

Abstract
Although many aspects of CD4+CD25+ T regulatory (Treg) cell development remain largely unknown, signaling through the IL-2R represents one feature for the production of Treg cells. Therefore, the present study was undertaken to further define early developmental steps in the production of Treg cells, including a more precise view on the role of interleukin (IL)-2 in this process. After adoptive transfer of wild-type Treg cells into neonatal IL-2Rβ−/− mice, only a small fraction of donor Treg cells selectively seeded the lymph node (LN). These donor Treg cells underwent rapid and extensive IL-2–dependent proliferation, followed by subsequent trafficking to the spleen. Thus, IL-2 is essential for Treg cell proliferation in neonatal LN. The number and distribution of Treg cells in the periphery of normal neonatal mice closely paralleled that seen for IL-2Rβ−/− mice that received Treg cells. However, for normal neonates, blockade of IL-2 decreased Treg cells in both the thymus and LN. Therefore, two steps of Treg cell development depend upon IL-2 in neonatal mice, thymus production, and subsequent expansion in the LN.
CD4+CD25+ T regulatory (Treg) cells represent a critical T cell subset that suppresses peripheral autoreactive T cells that escape thymic negative selection (1, 2). CD4+CD25+ Treg cells naturally arise in the thymus, in part through high-affinity interactions with self-peptide–MHC complexes (3, 4). The expression of the transcription factor Foxp3 is also required for thymic development of Treg cells (5–7). In the absence of functional Foxp3, Treg cells are not produced and such mice die within 3 wk of age due to a rapid lymphoproliferative disorder.
There is substantial evidence indicating that IL-2–IL-2R–dependent events play a role in the production of CD4+CD25+ Treg cells. Similar to mice with impaired Foxp3, IL-2–, IL-2Rα–, and IL-2Rβ–deficient mice exhibit an analogous lethal lymphoproliferative disorder accompanied with severe autoimmunity (8–10). IL-2 and IL-2Rβ knockout mice contain severely reduced numbers of CD4+CD25+ Treg cells (11, 12). More importantly, correcting the production of Treg cells in such mice, either in mixed bone marrow or T cell chimeric mice, led to inhibition of uncontrolled T cell proliferation and autoimmunity (13–15). In related experiments, the adoptive transfer of wild-type Treg cells into neonatal IL-2Rβ−/− mice resulted in substantial proliferation and stable engraftment of the donor Treg cells such that these mice lived a normal disease-free life-span. This protection was contingent upon donor Treg cells expressing a functional IL-2R and the host producing IL-2 (12). Furthermore, thymus-specific expression of IL-2Rβ in IL-2Rβ−/− mice restored the production of functional CD4+CD25+ Treg cells and prevented lethal autoimmunity, indicating that the IL-2–IL-2R interaction is necessary during thymic Treg cell development (12, 16).
Although several features for the production of Treg cells have been defined, many aspects of CD4+CD25+ Treg cell development remain largely unknown. Current data point to a role for IL-2 in both the thymus and periphery, but the relative importance and the relationship of thymic versus peripheral requirements for IL-2 are not yet apparent. Of special interest is a more detailed understanding of the initial steps for the expansion of Treg cells after transfer into neonatal IL-2Rβ−/− mice, as this leads to long-term engraftment and persistent suppression of autoimmunity by the donor cell inoculum. The most compelling data for IL-2 in Treg cell production relies on IL-2– and IL-2Rβ–deficient mice. The extent that these models recapitulate the normal process for Treg cell development remains an open issue. With these questions in mind, the present study was undertaken to further define early developmental steps in the production of Treg cells, including a more precise view on the role of IL-2 in this process. Our data support a model in which IL-2 critically acts within the thymus and neonatal lymph nodes to promote the expansion of Treg cells.
Results
Fate of CD4+CD25+ T regulatory cells in neonatal mice
Past studies have demonstrated that 4 weeks after adoptive transfer of normal CD4+CD25+ Treg cells into IL-2Rβ–deficient mice, donor Treg cells expanded at least 10–20-fold and were readily detected in the spleen and lymph nodes, but not in the thymus, of recipient mice (12). To evaluate early events during Treg cell engraftment, GFP+ CD4+ CD25+ Treg cells were adoptively transferred into neonatal IL-2Rβ−/− mice. FACS analysis 6 d later revealed that GFP+ CD4+ donor cells were readily seen in the lymph nodes whereas a very low, but a consistently detectable fraction, of GFP+ cells was found in the spleen (Fig. 1, A and B). Although there was a very small number of donor cells in the spleen, the fraction of donor cells within the CD4+ population was similar to that seen in the lymph nodes (Fig. 1 B). By contrast, essentially no GFP+ cells were found in the thymus and bone marrow when examined from 6–20 d after transfer (Fig. 1, A and B). Even as early as 2 d after transfer, essentially no donor cells were found within the thymus (unpublished data). For the lymph nodes, on day 6 ∼3–4% of the cells were GFP+ and this fraction remained relatively constant during the first 3 wk after birth, whereas the fraction GFP+ cells in the spleen noticeably increased on day 20 (Fig. 1, A and B). Furthermore, at each time point, essentially all GFP+ donor cells in the lymph nodes and most in the spleen were CD4+ T cells (Fig. 1 C), and a large majority of the donor CD4+ T cells coexpressed CD25 (Fig. 1 D).
Figure 1.
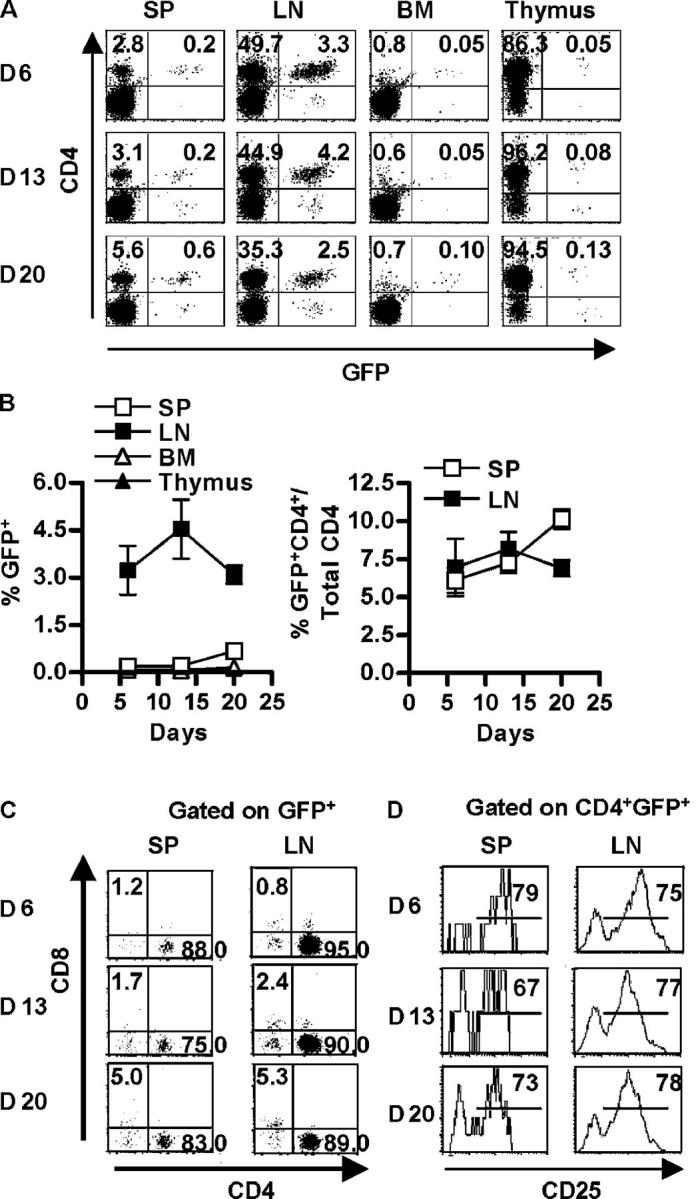
Initial appearance of CD4+CD25+ Treg cells in the lymph nodes of neonatal mice. Newborn C57BL/6 IL-2Rβ−/− mice received purified GFP-CD4+CD25+ T cells (4–5 × 105) and 6–20 d after transfer, spleen (SP), lymph node (LN), bone marrow (BM), and thymus cells from individual recipient mice were subjected to FACS analysis. (A) Dot plots for donor cells based on CD4 and GFP expression. The percent of cells expressing CD4 and/or GFP is indicated in appropriate quadrant. (B) The percent of donor GFP+ cells (left) or donor GFP+CD4+ of the total CD4+ cells (right) in the indicated organ. Data are mean ± SEM for three to five animals per day. (C) Dot plots for CD4 and CD8 expression for GFP+ donor cells. The percent of cells expressing CD4 or CD8 is indicated in the appropriate quadrant. (D) CD25 expression for CD4+ GFP+ donor cells. The percent of cells expressing CD25 is indicated in the upper right corner of each histogram. Data in A, C, and D are representative of three to five animals per day.
Expansion of Treg cells in neonatal mice
To directly assess the proliferative rate of Treg cells, 5-(and 6-) carboxyfluorescein diacetate, succinimidyl ester (CFSE)-labeled CD45.1+ CD4+CD25+ Treg cells were adoptively transferred into neonatal IL-2Rβ−/− mice (CD45.2). In the spleen, 2 d after the adoptive transfer, ∼1.8% of the spleen contained donor CD45.1+ cells with the majority of these cells remaining CFSEhi, indicating that these cells had not undergone proliferation (Fig. 2 A). At this time many of these donor cells had lost CD25 expression. At 4 and 6 d after transfer, there was nearly a 10-fold decrease in the fraction of CD45.1+ donor cells within the spleen (Fig. 2, A and B), and the CD4+ donor cells did not dilute CFSE. As we have been unable to detect donor cells in nonlymphoid tissue on day 6 (unpublished data), these data suggest that most of the donor cells that seeded the spleen may have undergone apoptosis.
Figure 2.

CD4+CD25+ Treg cells extensively proliferate in the lymph nodes of neonatal mice. Newborn C57BL/6 IL-2Rβ−/− mice received purified CD45.1+ CFSE-labeled CD4+CD25+ T cells (4–5 × 105) and 2–20 d after transfer, spleen (SP), and lymph node (LN) cells from individual recipient mice were subjected to FACS analysis. (A) Dot plots are shown for CD45.1 and CD4 expression (top) or CD25 and CFSE expression for CD45.1+CD4+ donor cells (bottom). The percent of donor CD4+ cells is indicated in the upper right corner of top dot plots. A total of 105 or 106 splenic cells were collected from 2- or 4-d-old mice, respectively. (B) Dot plots are shown for CD45.1 and CFSE expression. The percent of donor cells is indicated in the upper right corner of each dot plot. (C) Dot plots depicting CD4 and CD25 expression for CD45.1+ donor cells. The percent of cells expressing CD4 and CD25 of the gated populations is indicated in the upper right corner of the dot plots. Data in A and B are representative of two to five animals per day.
In marked contrast, within the lymph nodes 6 d after adoptive transfer, which was the earliest time point that lymph nodes could be collected from neonatal mice, ∼50% of the donor CD45.1+ cells were CFSE negative, indicating that these cells divided at least eight times (Fig. 2 B). These data demonstrate a very high proliferative potential of Treg cells in neonatal IL-2Rβ−/−mice. At this time, most of the lymph nodes were collected from these neonatal mice (total cellularity of ∼2 × 106) and on average ∼3% or 6 × 104 of these were donor Treg cells. Based on the mean fluorescent intensity of all donor cells, the engrafting Treg cells were on average the progeny of four to six cell divisions. Therefore, we estimate that the donor Treg cells within the lymph nodes were derived from ∼1,000 to 4,000 cells. At 2 and 3 wk after transfer, virtually all of the donor cells in the lymph nodes had lost CFSE staining (Fig. 2 B), indicating that these cells were progeny that had undergone substantial proliferation.
Approximately 85% of the donor CD45.1+ donor cells in the lymph nodes at day 6 were CD4+CD25+ T cells (Fig. 2 C) regardless of whether they had under gone extensive (CFSElo/neg) or minimal proliferation (CFSEhi; unpublished data). Most donor cells in the lymph nodes (80%) on days 13 and 20 after transfer coexpressed CD4 and CD25 (Fig. 2 C). With respect to the spleen, the few donor cells present at day 6 were contained within two subsets, CFSEneg and CFSEhi. For the donor cells that were found in the nondividing CFSEhi subpopulation, most coexpressed CD4 and CD25 whereas the majority of the rapidly proliferating splenic donor cells were CD4− and CD25− cells (unpublished data). Furthermore, a CFSE+ intermediate cell population was not detected in the spleen at any of the time points (Fig. 2, A and B), suggesting that the CFSEneg cells were not derived from dividing progeny within this organ. Importantly, only by 3 wk after transfer was a substantial population of donor (CD45.1+) cells detected within the spleen, which were CFSEneg (Fig. 2 B), and ∼70% of these donor cells were CD4+CD25+ T cells (Fig. 2 C). Collectively, these data indicate that donor CD4+CD25+ Treg cells first undergo extensive proliferation within the lymph nodes of neonatal mice and raise the possibility that these progeny may subsequently home and populate the spleen.
Treg cell production in peripheral lymphoid tissue of normal and Tg−/− mice
The extent that the engraftment and proliferation of Treg cells in this adoptive transfer model recapitulates the normal processes for Treg cells was first evaluated by quantifying Treg cells in the lymph nodes and spleen of normal C57BL/6 mice. As transgenic thymic expression of IL-2Rβ in IL-2Rβ–deficient mice (Tg−/− mice) results in the production of Treg cells (12), these results were compared with that obtained for donor CD4+CD25+ Treg cells transferred into neonatal IL-2Rβ−/− mice. In each mouse model, CD4+CD25+ T cells were first readily detected in the day 6 neonatal lymph nodes (Fig. 3 A). At this time very few CD4+CD25+ T cells were seen in the spleen, but both the percentage and number of splenic Treg cells increased to a readily measurable number by 20 d of age (Fig. 3 B). Moreover, the overall percentage and cell number of Treg cells in the lymph node and spleen from 6 to 20-d-old mice approximated that seen for the adoptively transferred IL-2Rβ−/− mice (Fig. 3, A and B). One exception was that the lymph nodes and spleen of neonatal Tg−/− mice contained a somewhat higher percentage and absolute number of CD4+CD25+ T cells (Fig. 3, A and B). This finding is consistent with previous data that showed more CD4+CD25+ T cells in the thymus, spleen, and lymph nodes of adult Tg−/− mice (12).
Figure 3.

Adoptive transfer of Treg cells into IL-2Rβ2/− mice largely follows the developmental scheme operative in normal mice. FACS analysis of lymph node and spleen cells was performed for 6–20-d-old mice, as indicated. The percent CD4- and CD25-positive cells (left) or the absolute number of CD4+CD25+ (right) in the (A) lymph node and (B) spleen. The absolute number of CD4+CD25+ T cells was calculated from the percent CD4+CD25+ cells in the lymph node and spleen times the number of cells in these organs. Data are mean ± SEM for three to seven mice per day per group. (C) CD69, CD62L, or CD103 expression for lymph node CD4+CD25+ T cells (n = 3–5 animals per group per day).
Phenotypic analysis of lymph node (Fig. 3 C) and splenic (unpublished data) derived CD4+CD25+ T cells from normal, Tg−/−, and adoptively transferred IL-2Rβ−/− neonatal mice indicated that the majority of these cells were CD69neg and CD62Lhigh, a phenotype associated with Treg cells rather than recently activated T cells. When compared with normal C57BL/6 mice, Tg−/− mice contained a somewhat higher fraction of CD62Lhigh cells whereas the adoptively transferred donor cells contained a lower fraction of these cells. Glucocorticoid-induced TNF receptor (GITR) was expressed on essentially all CD4+CD25+ cells from these three types of mice (unpublished data). In contrast, CD103, another marker associated with Treg cells (17), was expressed on a higher fraction of CD4+CD25+ donor cells from the adoptively transferred mice, especially 14–20 d after transfer. Collectively, these data indicate that the adoptive transfer of Treg cells into IL-2Rβ−/− mice largely follows the developmental scheme operative in normal and Tg−/− mice.
Role of IL-2 in early development of T regulatory cells
CFSE-labeled normal Treg cells were adoptively transferred into neonatal IL-2Rβ−/− mice in the absence or presence of anti–IL-2 mAb to test the extent that the initial engraftment and proliferation of the donor Treg cells in the lymph nodes depended upon IL-2 (Fig. 4). In control treated mice (n = 3), CD45.1 donor cells (4.4 ± 0.3%) were readily detected (Fig. 4 A), with the vast majority of the cells expressing CD4 and CD25 (Fig. 4 B). In contrast, for anti–IL-2–treated mice (n = 3), far fewer donor cells (1.0 ± 0.1%) were detected in the lymph nodes and many of these were not CD4+ and CD25+ cells (Fig.4, A and B). As lymph node cell yields were equivalent in both groups of mice, this lower percentage directly reflected differences in the absolute cell number. When correcting these data for the fraction of CD45.1+ donor cells that coexpressed CD4 and CD25, there was on average an 83.4% reduction of Treg cells in the lymph nodes of the anti–IL-2–treated recipients. However, for the few residual CD4+CD25+ T cells after treatment with anti–IL-2, many fully diluted CFSE (Fig. 4 C), suggesting that these cells were rapidly proliferating cells. It is not possible to discriminate whether this reflects IL-2–independent proliferation or some inefficiency of the anti–IL-2 blockade.
Figure 4.

The initial engraftment and proliferation of Treg cells in neonatal lymph nodes requires IL-2. Neonatal IL-2Rβ−/− mice were adoptively transferred with CD45.1+ CFSE-labeled CD4+CD25+ T cells (2 × 105) and received either PBS or α-IL-2 mAb (100 μg) on the day of transfer and 3 d later. 6 d after transfer, lymph node cells from individual recipient mice were subjected to FACS analysis. (A) Histograms of donor (CD45.1) engraftment. (B) Dot plots for CD4 and CD25 expression after gating on CD45.1+ donor cells. (C) Histograms of proliferating donor CD4+CD25+ T cells based on CFSE staining. n = 3 per group.
To further explore the role of IL-2 in early neonatal life, normal C57BL/6 mice were also treated with anti–IL-2 mAb within the first 24 h after birth. 1 wk later, the fraction of CD4+CD25+ T cells was substantially reduced in the lymph nodes of the anti–IL-2–treated neonates (Fig. 5 A). Anti–IL-2 also resulted in a lower fraction of CD4+ CD25+ GITR+ T cells (Fig. 5 B), another cell surface phenotype associated with Treg cells (17, 18). Furthermore, this decrease does not reflect a requirement for IL-2 to maintain expression of CD25 (19), as lymph nodes from control and anti–IL-2–treated mice lacked CD4+CD25-GITRhigh T cells. As lymph node cellularity was equivalent in both the control and anti–IL-2–treated groups, the reduced percentage of Treg cells by anti–IL-2 reflected the actual decrease in Treg cell number. Thus, these data indicate that IL-2 is normally required for production of Treg cells during early neonatal life and is not just a property seen upon transfer of Treg cells to IL-2Rβ−/− mice.
Figure 5.

A Role for IL-2 in early development of CD4+CD25+ Treg cells. B6 mice received either PBS (−) or α-IL2 mAb (+; 100 μg) on the day and 3 d after birth. 7 d after birth, lymph node cells from individual mice were subjected to FACS analysis. (A) The percent of CD4+CD25+ T cells. Data are mean ± SEM for 8–10 animals. (B) Dot plots of CD25 and GITR expression (left) and the absolute number of GITR+ cells for CD4+CD25+ cells (right). Data were mean ± SEM for three to four animals.
IL-2 regulates thymic Treg cell production
The preceding experiments indicate that IL-2 is critical for neonatal lymph node Treg cell expansion. However, our past work also indicates a role for IL-2 during thymic development of Treg cells (12, 16). Therefore, we further explored the contribution of thymic IL-2 for Treg cells. Anti–IL-2 treatment of normal C57BL/6 and Tg−/− neonatal mice resulted in a statistically significant reduction in CD4+ “single-positive” CD25+ T cells within the thymus (Fig. 6 A), which was much more striking for Tg−/− mice. The blockade by anti–IL-2 for Tg−/− mice provides direct data that the activity of the transgenic IL-2Rβ chain depends upon signaling through the reconstituted thymic IL-2R. These data also raise the possibility that the reduction of Treg cells within the lymph nodes of anti–IL-2–treated normal neonatal mice might in part be due to an effect on the thymic IL-2–IL-2R interaction. A decrease was also noted in the percentage of CD4+CD25+GITR+ thymocytes of anti–IL-2–treated normal C57BL/6 neonatal mice, confirming that Treg cells were reduced by inhibiting IL-2 (Fig. 6 B).
Figure 6.

IL-2 is required for thymic production of CD4+CD25+ Treg cells. All mice received either PBS (−) or α-IL2 mAb (+; 100 μg) on the day and 3 d after birth. 7 d after birth, thymocytes from individual mice were subjected to FACS analysis with 250,000 events collected. In the data shown, the analysis of the indicated markers was performed after gating on CD4+CD8− “single positive” thymocytes. (A) The percent of thymic CD4+CD25+ cells from B6 and Tg−/− treated mice. Data are mean ± SEM for six to eight animals per group. *P < 0.05 compared with B6, and #P < 0.001 compared with Tg−/− (one-way analysis of variance followed by Newman-Keuls Multiple Comparison). (B) Dot plots for CD25 and GITR expression for thymic CD4 single positive thymocytes from B6 mice (left) and the absolute number of GITR+ CD4 single positive thymocytes (right). Data are mean ± SEM for three to four animals per group.
A population of C57BL/6 thymocytes CD4+CD8− thymocytes was observed that expressed very high levels of CD25 and these cells were uniformly GITRneg (Fig. 6 B). The relationship of these cells to Treg cells is not known and were not included in analyses shown in Fig. 6 B. The extremely high levels of CD25 and lack of GITR expression distinguish these cells from Treg cells found in peripheral lymphoid tissue that express a somewhat lower level of CD25 and relatively high levels of GITR.
Discussion
Past work has established that CD4+CD25+ Treg cells develop within the thymus (11, 20, 21). Furthermore, the narrow window of time in which neonatal thymectomy renders mice susceptible to organ-specific autoimmune disease indicates that the early neonatal period is a critical time for seeding the periphery with Treg cells (21–23). However, very little is known concerning early developmental steps for the production of Treg cells. It is within this context that we have examined the trafficking and proliferation of CD4+CD25+ Treg cells, including an assessment of the contribution of IL-2. This paper demonstrates that neonatal Treg cell development occurs by rapid and extensive proliferation of these cells within the lymph nodes that is followed by trafficking to populate the spleen. Moreover, IL-2 drives the expansion of Treg cells within the thymus and neonatal lymph nodes.
Our main approach to study the early developmental events for Treg cell trafficking and proliferation was to adoptively transfer GFP+ or allotype-marked (CD45.1) CFSE-labeled purified CD4+CD25+ Treg cells into newborn IL-2Rβ−/− mice. In 1-wk-old recipient mice, a near normal complement of Treg cells, that had undergone considerable proliferation, was detected in the lymph nodes, whereas very few Treg cells were found in the spleen, and for these few cells, no observable proliferation occurred. However, by 3 wk after transfer, a readily measurable population of Treg cells were seen in both the lymph nodes and spleen, and these cells had completely diluted the CFSE label, indicating that they were progeny of the initial engrafting Treg cells. As no donor Treg cells were found in the thymus or bone marrow during this time, these experiments indicate that lymph nodes are the first secondary lymphoid organ where Treg cells traffic and proliferate, followed by population of the spleen.
Mice deficient in IL-2, IL-2Rα, and IL-2Rβ contain severely reduced numbers of CD4+CD25+ Treg cells, especially in peripheral lymphoid tissue (11, 12). An important advantage, therefore, of our adoptive transfer experiments is that we can follow the trafficking and proliferation of donor cells without competition from endogenously produced Treg cells. Such competition readily occurs because when a similar number of Treg cells were adoptively transferred to normal mice, very few donor cells were found in peripheral lymphoid tissues 4 wk later (12). It is highly likely that such competition reflects that there is constant Treg cell production from new thymic emigrants in normal recipient mice, which dilute the donor cells, whereas the IL-2Rβ−/− mice are solely dependent upon donor Treg cells. Importantly, the induction of autoimmunity by day 3 thymectomy (24) indicates that there are not sufficient Treg cells in these lymph nodes at this time to expand and effectively control autoimmune disease.
The appearance and number of Treg cells in the spleen and lymph nodes from both normal or Tg−/− mice were directly shown to closely parallel that seen using the adoptive transfer model. Therefore, the initial expansion of the Treg cells in the adoptively transferred IL-2Rβ−/− mice must approximate the normal process to produce these cells. We also confirmed that in all three of these models the phenotype of the CD4+ CD25+ cells was that expected for Treg cells, i.e., predominately CD69low, CD62Lhigh, and GITRhigh. Although our study did not directly address the homing of Treg cells upon exit from the thymus, these data support the idea that during the neonatal period Treg cells are generated in the thymus, exit to the lymph nodes, and then traffic and populate the spleen. Past work has shown that thymocytes that leave the thymus go mainly to T cell areas of the lymph nodes, Peyer's Patches, and spleen with little migration to the bone marrow, gut, or liver (25). The spleen is enriched in CD8+ and γδ TCR+ emigrants, whereas lymph nodes are enriched with CD4+ emigrants, with the majority of lymph node emigrants expressing L-selectin (26). CD4+CD25+ Treg cells, therefore, are included in this latter group of immediate thymic emigrants.
Based on their poor proliferative activity in vitro, Treg cells have been considered anergic. However, more recent studies indicate that it is possible to drive extensive proliferation of Treg cells in vitro, that in part requires high dose IL-2 (27, 28). Furthermore, adoptive transfer models demonstrate that antigen-responsive Treg cells readily proliferate in adult mice (29–33). Consistent with this idea, by enumerating the number of donor Treg cells in adult IL-2Rβ−/− recipients after neonatal adoptive transfer, we previously estimated that there was at least 10–20-fold expansion of Treg cells (12). Based on the current study, this degree of expansion is a substantial underestimation of the proliferation of Treg cells as they expand in peripheral immune tissue. By six days after adoptive transfer of CFSE-labeled Treg cells into IL-2Rβ−/− neonates, ∼50% of the donor CD4+CD25+ T cells fully diluted the CFSE. This is equivalent to at least six to eight cells divisions or a cycling rate of <24 h for these cells. Based on these data and the number of Treg cells in 6-d-old neonatal lymph nodes, it is now possible to estimate that the progeny of Treg cells in such adoptively transferred mice were derived by no more than 1,000–4,000 cells. It is conceivable that this number may even be lower because at the time point used for making this estimate many of the Treg cells fully diluted their CFSE, so the number of cell divisions may have been underestimated. In any case, we now estimate that to derive a near normal complement (1–2 × 106 cells) of donor-derived CD4+CD25+ T cells in adult mice, there may be as much as a 2,000-fold expansion of the Treg cells that initially seed the neonatal lymph nodes. Proliferation by T cells in neonatal lymph nodes is not a property unique for Treg cells. Substantial proliferation by both conventional CD4+ and CD8+ T cells has been noted within neonatal lymph nodes, the latter of which is dependent upon IL-7 (34, 35).
This large expansion of donor Treg cells raise two potentially important issues concerning the production of Treg cells. First, this process results in amplification of the Treg repertoire for those Treg cells that first seed the neonatal lymph node. Such amplification may represent a mechanism to rapidly generate suppressor cells to counteract autoreactive T cells that escape thymic negative selection. This may be especially meaningful during the neonatal period when thymic negative selection is thought to be less stringent (34, 36), as the time-frame for thymic development is much shorter. Second, only a relatively small fraction of injected donor-derived Treg cells engraft the neonatal lymph nodes. Based on the calculated number of Treg cells that initially engraft lymph nodes (1,000–4,000 as discussed above) and a requirement to transfer ∼105 Treg cells to prevent lethal autoimmunity in C57BL/6 IL-2Rβ−/− mice (12), it appears that no more than 1–4% of the donor inoculum is responsible for engrafting the neonatal lymph nodes. This finding raises the interesting possibility that only a particular subset of Treg cells are competent to engraft and expand in neonatal lymph nodes. Alternatively, this may simply reflect an inherent inefficiency for engraftment or a size limitation for the niche that accommodates Treg cells. Further study is required to distinguish between these possibilities.
Much recent data support an important role for IL-2 in the production of Treg cells in vivo (12–15). A very important aspect of this study is that we have much more precisely established the role of IL-2 in the production of Treg cells. There appears to be two levels in which IL-2 is required for Treg cells. One is to support Treg proliferation during development in the thymus and second is to support extensive proliferation as Treg cells seed the neonatal lymph nodes. This is in line with the notion that IL-2 is a potent T cell growth factor. An alternative idea is that IL-2 primarily provides a survival signal for the developing Treg cells and their growth depends upon other molecules. This possibility seems unlikely because providing IL-2−/− mice with survival signals by transgenic expression of Bcl-2 in T lineage cells did not mediate Treg cell production (37).
The interpretation of anti–IL-2 blockade for Treg production in the normal mice is somewhat complicated and most likely reflects effects within both the thymus and lymph nodes. As such, evaluation of normal neonatal mice reflects a composite of our other two models, which tend to discriminate developmental steps within the thymus versus the peripheral immune tissue. Thus, the low number of Treg cells in lymph nodes in the adoptive transfer model after administering anti–IL-2 directly reflects the need for IL-2 for the rapid growth of these cells in this immune organ as donor Treg cells are not detected within the thymus for 1-wk-old IL-2Rβ−/− recipient mice. In an analogous manner, Tg−/− mice are solely dependent upon thymic IL-2R expression for Treg cell production (12, 16). Tg−/− mice appear to somewhat exaggerate the role for IL-2 within the thymus as the most substantial inhibition for thymic Treg cells by anti–IL-2 treatment was observed for Tg−/− mice. This may partially account for why Treg cells are readily found in the periphery of Tg−/− mice without a need for expression of a high affinity IL-2R. The basis by which Tg−/− Treg cells accumulate in peripheral immune tissue in an IL-2–independent manner is not known. Studies are in progress to further explore the growth and homeostasis of Tg−/− Treg cells in peripheral immune tissues. In any case, this result provides direct data that the increase of Treg cells within the thymus of IL-2Rβ−/− mice after thymic-specific transgenic expression of IL-2Rβ was a consequence of IL-2–IL-2R signaling.
In conclusion, our data support a model in which initial production of thymic CD4+CD25+ Treg cells ensues without IL-2. However, IL-2 provides necessary signals within the thymus to increase their number. Upon trafficking to neonatal lymph nodes, IL-2 is normally required to drive substantial expansion of these cells. This does not exclude other immune molecules, such as TCR–MHC class II and B7–CD28 interactions, from contributing to this expansion, as indicated by other studies (30–32, 38, 39). Interestingly, in the latter case, the role for CD28–B7 appears to be due to its ability to induce IL-2 production (39). It is likely that in the absence of IL-2 in vivo, there are a few Treg cells or their precursors. Thus, it should be possible to generate Treg cells and potentially mediate suppression by IL-2−/− CD4+ T cells when IL-2 is available either directly or by transfer into an IL-2+/+ host. In fact, this has been observed several times experimentally (15, 40). A key outstanding issue is whether IL-2 is solely required for Treg cell growth or whether it more broadly regulates Treg cell function. Recent experiments indicate that IL-2 activation of Treg cells is necessary for their suppressor activity in vitro, which is consistent with a potential role for IL-2 in Treg cell function (28, 41).
Materials and Methods
Mice.
C57BL/6 were obtained from Jackson ImmunoResearch Laboratories. B6.SJL-Ptprc/BoAiTac mice, congenic for CD45, were obtained from Taconic and breed within our animal colony. IL-2Rβ−/− mice, backcrossed for at least 12 generations to C57BL/6 mice, have been described previously (10). These mice were maintained by using breeding pairs that were homozygous IL-2Rβ− / − that were rendered autoimmune-free by neonatal adoptive transfer of purified wild-type CD4+ or CD4+CD25+ Treg cells (12). The thymic-targeted transgenic wild-type IL-2Rβ expressed in IL-2Rβ−/− mice on the C57BL/6 genetic background (referred to as Tg−/− in this study) has been described previously (16). C57BL/6-Tg (ACTbEGFP)1Osb/J were provided by Dr. M. Okabe (Osaka University, Suita, Japan; referred to as GFP in this study; 42). Protocols were approved by the Institutional Animal and Use Committee at the University of Miami.
Reagents.
5-(and 6-)carboxyfluorescein diacetate, succinimidyl ester (CFSE) was purchased from Molecular Probes. Biotin-conjugated mAbs to CD69 and CD103, and Cy-chrome (cyc)–conjugated mAbs to CD4 and CD8, PE-conjugated anti-CD25 (PC61), PE-streptavidin, peridinin chlorophyll-α protein (PerCP)–conjugated CD8 and CD4 mAbs, PerCP-streptavidin, allophycocyanin (APC)-conjugated CD4 mAb, and APC-streptavidin were purchased from BD Biosciences. Biotin-conjugated GITR (also called TNFRSF18) mAb was purchased from R&D Systems. FITC–anti-CD4 (GK1.5), biotin-conjugated mAbs to CD45.1 (A20), CD62L (Mel14), CD25 (7D4), and CD44 (Pgp-1), and unlabeled anti–IL-2 mAb (α–IL-2, S4B6) were prepared in our laboratory.
Purification of CD4+CD25+ T regulatory cells.
Isolation of CD4+CD25+ T cells was described previously (12). In brief, splenic CD4 T cells were enriched by depletion of CD8+ T cells and B cells. Viable cells were obtained by centrifugation through lympholyte-M (Accurate Chemical and Scientific Corp.). The enriched CD4 (100 × 106/ml) were then incubated with biotin-conjugated anti-CD25 (10 μg/ml), washed, and positively selected with streptavidin MACS microbeads (Miltenyi Biotec). These cells were on average >90% CD4+CD25+ as assessed by flow cytometry. CFSE-labeled Treg cells (20 × 106cells/ml) were prepared by addition of CFSE to a final concentration of 2.5 μM/ml for 10 min at 37°C followed by one wash with RPMI 1640 containing 5% FBS, 1% penicillin-streptomycin, 1% L-glutamine, and 50 μM 2-mercaptoethanol and two washes with HBSS.
Experimental animals.
1–2-d-old neonatal C57BL/6 IL-2Rβ−/− mice were adoptively transferred by i.v. injection of 2–5 × 105 of purified CD4+CD25+ Treg cells. These mice are indicated by T → IL-2Rβ−/− in this study. In all cases, these mice were devoid of symptoms associated with IL-2Rβ–deficiency at the time of sacrifice. In some experiments, neonatal mice received an i.v. injection of either PBS or α–IL-2 mAb (100 μg S4B6) on the day they were born followed by an i.p. injection (100 μg) 3 d later. Inguinal, axillary, brachial, cervical, and mesenteric lymph nodes were collected at the time of sacrifice along with the spleen, thymus, and bone marrow and subjected to FACS analysis.
FACS analysis.
For three or four color analysis, cells were incubated for 20 min at 4°C with APC-CD4, PerCP-CD8, and PE-CD25 or biotin-conjugated mAbs to CD45.1, CD25, CD44, CD69, CD62L, CD103, GITR, or CD122 followed by two washes with HBSS containing 0.2% BSA and 150 mM NaN3. Cells were then either used in FACS analysis or incubated for 20 min at 4°C with APC-CD4, PE-CD25, and PerCP-streptavidin, or cyc-CD4 and PE-streptavidin, or APC-CD4, PerCP-CD8, and PE-Streptavidin, or PerCP-CD4, PE-CD25, and APC-streptavidin, or FITC-CD4, PerCP-CD8, PE-CD25, and APC-streptavidin, or FITC-CD4, cyc-CD8, and PE-streptavidin followed by a wash with same buffer, as above. Cells were analyzed using a Becton Dickinson LSR1 and CellQuest® software. GFP and CFSE fluorescence was detected using the FL1 channel. The total number of events collected for analysis was between 50,000 and 100,000, unless otherwise stated in the figure legends.
Acknowledgments
We thank Becky Adkins and Howard Petrie for critically reading this manuscript.
This work was supported by National Institutes of Health grants CA45957, AI 40114, and AI055815.
The authors have no conflicting financial interests.
Abbreviations used in this paper: APC, allophycocyanin; CFSE, 5-(and 6-)carboxyfluorescein diacetate, succinimidyl ester; cyc, Cy-chrome; GITR, glucocorticoid-induced TNF receptor; PerCP, peridinin chlorophyll-α protein; Treg, T regulatory.
References
- 1.Shevach, E.M. 2000. Regulatory T cells in autoimmmunity. Annu. Rev. Immunol. 18:423–449. [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi, S. 2000. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 101:455–458. [DOI] [PubMed] [Google Scholar]
- 3.Jordan, M.S., A. Boesteanu, A.J. Reed, A.L. Petrone, A.E. Holenbeck, M.A. Lerman, A. Naji, and A.J. Caton. 2001. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2:301–306. [DOI] [PubMed] [Google Scholar]
- 4.Apostolou, I., A. Sarukhan, L. Klein, and H. von Boehmer. 2002. Origin of regulatory T cells with known specificity for antigen. Nat. Immunol. 3:756–763. [DOI] [PubMed] [Google Scholar]
- 5.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 6.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 7.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 8.Sadlack, B., H. Merz, H. Schorle, A. Schimpl, A.C. Feller, and I. Horak. 1993. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 75:253–261. [DOI] [PubMed] [Google Scholar]
- 9.Willerford, D.M., J. Chen, J.A. Ferry, L. Davidson, A. Ma, and F.W. Alt. 1995. Interleukin-2 receptor α-chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 3:521–530. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki, H., T.M. Kundig, C. Furlonger, A. Wakeham, E. Timms, T. Matsuyama, R. Schmits, J.J. Simard, P.S. Ohashi, H. Griesser, et al. 1995. Deregulated T cell activation and autoimmunity in mice lacking interleulin 2 receptor β. Science. 268:1472–1476. [DOI] [PubMed] [Google Scholar]
- 11.Papiernik, M., M.L. de Moraes, C. Pontoux, F. Vasseur, and C. Penit. 1998. Regulatory CD4 T cells: expression of IL-2Rα chain, resistance to clonal deletion and IL-2 dependency. Int. Immunol. 10:371–378. [DOI] [PubMed] [Google Scholar]
- 12.Malek, T.R., A. Yu, V. Vincek, P. Scibelli, and L. Kong. 2002. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 17:167–178. [DOI] [PubMed] [Google Scholar]
- 13.Wolf, M., A. Schimpl, and T. Hunig. 2001. Control of T cell hyperactivation in IL-2-deficient mice by CD4+CD25− and CD4+CD25+ T cells: evidence for two distinct regulatory mechanisms. Eur. J. Immunol. 31:1637–1645. [DOI] [PubMed] [Google Scholar]
- 14.Almeida, A.R., N. Legrand, M. Papiernik, and A.A. Freitas. 2002. Homeostasis of peripheral CD4+ T cells: IL-2Rα and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. 169:4850–4860. [DOI] [PubMed] [Google Scholar]
- 15.Furtado, G.C., M.A. Curotto de Lafaille, N. Kutchukhidze, and J.J. Lafaille. 2002. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J. Exp. Med. 196:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malek, T.R., B.O. Porter, E.K. Codias, P. Scibelli, and A. Yu. 2000. Normal lymphoid homeostasis and lack of lethal autoimmunity in mice containing mature T cells with severely impaired IL-2 receptors. J. Immunol. 164:2905–2914. [DOI] [PubMed] [Google Scholar]
- 17.McHugh, R.S., M.J. Whitters, C.A. Piccirillo, D.A. Young, E.M. Shevach, M. Collins, and M.C. Byrne. 2002. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 16:311–323. [DOI] [PubMed] [Google Scholar]
- 18.Shimizu, J., S. Yamazaki, T. Takahashi, I. Y., and S. Sakaguchi. 2002. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 3:135–142. [DOI] [PubMed] [Google Scholar]
- 19.Malek, T.R., and J.D. Ashwell. 1985. Interleukin 2 upregulates expression of its receptor on a T cell clone. J. Exp. Med. 161:1575–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh, M., T. Takahashi, N. Sakaguchi, Y. Kuniyasu, J. Shimizu, F. Otsuka, and S. Sakaguchi. 1999. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 162:5317–5326. [PubMed] [Google Scholar]
- 21.Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor α-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151–1164. [PubMed] [Google Scholar]
- 22.Sakaguchi, S., M. Toda, M. Asano, M. Itoh, S.S. Morse, and N. Sakaguchi. 1996. T cell-mediated maintenance of natural self-tolerance: its breakdown as a possible cause of various autoimmune diseases. J. Autoimmun. 9:211–220. [DOI] [PubMed] [Google Scholar]
- 23.Asano, M., M. Toda, N. Sakaguchi, and S. Sakaguchi. 1996. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 184:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakaguchi, S., K. Fukuma, K. Kuribayashi, and T. Masuda. 1985. Organ-specific autoimmune disease induced in mice by elimination of T cell subsets. I. Evidence for the active participation of T cells in natural self-tolerance: deficit of a T cell subset as a possible cause of autoimmune disease. J. Exp. Med. 161:72–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scollay, R.G., E.C. Butcher, and I.L. Weissman. 1980. Thymus cell migration. Quantitative aspects of cellular traffic from the thymus to the periphery in mice. Eur. J. Immunol. 10:210–218. [DOI] [PubMed] [Google Scholar]
- 26.Holder, J.E., W.G. Kimpton, E.A. Washington, and R.N. Cahill. 1999. L-selectin expression on thymic emigrants defines two distinct tissue-migration pathways. Immunology. 98:422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang, Q., K.J. Henriksen, M. Bi, E.B. Finger, G.L. Szot, J. Ye, E. Masteller, H. McDevitt, M. Bonyhadi, and J.A. Bluestone. 2004. In vitro expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 199:1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thornton, A.M., C.A. Piccirillo, and E.M. Shevach. 2004. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur. J. Immunol. 34:366–376. [DOI] [PubMed] [Google Scholar]
- 29.Fisson, S., G. Darrasse-Jeze, E. Litvinova, F. Septier, D. Klatzmann, R. Liblau, and B.L. Salomon. 2003. Continuous activation of autoreactive CD4+CD25+ regulatory T cells in the steady state. J. Exp. Med. 198:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein, L., K. Khazaie, and H. von Boehmer. 2003. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proc. Natl. Acad. Sci. USA. 100:8886–8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker, L.S., A. Chodos, M. Eggena, H. Dooms, and A.K. Abbas. 2003. Antigen-dependent proliferation of CD4+CD25+ regulatory T cells in vivo. J. Exp. Med. 198:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cozzo, C., J. Larkin III, and A.J. Caton. 2003. Cutting edge: self-peptides drive the peripheral expansion of CD4+CD25+ regulatory T cells. J. Immunol. 171:5678–5682. [DOI] [PubMed] [Google Scholar]
- 33.Tarbell, K.V., S. Yamazaki, K. Olson, P. Toy, and R. Steinman. 2004. CD25+CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J. Exp. Med. 199:1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Min, B., R. McHugh, G.D. Sempowski, C. Mackall, G. Foucras, and W.E. Paul. 2003. Neonates support lymphopenia-induced proliferation. Immunity. 18:131–140. [DOI] [PubMed] [Google Scholar]
- 35.Schuler, T., G.J. Hammerling, and B. Arnold. 2004. Cutting edge: IL-7-dependent homeostatic proliferation of CD8+ T cells in neonatal mice allows the generation of long-lived natural memory T cells. J. Immunol. 172:15–19. [DOI] [PubMed] [Google Scholar]
- 36.Schneider, R., R.K. Lees, T. Pedrazzini, R.M. Zinkernagel, H. Hengartner, and H.R. MacDonald. 1989. Postnatal disappearance of self-reactive (Vβ6+) cells from the thymus of Mlsa mice. Implications for T cell development and autoimmunity. J. Exp. Med. 169:2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antov, A., L. Yang, M. Vig, D. Baltimore, and L. Van Parijs. 2003. Essential role for STAT5 signaling in CD25+CD4+ regulatory T cell homeostasis and the maintenance of self-tolerance. J. Immunol. 171:3435–3441. [DOI] [PubMed] [Google Scholar]
- 38.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]
- 39.Tang, Q., J.A. Smith, G.L. Szot, P. Zhou, M.L. Alegre, K.J. Henriksen, C.B. Thompson, and J.A. Bluestone. 2003. CD28/B7 regulation of anti-CD3-mediated immunosuppression in vivo. J. Immunol. 170:1510–1516. [DOI] [PubMed] [Google Scholar]
- 40.Klebb, G., I.B. Autenrieth, H. Haber, E. Gillert, B. Sadlack, K.A. Smith, and I. Horak. 1996. Interleukin-2 is indispensable for development of immunological self-tolerance. Clin. Immunol. Immunopathol. 81:282–286. [DOI] [PubMed] [Google Scholar]
- 41.Thornton, A.M., E.E. Donovan, C.A. Piccirillo, and E.M. Shevach. 2004. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J. Immunol. 172:6519–6523. [DOI] [PubMed] [Google Scholar]
- 42.Okabe, M., M. Ikawa, K. Kominami, T. Nakanishi, and Y. Nishimune. 1997. “Green mice” as a source of ubiquitous green cells. FEBS Lett. 407:313–319. [DOI] [PubMed] [Google Scholar]