

Abstract
Class switch recombination (CSR) occurs by an intrachromosomal deletion whereby the IgM constant region gene (Cμ) is replaced by a downstream constant region gene. This unique recombination event involves formation of double-strand breaks (DSBs) in immunoglobulin switch (S) regions, and requires activation-induced cytidine deaminase (AID), which converts cytosines to uracils. Repair of the uracils is proposed to lead to DNA breaks required for recombination. Uracil DNA glycosylase (UNG) is required for most CSR activity although its role is disputed. Here we use ligation-mediated PCR to detect DSBs in S regions in splenic B cells undergoing CSR. We find that the kinetics of DSB induction corresponds with AID expression, and that DSBs are AID- and UNG-dependent and occur preferentially at G:C basepairs in WRC/GYW AID hotspots. Our results indicate that AID attacks cytosines on both DNA strands, and staggered breaks are processed to blunt DSBs at the initiating ss break sites. We propose a model to explain the types of end-processing events observed.
Upon activation by antigen and accessory signals, naive IgM+IgD+ B cells undergo Ig class switching, which results in expression of a different heavy chain constant (CH) region gene, while maintaining expression of the same variable region gene. Because the CH region determines the antibody effector function, class switching allows the humoral immune system to respond adaptively to a variety of different infectious organisms to produce the best antibody for elimination of each pathogen. Class switching is mediated by DNA recombination between switch (S) region sequences residing 5′ to each CH gene, except Cδ (1), and is mediated by a type of nonhomologous end-joining (NHEJ; reference 2).
Class switch recombination (CSR) is completely dependent on activation-induced cytidine deaminase (AID; references 3, 4). AID is believed to initiate the formation of single-strand DNA breaks that are converted to the double-strand breaks (DSBs) that are necessary for the intrachromosomal deletions that result in CSR. γH2AX foci, which are indicative of DSBs, have been observed at the Ig locus in cells undergoing CSR, but not in cells deficient in AID (5); AID-dependent DSBs have been detected in Ig S regions by ligation-mediated (LM)-PCR in mice and humans (6, 7). However, the mechanism and site(s) of break formation are unknown. Most data support a model in which AID initiates DNA breaks by directly deaminating cytosines in S region DNA, thus generating uracils (8–12). Uracil is hypothesized to then be excised by uracil DNA glycosylase (UNG), leaving an abasic site (8, 13). Abasic sites can be recognized by an apurinic/apyrimidic (AP) endonuclease (APEX1 or 2) that nicks the DNA backbone (8). CSR is reduced severely in the absence of UNG (13, 14), but the function of UNG has been called into question. A recent study by Begum et al. (15) concluded that although UNG is required for CSR, the glycosylase activity of UNG is not required for γH2AX foci formation. It was suggested that UNG is not required for generating DNA breaks in S regions, but has another function in CSR. This study used UNG mutants that retain low levels of activity (16); thus, it is possible that a low level of uracil glycosylase activity is sufficient for CSR. It has not been reported whether AP endonuclease is involved in CSR.
Although the presence of uracils in Ig S regions has not been demonstrated directly, most data are in agreement with the direct deamination of cytosines in DNA by AID. AID has been shown to deaminate single-stranded (ss), but not double-stranded (ds), DNA (9–12) in vitro. Transcription of the S region is required for CSR (17, 18), and current results suggest that S region transcription generates an ss DNA substrate for AID (9, 19). Apparently as a result of the G-rich nature of the RNA transcript, RNA transcribed across S regions stably associates with the transcribed (bottom) DNA strand, forming R-loops that leave the nontranscribed (top) strand unpaired and vulnerable to attack by AID (20–22). However, it is not clear how nicks on both strands are made in order to generate DSBs. The bottom strand of the mouse Sμ region has ∼2.5-fold more C residues than the top strand, which could increase targeting of AID to the bottom strand if it were ss. Purified recombinant AID can deaminate both DNA strands within a supercoiled plasmid, which suggests that supercoiling might generate DNA with sufficient ss structure to form an AID substrate on both strands (23).
Understanding how the nicks are introduced into S regions and converted into DSBs is essential for understanding the mechanism of CSR. Previous investigators have used LM-PCR to detect S region breaks, but with varying results. Since the discovery of AID, two groups have detected AID-dependent blunt and staggered DSBs in Sμ segments in human peripheral blood B cells (6) and in mouse splenic B cells activated in culture (7). In addition, ss breaks were detected on the transcribed strand of Sα from the B lymphoma CH12F3-2, a cell line that switches inducibly to IgA (24). However, this group did not detect ss breaks on the nontranscribed strand, nor blunt or staggered DSBs, nor could they demonstrate AID-dependence of the breaks. There is also inconsistency in the sequences at which DSBs were detected among the studies (6, 7, 25), and the ss breaks detected in the B lymphoma occurred preferentially at C residues but not at the known AID hotspot, WRC (26–28).
To test the current model of AID-induced DSBs, we decided to investigate the dependence of S-region DSBs on AID and UNG, and to determine the site and structure of the breaks. We identify DNA breaks induced in Ig S regions during antibody CSR in mouse splenic B cells and show that they are dependent on AID and UNG. We show that the breaks are induced simultaneously with AID protein, and demonstrate that the blunt DSBs occur preferentially at G:C bp in WRC/GYW AID hotspots. The finding that DSBs in WT cells are almost always at G:C bp strongly implicates the glycosylase activity of UNG in DSB formation, whereas the few breaks detected in the absence of UNG do not occur preferentially at G:C bp; this suggests that they are not introduced by way of an alternative uracil glycosylase. The results allow us to conclude that AID-instigated breaks occur on both DNA strands, and then end-processing subsequently generates blunt DSBs at the sites of the ss breaks. We propose a model to explain the types of end-processing events observed, and suggest that certain types of end-processing are preferred.
RESULTS
To determine when AID-dependent breaks might appear, we examined the time course of induction of AID protein in splenic B cells induced to undergo CSR. B cells from WT and AID-deficient mice were stimulated with LPS plus IL-4 or anti-δ-dextran for various time periods before making cell extracts. BLyS/BAFF was added to all B cell cultures to reduce cell death (29), and to increase cell proliferation and CSR (Schmidt, M.R. and Woodland, R.T., unpublished data). AID protein was not detectable in resting splenic B cells and was barely detectable in the cytoplasm at 24 h, 26 h, and 30 h, but was greatly induced in cytoplasm and nuclei 48 h after activation; levels were even greater at 72 h (Fig. 1 A). AID was induced equally by LPS plus IL-4 or LPS plus anti-δ dextran, less well by LPS alone, and not at all by IL-4 plus anti-δ dextran (Fig. 1 B). These data are consistent with the finding that AID mRNA is not detected until 2 d after splenic B cell activation (reference 30 and unpublished data).
Figure 1.

AID protein is up-regulated 48 h after stimulation of cells to undergo CSR. Western blots of 40 μg cytoplasmic and 40 μg nuclear extracts prepared from WT or aid − / − splenic B cells cultured as indicated. Extracts from freshly isolated resting B cells are shown in the left-most lanes. The four blots were incubated with anti-AID antiserum simultaneously, and exposed to film simultaneously. A 42-kD protein in the nuclear extracts that cross-reacts with the AID antibody is shown to demonstrate nearly equivalent loading of protein in the different lanes. The 26-kD protein in cytoplasmic extracts is likely to be a cross-reacting protein, because a band this size also is observed sometimes in aid −/− cells (unpublished data). (A) Time course showing induction of AID in WT cells treated with LPS plus IL-4. (B) Blots of cytoplasmic and nuclear extracts from cells treated under the indicated conditions for 48 h. All conditions included BLyS.
We then used LM-PCR to detect blunt DSBs in Sμ. Resting splenic B cells from WT and AID-deficient mice were activated to switch for various times to determine the onset of breaks and whether they were dependent on AID. Genomic DNA was isolated, ligated to linker, and LM-PCR was performed using primers specific for Sμ or Cμ. A few breaks were detected at early time points in WT and AID-deficient B cells, but at 48 h, abundant breaks were detected in WT B cells stimulated with LPS plus IL-4 or with LPS plus anti-δ-dextran. This is consistent with the appearance of AID protein at 48 h (Fig. 2 A, 5′ end of Sμ; Fig. S1, 3′ end of Sμ, available at http://www.jem.org/cgi/content/full/jem.20050872/DC1). Very few breaks are observed in B cells from AID-deficient mice at this time point (Fig. 2 A, bottom). The DSBs detected are specific to cells undergoing CSR, because cells stimulated to proliferate with IL-4 plus anti-δ-dextran divide rapidly, but do not induce AID or undergo CSR (Fig. 1 B and not depicted); these cells do not develop AID-dependent DSBs in Sμ (Fig. 2 A). Very few breaks are detected in the Cμ gene, which is not used in CSR, and the few breaks observed are not AID-dependent (Fig. 2 B). AID-dependent DSBs also are introduced into Sγ3 segments at 48 h (Fig. 2 C). These data clearly indicate that AID is required for the introduction of blunt DSBs in the Sμ and Sγ3 regions during CSR.
Figure 2.
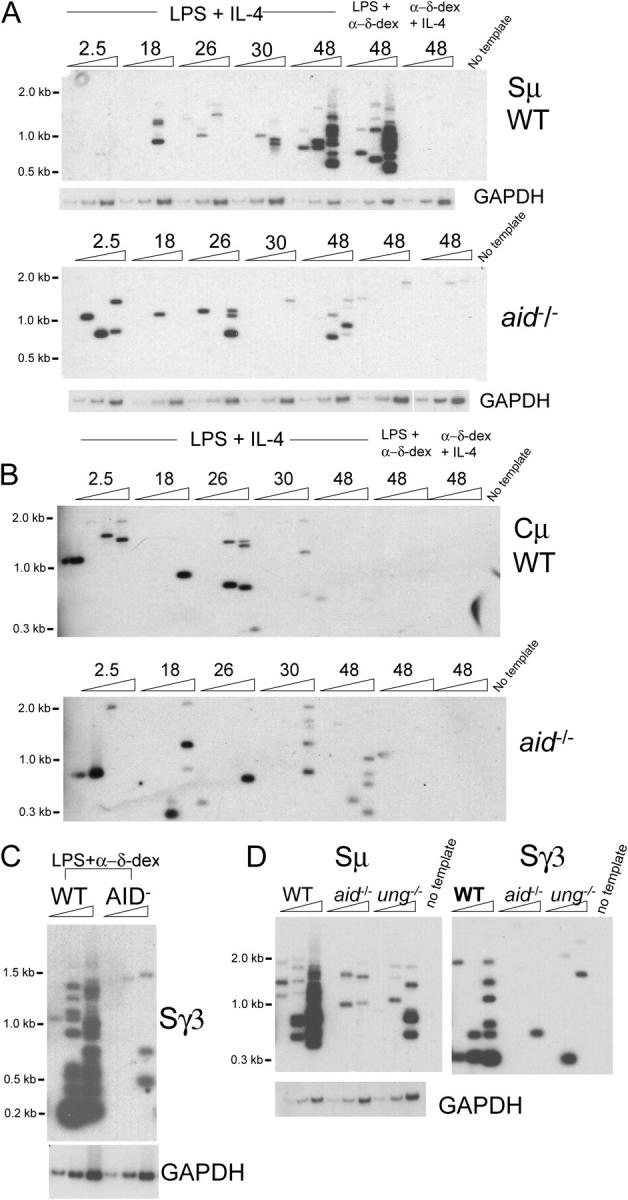
DSBs detected by LM-PCR are induced in Sμ and Sγ3 segments in wild type (WT) but not in aid −/− splenic B cells 48 h after induction of CSR. (A) Sμ LM-PCR products (amplified with the 5′Sμ primer) from WT and aid −/− cells treated as indicated for the indicated hours were blotted and hybridized with the Sμ probe. The PCR amplification of the GAPDH gene are shown below the blots as an internal control for template input: threefold dilutions of 1630 cell equivalents. (B) Cμ LM-PCR products from WT cells and aid −/− B cells detected with the Cμ probe. These blots are from the same experiment shown in (A). (C) Sγ3 LM-PCR products from WT and aid −/− B cells treated for 48 h with LPS plus anti-δ dextran detected with the Sγ3 probe. To detect Sγ3 breaks, the template input was increased by threefold (threefold dilutions of 4890 cell equivalents) over that used to detect Sμ and Cμ breaks. (D) Most DSBs in Sμ and Sγ3 are UNG-dependent. LM-PCR products from splenic B cells activated for 48 h with LPS plus IL-4 detected with the Sμ5′ probe or Sγ3 probe; threefold dilutions of 2770 cell equivalents. The finding of AID-dependent DSBs is reproducible and was obtained in all six of the six independent experiments performed, always tested in two or more ligations.
B cells from ung −/− mice have been shown to switch very poorly in culture (13, 15); however, whether UNG is required for introduction of S region breaks is controversial (14, 15). To examine this question, we activated ung −/− splenic B cells to undergo CSR, and analyzed surface Ig expression and blunt DSBs as described above. Consistent with the involvement of UNG in excision of uracil bases during CSR, we found a great reduction in CSR (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20050872/DC1) and the numbers of Sμ and Sγ3 DNA breaks detected in ung −/− B cells after 48 h of activation (Fig. 2 D).
Nucleotide sites of DNA breaks
The AID-UNG-APEX model for introduction of DNA breaks into S regions predicts that the initiating ss breaks will occur at dC residues in the AID hotspot motif WRC (where W = A or T, and R = purine). However, because blunt DSBs require end-processing from the ss nicks, it is unclear whether DSBs would occur at one of the original ss break sites. To address this question, we cloned the blunt DSB fragments, and analyzed the nucleotides at which they occurred. The base indicated in the first column of Table I refers to the site of the break (i.e., the nucleotide that is deleted and replaced by the linker [reading the top strand]). As shown in Table I, 89% of the breaks in the Sμ region from WT cells occurred at G:C bp—mostly at G residues; this is a highly significant preference in comparison with the sequence itself. Similarly, 88% of Sγ3 breaks occurred at G residues. By contrast, 67% of the Sμ breaks cloned from aid −/− cells were at G:C bp; this is not different from random, and is similar to the few Cμ breaks cloned from WT cells.
Table I.
Blunt DSBs in Sμ and Sγ3 from WT splenic B cells occur preferentially at G:C bp and at GYW/WRC AID hotspots
| Sμ
|
Sγ3
|
Cμ
|
||||||
|---|---|---|---|---|---|---|---|---|
| nt at break | WT | aid −/− | ung −/− | Sequencea | WT | Sequencea | WT | Sequencea |
| % (nbr) | % (nbr) | % (nbr) | % | % (nbr) | % | % (nbr) | % | |
| G | 83.9 (47) | 62.5 (15) | 53 (9) | 40.7 | 84 (21) | 48.7 | 57 (4) | 23.1 |
| C | 5.4 (3) | 4.2 (1) | 5.8 (1) | 16.1 | 4.0 (1) | 14.7 | 0 | 29.1 |
| A | 7.1 (4) | 20.8 (5) | 35.3 (6) | 21.4 | 8.0 (2) | 18.3 | 43 (3) | 25.4 |
| T | 3.6 (2) | 12.5 (3) | 5.8 (1) | 21.9 | 4.0 (1) | 18.4 | 0 | 22.4 |
| Total | 56 breaks | 24 breaks | 17 breaks | 2,000 nt | 25 breaks | 1,460 nt | 7 breaks | 2,058 nt |
| G+C | 89.3 | 66.7 | 58.8 | 56.8 | 88.8 | 63.4 | 57 | 52.2 |
| p-valueb | <0.001 | NS | NS | 0.039 | NS | |||
| Hotspots | 41.1 | 4.2 | 11.8 | 23.2c | 40 | 14.8 | 14.3 | 11.5c |
| p-valueb | 0.049 | NS | NS | <0.001 | NS | |||
The distribution of nucleotides in the genomic sequence analyzed.
Significance of difference from random.
Percentage of nucleotides in genomic sequence that are in G:C bp at the underlined nucleotide in WRC/GYW hotspots.
nbr, number of breaks.
Additionally, 41 and 40% of the Sμ and Sγ3 breaks, respectively, from WT cells occurred at the underlined C or G in WRC/GYW AID hotspots; this is a significant increase relative to random expectations (23.2 and 14.8%, respectively; Table I). These data are consistent with the level of preference for hotspots by AID in vitro and in vivo during somatic hypermutation of antibody variable region genes (26–28, 31). We also determined the sequences of 26 breaks by amplifying with a primer from the 3′ side of Sμ. Consistent with the break sites identified with the 5′ primer, 77% occurred at G:C bp, and 53.9% occurred in AID hotspots. The Sμ breaks cloned from aid −/− cells and Cμ breaks from WT cells did not occur at WRC/GYW motifs (Table I). These results are entirely consistent with the introduction of breaks into S regions by the AID-UNG-APEX pathway, and also suggest that the blunt DSBs are located at sites of ss nicks instigated by AID.
The Sμ break sites amplified with the 5′Sμ primer from WT cells are indicated on the Sμ sequence shown in Fig. 3. They are found at sites across the Sμ region, including the region 5′ to the tandem repeats, although most are found within the tandem repeat region. Breaks occurred at the same site very rarely, and because of the possibility of PCR contamination from previously cloned breaks, they were eliminated from our analysis.
Figure 3.

Mouse Sμ sequence with DSB sites from WT cells indicated. These breaks were obtained by LM-PCR using the indicated primer (5′Smu) located at the 5′ end of Sμ sequence. The arrows point to the base that was deleted, where the linker is ligated. Breaks occurring at G:C bp at the underlined base in WRC/GYW are indicated by bold arrows. Other breaks are indicated with gray arrows. Early experiments used the Smu probe (nt 453–480) to detect the cloned breaks, and thus, missed some of the breaks occurring upstream of the tandem repeats, which start at ∼820 in this sequence. Therefore, the distribution of break sites is not entirely representative. Furthermore, although additional long break fragments due to breaks occurring at the 3′ end of this segment were cloned, often the break site could not be determined because of difficulty in alignment with the tandem Sμ repeats. We did not include any break sites cloned more than once, because of concerns about amplification of previously cloned segments.
The low frequency of CSR in ung −/− B cells suggests that a few breaks can be introduced into S regions independently of UNG (13). To examine how these breaks are introduced, we determined the sites of Sμ DSBs from ung −/− cells, and found that they do not occur at G:C bp in WRC/GYW hotspots (Table I). This suggests that these few breaks are not introduced by an alternative uracil DNA glycosylase, followed by APEX action, because this would introduce DNA breaks at G:C bp in hotspots. Instead, it seems to be more likely that they are introduced by a pathway that introduces ss breaks at sites away from the initial lesion (e.g., nucleotide excision repair).
Most breaks are staggered
The AID-UNG-APEX pathway would be predicted to introduce ss breaks and staggered DSBs, rather than blunt DSBs. We determined whether treatment of DNA with T4 DNA polymerase to fill-in or excise ss overhangs before ligation of the linker-primer would increase the number of DNA breaks detected; we found that, indeed, it did increase the number of breaks detected (Fig. 4 A). Thus, similar to previous results (7), it seems that most Sμ breaks are staggered. Similar to the sites of blunt breaks, we found that 85% of the 26 cloned staggered breaks occurred at G:C bp.
Figure 4.

Most DSBs are staggered, and a model for conversion of staggered DSBs to blunt DSBs is shown. (A) Most DSB breaks are staggered, as demonstrated by an increase in Sμ LM-PCR products detectable after treatment of activated B cell DNA with 0.5 U T4 DNA polymerase and 200 μM dNTP at 11°C for 20 min before linker-ligation. Shown are threefold dilutions of 1,085 cell equivalents. The same DNA samples were analyzed plus and minus T4 DNA polymerase. (B) A model for the types of end-processing that might result in the pattern of DSBs observed after AID-UNG instigated ss break formation. The Cs on the flaps represent nt attacked by AID, and the G:C bp in the boxes indicates the first nucleotide of the deleted segment. This diagram is for DSBs detected using a primer at the 5′ end of the S region, as used in the experiments reported in this study, except in Fig. S1. Because of the finding that the blunt DSBs occur preferentially at G on the nontranscribed strand, it is likely that 5′ overhangs mostly are converted to blunt DSBs by fill-in DNA synthesis (left-side pathway), rather than by exonuclease or endonuclease activities (middle pathway). 3′ overhangs cannot be filled in and might be removed by exonuclease 1 and ERCC1-XPF (right-side pathway), both of which are known to participate in CSR (33, 34). This model also predicts that break sites detected by a primer located at the 3′ end of Sμ would occur preferentially at C on the top strand relative to the sequence itself. This is what we observed, because the G:C ratio of break sites detected with the 3′ primer (1.0) is much less than that for the 5′ primer (15.7). One of the many puzzles that remain is what removes the deoxyribose phosphate group left after AP endonuclease acts. To obtain blunt-end ligation at G:C bp, this group must be removed from the 5′ ends of the breaks.
DISCUSSION
The finding that the blunt DSBs are almost all at G:C bp was surprising. Staggered ss nicks resulting from AID deamination of C residues on opposite strands would require processing of DNA ends to generate blunt DSBs, and it seemed likely that end-processing activity would move the site of the break away from the original ss lesion. The additional finding that breaks occur preferentially at AID hotspots further demonstrates that the initial AID-dependent ss breaks become the site of blunt DSBs. These blunt DSBs would provide a favorable substrate for NHEJ. The lack of preference for S–S junctions using microhomologies is consistent with the use of blunt DSBs in recombination (32). Although some S–S junctions do show short microhomologies, they do not seem to be favored (i.e., their occurrence does not seem to be increased significantly relative to random expectations).
Fig. 4 B presents a model for how ss breaks and staggered DSBs that are introduced by the AID-UNG-APEX pathway could be converted to blunt DSBs. Because the blunt DSBs are detected at G residues on the nontranscribed strand, and AID attacks C residues, our data suggest that end-processing of the ss breaks could occur by fill-in DNA synthesis of 5′ overhangs (left-side pathway) and by removal of 3′ overhangs by exonuclease I and the strucutre-specific endonuclease ERCC1-XPF (right-side pathway). The lack of blunt DSBs at top-strand C residues suggests that 5′ overhangs are converted infrequently to DSBs by exonuclease or endonuclease activity (middle pathway). Exo1 and ERCC1-XPF are known to be involved in CSR (33, 34).
Our data also clearly indicate that C residues on the transcribed strand are targeted by AID. Although it has been shown that AID preferentially deaminates cytosines on the top, nontranscribed strand of plasmid DNA in vitro or in Escherichia coli (9, 19, 28), it also was shown that cytosines on both DNA strands of a nontranscribed supercoiled plasmid can be deaminated equally in vitro (23). Because of the apparent bias in types of processing events observed, the relative frequency of targeting the transcribed and nontranscribed strands cannot be determined from the sites of the blunt breaks. Consistent with this bias, breaks at C residues are distributed more equally on the transcribed and nontranscribed strands after T4 DNA polymerase treatment (Sμ breaks: G:C ratio is 3.4 for staggered breaks and 15.7 for blunt breaks). The breaks detected after T4 DNA polymerase treatment include blunt breaks and those that require T4 DNA polymerase processing to become ligatable. Thus, the finding that the G:C ratio of these breaks approaches the ratio in the sequence itself (which is 2.5) suggests that breaks occur approximately equally on both strands. This further suggests that the bottom transcribed strand exists as ss DNA—accessible to AID—at least transiently during CSR. Yu et al. (22) suggested that this might occur when R-loops collapse out-of-register because of the presence of tandem repeats in S regions.
The finding that the breaks in ung −/− cells do not occur preferentially at G:C bp in AID hotspots is consistent with recent results which showed that overexpression or deletion of the uracil DNA glycosylases, SMUG-1 or MBD4, respectively, have no effect on CSR (35, 36). Although it has been shown that a double-deficiency of UNG and the mismatch repair protein Msh2 completely eliminates CSR (36), it is not clear how mismatch repair could introduce the initiating DNA breaks. ERCC1-XPF is the only endonuclease that is known to be recruited by Msh2 (as a heterodimer with Msh3) (37). ERCC1-XPF does not seem to function with Msh2 in CSR (34), and Msh3 does not seem to be involved in CSR (38, 39). Although two endonucleases, ERCC1-XPF and XPG, participate in the nucleotide excision pathway in mammals—each of which introduce ss DNA breaks at sites distal to the lesion itself—this pathway is not known to recognize dU residues or abasic sites in vivo (40). However, nucleotide excision repair might recognize DNA that is distorted by multiple lesions that might accumulate in the absence of UNG.
Our data support a model in which most CSR is initiated by introduction of ss DNA breaks into the transcribed and nontranscribed strands of the S region by AID and UNG, likely followed by APEX activity. Single-strand breaks (SSBs) are converted into blunt DSBs that end at the site of one of the original ss lesions. If the SSBs on opposite strands are near each other, they could form staggered DSBs spontaneously that could be recombined by NHEJ. However, if the SSBs are more distal than a few bp, they are not likely to form a DSB. We propose that end-processing sometimes converts these distal SSBs to the blunt DSBs detected by LM-PCR.
MATERIALS AND METHODS
Mice.
Mice deficient in AID were obtained from T. Honjo (Kyoto University, Kyoto, Japan) and were backcrossed to C57BL/6 mice for four generations. They were housed in the Institutional Animal Care and Use Committee–approved specific pathogen-free facility at the University of Massachusetts Medical School. WT littermates were used as controls for all experiments shown. Some WT S region DSB sequences were obtained from (C57BL/6 × FVB) F1 mice. There was no difference in the break sites between these F1 mice and C57BL/6 mice. UNG-deficient mice were obtained from T. Lindahl and D. Barnes (London Research Institute, London, England). The mice were bred and used under guidelines formulated by the University of Massachusetts Animal Care and Use Committee.
Antibodies.
Antibody to AID was induced in rabbits by immunization with the COOH-terminal peptide (with an added NH2-terminal C) CEVDDLRDAFRMLGF-OH. Antibody was purified by ammonium persulfate precipitation of IgG, followed by immunoadsorption to the AID COOH-terminal peptide coupled to a Pierce Sulfo-link column, following the manufacturer's specifications. Isotype-specific antibodies, conjugated to FITC or PE, were purchased from Southern Biotechnology Associates, Inc.
Western blotting.
Cells were lysed in 0.6% NP-40, pelleted, and supernatants were taken as cytoplasmic extract. The nuclear pellet was washed once with low-salt buffer and extracted in high-salt buffer (41). 40 μg of extracts were electrophoresed on 12% polyacrylamide gels. The four blots shown were incubated with anti-AID antiserum simultaneously, followed by goat anti–rabbit horseradish peroxidase (Southern Biotechnology Associates, Inc.) and Western Lightning PLUS chemiluminescence reagent (PerkinElmer), and then were autoradiographed simultaneously.
B cell isolation and culture.
Single cell suspensions from spleens of 2–5-mo-old mice were subject to Percoll gradient centrifugation. Small cells from the 60/70% interface were collected and depleted of CD43+ cells by MACS (Miltenyi Biotech GmbH). CD43− small B lymphocytes were cultured at 4 × 105/ml in six-well plates in RPMI 1640 (BioWhittaker), with 10% FCS (Hyclone), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from GIBCO BRL), and 5 × 10−5 M 2-mercaptoethanol (Sigma-Aldrich). All cultures contained LPS (50 μg/ml, Sigma-Aldrich)—except where indicated in Fig. 2—and BLyS (recombinant, 100 ng/ml; Human Genome Sciences). IL-4 (800 U/ml; provided by W. Paul, National Institutes of Health, Bethesda MD) and anti-δ-dextran (0.3 ng/ml; gift from C. Snapper, Uniformed Services University of Health Sciences, Bethesda MD) were added where indicated. BLyS was added to increase cell viability; it also increased proliferation as assayed by CFSE monitoring (unpublished data) and by thymidine uptake (Schmidt, M.R., personal communication). The frequency of CSR in the cultures also increased (e.g., CSR to IgG3 after 4 d of culture increased from 10.1% in its absence to 23.5% in its presence; the average increase in five experiments was 1.9 fold). BLyS, in the absence of LPS, did not induce AID (Fig. 1, C and D), DNA breaks (Fig. 2 A), or CSR in splenic B cells (not depicted).
Genomic DNA preparation.
After culture for the indicated time periods, viable cells were isolated by flotation on Ficoll/Hypaque gradients (δ = 1.09). 106 cells in 25 μl PBS were mixed with an equal volume of 1% low-melt agarose in 40 mM EDTA in 1.5 ml Eppendorf tubes and put on ice. Agarose plugs were overlaid with proteinase K (1 mg/ml; Sigma-Aldrich) in 50 mM Tris-Cl, pH 8, 20 mM EDTA, and 1% SDS and incubated overnight at 55°C. Supernatant was removed and plugs were washed with 1 ml TE (50 mM Tris, 20 mM EDTA) rotating at room temperature for 1 h and the proteinase K treatment was repeated. Plugs were then washed three times in TE, treated with 2 mM PMSF for 30 min at 37°C, washed three more times in TE, and then overlaid with and stored in 1× ligase buffer (New England Biolabs, Inc.).
Linker LM-PCR.
Supernatant was removed from agarose plugs, replaced with 50 μl fresh 1× ligase buffer, and plugs were heated to 62°C to melt agarose. 20 μl DNA (∼10,000 cell equivalents) was added to 2 μl T4 DNA ligase (2 Weiss units, MBI Fermentis), 10 μl ds annealed linker in 1× ligase buffer, 3 μl 10× ligase buffer, and 30 μl dH20, and incubated overnight at 18°C. Linker was prepared by annealing 5 nmol each of LMPCR.1 (5′-GCGGTGACCCGGGAGATCTGAATTC-3′) and LMPCR.2 (5′-GAATTCAGATC-3′) in 300 μl 1× ligase buffer, which results in a ds oligo with a 14-nt ss overhang that can only ligate unidirectionally. Ligated DNA samples were heated to 70°C for 10 min, diluted fivefold in distilled H20, and assayed for GAPDH by PCR to adjust DNA input before LM-PCR. The following primers (Integrated DNA Technologies) were used in conjunction with linker primer (LMPCR.1) to amplify DNA breaks: 5′Sμ-GCAGAAAATTTAGATAAAATGGATACCTCAGTGG-3′ (used for all experiments except for data shown in Fig. S1); 3′Sμ: 5′-GCTCATCCCGAACCATCTCAACCAGG-3′ (used only for Fig. S1); Sg3-AP: 5′-AACATTTCCAGGGACCCCGGAGGAG-3′ (25); and CmuL2: 5′-CTGCGAGAGCCCCCTGTCTGATAAG-3′ (42).
Three-fold dilutions of input DNA (0.5, 1.5, and 4.5 μL) were amplified by Hotstar Taq (QIAGEN) using a touchdown PCR program. PCR products were run on 1.25% agarose gels and vacuum blotted (VacuGene XL, Pharmacia) onto nylon membranes (GeneScreen Plus, PerkinElmer). Blots were hybridized with oligonucleotide probes end-labeled with γ32P-ATP at 37°C overnight and washed at 55°C with 2X SSC/0.1%SDS. The following probes were used: μ probe 5′: 5′-AGGGACCCAGGCTAAGAAGGCAAT-3′; Sμ probe: 5′-GTTGAGAGCCCTAGTAAGCGAGGCTCTAAAAAGCACGCT-3′ (7); Sμ3′ probe: 5′-GGGCTGGCTGATGGGATGCCCC-3′ (used only for Fig. S1); Sγ3-LP: 5′-GGACCCCGGAGGAGTTTCCATGATCCTGGG-3′ (25); and Cμ: 5′-TGGCCATGGGCTGCCTAGCCCGGGACTTCCTG-3′ (42).
Cloning, identification, and sequence analysis of PCR products.
PCR products from cells cultured ≥48 h were cloned into the vector pCR4-TOPO (Invitrogen) and sequenced by Macrogen using T3 and T7 primers. Cloned breaks in Sμ were aligned with germline Sμ sequenced from C57BL/6 chromosome 12 (GenBank Accession #AC073553) with numbering starting at nt 136,645. This is the 5′Sμ primer binding site and ∼800 nt upstream of the beginning of the tandem repeats. Cloned breaks in Sγ3 were aligned with germline Sγ3 sequenced from BALB/c (MUSIGCD18), and cloned breaks in Cμ were aligned with germline Cμ from BALB/c (MUSIGCD10).
Statistics.
Fisher's exact test was used to determine the difference between the occurrence of breaks at hotspots (or G+C residues) and the occurrence of hotspots (or G+C residues) in the genomic sequence. The genomic sequence analyzed begins at the 5′ LM-PCR primer and extends to the most 3′ break cloned.
Online supplemental material
Fig. S1 shows DSBs detected using the 3′Sμ primer from WT and aid −/− cells. Fig. S2 shows flow cytometric analysis of surface Ig isotype expression in splenic B cells from WT, aid − / − and ung −/− mice induced to undergo CSR. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050872/DC1.
Acknowledgments
We thank D. Hilbert (Human Genome Sciences) for BLyS. We thank D. Schatz for information about the Sμ5′ probe before publication, and D. Kaminski for helpful comments on the text.
The research was supported by RO1s AI23283 and AI63026 from the National Institutes of Health (to J. Stavnezer) and RO1 AI65639 (to C.E. Schrader).
The authors have no conflicting financial interests.
Abbreviations used: AID, activation-induced cytidine deaminase; AP, apurinic/apyrimidic; CH, heavy chain constant; CSR, class switch recombination; ds, double-stranded; DSB, double-strand break; LM-PCR, ligation- mediated–PCR; NHEJ, nonhomologous end-joining; S, switch; ss, single-stranded; SSB, single-strand break; UNG, uracil DNA glycosylase.
References
- 1.Stavnezer, J. 2000. Molecular processes that regulate class switching. Curr. Top. Microbiol. Immunol. 245:127–168. [DOI] [PubMed] [Google Scholar]
- 2.Manis, J.P., M. Tian, and F.W. Alt. 2002. Mechanism and control of class-switch recombination. Trends Immunol. 23:31–39. [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. [DOI] [PubMed] [Google Scholar]
- 4.Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. [DOI] [PubMed] [Google Scholar]
- 5.Petersen, S., R. Casellas, B. Reina-San-Martin, H.T. Chen, M.J. Difilippantonio, P.C. Wilson, L. Hanitsch, A. Celeste, M. Muramatsu, D.R. Pilch, et al. 2001. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 414:660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catalan, N., F. Selz, K. Imai, P. Revy, A. Fischer, and A. Durandy. 2003. The block in immunoglobulin class switch recombination caused by activation-induced cytidine deaminase deficiency occurs prior to the generation of DNA double strand breaks in switch mu region. J. Immunol. 171:2504–2509. [DOI] [PubMed] [Google Scholar]
- 7.Rush, J.S., S.D. Fugmann, and D.G. Schatz. 2004. Staggered AID-dependent DNA double strand breaks are the predominant DNA lesions targeted to S mu in Ig class switch recombination. Int. Immunol. 16:549–557. [DOI] [PubMed] [Google Scholar]
- 8.Petersen-Mahrt, S.K., R.S. Harris, and M.S. Neuberger. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–104. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhuri, J., M. Tian, C. Khuong, K. Chua, E. Pinaud, and F.W. Alt. 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 422:726–730. [DOI] [PubMed] [Google Scholar]
- 10.Dickerson, S.K., E. Market, E. Besmer, and F.N. Papavasiliou. 2003. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 197:1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pham, P., R. Bransteitter, J. Petruska, and M.F. Goodman. 2003. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 424:103–107. [DOI] [PubMed] [Google Scholar]
- 12.Sohail, A., J. Klapacz, M. Samaranayake, A. Ullah, and A.S. Bhagwat. 2003. Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucleic Acids Res. 31:2990–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rada, C., G.T. Williams, H. Nilsen, D.E. Barnes, T. Lindahl, and M.S. Neuberger. 2002. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 12:1748–1755. [DOI] [PubMed] [Google Scholar]
- 14.Imai, K., G. Slupphaug, W.I. Lee, P. Revy, S. Nonoyama, N. Catalan, L. Yel, M. Forveille, B. Kavli, H.E. Krokan, et al. 2003. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 4:1023–1028. [DOI] [PubMed] [Google Scholar]
- 15.Begum, N.A., K. Kinoshita, N. Kakazu, M. Muramatsu, H. Nagaoka, R. Shinkura, D. Biniszkiewicz, L.A. Boyer, R. Jaenisch, and T. Honjo. 2004. Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch. Science. 305:1160–1163. [DOI] [PubMed] [Google Scholar]
- 16.Kavli, B., G. Slupphaug, C.D. Mol, A.S. Arvai, S.B. Peterson, J.A. Tainer, and H.E. Krokan. 1996. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J. 15:3442–3447. [PMC free article] [PubMed] [Google Scholar]
- 17.Jung, S., K. Rajewsky, and A. Radbruch. 1993. Shutdown of class switch recombination by deletion of a switch region control element. Science. 259:984–987. [DOI] [PubMed] [Google Scholar]
- 18.Chaudhuri, J., and F.W. Alt. 2004. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 4:541–552. [DOI] [PubMed] [Google Scholar]
- 19.Ramiro, A.R., P. Stavropoulos, M. Jankovic, and M.C. Nussenzweig. 2003. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 4:452–456. [DOI] [PubMed] [Google Scholar]
- 20.Daniels, G.A., and M.R. Lieber. 1995. RNA:DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination. Nucleic Acids Res. 23:5006–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian, M., and F.W. Alt. 2000. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J. Biol. Chem. 275:24163–24172. [DOI] [PubMed] [Google Scholar]
- 22.Yu, K., F. Chedin, C.L. Hsieh, T.E. Wilson, and M.R. Lieber. 2003. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 4:442–451. [DOI] [PubMed] [Google Scholar]
- 23.Shen, H.M., and U. Storb. 2004. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proc. Natl. Acad. Sci. USA.. 101:12997–13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arudchandran, A., R.M. Bernstein, and E.E. Max. 2004. Single-strand DNA breaks adjacent to cytosines occur during immunoglobulin class switch recombination. J. Immunol. 173:3223–3229. [DOI] [PubMed] [Google Scholar]
- 25.Wuerffel, R.A., J. Du, R.J. Thompson, and A.L. Kenter. 1997. Ig Sg3 DNA-specific double strand breaks are induced in mitogen-activated B cells and are implicated in switch recombination. J. Immunol. 159:4139–4144. [PubMed] [Google Scholar]
- 26.Yu, K., F.T. Huang, and M.R. Lieber. 2004. DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. J. Biol. Chem. 279:6496–6500. [DOI] [PubMed] [Google Scholar]
- 27.Rogozin, I.B., and M. Diaz. 2004. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J. Immunol. 172:3382–3384. [DOI] [PubMed] [Google Scholar]
- 28.Bransteitter, R., P. Pham, P. Calabrese, and M.F. Goodman. 2004. Biochemical analysis of hypermutational targeting by wild type and mutant activation-induced cytidine deaminase. J. Biol. Chem. 279:51612–51621. [DOI] [PubMed] [Google Scholar]
- 29.Mackay, F., P. Schneider, P. Rennert, and J. Browning. 2003. BAFF AND APRIL: a tutorial on B cell survival. Annu. Rev. Immunol. 21:231–264. [DOI] [PubMed] [Google Scholar]
- 30.Gonda, H., M. Sugai, Y. Nambu, T. Katakai, Y. Agata, K.J. Mori, Y. Yokota, and A. Shimizu. 2003. The balance between Pax5 and Id2 activities is the key to AID gene expression. J. Exp. Med. 198:1427–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shapiro, G.S., K. Aviszus, J. Murphy, and L.J. Wysocki. 2002. Evolution of Ig DNA sequence to target specific base positions within codons for somatic hypermutation. J. Immunol. 168:2302–2306. [DOI] [PubMed] [Google Scholar]
- 32.Dunnick, W., G.Z. Hertz, L. Scappino, and C. Gritzmacher. 1993. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 21:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bardwell, P.D., C.J. Woo, K. Wei, Z. Li, A. Martin, S.Z. Sack, T. Parris, W. Edelmann, and M.D. Scharff. 2004. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat Immunol. 5:224–229. [DOI] [PubMed] [Google Scholar]
- 34.Schrader, C.E., J. Vardo, E. Linehan, M.Z. Twarog, L.J. Niedernhofer, J.H. Hoeijmakers, and J. Stavnezer. 2004. Deletion of the nucleotide excision repair gene Ercc1 reduces Ig class switching and alters mutations near switch recombination junctions. J. Exp. Med. 200:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bardwell, P.D., A. Martin, E. Wong, Z. Li, W. Edelmann, and M.D. Scharff. 2003. Cutting edge: the G-U mismatch glycosylase methyl-CpG binding domain 4 is dispensable for somatic hypermutation and class switch recombination. J. Immunol. 170:1620–1624. [DOI] [PubMed] [Google Scholar]
- 36.Rada, C., J.M. Di Noia, and M.S. Neuberger. 2004. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 16:163–171. [DOI] [PubMed] [Google Scholar]
- 37.Paques, F., and J.E. Haber. 1997. Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 17:6765–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martomo, S.A., W.W. Yang, and P.J. Gearhart. 2004. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J. Exp. Med. 200:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li, Z., S.J. Scherer, D. Ronai, M.D. Iglesias-Ussel, J.U. Peled, P.D. Bardwell, M. Zhuang, K. Lee, A. Martin, W. Edelmann, and M.D. Scharff. 2004. Examination of Msh6- and Msh3-deficient mice in class switching reveals overlapping and distinct roles of MutS homologues in antibody diversification. J. Exp. Med. 200:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoeijmakers, J.H. 2001. Genome maintenance mechanisms for preventing cancer. Nature. 411:366–374. [DOI] [PubMed] [Google Scholar]
- 41.Lin, S.C., and J. Stavnezer. 1996. Activation of NF-κB/Rel by CD40 engagement induces the mouse germline immunoglobulin Cγ1 promoter. Mol. Cell. Biol. 16:4591–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papavasiliou, F.N., and D.G. Schatz. 2000. Cell-cycle-regulated DNA double-stranded breaks in somatic hypermutation of immunoglobulin genes. Nature. 408:216–221. [DOI] [PubMed] [Google Scholar]