R. Ballou, D.J. Conway, W.H. Reece, P. Gothard, et al. 2001. Efficacy of RTS,S/AS02 malaria vaccine against Plasmodium falciparum infection in semi-immune adult men in The Gambia: a randomised trial. Lancet. 358:1927–1934. [DOI] [PubMed] [Google Scholar]
- 21.Reece, W.H., M. Pinder, P.K. Gothard, P. Milligan, K. Bojang, T. Doherty, M. Plebanski, P. Akinwunmi, S. Everaere, K.R. Watkins, et al. 2004. A CD4(+) T-cell immune response to a conserved epitope in the circumsporozoite protein correlates with protection from natural Plasmodium falciparum infection and disease. Nat. Med. 10:406–410. [DOI] [PubMed] [Google Scholar]
- 22.Saul, A., V. Nussenzweig, and R.S. Nussenzweig. 2004. Rationale for malaria vaccine development. Novel Vaccination Strategies. S.H.E. Kaufmann, editor. Wiley-VCH, Weinheim. 479–503.
- 23.Wang, Q., S. Brown, D.S. Roos, V. Nussenzweig, and P. Bhanot. 2004. Transcriptome of axenic liver stages of Plasmodium yoelii. Mol. Biochem. Parasitol. 137:161–168. [DOI] [PubMed] [Google Scholar]
- 24.Hamilton, A.J., A. Suhrbier, J. Nicholas, and R.E. Sinden. 1988. Immunoelectron microscopic localization of circumsporozoite antigen in the differentiating exoerythrocytic trophozoite of Plasmodium berghei. Cell Biol. Int. Rep. 12:123–129. [DOI] [PubMed] [Google Scholar]
- 25.Moorthy, V.S., E.B. Imoukhuede, S. Keating, M. Pinder, D. Webster, M.A. Skinner, S.C. Gilbert, G. Walraven, and A.V. Hill. 2004. Phase 1 evaluation of 3 highly immunogenic prime-boost regimens, including a 12-month reboosting vaccination, for malaria vaccination in Gambian men. J. Infect. Dis. 189:2213–2219. [DOI] [PubMed] [Google Scholar]
- 26.Robertson, S. 1993. Module 8: Yellow fever. In The Immunological Basis for Immunization. WHO, Global Programme for Vaccines and Immunization, Geneva.
- 27.Chan, R.C., D.J. Penney, D. Little, I.W. Carter, J.A. Roberts, and W.D. Rawlinson. 2001. Hepatitis and death following vaccination with 17D-204 yellow fever vaccine. Lancet. 358:121–122. [DOI] [PubMed] [Google Scholar]
- 28.Galler, R., K.V. Pugachev, C.L. Santos, S.W. Ocran, A.V. Jabor, S.G. Rodrigues, R.S. Marchevsky, M.S. Freire, L.F. Almeida, A.C. Cruz, et al. 2001. Phenotypic and molecular analyses of yellow fever 17DD vaccine viruses associated with serious adverse events in Brazil. Virology. 290:309–319. [DOI] [PubMed] [Google Scholar]
- 29.Martin, M., T.F. Tsai, B. Cropp, G.J. Chang, D.A. Holmes, J. Tseng, W. Shieh, S.R. Zaki, I. Al-Sanouri, A.F. Cutrona, et al. 2001. Fever and multisystem organ failure associated with 17D-204 yellow fever vaccination: a report of four cases. Lancet. 358:98–104. [DOI] [PubMed] [Google Scholar]
- 30.WHO. 2003. Yellow fever vaccine: WHO position paper. Wkly. Epidemiol. Rec. 78:349–360. [PubMed] [Google Scholar]
- 31.Khromykh, A.A., A.N. Varnavski, P.L. Sedlak, and E.G. Westaway. 2001. Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA-based full-length cDNA clones of Kunjin virus. J. Virol. 75:4633–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bredenbeek, P.J., E.A. Kooi, B. Lindenbach, N. Huijkman, C.M. Rice, and W.J. Spaan. 2003. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J. Gen. Virol. 84:1261–1268. [DOI] [PubMed] [Google Scholar]
- 33.Amberg, S.M., and C.M. Rice. 1999. Mutagenesis of the NS2B-NS3-mediated cleavage site in the flavivirus capsid protein demonstrates a requirement for coordinated processing. J. Virol. 73:8083–8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rice, C.M., L. Dalgarno, R. Galler, Y.S. Hahn, E.G. Strauss, and J.H. Strauss. 1988. Molecular cloning of flavivirus genomes for comparative analysis and expression. Modern Trends in Virology. C. Scholtissek, editor. Springer-Verlag, Berlin. 83–97.
- 35.Bruna-Romero, O., J.C. Hafalla, G. Gonzalez-Aseguinolaza, G. Sano, M. Tsuji, and F. Zavala. 2001. Detection of malaria liver-stages in mice infected through the bite of a single Anopheles mosquito using a highly sensitive real-time PCR. Int. J. Parasitol. 31:1499–1502. [DOI] [PubMed] [Google Scholar]
- 36.Carvalho, L.H., J.C. Hafalla, and F. Zavala. 2001. ELISPOT assay to measure antigen-specific murine CD8(+) T cell responses. J. Immunol. Methods. 252:207–218. [DOI] [PubMed] [Google Scholar]
- 37.Nardin, E., R.W. Gwadz, and R.S. Nussenzweig. 1979. Characterization of sporozoite surface antigens by indirect immunofluorescence: detection of stage- and species-specific antimalarial antibodies. Bull. World Health Organ. 57:211–217. [PMC free article] [PubMed] [Google Scholar]