

Abstract
Tumor environment can be critical for preventing the immunological destruction of antigenic tumors. We have observed a selective accumulation of CD4+CD25+ T cells inside tumors. In a murine fibrosarcoma Ld-expressing Ag104, these cells made up the majority of tumor-infiltrating lymphocytes at the late stage of tumor progression, and their depletion during the effector phase, rather than priming phase, successfully enhanced antitumor immunity. We show here that CD4+CD25+ T cells suppressed the proliferation and interferon-γ production of CD8+ T cells in vivo at the local tumor site. Blockade of the effects of IL-10 and TGF-β partially reversed the suppression imposed by the CD4+ cells. Furthermore, local depletion of CD4+ cells inside the tumor resulted in a change of cytokine milieu and led to the eradication of well-established highly aggressive tumors and the development of long-term antitumor memory. Therefore, CD4+CD25+ T cells maintained an environment in the tumor that concealed the immunogenicity of tumor cells to permit progressive growth of antigenic tumors. Our study illustrates that the suppression of antitumor immunity by regulatory T cells occurs predominantly at the tumor site, and that local reversal of suppression, even at a late stage of tumor development, can be an effective treatment for well-established cancers.
Cancers often progress in immunocompetent hosts and are defined as having poor immunogenicity (1–4). It was thought that the failure to elicit an immune response was due to the lack of recognizable antigens. However, it is now evident that, in mice and humans, tumor cells can be antigenic due to encoded genes that are either normal, but aberrantly or overexpressed, or altered as the result of cancer-specific somatic mutations (3, 5–15). Although most of the tumor antigen-specific lymphocytes that recognize self antigens would normally be subjected to self tolerance processes (16, 17), lymphocytes recognizing mutated antigens, which are absolutely specific for the tumor cells, should not have undergone the same regulatory processes that accompany self tolerance (18). The notion that immune recognition of cancer occurs frequently, if not universally, in cancer patients, is consistent with the frequent observation of T cell infiltration into cancer tissues (19–22). However, it is exceptional for such tumor-infiltrating T cells to induce the spontaneous rejection of the established tumors. This curious paradox of antigenic and immune-recognized tumor cells that fail to elicit its rejection in immune competent hosts remains largely inexplicable.
Many studies present the possibility of a local environment at the tumor site may play critical roles for preventing the immunological destruction of antigenic tumors. One of the best examples is concomitant tumor immunity, a phenomenon observed in both animals and humans, in which tumor-bearing hosts fail to reject their primary tumor but generate systemic immunity to resist subsequent tumor implants of the same tumor cells at secondary sites (23). Even when the host bears a poorly immunogenic cancer, concomitant immunity can be rescued by systemic depletion of the CD4+CD25+ regulatory T cell subset (24). In other studies, mice that were incapable of rejecting established malignant tumor grafts were able to reject nonmalignant grafts that expressed the same strong, recognizable antigens (1, 25). The tumor antigen-specific T cells in these tumor-bearing hosts that failed to mediate rejection of the established tumor were neither clonally exhausted, nor systemically anergized, nor ignorant (1). Finally, the defect in TCR signal-transducing molecules in cancer patients was found to be much more pronounced in T cells infiltrating the tumor than the ones in the peripheral blood (26). All of these studies imply that a growing tumor harbors the ability to induce an antigen-specific destructive immune response while suppressing the effects of this immunity at the site of the established tumor. Stromal barrier inside the tumor has been proposed to be one of the important local factors to hinder T cell access to the established malignant tissues (1, 27, 28). However, other mechanisms may contribute to local suppression in the presence of infiltrating lymphocytes.
The CD4+CD25+ T cell subset (29, 30) has been established to be a powerful regulator of T cell responses in organ-specific autoimmunity and chronic infections (31–35). Earlier studies by Robert North and his colleagues have shown that cell-mediated suppression of antitumor immunity led to the progressive growth of an immunogenic tumor (36). The tumor-induced suppressor cells expressed the CD4+CD8− surface phenotype (37), and intravenous injection of a depleting anti-CD4 antibody led to CD8+ T cell–mediated tumor regression (38). The antibody was given at early stages of tumor growth and ineffective if T cell help was required to generate an immune response to the tumor. A major advance resulted from the discovery that the suppressive CD4+ T cell subset could be separated from CD4+ helper T cells by the expression of high levels of CD25 (29, 30) and that this subset had important regulatory functions in preventing autoimmunity to self antigens (31, 33–35). Importantly, it was also shown that elimination of these regulatory T cells, despite causing increased autoreactivity, could increase immune responses to tumors such as melanomas overexpressing self-antigens as targets (39–42). Here, we have found that CD4+CD25+ T cells selectively accumulated inside the tumor. These cells maintained a local cytokine environment that suppressed the effector function of tumor-infiltrating CD8+ T cells at the tumor site. Intratumoral depletion of these CD4+CD25+ T cells unmasked the immunogenicity of the tumors and reversed CTL tolerization leading to rapid rejection of well-established tumors. Therefore, we have revealed an important mechanism in the tumor environment that prevents immune destruction of the antigenic cancer. Our study shows that CD4+CD25+ T cells exert their suppressive effects predominantly in the tumor and abrogating their effects at the tumor site can be a highly efficient approach to enhance antitumor immunity.
Results
A strong antigen does not hinder tumor growth
Human cancers are often antigenic, and it is puzzling why most human cancers fail to induce their rejection, even when there is significant infiltration of lymphocytes into the tumor tissues (3, 14). In an attempt to further analyze what types of T lymphocytes infiltrate the tumor tissues, we found interestingly that very few CD4+ T cells infiltrated the breast cancer tissues we examined during the early stages of tumor progression. However, as the tumors progressed, the CD4+ T cell population increased dramatically inside the tumor (unpublished data). Because of the limitations in exploring the essential roles of these tumor-infiltrating CD4+ T cells for tumor progression in vivo with human cancers, we developed a murine tumor model that closely resembles human cancers with some infiltration of both CD4+ and CD8+ T cells. Ag104, a poorly differentiated solid tumor that arose spontaneously in C3H mouse, is highly progressive, poorly immunogenic, and resistant to immunotherapy (27, 43, 44). Ag104 cancer cells regularly cause tumors in both C3H and C3HXB6F1 (C3B6F1) mice when as few as 104 cells are inoculated. The low immunogenicity of this tumor, which leads to rapid growth in immunocompetent syngeneic wild-type mice in vivo, could be due to either a lack of or weak expression of tumor antigens. In an attempt to rule out this caveat, we transfected Ag104 tumor cell line with a gene encoding the Ld antigen, a strong antigen. The transfected tumor cell line expressed high levels of Ld in vitro. Unexpectedly, the Ld-expressing Ag104 (Ag104Ld) tumor grew with similar kinetics to the parental tumor at the dose of as low as 104. The tumor growth resulting from subcutaneous inoculation of 5 × 105 tumor cells into C3B6F1 mice in three experiments is shown in Fig.1 a. It has been reported that antigen-lost variant could be selected to avoid immune recognition (4, 45, 46). However, we found that Ag104Ld tumor cells isolated from the tumor-bearing mice expressed comparable level of Ld as the cells before injection (unpublished data). We concluded from this experiment that antigenic tumors can progress without losing their tumor antigens. Thus, it is unlikely that the poor immunogenicity of the Ag104Ld tumor, indicated by their rapid growth in immunocompetent syngeneic wild-type mice in vivo, was caused by low expression of MHC class I or by the loss of antigen.
Figure 1.


CD4+CD25+ T cells accumulate inside the tumor to suppress tumor rejection. (a) A strong antigen Ld does not hinder tumor growth. 5 × 105 Ag104 and Ag104Ld tumor cells were inoculated to C3B6F1 mice s.c. Tumor growth was monitored and volume was calculated as follows: volume = length × width × height/2. Three individual experiments (×3 mice per group) were pooled and shown. (b–e) 106 Ag104Ld tumor cells were inoculated to C3B6F1 mice s.c. at tail base and inguinal LN, spleen, and tumor tissue were isolated from the mice 7 or 16 d after tumor challenge. The percentage of CD4+CD25+ T cells among lymphocytes (b) and the expression of CD45RB on CD4+CD25+ or CD4+CD25− T cells (c) were determined by FACS. Results from pooled three experiments with three mice examined at each time point were shown. Spleen and tumor tissue were collected from these tumor-bearing mice 16 d after tumor challenge. CD4+ T cells were enriched from the spleen with a negative selective bead method using magnetic system. CD4+CD25− T cells from the spleen were further separated from CD4+CD25+ T cells with magnetic system. Tumor-infiltrating T cells were enriched with biotinylated anti-Thy1.2 antibody followed by antibiotin magnetic beads. CD4+ T cells infiltrating the tumor were further isolated with FACS sorting after stained with PE conjugated anti-CD4 antibody. (d) Total RNA was isolated from these purified CD4+ T cell populations and real-time RT-PCR for foxp3 was done on cDNA derived from the RNA. (e) 5 × 104 per well in a 96-well plate purified CD4+CD25− T cells from naive mice were stimulated with optimal dose (2 μg/ml) of coated anti-CD3 antibody for 72 h. Or 5 × 104 CD4+ T cells from tumor tissue or spleen of tumor-bearing mice were added to coculture with purified CD4+CD25− T cells and 3H was added for the last 24 h of culture and its incorporation was measured. Data shown were means and SD. (f) Anti-CD25 antibody was given to mice 14 d after tumor challenge, spleen and tumor tissues were collected 2 d later and the percentage of CD25+ or CD25− cells among CD4 or CD8 cells were compared with the one with control treatment. The representative data from one out of three experiments was shown. Tumor growth was monitored when CD4+ (g) or CD25+ (h) cells were depleted with antibody in vivo. The tumor growth pattern in one experiment representative of seven independent experiments with three to five mice in each group, either CD4-depleted or control group was shown in g. (h) Tumor volume at each time point was measured on 12 mice in each group, either CD25-depleted or control group, done in four independent experiments and data shown were means and SD. The difference in tumor growth between the two groups was significant (P < 0.001, the random effect models for longitudinal data). Tumor sizes were also significantly different between two groups after 20 d after tumor inoculation (P < 0.001, t test).
One reason why antigen Ld did not hinder tumor growth could be that this antigen was ignored and never seen by the immune system. To test this possibility, we surgically removed the Ag104Ld tumor 21 d after its inoculation and then rechallenged the mice with a larger dose of tumor cells 4 wk after the surgery. Surprisingly, all these mice rejected the second tumor challenge, although the naive mice died of the tumor burden (Table I). This result indicated that the T cells were primed to the Ld antigen and after the surgical removal of the tumor, these T cells were able to evolve into memory cells and confer protection against the second tumor challenge. We were able to show that these memory T cells were predominately Ld-specific because these mice could not reject the parental tumor Ag104 (Table I). These results suggested to us that antigen-specific T cells do exist, but do not function properly in the tumor-bearing animals. It raises a possibility that poor immunogenicity of Ag104Ld tumor may not be attributed to poor immune recognition but to aberrant immune responses in the presence of tumor, particularly within the tumor microenvironment.
Table I.
CD4 depletion induces immune responses against antigens other than Ld
| First tumor challengea | Rechallengea | ||||||
|---|---|---|---|---|---|---|---|
| Group | Tumor line | Dose | Treatment | Tumor line | Dose | Incidence of tumor growthb | Percent |
| 1 | Ag104Ld | 5 × 105 | Surgical removalc | Ag104Ld | 106 | 0/3 | 0 |
| 2 | 5 × 105 | Ag104 | 106 | 3/3 | 100 | ||
| 3 | 5 × 105 | CD4 depletiond | Ag104Ld | 106 | 0/10 | 0 | |
| 4 | 5 × 105 | Ag104 | 106 | 0/7 | 0 | ||
| 5 | None | None | Ag104Ld | 106 | 33/33 | 100 | |
| 6 | Ag104 | 106 | 20/20 | 100 | |||
Number of tumor cells as indicated were injected s.c. to C3B6F1 mice. Rechallenge was done 30 d after first tumor was surgically removed or rejected upon anti-CD4 antibody treatment.
The results were pooled from independent experiments. p-values calculated by chi-square test between groups were shown as follows: P < 0.001 for group 1 versus group 5, group 3 versus group 6, and group 4 versus group 6. P > 0.5 for group 2 versus group 5.
The tumors were surgically removed 21 d after first tumor challenge.
CD4+ cells were depleted by anti-CD4 antibody 14 d after first tumor challenge. Depletion was confirmed by checking peripheral blood with FACS.
CD4+CD25+ T cells accumulate inside the tumor
To investigate the cause of impaired tumor rejection even in the presence of immunogenic antigens, we first examined the tumor-infiltrating lymphocytes (TIL) in our animal model. Immunohistochemical and FACS stainings revealed that the Ag104Ld tumor tissues were infiltrated with both CD4+ and CD8+ cells that were colocalized. Very few tumor-infiltrating DX5+ NK or NK T cells were observed (unpublished data). Surprisingly, the lymphocytes that accumulated in the tumor mainly consisted of the CD4+CD25+ T cell subset (Fig. 1 b). We then compared the percentage of the CD4+CD25+ T cells among lymphocytes in the spleen, the draining lymph nodes (DLN), and the tumor on 0, 7, or 16 d after tumor challenge. We found that, with time, the percentage of the CD4+CD25+ T cells increased dramatically inside tumor although it remained constant in the spleen and DLN (Fig. 1 b). These data indicated that an increased percentage of CD4+CD25+ T cells was present at the effector site, but not in the lymphoid organs. These CD4+CD25+ T cells were of CD45RBlow phenotype (Fig. 1 c), which is consistent with what had been observed on CD4+CD25+ regulatory T cells (35). FOXP3 is a forkhead/winged helix transcription factor critical for mouse CD4+ CD25+ regulatory T cell differentiation and function (47–49). The real-time quantitative RT-PCR demonstrated much stronger expression of foxp3 mRNA in CD4+ T cells isolated from the tumor in comparison with CD4+ T cells isolated from the spleen of the tumor-bearing animals or CD4+CD25− T cells from the spleen of naive mice (Fig. 1 d). These observations raise the possibility that the CD4+ CD25+ cells that accumulate to comprise >70% of the CD4+ T cells at the effector site during tumor progression suppress the local immune response. We speculated that poor immunogenicity of the cancer might be attributed to immune regulation mechanisms by CD4+CD25+ T cells accumulating in the tumor environment.
CD4+CD25+ T cells suppress antitumor immunity
To examine whether tumor-infiltrating CD4+CD25+ T cells suppress T cell responses in vitro, we isolated the CD4+ T cells from the tumor tissues that had been established for 16 d in C3B6F1 mice and coculture them with purified CD4+ CD25− T cells from naive C3B6F1 mice in the presence of an optimal dose of plate-bound stimulatory anti-CD3 antibody. We observed that these tumor-infiltrating CD4+ T cells did significantly suppress the proliferation of CD4+CD25− T cells measured by 3H incorporation although the CD4+ T cells isolated from the spleen of tumor-bearing mice did not (Fig. 1 e). Because CD4+ and CD8+ cells can express CD25 during the early stage of T cell activation, it raises the concern that depletion of CD25+ cells in vivo would also eliminate the CD8+ T cells that are being activated. However, we observed that the majority of CD8+ T cells were CD25− in both spleen and tumor at 2 wk after tumor challenge (Fig. 1 f). Depletion of CD25+ cells did not reduce the CD8+ population because only a relatively few CD8+ cells express CD25 (Fig. 1 f). Either CD4+CD25+ cells preferentially accumulated or CD8+ cells failed to express CD25 inside tumor environment. To directly study the essential role of CD4+ T cells in tumor immunity in vivo, we depleted CD4+ cells and then observed the tumor growth. Impressively, the highly vascularized and poorly immunogenic Ag104Ld tumor was rejected rapidly in all of the 22 mice when the CD4+ cells were depleted (Fig. 1 g and Table II). Tumor rejection upon depletion of CD4+ cells was CD8+ cell dependent because tumor grew uncontrollably in the absence of both CD4+ and CD8+ cells (Table II). Depletion of NK1.1+ NK and NKT cells did not cause tumor rejection (Table II). To confirm that the subset of CD4+ T cells that express CD25 mainly contributes to the suppression of antitumor immunity in vivo, we treated mice bearing tumor for 2 wk with anti-CD25 antibody. Depletion of CD25+ cells with the antibody 14 d after tumor challenge dramatically suppressed the tumor growth in all of the 12 mice examined (Fig. 1 h). These data suggest that CD25+ T cells are mainly suppressive during the late phase of antitumor immune responses.
Table II.
Augmented rejection of tumor through elimination of CD4+ cells
| Tumor challengea | |||||
|---|---|---|---|---|---|
| Group | Tumor line | Dose | Treatment | Incidence of tumor growthb | Percent |
| 1 | Ag104Ld | 106 | None | 33/33 | 100 |
| 2 | 105 | 27/27 | 100 | ||
| 3 | 106 | CD4 depletionc | 0/9 | 0 | |
| 4 | 105 | 0/13 | 0 | ||
| 5 | 105 | CD4 and CD8 depletiond |
6/6 | 100 | |
| 6 | 106 | NK depletione | 3/3 | 100 | |
| 7 | 105 | NK and NKT depletionf | 6/6 | 100 | |
| 8 | Ag104 | 105 | None | 15/15 | 100 |
| 9 | 105 | CD4 depletionc | 4/8 | 50 | |
Number of tumor cells as indicated were injected s.c. to C3B6F1 mice.
The results were pooled from independent experiments. p-values calculated by chi-square test between groups were shown as follows: P < 0.001 for group 1 versus group 3, group 2 versus group 4, and group 4 versus group 5, P = 0.0015 for group 8 versus group 9.
CD4+ cells were depleted by anti-CD4 antibody 3 d before and 10 d after tumor challenge.
CD4+ cells were depleted by anti-CD4 antibody 3 d before and 10 d after tumor challenge. CD8+ cells were depleted by anti-CD8 antibody 14 d after tumor challenge.
NK cells were depleted by antiasialo GM-1 antibody 3 d before tumor challenge.
NK and NKT cells were depleted by anti-NK1.1 antibody 3 d before and 10 d after tumor challenge. Depletion of various cell populations was confirmed by checking peripheral blood by FACS.
Even in the absence of antigen Ld, depletion of CD4+ cells allowed 50% of the mice to reject the parental Ag104 tumor completely (Table II) and the remaining 50% of mice had delayed tumor growth kinetics (unpublished data). These observations suggest that other tumor antigens may also induce antitumor immunity once CD4-mediated suppression is removed. To test whether rejection leads to protective immunity against other tumor antigens, Ag104Ld tumor cells were initially inoculated and the CD4+ cells were depleted 14 d later. The rejection of tumor by depletion of CD4+ cells led to protection against future challenges not only of Ag104Ld tumor cells but also of the parental Ag104 tumor cells (Table I). Our data suggest that the removal of suppressive CD4+ cells allows an expanded repertoire of CD8+ cells to participate in protective immunity.
Regulatory T cells suppress CD8+ T cells at the effector phase
To test whether it is essential that CD4+ cells suppressed CD8+ T cell responses at the priming phase leading to progressive tumor growth, we first depleted CD4+ cells 3 d before tumor challenge. FACS analysis on the peripheral blood confirmed that the treatment with titrated dose of anti-CD4 antibody only eliminated CD4+ cells during the first 20 d of tumor growth. Delayed, yet progressive tumor development, observed in 11 out of 12 mice, resulted from CD4 depletion 3 d before tumor challenge, thus depleting CD4+ T cells during the early, predominantly priming phase of the antitumor response (Table III). However, we observed that CD4 depletion throughout the entire duration of tumor growth accomplished by injecting anti-CD4 antibody 3 d before and 10 d after tumor challenge, led to complete tumor rejection (Table III). This observation raised the possibility that eliminating CD4+CD25+ regulatory cells during the effector phase was essential for rescuing CD8+ T cell responses in the tumor. To test this hypothesis, tumor-bearing mice were treated with anti-CD4 antibody 20 d after tumor challenge when tumors were >1 cm in average diameter (or >500 mm3 in average volume). Complete tumor regression was observed in 29/29 tumor-bearing mice (Table III). We have yet to see any other immunoregulatory treatment that can result in the complete destruction of such a massive, well vascularized, highly progressive tumor. These data suggest that the absence of suppressive CD4+ T cells during the late stage of immune responses can be essential and sufficient to rescue antitumor immunity mediated by CD8+ T cells, which then causes tumor rejection.
Table III.
Suppression by CD4+ cells is in effector phase rather than priming phase
| Tumor challengea | |||||
|---|---|---|---|---|---|
| Group | Tumor line | Dose | Anti-CD4 treatment day relative to tumor challenge [0]b |
Incidence of tumor growthc | Percent |
| 1 | Ag104Ld | 105 | −3 and 10 | 0/13 | 0 |
| 2 | −3 | 11/12 | 92 | ||
| 3 | 20 | 0/21 | 0 | ||
| 4 | 5 × 105 | 20d | 0/8 | 0 | |
Number of tumor cells as indicated were injected s.c. to C3B6F1 mice.
Assume that tumor challenge is on day 0. CD4+ cells were depleted by anti-CD4 antibody. Days on which CD4+ cells were absent were confirmed by checking peripheral blood by FACS.
The results were pooled from multiple independent experiments. p-values calculated by chi-square test between groups were shown as follows: P < 0.001 for group 1 versus group 2 and group 2 versus group 3.
The average tumor size reached >500 mm3.
As described above, CD4 depletion-mediated tumor rejection was dependent on CD8+ T cells, suggesting that CD4+ cells might suppress CD8+ cells inside the tumor. We next examined how the CD4+ cells suppress CD8+ T cell function. First, we detected a dramatic increase of CD8+ T cells inside the tumor after CD4+ T cell depletion (Fig. 2 a) and increased IFN-γ production by the CD8+ T cells (Fig. 2 b). It suggested that CD4+ cells may inhibit the expansion of CD8+ cells responding to the tumor. To test this hypothesis, we adoptively transferred CFSE-labeled effector 2C T cells, activated in 2C/Rag-1 −/− mice by MC57-SIY tumor cells (50) into Ag104Ld-bearing C3B6F1 mice. 2C T cells were derived from TCR transgenic 2C mice, that can directly recognize the Ld antigen on Ag104Ld. These transferred effector 2C T cells expressed high levels of CD44 and down-regulated CD62L expression. However, adoptive transfer of 3 × 106 of these activated effector 2C T cells was not sufficient to cause tumor rejection (unpublished data). Before CD4+ T cell depletion, only a few CFSE-labeled effector 2C T cells were present inside the tumor, but after depletion, a dramatic increase in number and percentage of the effector cells was observed (Fig. 2 c). More importantly, proliferation of these 2C T cells occurred mainly at the effector site upon depletion of CD4+ T cells, suggesting that suppression predominantly occurs at the local tumor site (Fig. 2 c). Therefore, these CD4+ cells suppressed proliferation, maturation, and expansion of CD8+ T cells at the tumor site.
Figure 2.

CD4+ cells suppress the proliferation and IFN-γ production of tumor-infiltrating CD8+ T cells. 106 Ag104Ld tumor cells were inoculated to C3B6F1 mice s.c. and anti-CD4 antibody (GK1.5) was injected i.p. 14 d after tumor challenge. Spleen as well as tumor tissue were isolated from mice 1 wk after CD4 depletion. The percentage of CD8+ T cells in the tumor tissue was determined by FACS (a). The tumor-infiltrating T cells (TIL) were enriched with anti-Thy1.2 magnetic bead system. Spleen cells and purified TIL were stimulated in vitro with PMA and ionomycin in the presence of brefeldin A for 4 h. The percentage of IFN-γ–producing CD8+ T cells among total CD8+ T cells in the tumor was determined by FACS (b). 106 MC57-SIY tumor cells were injected at multiple sites s.c. to 2C TCR transgenic mice in Rag-1 − / − background to activate 2C T cells. 2C T cells were isolated 96 h after activation and of CD62LloCD44high phenotype. CFSE-labeled activated 2C T cells were adoptively transferred to these Ag104Ld tumor bearing mice with or without CD4 depletion. Tumor tissue was collected from mice 2 d after transfer of 2C T cell. The proliferation of CFSE-labeled 2C T cells was monitored by FACS (c). Results from one experiment representative of three were shown.
IL-10 and TGF-β are involved in the suppression mechanisms in vivo
To explore the mechanisms of how CD4+ cells suppressed antitumor immunity, we compared the levels of cytokines in the spleen and tumor before and after CD4+ cell depletion. In the spleen, there were no significant differences in the levels of cytokines that were detected in the presence or absence of CD4+ cells (Fig. 3 a). The Th2 cytokines, IL-4 and IL-5, did not show significant difference at the tumor site in the presence or absence of CD4+ T cells (Fig. 3 b). However, in the absence of CD4+ T cells, inflammatory cytokines including IFN-γ, TNF, IL-6, and MCP-1 were increased dramatically in the tumor tissues compared with the levels in the presence of CD4+ cells. The level of the antiinflammatory cytokine IL-10 was also much lower after the depletion of CD4+ cells (Fig. 3 b). These results suggested that the CD4+ cells maintained an antiinflammatory environment that inhibited antitumor immunity inside the tumor.
Figure 3.
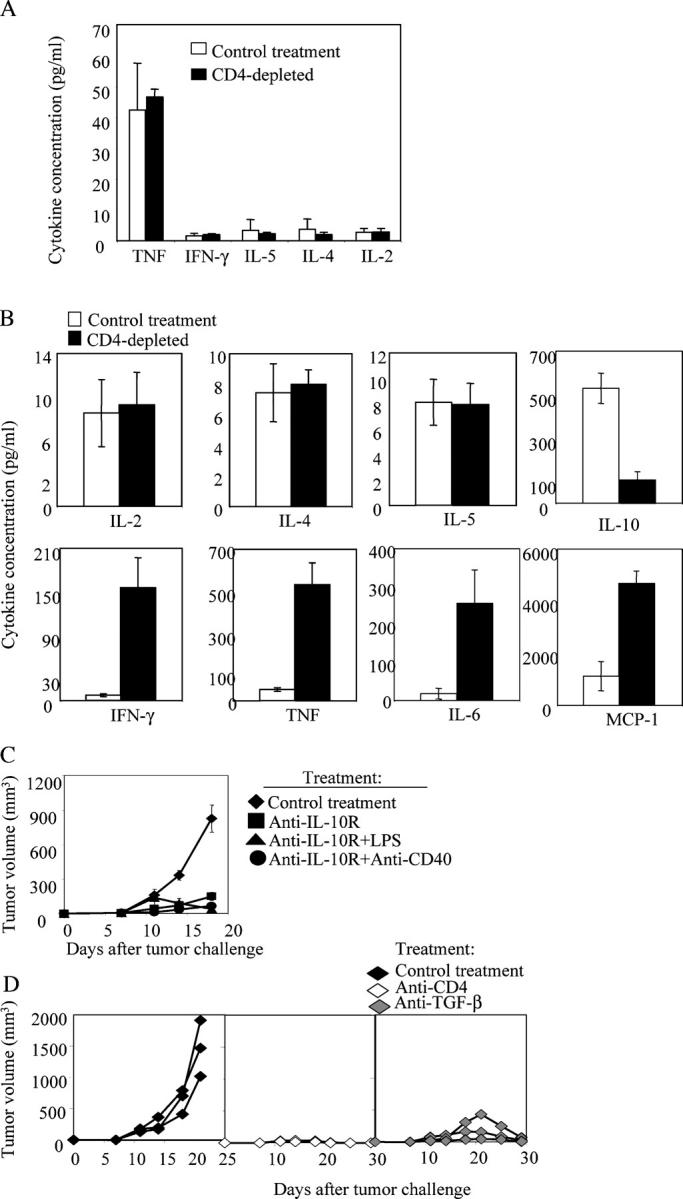
CD4+ cells maintain an antiinflammatory environment inside the tumor. (a and b) CD4+ cells were depleted by treating the mice with anti-CD4 antibody i.p.1 d before tumor challenge. 106 Ag104Ld tumor cells were inoculated to C3B6F1 mice s.c. and spleen (a) as well as tumor tissue (b) were collected from mice 2 wk after tumor challenge and homogenized. The debris was spun down and the supernatant was collected and subjected to CBA kit. Seven mice from CD4-depleted or control group were used. Data shown were means and SD. (c) 105 Ag104Ld tumor cells were inoculated to C3B6F1 mice s.c. and 100 μg of blocking anti–IL-10 receptor (IL-10R) antibody was injected to mice 1 d before and 7 d after tumor challenge. In combination with anti–IL-10R treatment, 100 μg of LPS or 100 μg of agonistic anti-CD40 antibody was given to mice i.p. 7 d after tumor challenge. Tumor growth was monitored. Data shown were means of tumor volume from 8 to 11 mice in each group and SD. The difference in tumor growth between the control group and all of the treated groups, as well as the anti–IL-10R–treated group with the ones in combination with LPS or anti-CD40 was significant by statistical analysis with the random effect models for longitudinal data (P < 0.001). Statistical analysis with t test also showed that the difference in tumor size between the control group and all of the treated groups was significant after 14 d after tumor inoculation (P < 0.001, t test). The difference in tumor size between group treated with anti–IL-10R antibody and the ones treated in combination with LPS or anti-CD40 groups was significant after 18 d after tumor inoculation (P < 0.001, t test). (d) 105 Ag104Ld tumor cells were inoculated to C3B6F1 mice s.c. and anti-CD4 antibody or 250 μg anti-TGF-β antibody was injected to mice 1 d before and 7 d after tumor challenge. The representative pattern of tumor growth in one experiment was shown.
We hypothesized that antitumor immunity could be promoted if we blocked the effects of the antiinflammatory cytokines. Indeed, tumor growth was dramatically inhibited in all of the 11 mice after the treatment with anti–IL-10 receptor (IL-10R) antibody (Fig. 3 c). The tumor growth was further inhibited in all of the 16 mice after the treatment with anti–IL-10R antibody in combination with inflammatory signals, such as LPS or agonistic anti-CD40 antibody (Fig. 3 c). Administration of LPS alone did not change tumor growth significantly, suggesting activation of DCs through toll-like receptor 4 (TLR4) alone may not be sufficient to reverse suppression (unpublished data). TGF-β is another cytokine that can suppress inflammatory responses. To investigate the role of TGF-β in this model, we blocked TGF-β at the time of challenge with Ag104Ld tumor cells. The tumors were completely rejected in all of the 6 mice after the blockade of TGF-β, whereas all of the 27 mice with control treatment died of tumor burden (P < 0.001, Chi-square test). The representative pattern of the tumor growth in one of the experiments was shown (Fig. 3 d). These results showed that the antiinflammatory environment inside the tumor correlates with the failure of tumor rejection. The CD4+ cells appear to actively maintain this antiinflammatory environment inside the tumor to suppress the antitumor immune response, thereby promoting the growth of the antigenic tumors.
Depletion of CD4+CD25+ T cells at the effector site eradicates established tumors
Considering CD4+CD25+ T cells are the main population of CD4+ T cells inside tumor during its progression, we propose that depletion of CD4+ cells at the tumor sites is sufficient to achieve tumor rejection. Selective depletion of CD4+ cells in the tumor-bearing hosts at the tumor site using a small amount of anti-CD4 antibody may efficiently reverse immune tolerance of CD8+ cells to the tumor while retaining normal numbers of CD4+ cells in the lymphoid tissues. To ensure that the depletion of CD4+-suppressive cells at the effector site after tumor's establishment can promote antitumor immunity, we used low doses of anti-CD4 antibody (12.5–50 μg/injection) for intratumor injection to assure depletion of CD4+ T cells inside the tumor but not systemically (Fig. 4 a). We found that intratumor depletion of CD4+ cells 14 d after tumor inoculation rapidly caused the complete rejection of well-established tumors in all of the 21 mice that were treated (Fig. 4 b). This result confirms that depletion of CD4+-suppressive cells inside the tumor is sufficient to cause the rejection of an established tumor and suggests that the active suppression of antitumor CD8+ T cells mainly occurs at the effector site and that it is reversible. Importantly, local treatment can become an affordable and effective treatment for tumor immunotherapy.
Figure 4.

Intratumor depletion of CD4+ cells leads to rejection of established tumor. 105 Ag104Ld tumor cells were inoculated to C3B6F1 mice s.c. and 12.5–50 μg anti-CD4 antibody (GK1.5) was injected intratumorally, once every 7 d, starting 14 d after tumor challenge. The arrow indicated the first treatment. Spleen as well as tumor tissue were isolated from mice 2 d after CD4 depletion. The percentage of CD4+ T cells in the spleen or tumor tissue was determined by FACS (a). Tumor growth was monitored and data shown was representative pattern of tumor growth in one of five experiments comprising 21 mice (b).
Discussion
Poor immune responses to weak tumor antigens is often believed to be the basis for the progressive growth of tumors in immunocompetent hosts, and is assumed to be a major hurdle for successful immunotherapy. In our study, we introduced a strong Ld alloantigen into the murine Ag104 fibrosarcoma cell line, a highly progressive, invasive, and vascularizing tumor that can kill the host in a few weeks. However, the presence of a strong antigen could not elicit an antitumor immune response that prevented or retarded the growth of the tumor. Although infiltration of lymphocytes is commonly observed in both human and animal cancers (19–22), immune rejection of cancer is rarely observed. In spite of obvious T cell infiltration, the failure of the host to eradicate the tumor is a confounding phenomenon that has not been well understood. A closer examination of TIL in our animal model revealed that the CD4+ subset, CD4+CD25+ T cells, selectively accumulate inside the tumor and comprise the majority of the total TIL. We further show that, during tumor progression, CD4+CD25+ T cells primarily mediate a suppressive environment inside the tumor and abrogate the effector function of CD8+ T cells. Local intratumoral depletion of these regulatory T cells unmasks the immunogenicity of the tumor and reverse CTL tolerization leading to the rapid rejection of well-established tumors. More importantly, in this study we demonstrated that T regulatory cells can be one important local factor to suppress immune responses against a strong tumor antigen leading to progressive growth of cancer in the immune-competent hosts.
The role of CD4+ T cells in tumor immunity has been extensively studied (51). CD4+ T cells have been shown to play an essential role in promoting CD8+ T cell responses (52–54) and the development of CD8 T cell memory (55–57). In antitumor immunity, CD4+ T cells also have been demonstrated to be important in sustaining the functions of adoptively transferred CD8+ T cells (50, 58, 59). Depletion of CD4+ cells in mice significantly diminished the ability of GM-CSF vaccine/anti-CTLA4 treatment to protect the animals from subsequent tumor challenge (60). On the other hand, early studies by Robert North have shown that intravenous injection of a depleting anti-CD4 antibody at early stages of tumor growth led to CD8+ T cell–mediated tumor regression (36–38). Recent studies strongly support the notion that a population of CD4+ cells, CD4+CD25+ T cells, significantly limits the efficacy of vaccine-induced antitumor immune responses, and their inhibition or elimination before tumor challenge could significantly enhance antitumor immunity(39–42, 61–66). For example, in a study, a combination of a GM-CSF–transduced tumor vaccine plus anti-CTLA4 was more effective at eliminating established tumors when animals were treated with anti-CD25 antibodies before tumor challenge (39). Another study revealed that splenic cells depleted of CD4+CD25+ T cells can mediate tumor regression presumably through promoting autoreactivity because autoimmune diseases were also induced (41). Although these studies suggest that regulatory T cells may inhibit initial priming of CD8+ T cells, some of which recognize self antigens, our data strongly demonstrate that CD4+CD25+ T cells selectively accumulate inside the tumor and play a greater role in suppressing the CD8+ T cell function at the effector phase of tumor progression and maintaining the local immune tolerance permissive of progressive growth of an antigenic tumor.
It has been a general concern that the depletion of CD4+CD25+ T cells in the tumor-bearing hosts may unwittingly eliminate the helper function of CD4+ T cells, which can also express the CD25+ marker after activation by tumor or by vaccination. We propose that CD4+ cells predominately play an enhancing helper role during the initial stages, but once tumors become chronically persistent and established, the increased accumulation of CD4+ regulatory T cells inhibit CD8+ cell function and mask the immunogenicity of tumor. Our study indeed establishes that the suppression of immunity against tumor by CD4+CD25+ T cells, chiefly CD8+ T cell responses, mainly occurs in the effector phase at tumor site and that depletion of the regulatory T cells at the late stage of tumor progression does not mitigate the possible T helper function. In fact, depletion of regulatory T cells unveils the immunogenicity of tumor cells and provides long-term protection against rechallenge of even the parental tumor cells lacking the strong antigen Ld. This result suggests that the induction of immunity was not exclusively against Ld, but rather, the depletion of regulatory T cells promoted immunity against previously poorly immunogenic tumor antigens and expanded the tumor-reactive CD8+ T cell repertoire. The parental Ag104 (without Ld), a highly progressive and poorly immunogenic tumor, fails to respond to various conventional immunotherapies but shows increased immunogenicity leading to tumor rejection or delayed tumor progression after depletion of regulatory T cells. It is conceivable that this strong antitumor immunity induced locally by depletion of CD4+ cells may also lead to the rejection of tumors at distal sites.
It has been shown that CD4+CD25+ T cell subset plays critical roles in down-regulating autoimmune inflammation and concomitant immunity during persistent infection (31–35). Tumor microenvironment may allow the activation or expansion of regulatory T cells after the progression of tumors. We speculate that chronic inflammation inside the tumor tissue may resemble some aspects of protective immunity to normal tissues during chronic autoimmune inflammation or persistent infection, which allows the accumulation of the CD4+CD25+ T cell subset to down-regulate immune responses, limit local inflammation, and reduce local tissue destruction. In addition, tumor-mediated suppressive factors may favor the survival of regulatory T cells over effector T cells. As a consequence, it is possible that the same regulatory mechanisms designed to prevent uncontrolled inflammatory responses in normal tissues are turned on to inhibit antitumor immunity. It is not uncommon to find that tumor-infiltrating T cells, which contain the CD4+CD25+ population, in various tumor tissues (22, 67–71). Our study revealed that the population of tumor-infiltrating cells is skewed to favor regulatory CD4+ T cells over the helper CD4+ T cells within the tumor tissue, especially as tumor progresses and gets established in the host. Therefore, understanding the nature of tumor-infiltrating CD4+ T cells by surface markers or cytokine profiles would not only avoid possible deleterious effects of regulatory CD4+ T cells in an adoptive transfer immunotherapy, but would also allow informed predictions about the prognosis of cancer patients.
Understanding the balance of T effector versus T regulatory cells may be more important to determine the outcome of immune responses inside tumors. Our recent study has demonstrated that rapid recruitment of naive lymphocytes and expansion of CD8+ T cells inside the tumor may be a way of creating a dominant proinflammatory environment, leading to the rejection of tumor at local and distal sites (72). Now, we have further demonstrated in this study that the depletion of regulatory T cells is another efficient way of converting the antiinflammatory environment inside tumor to proinflammatory one. The local treatment to eliminate regulatory T cells has certain critical advantages over systemic treatment. First, local treatment may avoid side effects induced by systemic depletion of all CD4+ T cells, which may abrogate T helper–mediated protective immunity against pathogens. Second, it would not hinder effective priming of CD8+ T cell in the lymphoid tissues by the helper CD4+ T cells, because depletion remains local. Third, the local treatment would be even expected to be more effective because the key suppression of CD4+CD25+ regulatory T cells resides inside tumor. Also intratumoral treatment would reduce the dose of antibody to be applied to the patients as well as be more affordable even for the developing countries. CD4+CD25+ T cells have been shown to be present in a variety of human cancer tissues (22, 67–71). A study published recently revealed that CD4+CD25+ T cells are negatively associated with the prognosis of the ovarian cancer patients (22). Therefore, regulatory T cells within the tumor environment represent an attractive target, and their depletion may lead to improvements in the current immunotherapy protocol in the future clinical trials. It is likely that a combination treatment, which would rapidly expand the effector cells at the tumor site while locally depleting the regulatory cells, could potentially provide a potent strategy for enhancing antitumor immunity and permitting a clinically desirable outcome for cancer patients.
Materials and Methods
Mice and cell lines.
Female C3B6F1 and C3H mice, aged 5–8 wk, were purchased from the Frederick Cancer Research Facility of the US National Cancer Institute (Frederick, MD) and were maintained in a specific pathogen-free facility at the University of Chicago (Chicago, IL). Animal care and experiments were done in accordance with institutional and National Institutes of Health (NIH) guidelines and were approved by an animal use committee at the University of Chicago. The Ag104 fibrosarcoma and Ag104 cell line (43) expressing murine H-2Ld (Ag104-Ld) has been described previously (1).
Measurement of cytokines in the spleen and tumor
We prepared tumor and spleen homogenates as described previously (72). In brief, comparable amounts of tumor or spleen tissues were collected, weighed, and homogenized in PBS containing protease inhibitors, and the supernatants were collected by centrifugation. The amount of cytokines in the supernatants was quantified using the cytometric bead array kit (CBA; BD Biosciences) on a FACSCaliber cytometer equipped with CellQuestPro and CBA software (Becton Dickinson) according to manufacture's instructions.
Flow cytometric analysis
FACS analyses were performed using FITC-, PE-, Cy-chrome– or biotin-conjugated antibodies to mouse CD45RB, CD4, CD8, CD25, and IFNγ (all BD Biosciences). Streptavidin–Cy-chrome conjugate was from BD Biosciences; biotin-conjugated clonotypic antibody (1B2) to 2C TCR and cell staining was performed as described previously (50). For intracellular cytokine staining, T cells purified from the tumor tissue were restimulated for 4 h with PMA (50 ng/ml; Sigma-Aldrich) and ionomycin (500 ng/ml; Sigma-Aldrich) in the presence of brefeldin A (Sigma-Aldrich). For analysis of CFSE-labeled 2C T cells, isolated tumor or spleen single-cell suspensions were stained with biotinylated 1B2 antibody, washed, and stained with a mixture of PE-coupled anti-CD8 and Cy-chrome–coupled streptavidin. FACS data were analyzed with FlowJo software (Becton Dickinson)
Cell isolation and in vitro assay.
To isolate CD4+ T cells from the tumor tissue, the mice were first bled to decrease the contamination of tumor tissue by blood. Tumor tissues were collected, washed in PBS, cut into pieces and resuspended in Dulbecco's modified Eagle's medium supplemented with 2% fetal calf serum and 1.5 mg/ml of collagenase D (collagenase D solution) for 25 min in a 37°C shaking incubator. The cell suspension was collected after 25 min, and the cell clumps were digested for another 25 min in collagenase D solution until all of the tumor tissue had resolved into a single-cell suspension. Tumor-infiltrating T cells were enriched from this single-cell suspension with biotin-conjugated Thy-1.2 antibody followed by antibiotin magnetic beads using the AutoMACS system (Miltenyi Biotech). Enriched tumor-infiltrating T cells were stained with PE-conjugated anti-CD4 and FACS-sorted into CD4+ population on a MoFlo. CD4+ or CD4+CD25+ and CD4+CD25− cells were purified from splenic single-cell suspensions with CD4+ T cells or CD4+CD25+ regulatory T cell isolation magnetic bead system (Miltenyi Biotech), respectively, using the AutoMACS system (Miltenyi Biotech). For all the cell isolations, >90% purity of desired population was routinely obtained. For T cell costimulation assay, we coated 96-well plates with 50 μl of 2 μg/ml anti-CD3 antibody overnight. The plates were then washed with PBS and 6 × 104 purified CD4+CD25− T cells were cultured per well with or without 6 × 104 isolated CD4+ T cells from the spleen or tumor tissue of the tumor bearing mice. In all assays, T cell proliferation was measured by adding 1 μCi of [3H]thymidine per well for the last 24 h of the 3-d culture. [3H]Thymidine incorporation was measured in a TopCount microplate scintillation counter (Packard Instrument Co.).
Adoptive transfer of 2C T cells
We first activated 2C T cells by inoculation of 106 MC57-SIY tumor cells s.c. at multiple sites to 2C mice in B6/Rag-1 −/− background (2C mice; 50). We then isolated lymph node cells and splenocytes from these challenged 2C mice and negatively selected for CD8+ T cells with a CD8+ T cell enrichment kit (Miltenyi Biotec) 5 d after tumor cell inoculation. When analyzed, majority of the 2C cells expressed CD44high CD62Llow phenotype. These 2C T cells were then labeled with CFSE as described previously (50). We injected 3 × 106 CFSE-labeled T cells in a 0.2-ml volume intravenously into the retro-orbital plexus of the tumor-bearing mice. Cells were isolated from the spleen and tumors 48 h after transfer and subjected to FACS analysis.
Cell depletions and cytokine blockade in vivo.
Mice were depleted of lymphocyte subsets systemically by standard procedures (50), using mAb GK1.5 for CD4+ cells, mAb 2.34 for CD8+ cells and PC61 for CD25+ cells. Examination of splenocytes and lymph node cells by flow cytometry showed that the depleted CD4+ or CD8+ subset represented <0.5% of the total lymphocytes, with normal levels of other subsets. Intratumor CD4+ cell depletion was done using 12.5 to 50 μg GK1.5 in 50 μl PBS. To block IL-10 or TGF-β, 100 μg anti–IL-10 receptor or 250 μg anti-TGF-β antibodies (A411) were injected to the mice i.p. 1 d before and 7 d after tumor challenge. 100 μg LPS or agonistic anti-CD40 antibody was given to the mice i.p. 7 d after tumor challenge.
Real-time quantitative RT-PCR assay.
The real-time quantitative RT-PCR assay for foxp3 was done as described previously (73). In brief, total RNA from tumors was isolated, and 5 μg of total RNA was reverse transcribed into cDNA by using the First Strand cDNA Synthesis Kit (Amersham Biosciences). Real-time quantitative PCR analysis was done on a SmartCycler real-time thermal cycler (Cepheid). Each cDNA sample was amplified for foxp3 and GAPDH by using the TaqMan universal PCR master mixture containing AmpliTaq gold DNA polymerase in accordance with the manufacturer's instructions (PerkinElmer). We determined the concentration of target gene by the comparative CT (threshold cycle number at the cross-point between amplification plot and threshold) method and normalized values to the internal GAPDH control. The primer sequences for foxp3 were 5′-CCCAGGAAAGACAGCAACCTT-3′ (forward primer) and 5′-TTCTCACAACCAGGCCACTTG-3′ (reverse primer), and the probe for foxp3 was 5′-ATCCTACCCACTGCTGGCAAATGGAGTC-3′.
Statistical analysis for difference in tumor growth.
Because the tumor growth was observed repeatedly over time on the same mouse, the random effect models for longitudinal data were used to analyze such data. For each experiment, the tumor growth was assumed to depend on treatment and to follow a linear growth rate over time. The model gave an overall estimate of the intercept and slope of the linear growth for each group. Both the intercept and slope were allowed to vary among individual mouse. We were interested mainly in comparing whether the slope, i.e., the growth rate, was different among different treatment groups. Because the actual tumor growth may not follow a linear growth trend over the entire follow up period. The increase of tumor growth was slow at the early stage and became rapid at the later stage in some experiments. We also investigated this by adding a quadratic term of the follow-up time in the above random effect models.
Acknowledgments
We thank Yang Wang for her technical assistance and Dr. Theodore Karrison (University of Chicago) for statistical analysis.
This research was supported by grants from NIH (R01-HD37104, R01-DK58897, R01-CA22677, R01-CA37156, and P01-CA09296-01). P. Yu is a recipient of NIH training grant (5T32DK07074).
The authors have no conflicting financial interests.
Abbreviations used: Ag104Ld, Ld-expressing Ag104; CBA, cytometric bead array kit; DLN, draining lymph nodes; TIL, tumor-infiltrating lymphocytes; TLR4, toll-like receptor 4.
References
- 1.Wick, M., P. Dubey, H. Koeppen, C.T. Siegel, P.E. Fields, L. Chen, J.A. Bluestone, and H. Schreiber. 1997. Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. J. Exp. Med. 186:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunn, G.P., L.J. Old, and R.D. Schreiber. 2004. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22:329–360. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg, S.A. 1999. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity. 10:281–287. [DOI] [PubMed] [Google Scholar]
- 4.Pardoll, D. 2003. Does the immune system see tumors as foreign or self? Annu. Rev. Immunol. 21:807–839. [DOI] [PubMed] [Google Scholar]
- 5.Monach, P.A., S.C. Meredith, C.T. Siegel, and H. Schreiber. 1995. A unique tumor antigen produced by a single amino acid substitution. Immunity. 2:45–59. [DOI] [PubMed] [Google Scholar]
- 6.Coulie, P.G., F. Lehmann, B. Lethe, J. Herman, C. Lurquin, M. Andrawiss, and T. Boon. 1995. A mutated intron sequence codes for an antigenic peptide recognized by cytolytic T lymphocytes on a human melanoma. Proc. Natl. Acad. Sci. USA. 92:7976–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolfel, T., M. Hauer, J. Schneider, M. Serrano, C. Wolfel, E. Klehmann-Hieb, E. De Plaen, T. Hankeln, K.H. Meyer zum Buschenfelde, and D. Beach. 1995. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 269:1281–1284. [DOI] [PubMed] [Google Scholar]
- 8.De Plaen, E., C. Lurquin, A. Van Pel, B. Mariame, J.P. Szikora, T. Wolfel, C. Sibille, P. Chomez, and T. Boon. 1988. Immunogenic (tum-) variants of mouse tumor P815: cloning of the gene of tum- antigen P91A and identification of the tum- mutation. Proc. Natl. Acad. Sci. USA. 85:2274–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lurquin, C., A. Van Pel, B. Mariame, E. De Plaen, J.P. Szikora, C. Janssens, M.J. Reddehase, J. Lejeune, and T. Boon. 1989. Structure of the gene of tum- transplantation antigen P91A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell. 58:293–303. [DOI] [PubMed] [Google Scholar]
- 10.Mandruzzato, S., F. Brasseur, G. Andry, T. Boon, and P. van der Bruggen. 1997. A CASP-8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J. Exp. Med. 186:785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robbins, P.F., M. El-Gamil, Y.F. Li, Y. Kawakami, D. Loftus, E. Appella, and S.A. Rosenberg. 1996. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 183:1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van den Eynde, B., B. Lethe, A. Van Pel, E. De Plaen, and T. Boon. 1991. The gene coding for a major tumor rejection antigen of tumor P815 is identical to the normal gene of syngeneic DBA/2 mice. J. Exp. Med. 173:1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beck-Engeser, G.B., P.A. Monach, D. Mumberg, F. Yang, S. Wanderling, K. Schreiber, R. Espinosa III, M.M. Le Beau, S.C. Meredith, and H. Schreiber. 2001. Point mutation in essential genes with loss or mutation of the second allele: relevance to the retention of tumor-specific antigens. J. Exp. Med. 194:285–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boon, T., and P. van der Bruggen. 1996. Human tumor antigens recognized by T lymphocytes. J. Exp. Med. 183:725–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houghton, A.N. 1994. Cancer antigens: immune recognition of self and altered self. J. Exp. Med. 180:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starr, T.K., S.C. Jameson, and K.A. Hogquist. 2003. Positive and negative selection of T cells. Annu. Rev. Immunol. 21:139–176. [DOI] [PubMed] [Google Scholar]
- 17.Palmer, E. 2003. Negative selection–clearing out the bad apples from the T-cell repertoire. Nat. Rev. Immunol. 3:383–391. [DOI] [PubMed] [Google Scholar]
- 18.Gilboa, E. 1999. The makings of a tumor rejection antigen. Immunity. 11:263–270. [DOI] [PubMed] [Google Scholar]
- 19.Clark, W.H., Jr., D.E. Elder, D.T. Guerry, L.E. Braitman, B.J. Trock, D. Schultz, M. Synnestvedt, and A.C. Halpern. 1989. Model predicting survival in stage I melanoma based on tumor progression. J. Natl. Cancer Inst. 81:1893-1904. [DOI] [PubMed] [Google Scholar]
- 20.Clemente, C.G., M.C. Mihm Jr., R. Bufalino, S. Zurrida, P. Collini, and N. Cascinelli. 1996. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 77:1303–1310. [DOI] [PubMed] [Google Scholar]
- 21.Zhang, L., J.R. Conejo-Garcia, D. Katsaros, P.A. Gimotty, M. Massobrio, G. Regnani, A. Makrigiannakis, H. Gray, K. Schlienger, M.N. Liebman, et al. 2003. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 348:203–213. [DOI] [PubMed] [Google Scholar]
- 22.Curiel, T.J., G. Coukos, L. Zou, X. Alvarez, P. Cheng, P. Mottram, M. Evdemon-Hogan, J.R. Conejo-Garcia, L. Zhang, M. Burow, et al. 2004. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10:942–949. [DOI] [PubMed] [Google Scholar]
- 23.Gorelik, E. 1983. Concomitant tumor immunity and the resistance to a second tumor challenge. Adv. Cancer Res. 39:71–120. [DOI] [PubMed] [Google Scholar]
- 24.Turk, M.J., J.A. Guevara-Patino, G.A. Rizzuto, M.E. Engelhorn, and A.N. Houghton. 2004. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J. Exp. Med. 200:771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perdrizet, G.A., S.R. Ross, H.J. Stauss, S. Singh, H. Koeppen, and H. Schreiber. 1990. Animals bearing malignant grafts reject normal grafts that express through gene transfer the same antigen. J. Exp. Med. 171:1205–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakagomi, H., M. Petersson, I. Magnusson, C. Juhlin, M. Matsuda, H. Mellstedt, J.L. Taupin, E. Vivier, P. Anderson, and R. Kiessling. 1993. Decreased expression of the signal-transducing zeta chains in tumor-infiltrating T-cells and NK cells of patients with colorectal carcinoma. Cancer Res. 53:5610–5612. [PubMed] [Google Scholar]
- 27.Singh, S., S.R. Ross, M. Acena, D.A. Rowley, and H. Schreiber. 1992. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J. Exp. Med. 175:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spiotto, M.T., P. Yu, D.A. Rowley, M.I. Nishimura, S.C. Meredith, T.F. Gajewski, Y.X. Fu, and H. Schreiber. 2002. Increasing tumor antigen expression overcomes “ignorance” to solid tumors via crosspresentation by bone marrow-derived stromal cells. Immunity. 17:737–747. [DOI] [PubMed] [Google Scholar]
- 29.Sakaguchi, S., K. Fukuma, K. Kuribayashi, and T. Masuda. 1985. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J. Exp. Med. 161:72–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugihara, S., Y. Izumi, T. Yoshioka, H. Yagi, T. Tsujimura, O. Tarutani, Y. Kohno, S. Murakami, T. Hamaoka, and H. Fujiwara. 1988. Autoimmune thyroiditis induced in mice depleted of particular T cell subsets. I. Requirement of Lyt-1 dull L3T4 bright normal T cells for the induction of thyroiditis. J. Immunol. 141:105–113. [PubMed] [Google Scholar]
- 31.Bach, J.F., and J. Francois Bach. 2003. Regulatory T cells under scrutiny. Nat. Rev. Immunol. 3:189–198. [DOI] [PubMed] [Google Scholar]
- 32.Belkaid, Y., C.A. Piccirillo, S. Mendez, E.M. Shevach, and D.L. Sacks. 2002. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 420:502–507. [DOI] [PubMed] [Google Scholar]
- 33.Sakaguchi, S. 2004. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22:531–562. [DOI] [PubMed] [Google Scholar]
- 34.Shevach, E.M. 2002. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389–400. [DOI] [PubMed] [Google Scholar]
- 35.Maloy, K.J., and F. Powrie. 2001. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2:816–822. [DOI] [PubMed] [Google Scholar]
- 36.Berendt, M.J., and R.J. North. 1980. T-cell-mediated suppression of antitumor immunity. An explanation for progressive growth of an immunogenic tumor. J. Exp. Med. 151:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bursuker, I., and R.J. North. 1984. Generation and decay of the immune response to a progressive fibrosarcoma. II. Failure to demonstrate postexcision immunity after the onset of T cell-mediated suppression of immunity. J. Exp. Med. 159:1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Awwad, M., and R.J. North. 1988. Immunologically mediated regression of a murine lymphoma after treatment with anti-L3T4 antibody. A consequence of removing L3T4+ suppressor T cells from a host generating predominantly Lyt-2+ T cell- mediated immunity. J. Exp. Med. 168:2193–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sutmuller, R.P., L.M. van Duivenvoorde, A. van Elsas, T.N. Schumacher, M.E. Wildenberg, J.P. Allison, R.E. Toes, R. Offringa, and C.J. Melief. 2001. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 194:823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steitz, J., J. Bruck, J. Lenz, J. Knop, and T. Tuting. 2001. Depletion of CD25(+) CD4(+) T cells and treatment with tyrosinase-related protein 2-transduced dendritic cells enhance the interferon alpha-induced, CD8(+) T-cell-dependent immune defense of B16 melanoma. Cancer Res. 61:8643–8646. [PubMed] [Google Scholar]
- 41.Shimizu, J., S. Yamazaki, and S. Sakaguchi. 1999. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J. Immunol. 163:5211–5218. [PubMed] [Google Scholar]
- 42.Jones, E., M. Dahm-Vicker, A.K. Simon, A. Green, F. Powrie, V. Cerundolo, and A. Gallimore. 2002. Depletion of CD25+ regulatory cells results in suppression of melanoma growth and induction of autoreactivity in mice. Cancer Immun. 2:1. [PubMed] [Google Scholar]
- 43.Ward, P.L., H. Koeppen, T. Hurteau, and H. Schreiber. 1989. Tumor antigens defined by cloned immunological probes are highly polymorphic and are not detected on autologous normal cells. J. Exp. Med. 170:217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Melero, I., W.W. Shuford, S.A. Newby, A. Aruffo, J.A. Ledbetter, K.E. Hellstrom, R.S. Mittler, and L. Chen. 1997. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 3:682–685. [DOI] [PubMed] [Google Scholar]
- 45.Urban, J.L., J.M. Holland, M.L. Kripke, and H. Schreiber. 1982. Immunoselection of tumor cell variants by mice suppressed with ultraviolet radiation. J. Exp. Med. 156:1025–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uyttenhove, C., J. Maryanski, and T. Boon. 1983. Escape of mouse mastocytoma P815 after nearly complete rejection is due to antigen-loss variants rather than immunosuppression. J. Exp. Med. 157:1040–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 48.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 49.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 50.Yu, P., M.T. Spiotto, Y. Lee, H. Schreiber, and Y.X. Fu. 2003. Complementary role of CD4+ T cells and secondary lymphoid tissues for cross-presentation of tumor antigen to CD8+ T cells. J. Exp. Med. 197:985–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ossendorp, F., R.E. Toes, R. Offringa, S.H. van der Burg, and C.J. Melief. 2000. Importance of CD4(+) T helper cell responses in tumor immunity. Immunol. Lett. 74:75–79. [DOI] [PubMed] [Google Scholar]
- 52.Houghton, A.N., J.S. Gold, and N.E. Blachere. 2001. Immunity against cancer: lessons learned from melanoma. Curr. Opin. Immunol. 13:134–140. [DOI] [PubMed] [Google Scholar]
- 53.Toes, R.E., F. Ossendorp, R. Offringa, and C.J. Melief. 1999. CD4 T cells and their role in antitumor immune responses. J. Exp. Med. 189:753–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang, R.F. 2001. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol. 22:269–276. [DOI] [PubMed] [Google Scholar]
- 55.Janssen, E.M., E.E. Lemmens, T. Wolfe, U. Christen, M.G. von Herrath, and S.P. Schoenberger. 2003. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 421:852–856. [DOI] [PubMed] [Google Scholar]
- 56.Sun, J.C., and M.J. Bevan. 2003. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 300:339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shedlock, D.J., and H. Shen. 2003. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 300:337–339. [DOI] [PubMed] [Google Scholar]
- 58.Dudley, M.E., J.R. Wunderlich, P.F. Robbins, J.C. Yang, P. Hwu, D.J. Schwartzentruber, S.L. Topalian, R. Sherry, N.P. Restifo, A.M. Hubicki, et al. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 298:850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, H.Y., J. Zhou, K. Zhu, A.I. Riker, F.M. Marincola, and R.F. Wang. 2002. Identification of a mutated fibronectin as a tumor antigen recognized by CD4+ T cells: its role in extracellular matrix formation and tumor metastasis. J. Exp. Med. 195:1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Elsas, A., R.P. Sutmuller, A.A. Hurwitz, J. Ziskin, J. Villasenor, J.P. Medema, W.W. Overwijk, N.P. Restifo, C.J. Melief, R. Offringa, and J.P. Allison. 2001. Elucidating the autoimmune and antitumor effector mechanisms of a treatment based on cytotoxic T lymphocyte antigen-4 blockade in combination with a B16 melanoma vaccine: comparison of prophylaxis and therapy. J. Exp. Med. 194:481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Casares, N., L. Arribillaga, P. Sarobe, J. Dotor, A. Lopez-Diaz de Cerio, I. Melero, J. Prieto, F. Borras-Cuesta, and J.J. Lasarte. 2003. CD4+/CD25+ regulatory cells inhibit activation of tumor-primed CD4+ T cells with IFN-gamma-dependent antiangiogenic activity, as well as long-lasting tumor immunity elicited by peptide vaccination. J. Immunol. 171:5931–5939. [DOI] [PubMed] [Google Scholar]
- 62.Golgher, D., E. Jones, F. Powrie, T. Elliott, and A. Gallimore. 2002. Depletion of CD25+ regulatory cells uncovers immune responses to shared murine tumor rejection antigens. Eur. J. Immunol. 32:3267–3275. [DOI] [PubMed] [Google Scholar]
- 63.Machiels, J.P., R.T. Reilly, L.A. Emens, A.M. Ercolini, R.Y. Lei, D. Weintraub, F.I. Okoye, and E.M. Jaffee. 2001. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 61:3689–3697. [PubMed] [Google Scholar]
- 64.Li, J., P. Hu, L.A. Khawli, and A.L. Epstein. 2003. Complete regression of experimental solid tumors by combination LEC/chTNT-3 immunotherapy and CD25(+) T-cell depletion. Cancer Res. 63:8384–8392. [PubMed] [Google Scholar]
- 65.Onizuka, S., I. Tawara, J. Shimizu, S. Sakaguchi, T. Fujita, and E. Nakayama. 1999. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 59:3128–3133. [PubMed] [Google Scholar]
- 66.Tawara, I., Y. Take, A. Uenaka, Y. Noguchi, and E. Nakayama. 2002. Sequential involvement of two distinct CD4+ regulatory T cells during the course of transplantable tumor growth and protection from 3-methylcholanthrene-induced tumorigenesis by CD25-depletion. Jpn. J. Cancer Res. 93:911–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dieckmann, D., H. Plottner, S. Berchtold, T. Berger, and G. Schuler. 2001. Ex vivo isolation and characterization of CD4(+) CD25(+) T cells with regulatory properties from human blood. J. Exp. Med. 193:1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liyanage, U.K., T.T. Moore, H.G. Joo, Y. Tanaka, V. Herrmann, G. Doherty, J.A. Drebin, S.M. Strasberg, T.J. Eberlein, P.S. Goedegebuure, and D.C. Linehan. 2002. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 169:2756–2761. [DOI] [PubMed] [Google Scholar]
- 69.Woo, E.Y., C.S. Chu, T.J. Goletz, K. Schlienger, H. Yeh, G. Coukos, S.C. Rubin, L.R. Kaiser, and C.H. June. 2001. Regulatory CD4(+) CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 61:4766–4772. [PubMed] [Google Scholar]
- 70.Woo, E.Y., H. Yeh, C.S. Chu, K. Schlienger, R.G. Carroll, J.L. Riley, L.R. Kaiser, and C.H. June. 2002. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J. Immunol. 168:4272–4276. [DOI] [PubMed] [Google Scholar]
- 71.Wang, H.Y., D.A. Lee, G. Peng, Z. Guo, Y. Li, Y. Kiniwa, E.M. Shevach, and R.F. Wang. 2004. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 20:107–118. [DOI] [PubMed] [Google Scholar]
- 72.Yu, P., Y. Lee, W. Liu, R.K. Chin, J. Wang, Y. Wang, A. Schietinger, M. Philip, H. Schreiber, and Y.X. Fu. 2004. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat. Immunol. 5:141–149. [DOI] [PubMed] [Google Scholar]
- 73.Kang, H.S., R.K. Chin, Y. Wang, P. Yu, J. Wang, K.A. Newell, and Y.X. Fu. 2002. Signaling via LTbetaR on the lamina propria stromal cells of the gut is required for IgA production. Nat. Immunol. 3:576–582. [DOI] [PubMed] [Google Scholar]