

Abstract
Microsomal triglyceride transfer protein (MTP), an endoplasmic reticulum (ER) chaperone that loads lipids onto apolipoprotein B, also regulates CD1d presentation of glycolipid antigens in the liver and intestine. We show MTP RNA and protein in antigen-presenting cells (APCs) by reverse transcription–polymerase chain reaction and by immunoblotting of mouse liver mononuclear cells and mouse and human B cell lines. Functional MTP, demonstrated by specific triglyceride transfer activity, is present in both mouse splenocytes and a CD1d-positive mouse NKT hybridoma. In a novel in vitro transfer assay, purified MTP directly transfers phospholipids, but not triglycerides, to recombinant CD1d. Chemical inhibition of MTP lipid transfer does not affect major histocompatibility complex class II presentation of ovalbumin, but considerably reduces CD1d-mediated presentation of α-galactosylceramide (α-galcer) and endogenous antigens in mouse splenic and bone marrow–derived dendritic cells (DCs), as well as in human APC lines and monocyte-derived DCs. Silencing MTP expression in the human monocyte line U937 affects CD1d function, as shown by diminished presentation of α-galcer. We propose that MTP acts upstream of the saposins and functions as an ER chaperone by loading endogenous lipids onto nascent CD1d. Furthermore, our studies suggest that a small molecule inhibitor could be used to modulate the activity of NKT cells.
Structurally homologous to MHC class I, the CD1 family of glycoproteins has evolved to present lipid antigens (1). The human group I CD1a, b, and c proteins present mycobacterial lipids and lipopeptides, but can also present host lipids to autoreactive CD1-restricted T cells (2–6). The intracellular localization of CD1 proteins is controlled by dileucine and tyrosine sorting motifs in their cytoplasmic tails (7, 8). The type of lipid each CD1 family member presents reflects both the shape of the CD1 hydrophobic antigen-binding pocket and the endosomal compartments through which the CD1 proteins traffic (9). Group II CD1d, which is found in humans and is the only CD1 protein in rodents, presents glycolipid antigens to NKT cells, which are defined as cells expressing NK surface markers and CD1d-restricted T cell receptors (1). Marine sponge–derived α-galactosylceramide (α-galcer) is an exogenous model antigen for NKT cells (10), and phosphatidyl inositol mannoside from mycobacteria, Leishmania donovani antigens, and sphingolipids from Sphingomonas capsulata, S. paucimobilis, and S. yanoikuyae have been shown to activate subsets of NKT cells in a CD1d-restricted manner (11–14). CD1d in vivo also presents endogenous glycolipid antigens (15, 16). Several host lipids have been proposed to associate with CD1d and activate subsets of NKT cells, including phosphatidyl inositol, phosphatidyl ethanolamine (PE), and isoglobotrihexosylceramide, which is required for NKT cell development (17–20). Activation of autoreactive NKT cells by CD1d-presenting host lipids can be beneficial during bacterial and viral infections, some antitumor responses, and regulation of autoimmune diseases such as diabetes (21–24). However, improper activation of NKT cells can lead to inflammatory bowel disease, asthma, and atherosclerosis (25–27).
The endoplasmic reticulum (ER) chaperones calnexin, calreticulin, and ERp57 associate with nascent CD1d and assist in folding and disulfide bond formation (28). Unlike MHC class I, which associates with β2-microglobulin (β2m) early in biogenesis, CD1d acquires β2m just before exiting the ER, and β2m is not essential for CD1d cell surface expression (28, 29). Phospholipids bind to the hydrophobic pocket of nascent CD1d and likely enable proper folding in a manner analogous to that of peptide in MHC class I assembly (15). NKT cell clones that recognize phosphatidyl inositol and PE have been characterized but likely represent a minority of NKT cells in vivo (15, 19). The ER phospholipids bound to CD1d may be replaced when CD1d recycles into endosomal compartments, and recent work on lysosomal lipid transfer proteins has shown that a family of lipid transfer proteins, including the saposins and GM2 activator, is capable of exchanging or editing the lipid cargo of CD1d (30, 31). In mice, saposin B can load isoglobotrihexosylceramide onto CD1d, which then activates invariant Vα14 NKT cells (20). The relative importance of ER lipids versus endosomal-derived lipids is unclear. Tail-deleted forms of CD1d that fail to traffic to endosomes activate Vα14− NK1.1− NKT cells but cannot present antigen to invariant NKT cells (32). Furthermore, mice that express only the tail-deleted form of CD1d support thymic development of diverse but not invariant NKT cells, indicating a distinction in the host lipids recognized by these two NKT cell populations (32).
Microsomal triglyceride transfer protein (MTP) is predominately found in the ER of hepatocytes and intestinal epithelial cells (IECs), where it loads triglycerides, cholesterol esters, and phospholipids onto apolipoprotein B (apoB) (33–35). In the absence of MTP-mediated lipid transfer, apoB is degraded and very low density lipoproteins or chylomicrons are not secreted from the liver or intestines, respectively (36–39). Humans with mutations in the gene encoding MTP develop abetalipoproteinemia, a disorder characterized by low serum lipoproteins and severe lipophilic vitamin deficiencies (40).
Recent work from our laboratory has shown the importance of MTP in CD1d antigen presentation by hepatocytes and IECs (41). We observed that MTP associates with CD1d in hepatocytes and that CD1d surface expression on hepatocytes was reduced in the absence of MTP. MTP silencing or gene deletion in hepatocytes or IECs resulted in diminished antigen presentation by CD1d in hepatocytes and IECs and protection from NKT cell–mediated hepatitis and colitis. However, whether MTP affects CD1d presentation in a broad range of tissues and cell types, and not in APCs, or whether MTP can mediate direct lipid transfer to CD1d was unclear. CD1d is expressed on DCs, monocytes, macrophages, cortical thymocytes, and most peripheral lymphocytes (42). We now show that MTP expression is also present in mouse and human professional APCs, that MTP lipid transfer activity is present in mouse splenocytes, and that MTP can directly transfer phospholipid to recombinant CD1d in vitro. We further show, by gene silencing and chemical inhibition, that the lack of MTP in such professional APCs results in a selective defect in CD1d presentation of exogenous and endogenous CD1d-restricted antigens. Importantly, MHC class II–restricted presentation of ovalbumin is unaffected by the loss of MTP, indicating that MTP plays a specific role in CD1d presentation. We therefore conclude that MTP is a broadly expressed ER chaperone that can lipidate CD1d and is involved in CD1d-restricted antigen presentation in professional APCs.
Results
MTP is present and functional in APCs
Because MTP plays a role in CD1d antigen presentation by hepatocytes and IECs, we hypothesized that MTP may serve as a chaperone for CD1d in all CD1d-positive cell types. MTP expression has never been detected in professional APCs, and most research in the field has focused on the functions of MTP in the liver and intestine (33). We therefore examined mttp transcript expression in a variety of potential APC types (Fig. 1 A; not depicted). mttp was detected in mouse primary tissues (liver, heart, lung, ovaries, peritoneal exudate cells, small intestine, colon, spleen, thymus, lymph node, B cells, liver DCs, and BM-derived DCs [BMDCs]); mouse thymus (RMAS), APC (RAW), and NKT cell (DN32) lines; and in human B, T, and monocyte cell lines (C1Rd, Jurkat, and U937). mttp was not present in DCs isolated from MTP gene–deleted mice, but was present in wild-type mice at low abundance in every other immunologic and nonimmunologic tissue examined. mttp expression in the intestine was notably increased relative to APCs, as would be expected from a tissue actively secreting lipoproteins.
Figure 1.

MTP is present in APCs. (A) cDNA was synthesized from 1 μg RNA from the indicated tissues. MTP transcripts were amplified by RT-PCR, and the volume of sample loaded was normalized to β-actin transcript levels. MTPΔ DCs were obtained from the liver of a pIpC-injected MTPmx1 mouse. (B) Protein was harvested from LMNCs isolated from MTPMx1 or C57BL/6 mice either injected or not injected with pIpC and B cell lines C1R and 721 transfected with human and mouse CD1d, respectively. 25 μg of cell lysate was loaded per lane, separated by reducing SDS-PAGE, and immunoblotted with polyclonal antisera to MTP:PDI. (C) Triglyceride transfer activity in 100 μg splenocyte lysate (▵), 50 μg splenocyte lysate (○), 100 μg splenocyte lysate with BMS197636 (▴), and 50 μg splenocyte lysate with BMS197636 (•) is shown. Assays were in triplicate with SDs less than 10% of the percent transfer at 3 h. Each well contained 100 picomoles of NBD-labeled triglyceride.
To verify the presence of MTP protein in APCs, we isolated liver mononuclear cells (LMNCs) from MTPMx1 mice that either had or had not been injected with poly-inositic:poly-cytodylic acid (pIpC) to induce MTP gene deletion. The LMNCs were then lysed, and proteins were separated by SDS-PAGE. Immunoblotting was performed using a polyclonal antisera raised against recombinant human MTP:protein disulfide isomerase (PDI), which has been demonstrated to recognize human and mouse MTP large subunits and PDI. As shown in Fig. 1 B, we were able to detect a 97-kD protein in the nondeleted mice that was not present in the MTP gene–deleted mice. To show that the lack of MTP protein was not caused by pIpC injection, we also analyzed LMNC lysates from wild-type mice treated or not treated with pIpC. In addition, we were able to detect evidence of MTP protein in lysates of the human B cell line C1Rd and the mouse B cell line 721d.
To demonstrate that MTP is functional in APCs, we used a previously described triglyceride transfer assay (43). In this assay, MTP function is assessed by studying the transfer of fluorescently labeled triglycerides embedded in donor phospholipid vesicles to acceptor vesicles. During the transfer, the amount of triglyceride associated with MTP can be measured as an increase in fluorescence over time. Splenocyte lysates contained measurable triglyceride transfer activity, which could be abrogated by the addition of 200 nM BMS197636, a known chemical inhibitor of MTP lipid transfer (Fig. 1 C; references 44, 45). Specific activity in the splenocyte lysates was 0.04% triglyceride being transferred per microgram of lysate protein per hour. Although the MTP-specific activity in primary splenocytes was low, the activity was abolished by the addition of BMS197636, which is consistent with MTP being present in these cells. The splenocytes used were an unfractionated population containing CD1d-positive and -negative cells. When we examined a homogenous population of CD1d-positive cells, the mouse NKT cell line DN32, a specific activity of 0.22 was observed. Athar et al. calculated MTP-specific activity from whole cell lysates of the hepatocyte cell line HepG2 to be 0.982, and, in our own assays, purified rat liver microsomes exhibited a specific activity of 4.78 (43). Thus, MTP lipid transfer activity is present in APCs but less abundant than that observed in tissues such as the liver, which actively secrete lipoproteins.
MTP can transfer a lipid to recombinant CD1d in vitro
MTP could directly transfer lipids to CD1d, or it could indirectly influence CD1d presentation, such as through a lipoprotein intermediate. To test for direct lipid transfer from MTP to CD1d, we used a reductionist in vitro approach using PE as a model lipid antigen. PE has been shown to bind to CD1d and can be recognized by several invariant NKT cell hybridomas (15, 19). PE is also a known substrate for MTP (34, 35).
Recombinant β2m-associated CD1d was coated onto a 96-well plate and incubated with liposomes containing a 6:1 ratio of unlabeled PE/7-nitro-2,1,3-benzoxadiazol-4-yl (NBD)–labeled PE. The fluorescent NBD label was conjugated to the PE headgroup in order to avoid steric interference between PE acyl tails and CD1d hydrophobic pockets. MTP purified from rat liver homogenates was added to the CD1d- or BSA-coated wells at a 1:10 molar ratio of MTP/CD1d, incubated at 37°C for 2 h, and washed with PBS. Lipids bound to CD1d were eluted with isopropanol, transferred to a black microtiter plate, and the level of fluorescence was determined. As shown in Fig. 2 A, PE was transferred to CD1d in the presence of MTP, but not to BSA, as seen by a threefold increase in fluorescence above background. To show that MTP transfer to CD1d is specific for phospholipid, the same assay was performed using NBD-labeled triglyceride vesicles. No transfer of fluorescence was observed under these conditions, indicating that, as expected, phospholipid but not triglyceride binds to CD1d and, furthermore, that MTP itself is not remaining in the wells during the elution phase of the assay. As can be seen in Fig. 2 B, the quantity of fluorescent PE transferred to CD1d increased with increasing concentrations of purified MTP.
Figure 2.
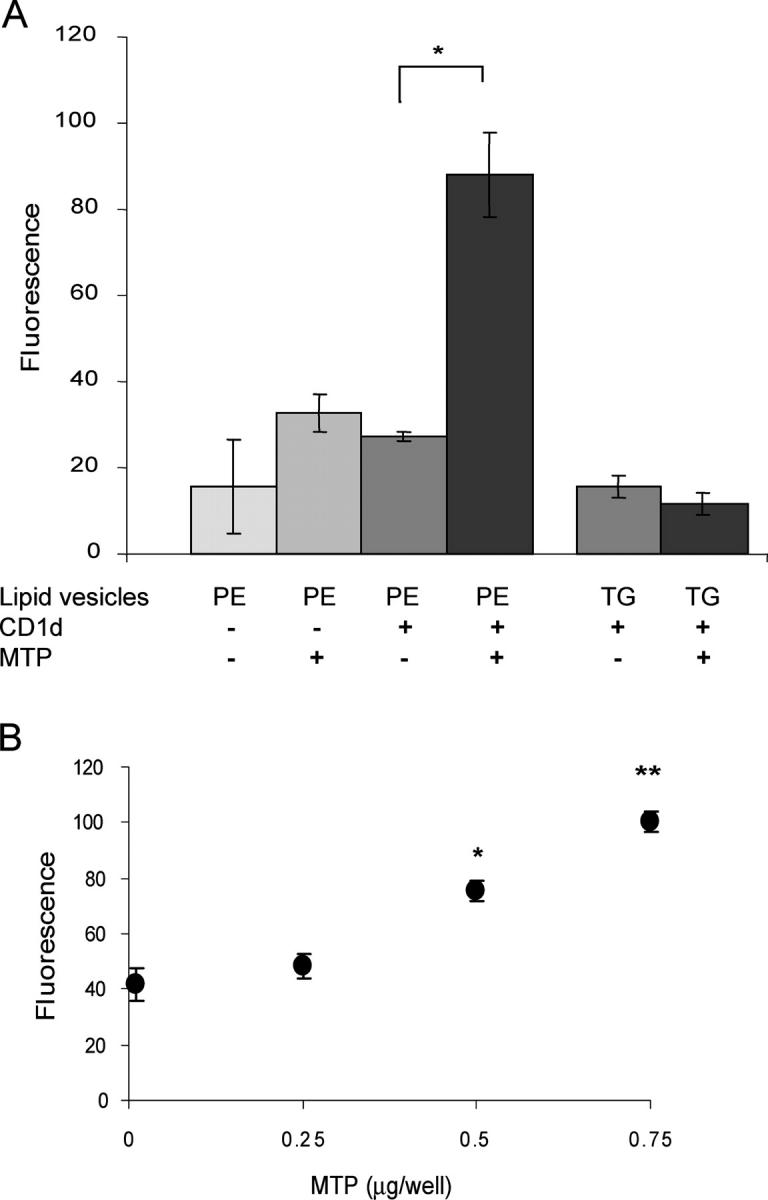
Purified MTP transfers a lipid to recombinant CD1d in vitro. (A) 2 μg BSA or recombinant CD1d was coated onto microtiter wells and incubated with either lipid vesicles alone or lipid vesicles plus 0.5 μg purified MTP. Lipid vesicles contained either PE:NBD or TG:NBD. Donor PE vesicles were composed of 450 nmol unlabeled PE and 14 nmol PE:NBD per milliliter. Results are representative of six independent experiments. *, P < 0.01. (B) 2 μg of recombinant CD1d was coated onto microtiter wells and incubated with PE:NBD vesicles and increasing concentrations of purified MTP. *, P < 0.005; **, P < 0.001 compared with 0 μg MTP. Values are ±SD.
In vitro inhibition of MTP reduces CD1d-mediated antigen presentation of mouse DCs
BMS212122 is a specific chemical inhibitor of MTP-mediated lipid transfer that functions in vitro at concentrations as low as 0.03 nM and in vivo in primates (46). CD1d presentation in the mouse epithelial cell line, MODE-K, is regulated by MTP (41). As shown in Fig. 3 A, incubation of MODE-K cells with 13 μM BMS212122 decreased the presentation of α-galcer to DN32 cells. These experiments indicate that the MTP inhibitor BMS212122 blocks CD1d function, similar to its previously established role in inhibiting apoB secretion (46).
Figure 3.

Chemical inhibition of mouse MTP causes a selective defect in CD1d antigen presentation. (A) MODE-K cells cultured in the presence of BMS212122 (MTPi) or vehicle were incubated with α-galcer for 3 h, washed, and co-cultured with DN32 cells. (B) Splenocytes were isolated from wild-type mice, incubated for 24 h with BMS212122, and washed. The exogenous antigens ovalbumin or α-galcer were added as indicated. Splenocytes were then co-cultured with autoreactive 24.8 NKT cells (left), DN32 NKT cells (center), or CD4+ cells from an OT-II transgenic mouse (right). *, P < 0.05. Results are representative of two independent experiments. (C) CD11c+ splenocytes were incubated for 24 h with BMS212122, DMSO, or 9-fluorenyl carboxylic acid, washed, and co-cultured with 50,000 24.8 NKT cells per well. *, P < 0.005; **, P < 0.05 compared with DMSO values. Results are representative of five independent experiments. (D) CD11c+ splenocytes were incubated for 4 d with BMS212122 or DMSO, pulsed with the indicated concentrations of α-galcer, washed, and co-cultured with DN32 NKT cells (E/T = 100,000:30,000). *, P < 0.05. (E) 30,000 CD11c+ splenocytes incubated for 24 h with BMS212122 or DMSO were washed and co-cultured with 100 μg/ml ovalbumin and 100,000 CD4+ cells from an OT-II transgenic mouse. Values are ±SD.
We next examined the role of MTP in CD1d presentation by primary mouse APCs. Splenocytes were isolated from wild-type mice and assayed for their ability to present an exogenous antigen, α-galcer, to DN32 NKT cells and their ability to stimulate the autoreactive NKT cell line 24.8. When splenocytes were incubated with BMS212122 for 24 h before the addition of NKT cells, their ability to present exogenous α-galcer and endogenous CD1d-restricted antigens was reduced (Fig. 3 B). BMS212122 exhibited no effect, however, on splenocyte presentation of ovalbumin to the MHC class II–restricted T cell line, OT-II (Fig. 3 B). DCs were then isolated from wild-type splenocytes by positive selection on CD11c magnetic beads and cultured with DMSO, BMS212122, or 9-fluorenyl carboxylic acid, a negative control compound of similar structure to BMS212122, but with no MTP inhibitory activity at concentrations as high as 30 μM in an in vitro triglyceride transfer assay (Harrity, T.W., personal communication). The effect of MTP inhibition on presentation of an endogenous ligand to the 24.8 NKT cells was highly pronounced with BMS212122, but not 9-fluorenyl carboxylic acid, in the CD11c-positive DC population (Fig. 3 C). CD11c-positive cells were also pulsed with titrated amounts of α-galcer, washed, and co-cultured with DN32 iNKT cells. At every concentration of α-galcer, the MTP inhibitor–treated cells showed reduced ability to activate iNKT cells compared with vehicle- or control compound–treated splenic DCs (Fig. 3 D). BMDCs cultured with BMS212122 or DMSO were also pulsed with titrated amounts of α-galcer up to 1000 ng/ml, with a similar inhibitory effect of BMS212122 observed at all concentrations of α-galcer (unpublished data). CD11c-positive DCs cultured with BMS212122 or vehicle were cultured with ovalbumin and CD4 T cells isolated from OT-II transgenic mice (Fig. 3 E). MTP inhibition showed no effect on MHC class II presentation of ovalbumin implying a specific role for MTP in the CD1d pathway.
BMDCs were cultured with BMS212122 or vehicle control after 6 d of differentiation in the presence of GM-CSF. On days 10–13, BMDCs were collected and co-cultured with NKT cells. BMS212122 had a significant effect on both the presentation of an endogenous ligand to autoreactive 24.8 cells (P < 0.01; Fig. 4 A) and on the presentation of α-galcer to DN32 cells (P < 0.05; Fig. 4 B). >90% of BMDCs were CD11c-positive on day 13, and viability was unaffected by MTP inhibition. Surface expression of CD11b, CD11c, CD40, and CD80 was unaffected by BMS212122, indicating that no deleterious effects on the maturation of BMDCs had occurred (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20050183/DC1). In contrast, a decrease in CD1d surface expression on BMDCs was observed over time in the presence of BMS212122. One day after the addition of BMS212122, CD1d expression on BMDCs was 86% of the level observed in vehicle-treated controls, and after 4 d of BMS212122 treatment, CD1d expression was decreased to 66% of the vehicle controls. In a total of three independent experiments on BMDCs, extended culture with BMS212122 reduced surface CD1d expression by an average of 68 ± 2.6%. The negative control compound 9-fluorenyl carboxylic acid had no effect on CD1d surface expression (Fig. 4 C). The MHC II pathway was intact in BMS212122-treated cells; like CD11c-positive splenic DCs, BMDCs exhibited efficient presentation of ovalbumin to CD4+, MHC class II–restricted, ovalbumin-specific T cells (Fig. 4 D). Thus, MTP inhibition results in a selective defect in CD1d-mediated antigen presentation.
Figure 4.

Chemical inhibition of MTP in mouse BMDCs causes a selective defect in CD1d antigen presentation. (A) BMDCs cultured for 6 d with BMS212122 were washed and co-cultured with 50,000 24.8 NKT cells per well. *, P < 0.01. Results are representative of three experiments. (B) BMDCs cultured for 4 d with BMS212122 were pulsed with α-galcer, washed, and co-cultured with 100,000 DN32 NKT cells. *, P < 0.05. Results are representative of three experiments. (C) BMDCs cultured for 4 d with BMS212122 or 9-fluorenyl carboxylic acid were stained with the FITC-conjugated mAb 1B1 and analyzed by flow cytometry. The dashed line represents the isotype control. Results are representative of three independent experiments. (D) 50,000 BMDCs incubated for 3 d with BMS212122 or DMSO were washed and co-cultured with 100 μg/ml ovalbumin and 100,000 CD4+ cells from an OT-II transgenic mouse. Values are ±SD.
MTP is critical for CD1d-restricted antigen presentation in human cells
MTP is highly conserved among mammalian species with an 86% identity between mouse and human MTP at the amino acid level. We therefore hypothesized that MTP inhibition in human APCs would affect CD1d antigen presentation. To test this, MTP expression was silenced in the human monocyte cell line U937. U937 cells express MTP (Fig. 1 A) and low levels of surface CD1d but are highly potent APCs for CD1d-restricted NKT cells (10). U937 cells that were transfected with human mtp-specific small interfering (si) RNA oligomers displayed a 48% reduction in mtp transcript levels compared with cells transfected with irrelevant oligomers (mock) after 48 h, as shown in Fig. 5 A. The silenced and mock-silenced U937 cells were incubated with α-galcer for 3 h, washed, and co-cultured with the iNKT cell line DN32. NKT cell activation was measured by IL-2 production, and, as shown in Fig. 5 B, the mtp-silenced U937 cells exhibited a significant (P < 0.001) reduction in their ability to present the model CD1d-restricted exogenous antigen. A similar reduction in IL-2 release from DN32 cells was observed when U937 cells were cultured with α-galcer and BMS212122, as compared with the vehicle control (Fig. 5 C).
Figure 5.

MTP in human cells is critical for CD1d antigen presentation. (A) U937 cells were treated with irrelevant or MTP-specific siRNA oligomers. RNA was isolated 48 h after silencing, and transcript levels of mtp and β-actin were determined by RT-PCR. (B) Silenced and mock-silenced U937 cells were incubated with α-galcer for 3 h, washed, and co-cultured with DN32 cells (E/T = 1:1). *, P < 0.001. Results are representative of two independent experiments. (C) U937 cells cultured with BMS212122 (MTPi) or vehicle for 3 d were incubated with α-galcer for 3 h, washed, and co-cultured with DN32 cells (E/T = 1:1). *, P < 0.001. (D) C1Rd cells cultured with BMS212122 (MTPi) or vehicle for 4 d were incubated with α-galcer for 4 h, fixed with 0.05% glutaraldehyde for 30 s, washed, and co-cultured with DN32 cells (E/T = 2:1). *, P < 0.02. Results are representative of three independent experiments. (E) C1Rd cells cultured with BMS197636 or vehicle were incubated with NKT cell lines derived from human peripheral blood in the presence of 1 ng/ml PMA. NKT cells co-cultured with C1R mock transfected cells in the same assay yielded 33.6 ± 9.8 pg/ml IFN-γ. (F) Human monocyte-derived DCs were stained with 42.1 (CD1d), anti–HLA-A,B,C (MHC class I), or isotype control antibodies (dashed lines) after 6 d of differentiation in the presence of BMS212122 (gray lines, MTPi) or vehicle control (black lines). Results are representative of two independent experiments. (G) Day 6 human moDCs differentiated in the presence of BMS212122 (○, MTPi) or vehicle (•) were incubated with α-galcer for 3 h, washed, and co-cultured with 50,000 DN32 cells. *, P < 0.05. Results are representative of two independent experiments. Values are ±SD.
C1Rd is a human B cell line transfected with human CD1d (7). C1Rd cells were cultured with MTP inhibitor or vehicle for 4 d, pulsed with α-galcer, fixed with glutaraldehyde, and washed before co-culture with DN32 NKT cells. Incubation of C1Rd cells with BMS212122, but not the vehicle, inhibited the presentation of α-galcer, as determined by a reduction in IL-2 secretion (Fig. 5 D). To exclude nonspecific effects of BMS212122, C1Rd cells were incubated with a second MTP chemical inhibitor, BMS197636, which resulted in dose-dependent inhibition of CD1d-mediated activation of a human peripheral blood–derived iNKT cell line in the presence of a limiting concentration of PMA (Fig. 5 E). Thus, MTP inhibition in C1Rd cells resulted in a diminished ability to present CD1d-restricted antigens to mouse and human iNKT cells.
To further determine the effect of MTP inhibition in primary human APCs, we differentiated monocyte-derived DCs (moDC) from CD14-positive peripheral blood mononuclear cells obtained from healthy volunteers. Monocytes were selected on CD14 magnetic beads and cultured in media supplemented with autologous plasma, GM-CSF, IL-4, and either BMS212122 or vehicle. >95% of cells expressed high levels of DC surface markers (unpublished data). After 6 d in the presence of BMS212122, the moDCs had up-regulated MHC class I to an equal extent in comparison to vehicle treatment (mean fluorescence intensity [MFI]: 129 vs. 100, respectively); however, moDCs cultured in the presence of BMS212122 expressed significantly lower levels of surface CD1d (MFI: 4.8 vs. 6.3; P < 0.01; Fig. 5 F). As can be seen in Fig. 5 G, the MTP-inhibited moDCs also displayed a reduced capacity to present α-galcer to DN32 iNKT cells.
Discussion
We show the presence of MTP transcripts, protein, and triglyceride transfer ability in APCs and demonstrate the ability of MTP to transfer a phospholipid antigen directly to CD1d in vitro. We propose that MTP modulates CD1d function in APCs because chemical inhibition of MTP lipid transfer and/or silencing of MTP expression results in diminished CD1d-restricted presentation of exogenous and endogenous antigens to NKT cells. Notably, presentation via MHC class II is unaffected by the absence of MTP, indicating that the lack of MTP specifically affects CD1d function.
We therefore hypothesize that the ER-resident MTP serves as a chaperone during CD1d biogenesis and could be responsible for transferring lipids to the nascent CD1d pocket within the ER. We have provided evidence that purified MTP can transfer phosphatidylethanolamine to recombinant immobilized CD1d in vitro. Given that we detected MTP lipid transfer activity in primary splenocytes, that MTP can directly lipidate CD1d in vitro, and that treatment of APCs with small molecule inhibitors of MTP lipid transfer function caused considerable impairment in CD1d function, we predict that MTP transfers lipids to CD1d in vivo. Although the endogenous ligands associated with nascent CD1d are not well characterized, previous studies (17–19) have isolated endogenous PE and glycophosphatidyl inositol from the CD1d pocket; thus, PE could be one of several host lipids that MTP might transfer to nascent CD1d. The reductionist system described here can be used to test a variety of lipids to determine potential substrates for MTP, which should provide valuable insights into the role of MTP in loading endogenous glycolipid ligands.
Recent work has shown that lysosomal saposins edit the antigens presented on CD1d by replacing presumed ER-derived lipids with lipids present in endosomes and lysosomes (30, 31). We propose that MTP could act upstream of the saposins as an ER lipid transfer protein and chaperone for CD1d by loading endogenous lipids into the nascent antigen binding pocket. The types of lipids loaded initially onto CD1d could affect the ability of the CD1d antigen to be edited by saposins or other endosomal lipid transfer proteins. Further studies are needed to reveal the precise lipids transferred to CD1d, how the lipid profile varies by cell type or activation state, and the downstream effects of MTP on the loading of endosomal and lysosomal antigens.
Although α-galcer presentation is enhanced by entry into the endosomal pathway where saposins facilitate loading onto lysosomal CD1d, α-galcer is also capable of direct binding to cell surface CD1d (47). MTP inhibition caused a decrease in α-galcer presentation that could be explained by two nonexclusive hypotheses. First, the absence of MTP chaperone function could result in fewer molecules of CD1d egressing from the ER, as supported by the fact that, as we previously observed in hepatocytes (41) and in some APCs examined here, MTP inhibition resulted in decreased surface expression of CD1d. Although important, this diminution in MFI was a modest 34% decrease on BMS212122-treated murine BMDCs and a 23% decrease on BMS212122-treated human moDCs. The human monocyte line U937, despite being a potent stimulator of NKT cells, expresses such low endogenous levels of CD1d (48, 49) that we were unable to observe a change in CD1d expression in the absence of MTP function. On the other hand, C1Rd cells transfected with human CD1d expressed equally high levels of CD1d in the presence and absence of BMS212122 (unpublished data). Because the number of molecules of CD1d required for optimal NKT cell activation has been previously reported to vary by cell type but is usually considered to be quite low (48, 49), it is likely that alterations in surface expression contribute to, but might not completely account for, the reduction in α-galcer presentation observed. As a second consideration, MTP-loaded lipids could be preferentially replaced by α-galcer or be optimal targets for saposin-mediated exchange. In the absence of MTP-loaded lipids, the CD1d pocket could thus be aberrantly folded or refractory to α-galcer loading. Precise quantitation of the assembly, folding, and glycosylation of CD1d, as well as the subsequent editing of CD1d by saposins, in the absence of MTP would be needed to answer this question.
MTP inhibition causes a reduction in the surface expression of CD1d consistent with MTP being an ER chaperone for CD1d. MTP inhibition did not, however, completely eradicate cell surface expression of CD1d on primary APCs, possibly because of incomplete blockade of MTP or the long half-life of CD1d, which could account for some residual persistence (50). Furthermore, CD1d is less reliant on ER chaperones than MHC I because CD1d can escape from the ER without associated β2m (28, 29). Determining, therefore, the relative location of MTP-mediated lipidation within the cascade of chaperoning events in the ER associated with CD1d folding will be important to define (28, 29). Because MTP lipidation of apoB likely occurs co-translationally as the nascent apoB protein emerges into the ER (33), it might be predicted that MTP lipidation of CD1d is an early event in CD1d biogenesis. Nevertheless, it will be interesting to determine if CD1d, in the absence of MTP-transferred lipids, is unstable in a manner analogous to MHC class I molecules in the absence of peptide loading (51).
Although MTP is known to transfer a wide range of lipids, kinetics studies have shown that triglycerides are the preferred substrate and are transferred much faster than phospholipids, suggesting at least two different lipid binding sites on MTP (34, 35). Importantly, the ability of MTP to transfer lipids depends on the number and length of the lipid acyl chains and does not depend on the head group, as MTP has been shown to transfer all types of phospholipids tested with equal efficacy (34, 35). We have shown for the first time that a series of small molecules that specifically inhibit MTP-mediated lipid transfer and lipidation of apoB (44–46) are also capable of inhibiting MTP-mediated regulation of CD1d function. However, these inhibitors were developed to block transfer of triglycerides and are only partially effective at blocking transfer of phospholipids, as shown by transfer of radiolabeled lipids from vesicles in vitro. For example, BMS200150, an MTP inhibitor with published lipid transfer values, exhibited IC50 = 0.6 μM for triglyceride transfer, yet only achieved 30% inhibition of phosphatidylcholine transfer at the highest concentration tested (30 μM), presumably because of differential effects of the drug on multiple MTP lipid transfer domains or different affinities of MTP for the various lipid classes (44). In this study, BMS197636 was used at a concentration (200 nM) known to specifically inhibit MTP-mediated triglyceride transfer with no effect on other common lipid transfer proteins. At this concentration, BMS197636 inhibits 95% of MTP-mediated triglyceride transfer, but only 60% of MTP-mediated phospholipid transfer, which again implies that compounds binding to different sites on MTP can differentially affect classes of lipid transfer (52). In the assays reported here involving the inhibition of MTP transfer of lipid to CD1d, BMS212122 was used at a concentration of 13 μM despite having IC50 = 1 nM for triglyceride transfer (46). Although the current inhibitors are not optimal for inhibiting phospholipid transfer, our data suggest that CD1d function could potentially be inhibited in vivo by the use of small molecule inhibitors targeted to the putative phospholipid transfer site of MTP. In this case, diseases such as in ulcerative colitis, lupus, atherosclerosis, and airway hypersensitivity, in which CD1d-mediated antigen presentation has been shown to contribute to pathogenesis (1, 25-27), may be particularly benefited.
Before this study, apoB was the only known recipient of MTP-transferred lipids (33), yet we were unable to detect apoB transcripts by RT-PCR in MTP-positive APCs (unpublished data). MTP protein has also been recently reported in adipocytes that do not secrete lipoproteins (53). We have also observed MTP expression and function in the absence of lipoprotein production and suggest that MTP may have evolved as a general ER chaperone that developed additional importance in the liver and intestine for its distinct functions in lipid absorption and the distribution of essential lipid nutrients. As such, it might be surmised that DCs and other APC types have coopted MTP for a role in CD1d presentation of lipid antigens. The possibility that MTP is a general mediator of CD1d acquisition of lipids in APCs rather than being restricted to hepatocytes and IECs predicts a role for MTP in immune responses associated with infections, cancers, immune tolerance, allergy, and autoimmunity, given the known roles of CD1d presentation and NKT cell function in these contexts (1). In addition, given the similarities between CD1d and the type 1 CD1 proteins (CD1a–c; reference 1), the knowledge that MTP regulates human CD1d function suggests that MTP could play a role in lipidation and function of human type 1 CD1 molecules.
We provide direct evidence of MTP-mediated lipid transfer to CD1d in vitro and show the results of chemical inhibition and gene silencing of MTP in CD1d-mediated endogenous and exogenous antigen presentation by primary murine and human APCs and APC lines to murine, as well as human, NKT cells. Further studies are needed to characterize the lipids transferred from MTP to CD1d, and understanding how endogenous lipids are presented to nascent CD1d in vivo will provide important insights into the regulation of NKT cells and the mechanisms of NKT cell–mediated disease.
Materials and Methods
Cell cultures.
Cells were maintained in R10 (RPMI 1640 with 2 mM l-glutamine; 10% FBS) unless otherwise indicated in the figures. MODE-K, a mouse IEC line, has been previously described (54). DN32.D3 (referred to as DN32), a mouse Va14Ja18 invariant TCR-positive T cell hybridoma, was provided by A. Bendelac (University of Chicago, Chicago, IL; reference 55). The autoreactive mouse Vα14Jα18 invariant TCR-positive hybridoma 24.8 was provided by S. Behar (Brigham and Women's Hospital, Boston, MA; references 15, 19). RMAS is a transporter associated with antigen processing–deficient mouse T cell lymphoma (51); RAW is an Abelson virus–transformed mouse macrophage cell line (American Type Culture Collection). Jurkat is a human T lymphocyte line (10); U937 is a human histocytic lymphoma cell line (10). C1R, a human B cell line which lacks expression of MHC class I, was transfected with human CD1d, as previously described, to generate C1Rd (7). 721, a mouse B cell line that lacks expression of MHC class I, was transfected with mouse CD1d, as previously described, to generate 721d (7).
Antigen presentation assay.
APCs were incubated with 100 ng/ml α-galcer (provided by H. Ploegh, Harvard Medical School, Boston, MA) for 3 h, washed three times in PBS, and aliquoted into a 96-well plate at 106 cells/ml. DN32 cells were added at a 1:1 ratio unless otherwise indicated in the figures. CD4+ cells were isolated from spleens of OT-II transgenic mice using positive selection on CD4 magnetic beads (Miltenyi Biotec) and incubated with APCs and 100 μg/ml ovalbumin. The 24.8 NKT cell line was incubated with APCs in the absence of added antigen at a 2:1 E/T ratio unless otherwise indicated in the figures. Mouse IL-2 production was assessed by ELISA (OptEIA; BD Biosciences) of 24- or 40-h culture supernatants. APCs were treated with 13 μM BMS212122 (provided by Bristol Myers Squibb, Princeton, NJ), dissolved in DMSO, in all assays or 13 μM 9-fluorenyl carboxylic acid (Acros Chemicals) dissolved in DMSO as indicated in the figures and washed before the addition of T or NKT cells. Human NKT cell lines generated from healthy donor leukopaks (>90% pure) were incubated with 1 ng/ml PMA and C1Rd or C1R mock-transfected cells in the presence of BMS197636 (provided by Bristol Myers Squibb, Princeton, NJ) or DMSO control at the concentrations indicated in the figures.
Animals.
Mice with two “floxed” mttp alleles were bred with mice transgenic for Mx1 promoter–driven Cre recombinase to generate MTPmx1 (previously referred to as Mttp flox/flox/Mx1-Cre) mice (56). C57BL/6 mice were purchased from Charles River Laboratories. All animal experimentation was done in accordance with institutional guidelines and the review board of Harvard Medical School, which granted permission for this study.
RT-PCR.
Total RNA was extracted using Trizol (Invitrogen), according to the manufacturer's instructions, and cDNA was synthesized with Powerscript reverse transcriptase (CLONTECH Laboratories, Inc.). mttp transcripts were amplified by PCR using 5 μl cDNA per reaction and primers 5′-GGACTTTTTGGATTTCAAAAGTGAC-3′ and 5′-GGAGAAACGGTCATAATTGTG-3′, which amplify both mouse (696 bp) and human (699 bp) transcripts. PCR reactions were heated at 94°C for 3 min, followed by 35 cycles of 94°C for 40 s, 53°C for 90 s, and 72°C for 60 s. Samples were loaded onto a 1.2% agarose gel and visualized by ethidium bromide staining. The volumes of the PCR products loaded were normalized to β-actin transcripts amplified using 2 μl cDNA and primers 5′-ATCTGGCACCACACCTTCTACATTGAGCTGCG-3′ and 5′-CGTCATACTCCTGCTTGCTGATCCACATCTGC-3′.
LMNC isolation and Western blotting.
Livers were perfused with 10 ml PBS, excised, and crushed using the back of a 3-ml syringe plunger. Liver suspensions were incubated in PBS containing 1 mg/ml collagenase IV (Sigma-Aldrich) at 37°C with shaking for 30 min. Cell suspensions were then passed through a 70-μM cell strainer and centrifuged at 1,500 g for 10 min. Pellets were resuspended in 40% Percoll, layered onto 70% Percoll, and centrifuged at 1,500 g for 25 min. LMNCs were collected from the interface, washed, and depleted of erythrocytes using hypotonic lysis buffer. Purified LMNCs or B cell lines were lysed in radioimmunoprecipitation assay buffer, and the protein concentration was quantified using a B cell–attracting chemokine assay (Pierce Chemical Co.). Separation by SDS-PAGE was done by standard methods, followed by immunoblotting with polyclonal antisera raised in rabbits against recombinant human MTP:PDI complexes. The antiserum was provided by C. Shoulders (Imperial College London, London, UK) and recognizes both human and mouse MTP and PDI.
Triglyceride and PE transfer assays.
The triglyceride transfer activity of MTP was measured using donor phospholipid vesicles (Chylos Inc.) as described previously (43). In brief, donor vesicles comprising 100 nmol of NBD-labeled triglycerides per assay embedded in a phosphatidylcholine bilayer were added with an equal volume of phosphatidylcholine acceptor vesicles. NBD fluorescence is quenched when embedded in a lipid bilayer, but it is not quenched when bound to MTP such that MTP transfer is recorded as an increase in fluorescence over time. BMS197636 (IC50 = 0.5 nM) was used at 200 nM to inhibit MTP triglyceride transfer (44, 45). PE transfer was done by overnight coating of a microtiter plate with 2 μg per well of recombinant murine CD1d purified by baculovirus expression system (57). Wells were washed with PBS, incubated with PBS containing 0.5% isopropanol for 2 h at 37°C, and washed again with PBS. MTP purified from rat liver (>95% pure by SDS-PAGE) and PE vesicles containing 6:1 PE/NBD-PE were resuspended in transfer buffer (1 mM Tris-HCl, pH 7.4, 0.2 mM EDTA, 15 mM NaCl, and 0.1% fatty acid–free BSA; Sigma-Aldrich), added to the wells, and incubated at 37°C for 2 h. Wells were washed three times with PBS. 100 μl isopropanol was added to each well for 60 s and transferred to a black microtiter plate (Thermo Labsystems). Isopropanol elutes were from both unlabeled and NBD-labeled PE. Fluorescence was read with a fluorescence plate reader (7620 Microplate Fluorimeter; Cambridge Technology) using 460-nm excitation and 530-nm emission wavelengths.
DC cultures.
BM was extracted from the femurs of C57BL6 or MTPmx1cre mice, washed once in PBS, and resuspended in R10 supplemented with culture supernatant from murine GM-CSF–transfected cells for a final concentration of 200 U/ml GM-CSF. BMDCs were then cultured in bacteriological plates for 12–14 d as described previously (58). Differentiated cells were harvested by gentle pipetting, washed in PBS, and analyzed by flow cytometry. Human monocytes were harvested from the peripheral blood of healthy volunteers by positive selection on CD14 magnetic beads (Miltenyi Biotec) and cultured in R10 medium at 106 cells/ml, supplemented with 200 U/ml recombinant hIL-4 and 300 U/ml recombinant hGM-CSF (PeproTech). After 5–6 d, cells were dislodged by gentle pipetting, analyzed by flow cytometry, and assayed for antigen presentation.
Flow cytometry.
Mouse cells were stained using the following antibodies: FITC-conjugated α-CD1d (1B1; BD Biosciences), PE-conjugated α-CD11c (HL3; BD Biosciences), FITC-conjugated α-I-Ad (AMS-32.1; BD Biosciences), FITC-conjugated α-CD11b (M1/70; BD Biosciences), FITC-conjugated α-CD40 (HM40-3; BD Biosciences), and FITC-conjugated α-CD80 (16-10A1; BD Biosciences). Human cells were stained using PE-conjugated α-CD83 (550634; BD Biosciences), FITC-conjugated α-HLA-A,B,C (557348; BD Biosciences), α-HLA DR,DP,DQ (32381A; BD Biosciences), α-CD1d (42.1 or 51.1.3), and α-mouse IgG+IgM (AMI1708; Biosource International). Staining was performed in the presence of Via-Probe (BD Biosciences) and analyzed with a flow cytometer (FACSort; Becton Dickinson).
Silencing.
U937 cells in American Type Culture Collection complete media (RPMI 1640 with 2 mM l-glutamine adjusted to contain 1.5 g/liter sodium bicarbonate, 4.5 g/liter glucose, 10 mM Hepes, and 1.0 mM sodium pyruvate [90%]; FBS [10%]) were aliquoted into a six-well plate and simultaneously transfected with two human mtp-specific siRNA oligos at 25 nM each (target sequences: NNUUAUGACCGUUUCUCCAGG and AAGCUCACGUACUCCACUGAA) or transfected with mock siRNA (specific for mouse but not human MTP transcripts) at 50 nM. The gene encoding MTP is mttp in mice and mtp in humans. Oligos were complexed with siPORT amine (Ambion), according to the manufacturer's instructions, and added to U937 cells for a final culture volume of 2 ml. Fresh media was added after 24 hr. Cells were harvested 72 h later, incubated with 100 ng/ml α-galactosylceramide (α-galcer) or vehicle, washed, and co-cultured with DN32 cells (E/T = 1:1) for 24 h. Culture supernatants were collected and assayed for IL-2 by sandwich ELISA. A portion of the 72-h silenced cells were used for RNA extraction, and mtp transcript knockdown was measured by RT-PCR.
Online supplemental material.
Fig. S1 shows a flow cytometric analysis of mouse BMDCs. No effect of BMS212122 on surface expression of CD11c, CD11b, CD40, or CD80 was observed. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050183/DC1.
Acknowledgments
We thank M. Brenner, G. Dranoff, H. Ploegh, R. Gregg, J. Wetterau, and M. Dougan for their advice.
M. Kronenberg was supported by National Institutes of Health (NIH) grant RO1 AI 40617; M. Exley was supported by NIH grant DK66917; M.M. Hussain was supported by NIH grants DK46900 and HL64272; and R.S. Blumberg was supported by NIH grants DK44319, DK53056, and DK51362 and by Harvard Digestive Diseases Center grant P30 DK034854-21.
The authors have no conflicting financial interests.
Abbreviations used: α-galcer, α-galactosylceramide; apoB, apolipoprotein B; β2m, β2- microglobulin; BMDC, BM- derived DC; ER, endoplasmic reticulum; IEC, intestinal epithelial cell; LMNC, liver mononuclear cell; MFI, mean fluorescence intensity; moDC, monocyte- derived DC; MTP, microsomal triglyceride transfer protein; NBD, 7-nitro-2,1,3-benzoxadiazol-4-yl; PDI, protein disulfide isomerase; PE, phosphatidyl ethanolamine; pIpC, poly-inositic:poly-cytodylic acid; si, small interfering.
References
- 1.Brigl, M., and M.B. Brenner. 2004. CD1: antigen presentation and T cell function. Annu. Rev. Immunol. 22:817–890. [DOI] [PubMed] [Google Scholar]
- 2.Beckman, E.M., S.A. Porcelli, C.T. Morita, S.M. Behar, S.T. Furlong, and M.B. Brenner. 1994. Recognition of a lipid antigen by CD1-restricted alpha beta+ T cells. Nature. 372:691–694. [DOI] [PubMed] [Google Scholar]
- 3.Moody, D.B., D.C. Young, T.Y. Cheng, J.P. Rosat, C. Roura-Mir, P.B. O'Connor, D.M. Zajonc, A. Walz, M.J. Miller, S.B. Levery, et al. 2004. T cell activation by lipopeptide antigens. Science. 303:527–531. [DOI] [PubMed] [Google Scholar]
- 4.Porcelli, S., C.T. Morita, and M.B. Brenner. 1992. CD1b restricts the response of human CD4-8- T lymphocytes to a microbial antigen. Nature. 360:593–597. [DOI] [PubMed] [Google Scholar]
- 5.Moody, D.B., T. Ulrichs, W. Muhlecker, D.C. Young, S.S. Gurcha, E. Grant, J.P. Rosat, M.B. Brenner, C.E. Costello, G.S. Besra, and S.A. Porcelli. 2000. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature. 404:884–888. [DOI] [PubMed] [Google Scholar]
- 6.Vincent, M.S., D.S. Leslie, J.E. Gumperz, X. Xiong, E.P. Grant, and M.B. Brenner. 2002. CD1-dependent dendritic cell instruction. Nat. Immunol. 3:1163–1168. [DOI] [PubMed] [Google Scholar]
- 7.Sugita, M., R.M. Jackman, E. van Donselaar, S.M. Behar, R.A. Rogers, P.J. Peters, M.B. Brenner, and S.A. Porcelli. 1996. Cytoplasmic tail-dependent localization of CD1b antigen-presenting molecules to MIICs. Science. 273:349–352. [DOI] [PubMed] [Google Scholar]
- 8.Jayawardena-Wolf, J., K. Benlagha, Y.H. Chiu, R. Mehr, and A. Bendelac. 2001. CD1d endosomal trafficking is independently regulated by an intrinsic CD1d-encoded tyrosine motif and by the invariant chain. Immunity. 15:897–908. [DOI] [PubMed] [Google Scholar]
- 9.Dascher, C.C., K. Hiromatsu, X. Xiong, M. Sugita, J.E. Buhlmann, I.L. Dodge, S.Y. Lee, C. Roura-Mir, G.F. Watts, C.J. Roy, et al. 2002. Conservation of CD1 intracellular trafficking patterns between mammalian species. J. Immunol. 169:6951–6958. [DOI] [PubMed] [Google Scholar]
- 10.Nicol, A., M. Nieda, Y. Koezuka, S. Porcelli, K. Suzuki, K. Tadokoro, S. Durrant, and T. Juti. 2000. Human invariant Vα24+ natural killer T cells activated by α-galactoslceramide (KRN7000) have cytotoxic anti-tumor activity through mechanisms distinct from T cells and natural killer cells. Immunology. 99:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer, K., E. Scotet, M. Niemeyer, H. Koebernick, J. Zerrahn, S. Maillet, R. Hurwitz, M. Kursar, M. Bonneville, S.H. Kaufmann, and U.E. Schaible. 2004. Mycobacterial phosphatidylinositol mannoside is a natural antigen for CD1d-restricted T cells. Proc. Natl. Acad. Sci. USA. 101:10685–10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amprey, J.L., J.S. Im, S.J. Turco, H.W. Murray, P.A. Illarionov, G.S. Besra, S.A. Porcelli, and G.F. Spath. 2004. A subset of liver NKT cells is activated during Leishmania donovani infection by CD1d-bound lipophosphoglycan. J. Exp. Med. 200:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinjo, Y., D. Wu, G. Kim, G.W. Xing, M.A. Poles, D.D. Ho, M. Tsuji, K. Kawahara, C.H. Wong, and M. Kronenberg. 2005. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. 434:520–525. [DOI] [PubMed] [Google Scholar]
- 14.Mattner, J., K.L. Debord, N. Ismail, R.D. Goff, C. Cantu III, D. Zhou, P. Saint-Mezard, V. Wang, Y. Gao, N. Yin, et al. 2005. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. 434:525–529. [DOI] [PubMed] [Google Scholar]
- 15.Gumperz, J.E., C. Roy, A. Makowska, D. Lum, M. Sugita, T. Podrebarac, Y. Koezuka, S.A. Porcelli, S. Cardell, M.B. Brenner, and S.M. Behar. 2000. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity. 12:211–221. [DOI] [PubMed] [Google Scholar]
- 16.Brigl, M., L. Bry, S.C. Kent, J.E. Gumperz, and M.B. Brenner. 2003. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat. Immunol. 4:1230–1237. [DOI] [PubMed] [Google Scholar]
- 17.De Silva, A.D., J.J. Park, N. Matsuki, A.K. Stanic, R.R. Brutkiewicz, M.E. Medof, and S. Joyce. 2002. Lipid protein interactions: the assembly of CD1d1 with cellular phospholipids occurs in the endoplasmic reticulum. J. Immunol. 168:723–733. [DOI] [PubMed] [Google Scholar]
- 18.Park, J.J., S.J. Kang, A.D. De Silva, A.K. Stanic, G. Casorati, D.L. Hachey, P. Cresswell, and S. Joyce. 2004. Lipid-protein interactions: biosynthetic assembly of CD1 with lipids in the endoplasmic reticulum is evolutionarily conserved. Proc. Natl. Acad. Sci. USA. 101:1022–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rauch, J., J. Gumperz, C. Robinson, M. Skold, C. Roy, D.C. Young, M. Lafleur, D.B. Moody, M.B. Brenner, C.E. Costello, and S.M. Behar. 2003. Structural features of the acyl chain determine self-phospholipid antigen recognition by a CD1d-restricted invariant NKT (iNKT) cell. J. Biol. Chem. 278:47508–47515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou, D., J. Mattner, C. Cantu, N. Schrantz, N. Yin, Y. Gao, Y. Sagiv, K. Hudspeth, Y.P. Wu, T. Yamashita, et al. 2004. Lysosomal glycosphingolipid recognition by NKT cells. Science. 306:1786–1789. [DOI] [PubMed] [Google Scholar]
- 21.Nieuwenhuis, E.E., T. Matsumoto, M. Exley, R.A. Schleipman, J. Glickman, D.T. Bailey, N. Corazza, S.P. Colgan, A.B. Onderdonk, and R.S. Blumberg. 2002. CD1d-dependent macrophage-mediated clearance of Pseudomonas aeruginosa from lung. Nat. Med. 8:588–593. [DOI] [PubMed] [Google Scholar]
- 22.Exley, M.A., N.J. Bigley, O. Cheng, A. Shaulov, S.M. Tahir, Q.L. Carter, J. Garcia, C. Wang, K. Patten, H.F. Stills, et al. 2003. Innate immune response to encephalomyocarditis virus infection mediated by CD1d. Immunology. 110:519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gillessen, S., Y.N. Naumov, E.E. Nieuwenhuis, M.A. Exley, F.S. Lee, N. Mach, A.D. Luster, R.S. Blumberg, M. Taniguchi, S.P. Balk, et al. 2003. CD1d-restricted T cells regulate dendritic cell function and antitumor immunity in a granulocyte-macrophage colony-stimulating factor-dependent fashion. Proc. Natl. Acad. Sci. USA. 100:8874–8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang, B., Y.B. Geng, and C.R. Wang. 2001. CD1-restricted NK T cells protect nonobese diabetic mice from developing diabetes. J. Exp. Med. 194:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heller, F., I.J. Fuss, E.E. Nieuwenhuis, R.S. Blumberg, and W. Strober. 2002. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity. 17:629–638. [DOI] [PubMed] [Google Scholar]
- 26.Akbari, O., P. Stock, E. Meyer, M. Kronenberg, S. Sidobre, T. Nakayama, M. Taniguchi, M.J. Grusby, R.H. DeKruyff, and D.T. Umetsu. 2003. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat. Med. 9:582–588. [DOI] [PubMed] [Google Scholar]
- 27.Tupin, E., A. Nicoletti, R. Elhage, M. Rudling, H.G. Ljunggren, G.K. Hansson, and G.P. Berne. 2004. CD1d-dependent activation of NKT cells aggravates atherosclerosis. J. Exp. Med. 199:417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang, S.J., and P. Cresswell. 2002. Calnexin, calreticulin, and ERp57 cooperate in disulfide bond formation in human CD1d heavy chain. J. Biol. Chem. 277:44838–44844. [DOI] [PubMed] [Google Scholar]
- 29.Kim, H.S., J. Garcia, M. Exley, K.W. Johnson, S.P. Balk, and R.S. Blumberg. 1999. Biochemical characterization of CD1d expression in the absence of beta2-microglobulin. J. Biol. Chem. 274:9289–9295. [DOI] [PubMed] [Google Scholar]
- 30.Zhou, D., C. Cantu, Y. Sagiv, N. Schrantz, A.B. Kulkarni, X. Qi, D.J. Mahuran, C.R. Morales, G.A. Grabowski, K. Benlagha, et al. 2004. Editing of CD1d-bound lipid antigens by endosomal lipid transfer proteins. Science. 303:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang, S.J., and P. Cresswell. 2004. Saposins facilitate CD1d-restricted presentation of an exogenous lipid antigen to T cells. Nat. Immunol. 5:175–181. [DOI] [PubMed] [Google Scholar]
- 32.Chiu, Y.H., S.H. Park, K. Benlagha, C. Forestier, J. Jayawardena-Wolf, P.B. Savage, L. Teyton, and A. Bendelac. 2002. Multiple defects in antigen presentation and T cell development by mice expressing cytoplasmic tail-truncated CD1d. Nat. Immunol. 3:55–60. [DOI] [PubMed] [Google Scholar]
- 33.Hussain, M.M., J. Shi, and P. Dreizen. 2003. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lipid Res. 44:22–32. [DOI] [PubMed] [Google Scholar]
- 34.Jamil, H., J.K. Dickson Jr., C.H. Chu, M.W. Lago, J.K. Rinehart, S.A. Biller, R.E. Gregg, and J.R. Wetterau. 1995. Microsomal triglyceride transfer protein. Specificity of lipid binding and transport. J. Biol. Chem. 270:6549–6554. [DOI] [PubMed] [Google Scholar]
- 35.Atzel, A., and J.R. Wetterau. 1994. Identification of two classes of lipid molecule binding sites on the microsomal triglyceride transfer protein. Biochemistry. 33:15382–15388. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell, D.M., M. Zhou, R. Pariyarath, H. Wang, J.D. Aitchison, H.N. Ginsberg, and E.A. Fisher. 1998. Apoprotein B100 has a prolonged interaction with the translocon during which its lipidation and translocation change from dependence on the microsomal triglyceride transfer protein to independence. Proc. Natl. Acad. Sci. USA. 95:14733–14738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gordon, D.A., H. Jamil, D. Sharp, D. Mullaney, Z. Yao, R.E. Gregg, and J. Wetterau. 1994. Secretion of apolipoprotein B-containing lipoproteins from HeLa cells is dependent on expression of the microsomal triglyceride transfer protein and is regulated by lipid availability. Proc. Natl. Acad. Sci. USA. 91:7628–7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang, J., and H.N. Ginsberg. 2001. Microsomal triglyceride transfer protein binding and lipid transfer activities are independent of each other, but both are required for secretion of apolipoprotein B lipoproteins from liver cells. J. Biol. Chem. 276:28606–28612. [DOI] [PubMed] [Google Scholar]
- 39.Bakillah, A., N. Nayak, U. Saxena, R.M. Medford, and M.M. Hussain. 2000. Decreased secretion of apoB follows inhibition of apoB-MTP binding by a novel antagonist. Biochemistry. 39:4892–4899. [DOI] [PubMed] [Google Scholar]
- 40.Berriot-Varoqueaux, N., L.P. Aggerbeck, M. Samson-Bouma, and J.R. Wetterau. 2000. The role of the microsomal triglyceride transfer protein in abetalipoproteinemia. Annu. Rev. Nutr. 20:663–697. [DOI] [PubMed] [Google Scholar]
- 41.Brozovic, S., T. Nagaishi, M. Yoshida, S. Betz, A. Salas, D. Chen, A. Kaser, J. Glickman, T. Kuo, A. Little, et al. 2004. CD1d function is regulated by microsomal triglyceride transfer protein. Nat. Med. 10:535–539. [DOI] [PubMed] [Google Scholar]
- 42.Exley, M., J. Garcia, S.B. Wilson, F. Spada, D. Gerdes, S.M. Tahir, K.T. Patton, R.S. Blumberg, S. Porcelli, A. Chott, and S.P. Balk. 2000. CD1d structure and regulation on human thymocytes, peripheral blood T cells, B cells and monocytes. Immunology. 100:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Athar, H., J. Iqbal, X.C. Jiang, and M.M. Hussain. 2004. A simple, rapid, and sensitive fluorescence assay for microsomal triglyceride transfer protein. J. Lipid Res. 45:764–772. [DOI] [PubMed] [Google Scholar]
- 44.Jamil, H., D.A. Gordon, D.C. Eustice, C.M. Brooks, J.K. Dickson Jr., Y. Chen, B. Ricci, C.H. Chu, T.W. Harrity, C.P. Ciosek Jr., et al. 1996. An inhibitor of the microsomal triglyceride transfer protein inhibits apoB secretion from HepG2 cells. Proc. Natl. Acad. Sci. USA. 93:11991–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wetterau, J.R., R.E. Gregg, T.W. Harrity, C. Arbeeny, M. Cap, F. Connolly, C.H. Chu, R.J. George, D.A. Gordon, H. Jamil, et al. 1998. An MTP inhibitor that normalizes atherogenic lipoprotein levels in WHHL rabbits. Science. 282:751–754. [DOI] [PubMed] [Google Scholar]
- 46.Robl, J.A., R. Sulsky, C.Q. Sun, L.M. Simpkins, T. Wang, J.K. Dickson Jr., Y. Chen, D.R. Magnin, P. Taunk, W.A. Slusarchyk, et al. 2001. A novel series of highly potent benzimidazole-based microsomal triglyceride transfer protein inhibitors. J. Med. Chem. 44:851–856. [DOI] [PubMed] [Google Scholar]
- 47.Prigozy, T.I., O. Naidenko, P. Qasba, D. Elewaut, L. Brossay, A. Khurana, T. Natori, Y. Koezuka, A. Kulkarni, and M. Kronenberg. 2001. Glycolipid antigen processing for presentation by CD1d molecules. Science. 291:664–667. [DOI] [PubMed] [Google Scholar]
- 48.Spada, F.M., F. Borriello, M. Sugita, G.F.M. Watts, Y. Koezuka, and S.A. Porcelli. 2000. Low expression level but potent antigen presenting function of CD1d on monocyte lineage cells. Eur. J. Immunol. 30:3468–3477. [DOI] [PubMed] [Google Scholar]
- 49.Metelitsa, L.S., O.V. Naidenko, A. Kant, H.W. Wu, M.J. Loza, B. Perussia, M. Kronenberg, and R.C. Seeger. 2001. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J. Immunol. 167:3114–3122. [DOI] [PubMed] [Google Scholar]
- 50.Kim, H.S., S.P. Colgan, R. Pitman, R.M. Hershberg, and R.S. Blumberg. 2000. Human CD1d associates with prolyl-4-hydroxylase during its biosynthesis. Mol. Immunol. 37:861–868. [DOI] [PubMed] [Google Scholar]
- 51.Townsend, A., T. Elliott, V. Cerundolo, L. Foster, B. Barber, and A. Tse. 1990. Assembly of MHC class I molecules analyzed in vitro. Cell. 62:285–295. [DOI] [PubMed] [Google Scholar]
- 52.Rava, P., H. Athar, C. Johnson, and M.M. Hussain. 2005. Measuring cholesterol ester and phospholipid transfer and net deposition by microsomal triglyceride transfer protein using fluorescent lipids. J. Lipid Res. 10.1194/jlr.D400043-JLR200. [DOI] [PubMed]
- 53.Swift, L.L., B. Kakkad, C. Boone, A. Jovanovska, W.G. Jerome, P.J. Mohler, and D.E. Ong. 2005. Microsomal triglyceride transfer protein expression in adipocytes: a new component in fat metabolism. FEBS Lett. 579:3183–3189. [DOI] [PubMed] [Google Scholar]
- 54.Van de Wal, Y., N. Corazza, M. Allez, L.F. Mayer, H. Iijima, M. Ryan, S. Cornwall, D. Kaiserlian, R. Hershberg, Y. Koezuka, et al. 2003. Delineation of a CD1d-restricted antigen presentation pathway associated with human and mouse intestinal epithelial cells. Gastroenterology. 124:1420–1431. [DOI] [PubMed] [Google Scholar]
- 55.Bendelac, A., O. Lantz, M.E. Quimby, J.W. Yewdell, J.R. Bennink, and R.R. Brutkiewicz. 1995. CD1 recognition by mouse NK1+ T lymphocytes. Science. 268:863–865. [DOI] [PubMed] [Google Scholar]
- 56.Raabe, M., M.M. Veniant, M.A. Sullivan, C.H. Zlot, J. Bjorkegren, L.B. Nielsen, J.S. Wong, R.L. Hamilton, and S.G. Young. 1999. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J. Clin. Invest. 103:1287–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naidenko, O.V., J.K. Maher, W.A. Ernst, T. Sakai, R.L. Modlin, and M. Kronenberg. 1999. Binding and antigen presentation of ceramide-containing glycolipids by soluble mouse and human CD1d molecules. J. Exp. Med. 190:1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lutz, M.B., N. Kukutsch, A.L. Ogilvie, S. Rossner, F. Koch, N. Romani, and G. Schuler. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods. 223:77–92. [DOI] [PubMed] [Google Scholar]