

Abstract
A long-standing paradox in the study of T cell antigen recognition is that of the high specificity–low affinity T cell receptor (TCR)–major histocompatibility complex peptide (MHCp) interaction. The existence of multivalent TCRs could resolve this paradox because they can simultaneously improve the avidity observed for monovalent interactions and allow for cooperative effects. We have studied the stoichiometry of the TCR by Blue Native–polyacrylamide gel electrophoresis and found that the TCR exists as a mixture of monovalent (αβγɛδɛζζ) and multivalent complexes with two or more ligand-binding TCRα/β subunits. The coexistence of monovalent and multivalent complexes was confirmed by electron microscopy after label fracture of intact T cells, thus ruling out any possible artifact caused by detergent solubilization. We found that although only the multivalent complexes become phosphorylated at low antigen doses, both multivalent and monovalent TCRs are phosphorylated at higher doses. Thus, the multivalent TCRs could be responsible for sensing low concentrations of antigen, whereas the monovalent TCRs could be responsible for dose-response effects at high concentrations, conditions in which the multivalent TCRs are saturated. Thus, besides resolving TCR stoichiometry, these data can explain how T cells respond to a wide range of MHCp concentrations while maintaining high sensitivity.
The TCR contains a ligand-binding moiety, formed by a TCRα/β (or TCRγ/δ) heterodimer noncovalently bound to the signal-transducing subunits CD3ɛ, CD3γ, CD3δ, and ζ (CD3ζ/CD247). The TCR serves a critical role in the differentiation and activation of T cells and is therefore a key element in the initiation of the adapted immune response. Existing models of the changes undergone by the TCR on MHC peptide (MHCp) binding involve TCR clustering and conformational changes (1–3). TCR clustering by multimeric MHCp would bring individual TCRs into close proximity, enabling transphosphorylation of the receptors by associated tyrosine kinases. Indeed, soluble monomeric MHCp is not capable of activating the TCR in CD4+ T cells, unlike dimeric or oligomeric MHCp (4–6). Nevertheless, monomeric MHCp can activate the TCR in CD8+ T cells (7), possibly via heteroclustering of the TCR with CD8.
The cytoplasmic tail of CD3ɛ undergoes a ligand-induced conformational change that results in exposure of a proline-rich sequence and recruitment of the adaptor Nck (8). However, with one exception (9), crystallographic data exclude that one α/β heterodimer might undergo major structural changes on MHCp engagement (10, 11). It is therefore difficult to envision how a conformational change can be transmitted from the α/β heterodimer to the CD3 tails.
The TCR provides T cells with the capacity to discriminate between subtle changes in the MHCp (2). Moreover, APCs often express a 103-fold higher concentration of self-MHCp than antigenic MHCp, but self-MHCp complexes do not elicit a response. Indeed, it is difficult to understand how the low affinity for antigenic MHCp can simultaneously lead to the high specificity observed (5). Therefore, it is clear that a deeper understanding of the architecture of the TCR is required to resolve these fundamental questions.
Studies into TCR stoichiometry have not completely clarified our understanding. In T cells that simultaneously express human and murine CD3ɛ, the TCR contains at least two CD3ɛ subunits (12, 13); because CD3ɛ pairs with CD3γ or CD3δ, the TCR might contain one γɛ and one δɛ. Furthermore, attempts to demonstrate that more than one α/β heterodimer might be present in the complex, which is critical to determine whether the interaction with MHCp is monovalent or multivalent, have led to the formulation of two apparently irreconcilable models. Immunoprecipitation from cells expressing two different α/β heterodimers (14) and the construction of the TCR in an in vitro translation system (15) have indicated that the TCR is monovalent (i.e., containing one α/β). However, immunoprecipitation, sucrose density centrifugation, and fluorescence resonance energy transfer between two different TCRβ subunits have been used to show that the TCR contains at least two α/β heterodimers (16, 17). Analogy between the TCR and the B cell antigen receptor (BCR) led to the hypothesis that the TCR might also be oligovalent (18, 19).
Given the importance of the TCR arrangement for understanding the TCR triggering mechanisms, we have used Blue Native (BN)–PAGE to study the TCR under nondenaturing conditions. Our results show that monovalent (αβγɛδɛζζ) and multivalent high molecular weight TCR species coexist in T cells, findings that were confirmed by immunoelectron microscopy on intact T cells. Finally, we show that the multivalent TCR endows the T cell with its acute sensitivity, whereas the monovalent TCR allows the T cell to show concentration-dependent responses at high antigen concentrations.
Results
The TCR appears as a distinct complex with a αβγɛδɛζζ stoichiometry when solubilized in digitonin
To distinguish between monovalent and bivalent/multivalent formations of the TCR complex using BN-PAGE (Fig. 1 A), we established gentle purification procedures to isolate the TCR complex in its native form. A nitro-hydroxy-phenylacetate (NP)–specific single chain (sc) TCRβ protein was constructed and stably transfected into wild-type Jurkat (Jk) T cells, as well as into a TCRβ-deficient Jk mutant line (31-13). This scTCR complex can be immobilized on NP columns and eluted with the higher affinity iodo-NP (NIP) hapten. Furthermore, membrane preparations solubilized in digitonin were analyzed directly by BN-PAGE. Finally, an antiphosphotyrosine antibody was used to immunoprecipitate proteins from detergent lysates of pervanadate-stimulated cells, followed by elution of phosphoproteins with phenylphosphate and dephosphorylation. Purified TCR complexes isolated from digitonin-solubilized cell lysates by the above procedures were subjected to BN-PAGE, and immunoblotting was performed with an anti-ζ antiserum (Fig. 1 B). Because ζ is the last subunit to be added to the TCR complex during assembly (20, 21), the use of this anti-ζ antiserum in immunoblotting permits the detection of completely assembled complexes. Irrespective of the method or the cells used, the TCR appeared as a single band that characteristically migrated near the ferritin f1 marker (Fig. 1 B). Although the mass of a complex cannot be accurately deduced from its mobility in BN-PAGE (19), it is a valid method to measure relative molecular sizes. Hence, the complexes containing scTCRβ were larger than wild-type complexes because of the addition of the sc domains (Fig. 1 B, compare lane 4 with 5). Furthermore, the TCR complex was smaller than the BCR (lane 10). The use of other antibodies for inmmunoblotting demonstrated that the digitonin-solubilized complex contained CD3ɛ and TCRα/β, confirming that the complex recognized by the anti-ζ antiserum is indeed the TCR (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20042155/DC1).
Figure 1.

Digitonin-solubilized TCR has an αβγɛδɛζζ stoichiometry. (A) Current structural models of the TCR complex. The minimal hexameric complex, which comprises one TCRα/β dimer (monovalent), and an alternative model with two TCRα/β dimers (bivalent) are shown. (B) The digitonin-solubilized TCR complex is of a defined size. TCRs from the sources indicated were prepared either by NIP elution from NP-Sepharose columns, from membrane fractions, or by phenylphosphate elution from antiphosphotyrosine affinity columns. After separation by BN-PAGE, immunoblotting was performed with an anti-ζ serum. As a control, the BCR was run in parallel and immunoblotted with an antilight chain serum. The marker protein in this and all subsequent BN-PAGEs is ferritin in its 24-mer, 48-mer, and 72-mer forms (f1, 440 kD; f2, 880 kD; and f3, 1,320 kD). (C) The digitonin-solubilized TCR contains two CD3 dimers. TCRs contained in total membrane fractions from human PBMCs (lanes 1–3) or in antiphosphotyrosine immunoprecipitates from CH7C17 T cells (lanes 4–6) were incubated with the amounts of the anti-CD3 (UCHT1) Fab fragments indicated and resolved by BN-PAGE. (D) The digitonin-solubilized complex contains one TCRβ subunit. TCRs prepared from antiphosphotyrosine immunoprecipitates of human PBMCs (lanes 1–3) or CH7C17 cells (lanes 4–5) were incubated with the anti-TCRβ antibodies Jovi1 (lanes 1–3) or Jovi3 (lanes 4–6) and resolved by BN-PAGE.
To ascertain the stoichiometry of the digitonin-solubilized TCR complex, we developed a gel shift assay. TCR complexes from primary human T cells (Fig. 1 C, lanes 1–3) or the Jk-derived T cell line CH7C17 (lanes 4–6) were incubated with different amounts of the Fab fragment of the anti-CD3 antibody UCHT1 (specific for γɛ and δɛ dimers; reference 22) and subjected to BN-PAGE. The binding of the Fab fragment to the TCR was reflected by the appearance of two shifted bands indicative of two UCHT1 binding sites. Thus, we concluded that the digitonin-solubilized TCR contains two CD3 dimers. In contrast, only one shift was produced by binding an anti-TCRβ antibody (Fig. 1 D, lanes 1–3 using PBMCs and lanes 4–6 using CH7C17), indicating that the digitonin-solubilized complex contains one α/β heterodimer. For the anti-TCRβ shift assay (Fig. 1 D) we used a whole antibody, thus explaining the reduced mobility of the shifted complex compared with the TCR complex bound to one Fab fragment of the anti-CD3 antibody (Fig. 1 C). Nevertheless, the use of a Fab fragment of a different anti-TCRβ antibody in another T cell system also resulted in a single shifted band (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20042155/DC1). The shift in Fig. 1 D, lanes 1–3, was not complete, possibly because the shifting antibody only recognizes Cβ1-TCRs, thus leaving the Cβ2-TCRs unbound. Collectively, these results and the use of a battery of other TCR-specific and control antibodies (unpublished data) allowed us to conclude that in digitonin the TCR complex has a stoichiometry of αβγɛδɛζζ and is therefore monovalent.
The TCR is detected in high molecular weight complexes when solubilized in Brij96
To investigate whether the detergent used for solubilization influences the size and stoichiometry of the TCR complex, we analyzed the TCR in several detergents known to respect the interactions between TCR and CD3 subunits. In contrast to TCRs solubilized in digitonin, TCRs solubilized in Brij58, Brij96, Brij97, and Brij98 did not migrate as a discrete band in BN-PAGE (Fig. 2 A and not depicted). Indeed, only a portion of the TCR complex isolated with Brij96 had the same mobility as the digitonin-solubilized monovalent complex (Fig. 2 A); the rest of the TCR was found in complexes of higher molecular weights. The larger complexes did not appear to result simply from incomplete solubilization of membrane proteins because higher and lower doses of Brij96 did not alter the mobility of the complex (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20042155/DC1). Furthermore, the larger complexes did not result from artifactual aggregation provoked by Brij96 because the digitonin-solubilized complex was not aggregated when transferred to Brij96 (Fig. 2 B, lane 3). Further proof against a possible aggregation effect of Brij96 was obtained in two-dimensional BN/BN-PAGE in which the mobility of each molecular species remained unaltered in the second dimension, thus forming a perfect diagonal (Fig. 2 C). Most important, when a battery of membrane proteins was analyzed by two-dimensional BN/SDS-PAGE, the mobility of the other membrane proteins (CD45, CD59, LAT, and Lck) in Brij96 and digitonin varied little in comparison with the large differences in mobility observed for the TCR (Fig. 2 D). Therefore, the varying mobility of the TCR when extracted in Brij96 versus digitonin is rather a specific property of the TCR and not a general property of the detergent.
Figure 2.
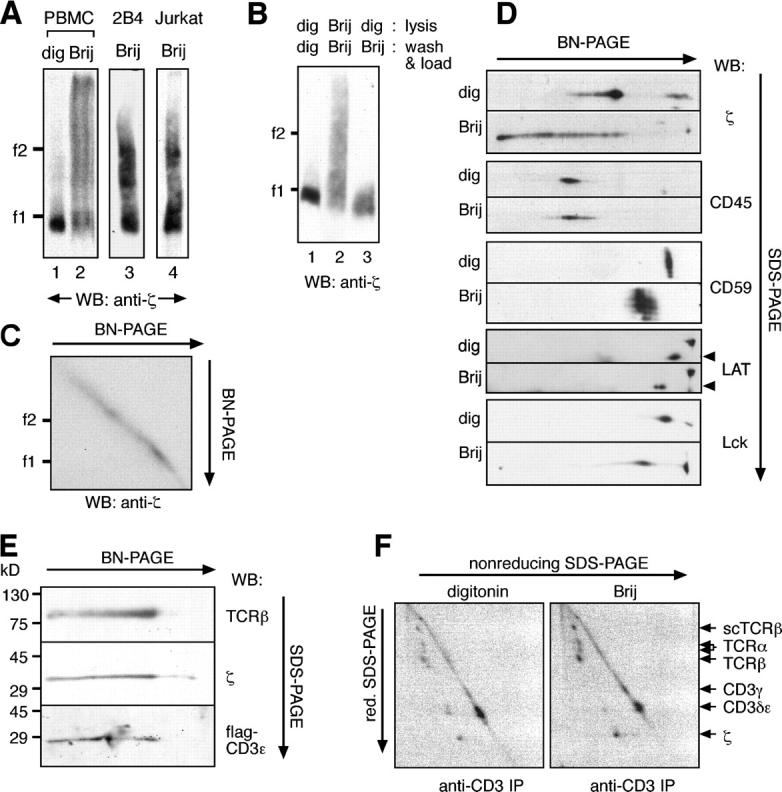
The Brij96-solubilized TCR appears in distinct and stable high molecular weight complexes. (A) Part of the Brij96-solubilized TCR has a low mobility in BN-PAGE. TCRs, prepared from total membranes of human PBMCs (lanes 1 and 2) or by antiphosphotyrosine immunoprecipitation from 2B4 and Jk cells (lanes 3 and 4), were solubilized either in digitonin or in Brij96 and analyzed by BN-PAGE. (B) The TCR does not aggregate in Brij96. Jk cells were lysed, and the TCR was immunoprecipitated with antiphosphotyrosine in the continuous presence of either digitonin (lane 1) or Brij96 (lane 2). Alternatively, they were lysed in digitonin, and immunoprecipitation and loading were performed in Brij96 (lane 3). (C) The TCR does not aggregate during a second BN-PAGE in Brij96. TCRs were isolated from Jk by antiphosphotyrosine immunoprecipitation and analyzed by two-dimensional BN/BN-PAGE. (D) Other cell surface proteins did not appear in high molecular weight complexes when the cells were lysed in Brij96. Total membrane fractions of Jk cells were solubilized in digitonin or Brij96 and separated by two-dimensional BN/SDS-PAGE. Immunoblotting was performed with the antibodies indicated. The arrowheads indicate the LAT spot. (E) The Brij96-extracted TCR contains the α/β, ζ, and CD3 subunits. TCR from Jk.flagɛ cells was analyzed by two-dimensional gel BN/SDS-PAGE. Immunoblotting was performed with antibodies to the indicated TCR subunits. (F) The Brij96-solubilized TCR is not found associated to cell surface proteins other than the TCR subunits. Jk.scTCRβ cells were surface iodinated and lysed in the detergent indicated. After immunoprecipitation with the anti-CD3 antibody UCHT1, TCR complexes were resolved by two-dimensional SDS-PAGE (nonreducing/reducing) and visualized by autoradiography.
When the composition of these high molecular weight complexes was analyzed, the TCRβ, CD3ɛ, and ζ chains were detected (Fig. 2 E). Although this suggests that their large size was not caused by the association of ζ with receptors other than the TCR, the possibility remained that when solubilized in Brij96 the TCR might remain associated to membrane proteins that are released in digitonin. Two-dimensional SDS-PAGE was performed on anti-CD3 immunoprecipitates obtained from surface 125I-labeled Jk.scTCRβ cells. All the TCR subunits were isolated when solubilized in both detergents, including heterodimers of TCRα with endogenous TCRβ and scTCRβ (Fig. 2 F). Indeed, no essential differences could be observed in the 2D profiles of either complex. Moreover, we were unable to detect any additional proteins in the Brij96-solubilized TCR isolated from metabolically labeled cells when compared with that isolated from cells lysed in digitonin (unpublished data). Therefore, the size heterogeneity of the TCR complex in Brij96 was not caused by the association of other proteins. Although non-TCR proteins (e.g., signaling proteins) might be associated to the low and high molecular weight TCRs in resting T cells, these associations would be at such low stoichiometric ratios that they could not explain the observed size heterogeneity.
Brij96 extraction, as opposed to digitonin extraction, preserves a cholesterol-dependent multivalent TCR
To determine whether the size heterogeneity detected by BN-PAGE corresponded to the coexistence of different oligomeric forms of the TCR complex, Jk.scTCRβ cells were lysed in either Brij96 or digitonin, and the scTCRβ-containing TCR was precipitated with NIP-Sepharose (Fig. 3 A, lanes 2 and 4). The precipitate was analyzed by immunoblotting with an anti-Vβ8 antibody that recognizes endogenous (TCR-Vβ8) but not transfected (scTCR-Vβ3) TCRβ chains. The endogenous TCRβ coprecipitated with scTCRβ from Brij96 but not from digitonin lysates. Furthermore, endogenous TCRβ could not be coprecipitated from Jk and 31-13.scTCRβ cell mixtures (lane 3), thus excluding the possibility that these TCR complexes associated after lysis of the cells. The presence of scTCRβ in all the immunoprecipitates was confirmed by reprobing the membrane with an anti-Cβ antibody that recognizes both TCRβ chains (Fig. 3 A). Although these results further support a monovalent stoichiometry for the complex isolated in digitonin, the coprecipitation of two different TCRβs in Brij96 shows that the TCR can contain at least two α/β heterodimers in this detergent.
Figure 3.
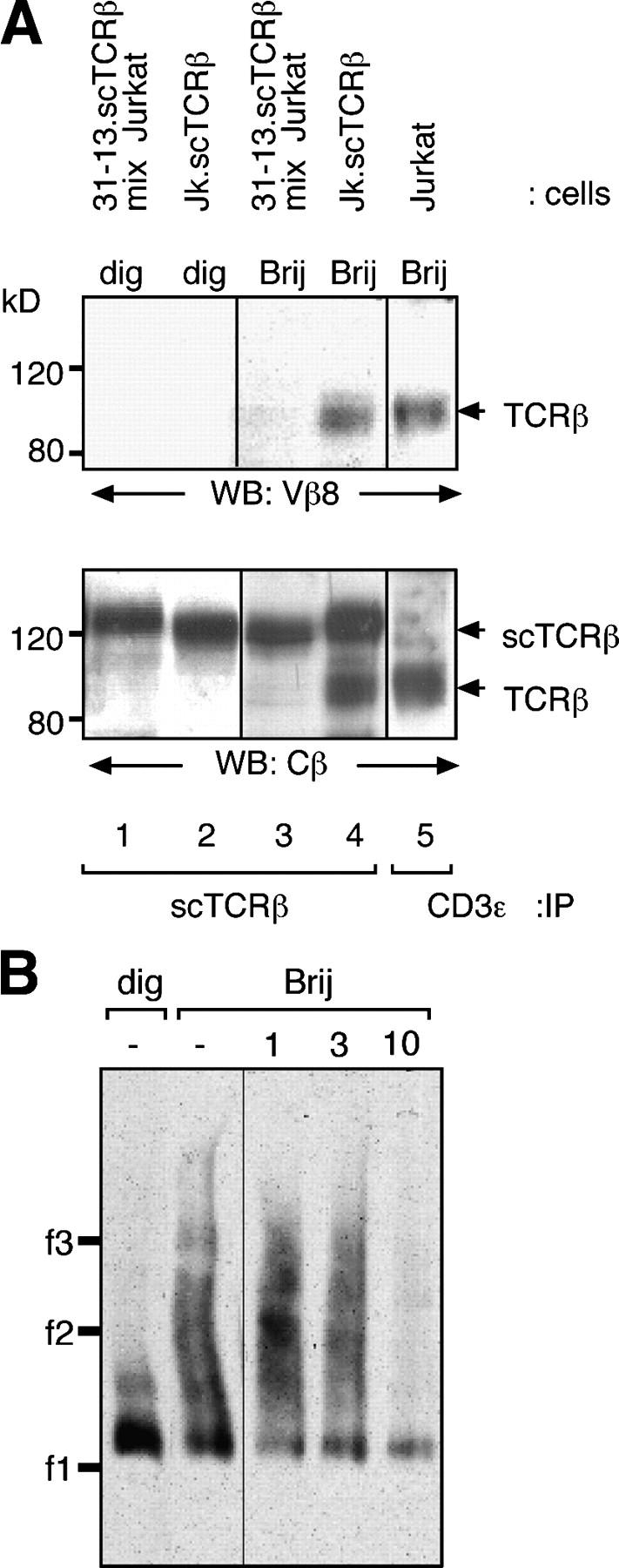
Presence of a cholesterol-dependent multivalent TCR in Brij96 lysates. (A) The Brij96-solubilized TCR contains at least two α/β heterodimers. Jk.scTCRβ cells coexpressing TCRβ and scTCRβ were lysed in digitonin or Brij96 (lanes 2 and 4). scTCRβ-containing complexes were isolated using NP-Sepharose and resolved by nonreducing SDS-PAGE. As a control, the same procedure was applied to a mixture of Jk and 31-13.scTCRβ cells (lanes 1 and 3). In addition, the TCR of Brij96-lysed Jk cells was immunoprecipitated using an anti-CD3 antibody. Immunoblotting was performed using an anti-TCR–Vβ8 antibody, which only recognizes the endogenous TCRβ (top). The membrane was reprobed with an anti-TCR–Cβ antibody (bottom), which recognizes both the endogenous TCRβ and the transfected scTCRβ. (B) The high molecular weight TCR complexes disappear on treatment with MβCD. 31-13.scTCRβ cells were lysed in either digitonin or in Brij96 in the presence of the concentrations of MβCD indicated, and the TCR was purified by NP-Sepharose precipitation.
The size heterogeneity of the TCR complex in Brij96 could be explained if it comprised a mixture of monovalent, bivalent, and higher order multivalent TCR structures. Because digitonin is a cholesterol-solubilizing detergent, it is possible that the sequestering of cholesterol caused the disruption of the high multivalent TCR complexes. This indeed seems to be the case because incubating the TCRs isolated from Brij96-solubilized lysates with methyl-β-cyclodextrin (MβCD), a cholesterol-extracting agent, reduced the size of the TCR to that of the monovalent complex (Fig. 3 B). This result suggests that cholesterol is required to maintain the structure of the multivalent TCR complexes, but not of the monovalent form.
Multivalent and monovalent TCRs coexist on the plasma membrane of intact nonstimulated T cells
To confirm that multivalent TCRs exist at the T cell surface, fixed nonstimulated 2B4 T cells were immunolabeled with an anti-CD3 antibody visualized with 10 nm gold–protein A. In label fracture and transmission electron microscopy, a mixture of TCR complexes of different size was apparent in the plasma membrane (Fig. 4, A and E). Indeed, a significant (P < 0.0001) proportion of the TCR complexes was labeled with more than two gold particles, indicating that at least these complexes were multivalent (Fig. 4 A, closed arrows). However, monovalent complexes, labeled with one gold particle where the bivalent anti-CD3 antibody binds simultaneously to the two CD3 dimers of the complex, were also observed (open arrows). Cholesterol extraction with MβCD disrupted the large complexes favoring the appearance of the smaller complexes without diminishing the total amount of TCR on the cell surface (Fig. 4, A and C). This confirms the data obtained by BN-PAGE (Fig. 3 B) and excludes that the accumulation of gold particles is caused by antibody aggregation. The coexistence of multivalent and monovalent TCR complexes and the cholesterol dependence were also demonstrated in cells labeled with an anti-TCRβ antibody (Fig. 4 B). Of note, we often were unable to detect a gap between two adjacent gold particles (Fig. 4, A, B, E, and F). Considering the size of these particles (10 nm) and the resolution of the microscope, one can calculate that the gap must be smaller than 1 nm, indicating a very close interaction between the TCR complexes (see Discussion). An estimate of the distribution of gold particles in isolated TCRs and TCR clusters indicates that more than half (55%) of the TCR in untreated 2B4 cells is in the form of multivalent complexes (Fig. 4, C and D). Interestingly, the large TCR complexes were not present as unorganized clusters but had a propensity to assemble into linear structures (Fig. 4 E). Linear multivalent TCR complexes were also detected in CH7C17 and in primary human T cells (Fig. 4 F) in which >45% of the TCR was found as multivalent complexes (unpublished data). These results are therefore in agreement with the BN-PAGE and coimmunoprecipitation data and show that TCR monovalent and multivalent complexes are coexpressed on the surface of intact T cells.
Figure 4.
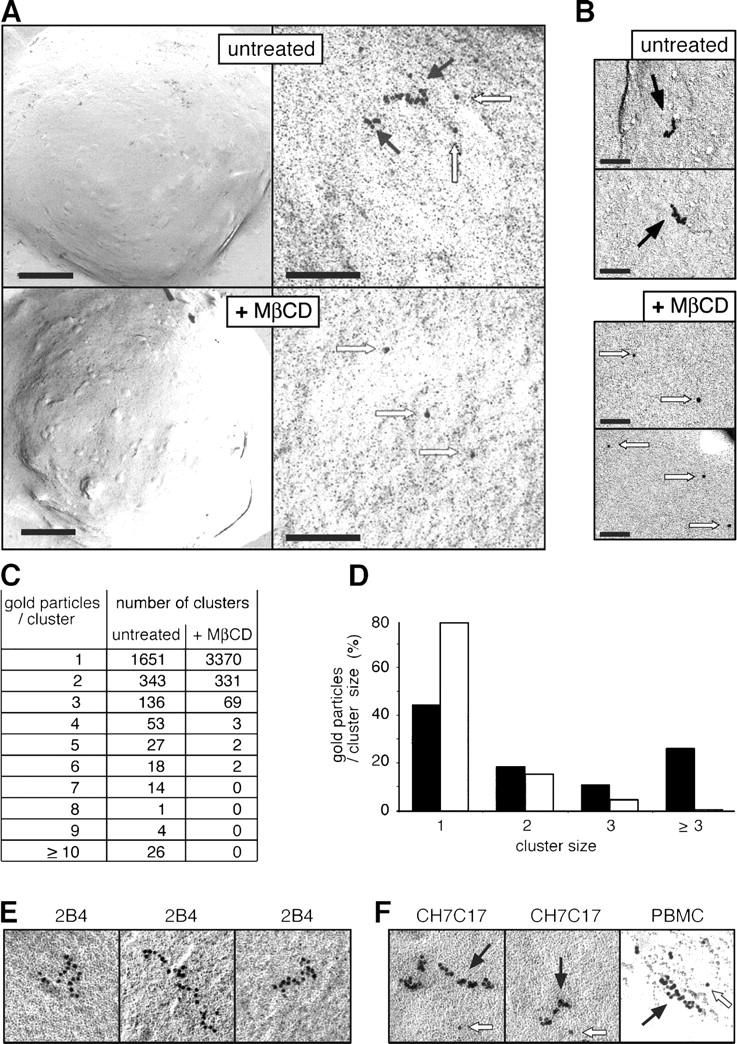
Monovalent and multivalent TCRs coexist on the intact cell surface. (A) Large TCR complexes exist on the T cell surface. 2B4 cells labeled after fixation with the anti-CD3 dimer antibody 145-2C11 and 10 nm gold–protein A are shown at low (left) and high magnification (right). Bars: (left) 2 μm; (right) 100 nm. Before immunolabeling, cells were incubated with 5 mM MβCD or left untreated. Photographs were taken after label fracture by transmission electron microscopy and show the immunogold labeling from the internal side of the plasma membrane. Closed arrows, multivalent complexes; open arrows, monovalent TCRs. (B) The large TCRs are multivalent. Anti-TCRβ immunogold-labeled TCRs from untreated or MβCD-treated 2B4 cells are shown. Closed arrows, multivalent complexes; open arrows, monovalent TCRs. Bar, 60 nm. (C and D) The large TCRs are disassembled by cholesterol extraction. The total number of clusters of each size in untreated and MβCD-treated 2B4 cells are shown (C). Distribution of gold particles according to the cluster size is shown as a percentage of the total number of gold particles (D). Black bars, untreated cells; white bars, MβCD-treated cells. (E) Multivalent TCRs have a propensity to form linear arrays. Various representative individual large TCR complexes stained by the 145-2C11 antibody are shown. (F) Multivalent and monovalent TCRs are coexpressed in different T cell lines and primary T cells. The Jk-derived CH7C17 cell line and human PBMCs were labeled after fixation with anti-CD3 dimer antibody OKT3 and 10 nm gold–protein A as in panel A. Closed arrows, multivalent complexes; open arrows, monovalent TCRs.
Only multivalent TCR complexes are tyrosine phosphorylated when stimulated with low concentrations of antigen
There is a large body of evidence demonstrating that T cells respond to a wide range of antigen concentrations (6, 23–25). Moreover, although T cells can respond to a few MHCp molecules, they still show increasing responses to concentrations that exceed by 5 or more orders of magnitude the minimal effective concentration (26, 27). To determine whether the multivalent and monovalent TCRs are differentially involved in the response to low and high antigen concentrations, we used the CH7C17 Jk T cell line (specific for the hemagglutinin HA307-319 peptide in combination with HLA-DR1; reference 28). When these cells were incubated with HA peptide–loaded DR1-expressing cells, the activation marker CD69 was up-regulated in a dose-dependent manner over a concentration range of nearly four orders of magnitude (Fig. 5 A). We stimulated CH7C17 cells with different concentrations of HA peptide loaded onto the presenting cells and monitored the appearance of the 23-kD phosphorylated ζ form, indicative of active TCRs, by two-dimensional BN/SDS-PAGE followed by antiphosphotyrosine immunoblotting (Fig. 5 B). Clearly, at low antigen doses (4–10 μM) only the multivalent complexes were phosphorylated, whereas the monovalent complexes became phosphorylated at higher concentrations (40–100 μM). In contrast, both types of TCR complexes, monovalent and multivalent, were phosphorylated after pharmacological stimulation with pervanadate (Fig. 5 B). Therefore, at low concentrations of antigen, only the ζ chains present in multivalent TCRs became phosphorylated, whereas the monovalent TCRs required higher antigen concentrations, a situation in which the multivalent complexes might already be saturated.
Figure 5.
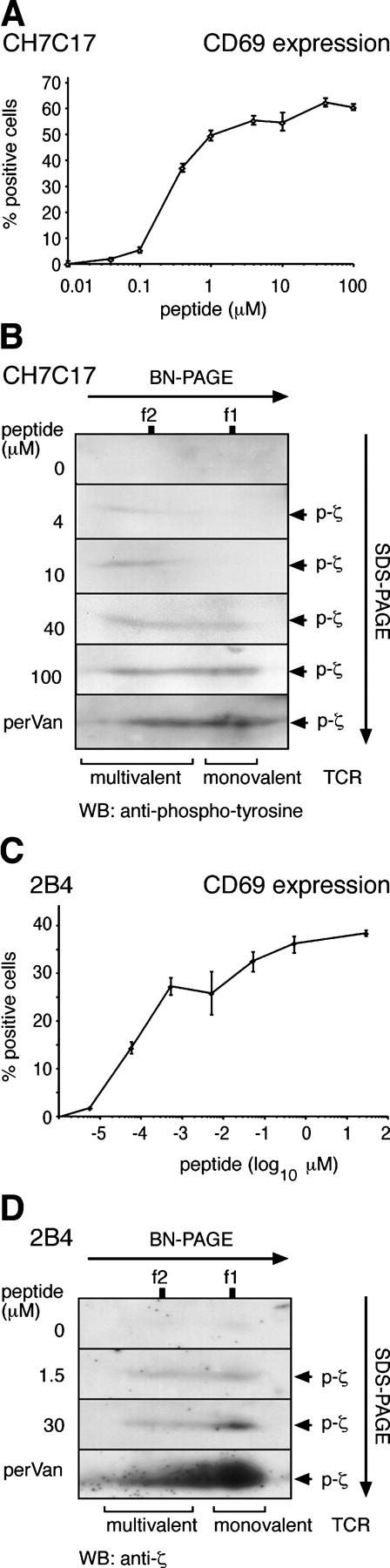
The monovalent and multivalent TCR complexes are differentially sensitive to antigen stimulation. (A and C) T cells respond differentially over a wide range of antigen concentrations. The HA peptide–specific T cell line CH7C17 and the PCC peptide–specific T cell hybridoma 2B4 were stimulated for 24 h with varying concentrations of peptide loaded onto either DAP-DR1 or DCEK APCs. Surface staining of the T cells with anti-CD69 antibodies was measured using a flow cytometer and the percentage of positive cells is depicted in semilogarithmic scale. Values are mean ± SD. (B and D) At low antigen doses, only multivalent TCRs become tyrosine phosphorylated. CH7C17 and 2B4 cells were antigen stimulated for 5 min as in A and C or stimulated with pervanadate. Cells were lysed in Brij96, and phosphorylated TCRs were affinity purifed with immobilized antiphosphotyrosine antibody and resolved by two-dimensional BN/reducing SDS-PAGE. Immunoblotting was performed using an antiphosphotyrosine antibody (B) or anti-ζ (D). The position of phosphorylated ζ (p-ζ) is indicated.
This type of experiment was repeated using 2B4 as responding T cells and DCEK primed with pigeon cytochrome c (PCC) peptide as APCs. 2B4 cells already responded by increasing CD69 expression at a concentration as low as 5 pM and still showed dose-dependent responses at concentrations of 0.5–30 μM (i.e., six to seven orders of magnitude higher; Fig. 5 C). Our biochemical approach was not sensitive enough to detect induced tyrosine phosphorylation of ζ at concentrations <1.5 μM. At this concentration of antigen, we could detect tyrosine phosphorylation of the multivalent TCRs and also some phosphorylation of the monovalent TCR (Fig. 5 D). However, a 20-fold increase in antigen concentration (30 μM) led to increased tyrosine phosphorylation of the monovalent but not the multivalent TCRs. These results confirm the CH7C17 data by showing that at low concentrations of antigen, the multivalent TCRs are already saturated, whereas the monovalent TCR is still increasingly phosphorylated at increasing antigen concentrations.
Discussion
We show that multivalent and monovalent forms of the TCR coexist in the T cell membrane. Other studies have failed to detect the multivalent complexes (14, 15), probably because they relied on the use of digitonin, a detergent that disassembles the multivalent TCR. Furthermore, although one of these studies (15) is limited to an in vitro translation system, we have studied TCR stoichiometry in both digitonin and Brij96 lysates of T cells as well as in intact T cells. Thus, the picture that emerges from our data is that there is not a single stoichiometry of the TCR but a combination of monovalent and multivalent complexes, the latter being formed by a heterogenous combination of dimers, trimers, et cetera, including TCR complexes with >20 TCR units. Using a novel method, we demonstrate that the TCR has a stoichiometry of αβɛγɛδζζ when extracted from T cells in digitonin, possessing only one ligand-binding moiety (monovalent TCR). Although this is in agreement with the stoichiometry found in the in vitro translation system (15), the stoichiometry of the minimal TCR unit (i.e., in the absence of digitonin) is still unknown. The multivalent TCR complexes identified in Brij96 do not migrate as discrete bands in BN-PAGE, raising the concern that they are artifacts produced by the detergent. We have, however, excluded this possibility because Brij96 did not cause the aggregation of TCRs, in agreement with a recent study in which Brij98 did not produce a mixing of distinct membrane microdomains (29). Furthermore, our study shows that other membrane proteins extracted in Brij96 are not present in high molecular weight complexes. Finally, and most important, small and large TCR complexes were seen to coexist on the cell surface of nonstimulated immunogold-labeled T cells, reflecting the monovalent and multivalent stoichiometries. Interestingly, a variety of multivalent TCRs appear to be generated, ranging from complexes that comprise only a few CD3 and TCRα/β dimers to huge complexes consisting of ≥20 CD3 and TCRα/β dimers. Thus, the different molecular weights found in BN-PAGE possibly reflect the distinct stoichiometries found by electron microscopy. Because proteins in general did not run as sharp bands when Brij96 is present during BN-PAGE separation, the mixture of molecularly distinct TCR complexes overlap, producing a continuous smeary band.
The multivalent complexes were predominantly found as linear structures (Fig. 4), possibly indicating that the large TCR multimers are assembled rather than randomly aggregated. The distance between two 10-nm gold particles is often smaller than 1 nm (Fig. 4). Because the widths of TCRα/β and the CD3 dimers are in the range of 50 Å, the maximal width of a monovalent TCR complex is 150 Å (30), larger than the 10-nm gold particles. Indeed, our preliminary electron microscopy data on purified TCR complexes indicate that the diameter of a single TCR unit is slightly larger than 100 Å (unpublished data). Therefore, the proximity between gold particles in the electron microscopy experiments strongly suggests the presence of protein–protein interactions within the multivalent complexes. Clearly, this hypothesis will have to be addressed experimentally. The organization of the multivalent complexes in linear arrays could indicate the existence of a monospecific interaction between TCRs (i.e., between one subunit of a monomeric complex and another subunit of the next complex). Indeed, in an earlier study we suggested the possible existence of linear arrays of the TCR maintained by Vα–Vα interactions (16). However, other interactions are possible, even if the minimal TCR complex is indeed the monovalent complex (e.g., a CD3γ–CD3δ, a CD3ɛ–CD3ɛ, or a ζ2–ζ2 interaction).
Similarly, the proximity between two TCRβ subunits has been shown using fluorescence resonance energy transfer, an approach that does not depend on the detergent extraction of the TCR, indicating that at least two α/β heterodimers are present in the complex (16). However, the coexistence of monovalent with bivalent and multivalent TCRs was not excluded. This study allows us to conclude that monovalent and multivalent TCR complexes exist simultaneously in the plasma membrane of unstimulated T cells (Fig. 6 A). This conclusion is supported by a previous report showing a decrease but not a complete abrogation of MHCp tetramer binding in T cells treated with MβCD, suggesting that two different TCR populations might exist on the surface of living cells (31).
Figure 6.

A model of TCR signaling via mono- and multivalent complexes can account for high sensitivity and wide response range of T cells to antigen. (A) Multivalent and monovalent TCR complexes coexist on the surface of nonstimulated T cells. For simplicity, a single TCR complex is depicted as a ligand-binding ectodomain (corresponding to the TCRα/β heterodimer) and signal-transducing cytoplasmic tails (representing the CD3 subunits). (B) At low MHCp concentrations, the multimeric complexes become preferentially phosphorylated. This might be caused by their higher avidity for dimeric MHC/agonistic peptide (blue, MHCp molecules). On ligand binding, the triggering signal might propagate to neighboring TCRs of the same multimer by a spreading of the conformational change via cooperative interactions and by the spreading of tyrosine phosphorylation (red, tails of activated CD3). Alternatively, the TCRα/β heterodimers of a multivalent TCR that are not engaged by MHC/agonistic peptide might bind to MHC/self-peptide (B′, green). By amplifying the effect of a few MHCp binding events to neighboring receptors within a TCR multimer, the T cell might be able to respond to very low MHCp doses. (C) At higher MHCp concentrations, the monovalent TCRs begin to be phosphorylated, indicating that they now also bind to MHCp, which dimerizes or clusters the monovalent TCRs. This allows the T cell to sense high doses of antigen, a situation in which the multivalent TCRs are already saturated.
It has already been pointed out that a bivalent/multivalent TCR might improve the high off rate found for monovalent MHCp binding and can resolve the paradox of the high specificity–low affinity TCR–MHCp interaction (32). However, this does not readily explain how T cells manage to respond to a wide range of MHCp concentrations (6, 23–25). The coexistence of monovalent and multivalent TCRs may clarify this issue. Theoretical calculations suggest that cells coexpressing monovalent and multivalent forms of a given receptor can show high sensitivity to low concentrations of a ligand and concentration-dependent responses to high ligand concentrations (33). Indeed, by analyzing antigen-stimulated TCRs in BN-PAGE, we show that the multivalent TCR might facilitate signaling at low MHCp doses because only these complexes are phosphorylated at low peptide concentrations. On the other hand, the monovalent TCRs are only phosphorylated at high concentrations of peptide antigen, a situation in which the multivalent TCRs might already be saturated. Thus, the presence of monovalent TCRs might endow the T cell with the capacity to produce a concentration-dependent response even at high MHCp doses. In contrast, the multivalent complexes may augment the sensitivity of the T cell to antigen simply by increasing the avidity for an MHCp dimer or, additionally, by spreading the signal (i.e., the ligand-induced signaling activity of one receptor propagates to neighboring receptors of the same multimer), thereby amplifying the effect of binding. This spreading of activity could be accomplished by cooperative interactions between the different receptors (Fig. 6 B) or by very low affinity binding of MHC/self-peptides to the neighboring receptors (Fig. 6 B; references 34, 41). With respect to the molecular mechanisms that might propagate the triggering signal from engaged to nonengaged TCRs, one could hypothesize that the linear arrangement of the multivalent TCRs could serve to transmit the signal along the multimer axis as a “domino effect.” Recently, spread of activity from engaged to nonengaged receptors within an oligomer has been described for bacterial chemotaxis receptors (34). Alternatively, the linear arrays of multivalent complexes could help to serially trigger several TCRs by a single MHCp (35).
The existence of multivalent TCRs could also shed light on how the receptor transmits signals across the membrane. On antigen binding, the TCR undergoes a conformational change that makes the proline-rich sequence of the CD3ɛ tail more accessible to binding of the Nck adaptor (3, 8). Crystallography of the soluble part of the α/β heterodimer alone or together with MHCp demonstrated that, with one exception (9), the α/β ectodomains do not change their structure appreciably on antigen binding, making it difficult to imagine how a change in conformation could be induced in the CD3 subunits (10, 11). However, in the multivalent complexes, the simultaneous binding of both α/βs to MHCp could alter the distance or the orientation of one of the α/β heterodimers with respect to the other (36, 37) without changing the structure of either individual α/β unit (Fig. 6). This could lead to a rearrangement of the whole TCR complex, including its cytoplasmic tails. Indeed, it has been shown that, at the cell surface, MHC class I and II are present as dimers or in even larger arrays (38–40). Thus, in principle, two identical agonist peptides could simultaneously bind to one multivalent TCR complex or, alternatively, one agonist peptide could bind together with a self-peptide to the second MHC molecule (41, 42). Such a scenario would also explain why, unlike dimeric MHCp, monomeric soluble MHCp does not activate T cells, providing an alternate explanation to the proposed need for TCR clustering (4–6).
Lipid rafts are thought to be cholesterol-rich microdomains in the plasma membrane. We show that the extraction of cholesterol disrupts the multivalent TCR complexes but leaves the monovalent complexes intact. This finding suggests that cholesterol molecules might contribute to the TCR multimers and that these multimers might assemble raft-like structures around them. In agreement with this idea, several reports have proposed that part of the TCR is present in lipid rafts in nonstimulated T cells (29, 43). Monovalent complexes seem not to depend on cholesterol and, thus, they might be located in a different lipid environment than the multivalent complexes, opening the possibility that both receptor forms could couple to different signaling molecules. Cholesterol extraction of T cells impairs TCR-induced signaling, suggesting that lipid rafts are important for the transduction of signals via the TCR (44–46). This finding can now be reinterpreted in the light of the deleterious effect of cholesterol depletion on the integrity of multivalent TCR complexes.
Our findings are also in line with others that demonstrate the effect of cholesterol on soluble MHCp binding to T cells. Flow cytometry studies have shown that the avidity of MHCp tetramer binding to T cells diminishes after cholesterol extraction without reducing the number of TCR molecules (31, 47, 48). Because cholesterol depletion disrupts multivalent TCRs, the picture that emerges is that these TCRs would be responsible for high-avidity MHCp tetramer-binding, whereas the monovalent TCR independent of cholesterol binds MHCp tetramers with lower avidity. Furthermore, a cholesterol-dependent increase in TCR avidity for MHCp tetramers has been shown in activated versus naive T cells (47). We suggest that T cells might regulate their sensitivity to antigen recognition by altering the ratio of monovalent to multivalent TCRs, although this remains to be demonstrated.
MATERIALS AND METHODS
Expression vector.
The plasmid pSRα-scTCRVβ3 encoding the scTCRVβ3 fusion protein has been described previously (8). In brief, the NP-specific sc containing the fused variable Ig domains of the light and heavy chain of an NP-specific antibody was fused to the NH2 terminus of a mature TCRβ protein to yield the chimera scTCRβ.
Cells.
The Jk.scTCRβ cell line (clone 2D10) was obtained by transfecting Jk cells with the pSRα-scTCRVβ3 expression vector. The transfectants 31-13.scTCRβ and Jk.flagɛ have been described previously (8). scTCRβ reconstituted the expression of the TCR complex in the TCRβ-deficient mutant (unpublished data), showing that the chimera assembles into the complete TCR complex. The Jk HA1.7 TCRα-TCRβ transfectant (CH7C17) was a gift from A. Alcover (Institut Pasteur, Paris, France) and the B cell line J558Lμm3-11 was a gift from M. Reth (Max-Planck-Institut für Immunbiologie, Freiburg, Germany). The murine citochrome c–responsive T cell hybridoma 2B4 was provided by J. Ashwell (National Cancer Institute, Bethesda, MD). Fibroblast cell lines DAP DR1+, stably transfected with HLA class II DR1 and ICAM-1 molecules, and DCEK, stably transfected with IEk and CD80 expression vectors, were provided by M. Owen (Cancer Research UK, London, UK) and R. Germain (National Institutes of Health, Bethesda, MD), respectively. Human PBMCs were isolated from healthy donors using a Ficoll gradient. All cells were maintained in complete RPMI 1640 with 5% FBS.
Antibodies and reagents.
The rabbit anti-ζ antiserum 448 has been described previously (8). The following TCR antibodies were used: 17-34-14 (anti–human Vβ8; G. Spagnoli, University of Basel, Basel, Switzerland), RP221 (anti-CD45; F. Sánchez-Madrid, Hospital de la Princesa, Madrid, Spain), Jovi1 (Cβ1) and Jovi3 (Vβ3; M. Owen), UCHT1 (anti-CD3; P. Beverly, The Edward Jenner Institute for Vaccine Research, Berkshire, UK), and MEM-43/5 (anti-CD59) and anti-Lck antibodies (V. Horejsi, Institute of Molecular Genetics, Prague, Czech Republic). The antiphosphotyrosine (4G10) and anti-LAT antibodies were purchased from Upstate Biotechnology, the anti-Flag (M2) was purchased from Sigma-Aldrich, the anti-CD3 OKT3 hybridoma was purchased from the American Type Culture Collection, and the anti-CD69–PE was purchased from Caltag. Fab fragments were prepared using the Immunopure IgG1 Fab Preparation Kit (Pierce Chemical Co.) and confirmed by SDS-PAGE and silver staining. Secondary antibodies were obtained from Southern Biotechnology Associates, Inc. NP-conjugated Sepharose and NIP-conjugated Sepharose, as well as free NIP (NIPcap), were purchased from Biosearch Technologies. MβCD was obtained from Sigma-Aldrich.
TCR purifications.
The scTCR was purified from cell lysates using NP-conjugated Sepharose that was washed three times, and the scTCR was eluted in BN buffer containing the appropriate detergent and 0.5 mM free NIP. Alternatively, membrane fractions were prepared by disrupting 107 cells in 1 ml of a hypotonic buffer (10 mM Hepes, pH7.4, 42 mM KCl, 5 mM MgCl2, and protease inhibitors) using a Dounce homogenizer and pelleting the membranes in an ultracentrifuge (Sorvall; Combi) at 150,000 g. Membranes were lysed in 150 μl BN buffer containing the appropriate detergent. The third method used to isolate TCR involved incubating 107 cells in 200 μM pervanadate (8.8 μl of 23 mM sodium orthovanadate plus 1.2 μl of 30% H2O2) at 37°C for 5 min in 1 ml RPMI 1640. Cells were lysed in 1 ml of lysis buffer containing 20 mM TrisHCl, pH 8, 137 mM NaCl, 2 mM EDTA, 10% glycerol, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM PMSF, 500 μM sodium orthovanadate, 1 mM NaF, and the appropriate detergent (1% digitonin or 0.5% Brij96 unless otherwise indicated). Phosphorylated proteins were purified with 1 μg 4G10 and 3 μl protein G–Sepharose, washed three times, and subsequently eluted in BN buffer (500 mM ɛ-aminocaproic acid, 20 mM NaCl, 10% glycerol, 2 mM EDTA, and 20 mM BisTris, pH 7.0), including 50 mM phenylphosphate, 1 unit alkaline phosphatase, and the appropriate detergent.
For peptide antigen stimulation, DAP-DR1 cells were loaded overnight with concentrations of HA307-319 peptide PKYVKQNTLKLAT in RPMI 1640 with 1% FCS, as indicated in the figures. Cells were removed from the dish with 0.5 mM EDTA and washed twice in PBS. CH7C17 (or Jk) cells were starved overnight in RPMI 1640 without FCS. For each stimulation, 2.5 × 107 T cells were mixed with 2 × 107 APCs, centrifuged briefly, and maintained as a pellet for 5 min at 37°C before lysis in the lysis buffer mentioned above containing 0.5% Brij96. After affinity purification with antiphosphotyrosine antibodies, proteins were eluted as above, with the addition of sodium orthovanadate, and in the absence of phosphatases. For stimulation of 2B4 cells, DCEK (15 × 107 per point) was loaded for 2 h with the PCC peptide KAERADLIAYLKQATK in RPMI 1640 without serum. 2B4 cells (50 × 107 per point) were mixed with the antigen-loaded DCEK and stimulated as for CH7C17 cells. Anti-CD3 (UCHT1) immunoprecipitations were performed as described above for the antiphosphotyrosine purification.
Antibody shift assay, BN-PAGE, and SDS-PAGE.
The antibody or Fab fragment amounts indicated were added to the samples (15 to 20 μl) and incubated for 30 min on ice before BN-PAGE loading. When >2 μl of antibody had to be added (because of low antibody concentrations), the antibody was dialyzed against BN buffer to prevent introducing an excess of NaCl. Samples were separated by BN-PAGE (5.5–15%) at 4°C as previously described (19, 49). Ferritin was used as the marker protein in its 24-mer, 48-mer, and 72-mer forms (f1, 440 kD; f2, 880 kD; and f3, 1,320 kD). SDS-PAGE and Western blotting were performed using standard protocols.
Immunogold detection of surface TCR by label fracture and electron microscopy.
2B4 cells, untreated or treated with 5 mM MβCD at 37°C for 30 min in RPMI 1640 were fixed with ice-cold 2% paraformaldehyde in PBS for 30 min at 0°C and washed with RPMI 1640. Cells were incubated with 20 μg/ml 145-2C11 or H57-597 antibodies for 30 min at 0°C, followed by 10 nm gold-conjugated protein A (vol/vol, 1:100; Sigma-Aldrich). Samples were adsorbed on mica sheets pretreated with 100 μg/ml polylysine and kept at 0°C for 90 min. After washing, 0.1% (wt/vol) glutaraldehyde was added for 20 min. The samples were processed for label fracture and metal replication as described previously (50). A sandwich was made with another mica sheet, fast frozen in liquid ethane (104 °C/s), and transferred into a freeze fracture unit (BAF 060; BAL-TEC). Etching was performed for 15 min after switching temperatures from –150 to –90°C. Metal replicas of the exposed surfaces were obtained by evaporating 2 nm of platinum with an electron gun at an angle of 45° and 20 nm of carbon with an electron gun at an angle of 90°. Replicas were floated in commercial bleach and incubated overnight for the digestion of cellular material. After washing, replicas were picked up in uncoated copper electron microscopy grids and examined in a transmission electron microscope (1200-EX II; JEOL) operating at 100 kV. Individual and groups of gold particles were counted in 20 cells per sample. Human PBMCs and CH7C17 cells were prepared for label fracture and electron microscopy as above, except that OKT3 was used for T cell staining after fixation in paraformaldehyde.
CD69 up-regulation and surface iodination.
105 DAP-DR1 or DCEK APCs were incubated overnight with the concentrations of the HA307-319 peptide indicated in the figures. To stimulate the CH7C17 T cell line, the supernatant of the DAP-DR1 cells was removed and 105 CH7C17 cells were added for 24 h at 37°C. Cells were stained with PE-labeled anti-CD69 antibodies (1:100), washed, and analyzed with a FACSCalibur (Becton Dickinson). Surface iodination was performed using the lactoperoxidase method as described previously (51).
Online supplemental material.
Fig. S1 shows a two-dimensional BN/SDS-PAGE electrophoresis of TCR isolated from flag CD3ɛ-transfected Jk cells immunoblotted with anti-TCRβ, anti-flag, and anti-ζ antibodies. Fig. S2 shows a gel shift assay on TCR isolated from 2B4 cells and incubated with different doses of a Fab fragment of anti-TCRβ antibody H57-597. Fig. S3 shows a BN-PAGE of TCR extracted from 2B4 and Jk cells with the limiting concentrations of Brij96. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20042155/DC1.
Acknowledgments
We thank Drs. M. Alonso, K. Karjalainen, G. Siegers, and M. Toribio for critical readings of the manuscript. We are indebted to Dr. M. Reth for continuous discussion and support and to Maite Rejas for technical assistance.
This work was supported by grant SAF2002-03589 from the Centro de Investigación Científica y Tecnólogica and grant 08.3/0030/2001 from the Comunidad de Madrid (both to B. Alarcon), and by German-Spanish joint grants HA20020094 and D/0232957 from the Deutscher Akademischer Austausch Dienst. W.W.A. Schamel was in part supported by Marie Curie individual fellowship HPMF-CT-1999-00177 from the European Union and in part by Emmy Noether grant SCHA 976/1 from the Deutsche Forschungsgemeinschaft. The institutional support of Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa is also acknowledged.
The authors have no conflicting financial interests.
Abbreviations used: BCR, B cell antigen receptor; BN, Blue Native; Jk, Jurkat; MβCD, methyl-β-cyclodextrin; NIP, iodo-NP; NP, nitro-hydroxy-phenylacetate; MHCp, MHC peptide; PCC, pigeon cytochrome c; sc, single chain.
References
- 1.Germain, R.N. 2001. The T cell receptor for antigen: signaling and ligand discrimination. J. Biol. Chem. 276:35223–35226. [DOI] [PubMed] [Google Scholar]
- 2.Malissen, B. 2003. An evolutionary and structural perspective on T cell antigen receptor function. Immunol. Rev. 191:7–27. [DOI] [PubMed] [Google Scholar]
- 3.Alarcon, B., D. Gil, P. Delgado, and W.W. Schamel. 2003. Initiation of TCR signaling: regulation within CD3 dimers. Immunol. Rev. 191:38–46. [DOI] [PubMed] [Google Scholar]
- 4.Abastado, J.P., Y.C. Lone, A. Casrouge, G. Boulot, and P. Kourilsky. 1995. Dimerization of soluble major histocompatibility complex–peptide complexes is sufficient for activation of T cell hybridoma and induction of unresponsiveness. J. Exp. Med. 182:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boniface, J.J., J.D. Rabinowitz, C. Wulfing, J. Hampl, Z. Reich, J.D. Altman, R.M. Kantor, C. Beeson, H.M. McConnell, and M.M. Davis. 1998. Initiation of signal transduction through the T cell receptor requires the multivalent engagement of peptide/MHC ligands. Immunity. 9:459–466. (published erratum appears in Immunity. 1998. 9:891) [DOI] [PubMed] [Google Scholar]
- 6.Cochran, J.R., T.O. Cameron, and L.J. Stern. 2000. The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity. 12:241–250. [DOI] [PubMed] [Google Scholar]
- 7.Delon, J., N. Bercovici, R. Liblau, and A. Trautmann. 1998. Imaging antigen recognition by naive CD4+ T cells: compulsory cytoskeletal alterations for the triggering of an intracellular calcium response. Eur. J. Immunol. 28:716–729. [DOI] [PubMed] [Google Scholar]
- 8.Gil, D., W.W. Schamel, M. Montoya, F. Sanchez-Madrid, and B. Alarcon. 2002. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 109:901–912. [DOI] [PubMed] [Google Scholar]
- 9.Kjer-Nielsen, L., C.S. Clements, A.W. Purcell, A.G. Brooks, J.C. Whisstock, S.R. Burrows, J. McCluskey, and J. Rossjohn. 2003. A structural basis for the selection of dominant alphabeta T cell receptors in antiviral immunity. Immunity. 18:53–64. [DOI] [PubMed] [Google Scholar]
- 10.Reiser, J.B., C. Gregoire, C. Darnault, T. Mosser, A. Guimezanes, A.M. Schmitt-Verhulst, J.C. Fontecilla-Camps, G. Mazza, B. Malissen, and D. Housset. 2002. A T cell receptor CDR3beta loop undergoes conformational changes of unprecedented magnitude upon binding to a peptide/MHC class I complex. Immunity. 16:345–354. [DOI] [PubMed] [Google Scholar]
- 11.Ding, Y.H., B.M. Baker, D.N. Garboczi, W.E. Biddison, and D.C. Wiley. 1999. Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signals are nearly identical. Immunity. 11:45–56. [DOI] [PubMed] [Google Scholar]
- 12.de la Hera, A., U. Muller, C. Olsson, S. Isaaz, and A. Tunnacliffe. 1991. Structure of the T cell antigen receptor (TCR): two CD3ɛ subunits in a functional TCR–CD3 complex. J. Exp. Med. 173:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blumberg, R.S., B. Alarcon, J. Sancho, F.V. McDermott, P. Lopez, J. Breitmeyer, and C. Terhorst. 1990. Assembly and function of the T cell antigen receptor. Requirement of either the lysine or arginine residues in the transmembrane region of the alpha chain. J. Biol. Chem. 265:14036–14043. [PubMed] [Google Scholar]
- 14.Punt, J.A., J.L. Roberts, K.P. Kearse, and A. Singer. 1994. Stoichiometry of the T cell antigen receptor (TCR) complex: each TCR–CD3 complex contains one TCRα, one TCRβ, and two CD3ɛ chains. J. Exp. Med. 180:587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Call, M.E., J. Pyrdol, M. Wiedmann, and K.W. Wucherpfennig. 2002. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 111:967–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez-Miguel, G., B. Alarcon, A. Iglesias, H. Bluethmann, M. Alvarez-Mon, E. Sanz, and A. de la Hera. 1999. Multivalent structure of an alphabeta T cell receptor. Proc. Natl. Acad. Sci. USA. 96:1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Exley, M., T. Wileman, B. Mueller, and C. Terhorst. 1995. Evidence for multivalent structure of T-cell antigen receptor complex. Mol. Immunol. 32:829–839. [DOI] [PubMed] [Google Scholar]
- 18.Reth, M. 2001. Oligomeric antigen receptors: a new view on signaling for the selection of lymphocytes. Trends Immunol. 22:356–360. [DOI] [PubMed] [Google Scholar]
- 19.Schamel, W.W., and M. Reth. 2000. Monomeric and oligomeric complexes of the B cell antigen receptor. Immunity. 13:5–14. [DOI] [PubMed] [Google Scholar]
- 20.Minami, Y., L.E. Samelson, and R.D. Klausner. 1987. Internalization and cycling of the T cell antigen receptor. Role of protein kinase C. J. Biol. Chem. 262:13342–13347. [PubMed] [Google Scholar]
- 21.De Waal Malefyt, R., B. Alarcon, H. Yssel, J. Sancho, A. Miyajima, C.P. Terhorst, H. Spits, and J.E. De Vries. 1989. Introduction of T cell receptor (TCR)-alpha cDNA has differential effects on TCR-gamma delta/CD3 expression by PEER and Lyon-1 cells. J. Immunol. 142:3634–3642. [PubMed] [Google Scholar]
- 22.Salmeron, A., F. Sanchez-Madrid, M.A. Ursa, M. Fresno, and B. Alarcon. 1991. A conformational epitope expressed upon association of CD3-epsilon with either CD3-delta or CD3-gamma is the main target for recognition by anti-CD3 monoclonal antibodies. J. Immunol. 147:3047–3052. [PubMed] [Google Scholar]
- 23.Reay, P.A., K. Matsui, K. Haase, C. Wulfing, Y.H. Chien, and M.M. Davis. 2000. Determination of the relationship between T cell responsiveness and the number of MHC-peptide complexes using specific monoclonal antibodies. J. Immunol. 164:5626–5634. [DOI] [PubMed] [Google Scholar]
- 24.Irvine, D.J., M.A. Purbhoo, M. Krogsgaard, and M.M. Davis. 2002. Direct observation of ligand recognition by T cells. Nature. 419:845–849. [DOI] [PubMed] [Google Scholar]
- 25.Holler, P.D., and D.M. Kranz. 2003. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 18:255–264. [DOI] [PubMed] [Google Scholar]
- 26.Hemmer, B., I. Stefanova, M. Vergelli, R.N. Germain, and R. Martin. 1998. Relationships among TCR ligand potency, thresholds for effector function elicitation, and the quality of early signaling events in human T cells. J. Immunol. 160:5807–5814. [PubMed] [Google Scholar]
- 27.Martin, S., and M.J. Bevan. 1998. Transient alteration of T cell fine specificity by a strong primary stimulus correlates with T cell receptor down-regulation. Eur. J. Immunol. 28:2991–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hewitt, C.R., J.R. Lamb, J. Hayball, M. Hill, M.J. Owen, and R.E. O'Hehir. 1992. Major histocompatibility complex independent clonal T cell anergy by direct interaction of Staphylococcus aureus enterotoxin B with the T cell antigen receptor. J. Exp. Med. 175:1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drevot, P., C. Langlet, X.J. Guo, A.M. Bernard, O. Colard, J.P. Chauvin, R. Lasserre, and H.T. He. 2002. TCR signal initiation machinery is pre-assembled and activated in a subset of membrane rafts. EMBO J. 21:1899–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun, Z.Y., S.T. Kim, I.C. Kim, A. Fahmy, E.L. Reinherz, and G. Wagner. 2004. Solution structure of the CD3epsilondelta ectodomain and comparison with CD3epsilongamma as a basis for modeling T cell receptor topology and signaling. Proc. Natl. Acad. Sci. USA. 101:16867–16872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uhlin, M., M.G. Masucci, and V. Levitsky. 2003. Pharmacological disintegration of lipid rafts decreases specific tetramer binding and disrupts the CD3 complex and CD8 heterodimer in human cytotoxic T lymphocytes. Scand. J. Immunol. 57:99–106. [DOI] [PubMed] [Google Scholar]
- 32.Jacobs, H. 1997. Pre-TCR/CD3 and TCR/CD3 complexes: decamers with differential signalling properties? Immunol. Today. 18:565–569. [PubMed] [Google Scholar]
- 33.Bray, D., M.D. Levin, and C.J. Morton-Firth. 1998. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 393:85–88. [DOI] [PubMed] [Google Scholar]
- 34.Sourjik, V., and H.C. Berg. 2004. Functional interactions between receptors in bacterial chemotaxis. Nature. 428:437–441. [DOI] [PubMed] [Google Scholar]
- 35.Valitutti, S., S. Muller, M. Cella, E. Padovan, and A. Lanzavecchia. 1995. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 375:148–151. [DOI] [PubMed] [Google Scholar]
- 36.Cochran, J.R., D. Aivazian, T.O. Cameron, and L.J. Stern. 2001. Receptor clustering and transmembrane signaling in T cells. Trends Biochem. Sci. 26:304–310. [DOI] [PubMed] [Google Scholar]
- 37.Karjalainen, K. 1994. High sensitivity, low affinity–paradox of T-cell receptor recognition. Curr. Opin. Immunol. 6:9–12. [DOI] [PubMed] [Google Scholar]
- 38.Kropshofer, H., S. Spindeldreher, T.A. Rohn, N. Platania, C. Grygar, N. Daniel, A. Wolpl, H. Langen, V. Horejsi, and A.B. Vogt. 2002. Tetraspan microdomains distinct from lipid rafts enrich select peptide-MHC class II complexes. Nat. Immunol. 3:61–68. [DOI] [PubMed] [Google Scholar]
- 39.Triantafilou, K., M. Triantafilou, and N. Fernandez. 2000. Molecular associations and microdomains in antigen-presenting cell-T-cell interactions. Crit. Rev. Immunol. 20:359–373. [PubMed] [Google Scholar]
- 40.Anderson, H.A., E.M. Hiltbold, and P.A. Roche. 2000. Concentration of MHC class II molecules in lipid rafts facilitates antigen presentation. Nat. Immunol. 1:156–162. [DOI] [PubMed] [Google Scholar]
- 41.Krogsgaard, M., Q.J. Li, C. Sumen, J.B. Huppa, M. Huse, and M.M. Davis. 2005. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 434:238–243. [DOI] [PubMed] [Google Scholar]
- 42.Wulfing, C., C. Sumen, M.D. Sjaastad, L.C. Wu, M.L. Dustin, and M.M. Davis. 2002. Costimulation and endogenous MHC ligands contribute to T cell recognition. Nat. Immunol. 3:42–47. [DOI] [PubMed] [Google Scholar]
- 43.Janes, P.W., S.C. Ley, and A.I. Magee. 1999. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 147:447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Horejsi, V. 2003. The roles of membrane microdomains (rafts) in T cell activation. Immunol. Rev. 191:148–164. [DOI] [PubMed] [Google Scholar]
- 45.Montixi, C., C. Langlet, A.M. Bernard, J. Thimonier, C. Dubois, M.A. Wurbel, J.P. Chauvin, M. Pierres, and H.T. He. 1998. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 17:5334–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xavier, R., T. Brennan, Q. Li, C. McCormack, and B. Seed. 1998. Membrane compartmentation is required for efficient T cell activation. Immunity. 8:723–732. [DOI] [PubMed] [Google Scholar]
- 47.Fahmy, T.M., J.G. Bieler, M. Edidin, and J.P. Schneck. 2001. Increased TCR avidity after T cell activation: a mechanism for sensing low-density antigen. Immunity. 14:135–143. [PubMed] [Google Scholar]
- 48.Drake, D.R., III, and T.J. Braciale. 2001. Cutting edge: lipid raft integrity affects the efficiency of MHC class I tetramer binding and cell surface TCR arrangement on CD8+ T cells. J. Immunol. 166:7009–7013. [DOI] [PubMed] [Google Scholar]
- 49.Schagger, H., and G. von Jagow. 1991. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199:223–231. [DOI] [PubMed] [Google Scholar]
- 50.Risco, C., and P. Pinto da Silva. 1998. The fracture-flip technique reveals new structural features of the Escherichia coli cell wall. J. Microsc. 189:213–218. [DOI] [PubMed] [Google Scholar]
- 51.Alarcon, B., S.C. Ley, F. Sanchez-Madrid, R.S. Blumberg, S.T. Ju, M. Fresno, and C. Terhorst. 1991. The CD3-gamma and CD3-delta subunits of the T cell antigen receptor can be expressed within distinct functional TCR/CD3 complexes. EMBO J. 10:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]